

Abstract
In this study, Pt nanoparticles on zeolite/γ-Al2O3 composites (50/50 wt) were located either in the zeolite or on the γ-Al2O3 binder, hereby varying the average distance (intimacy) between zeolite acid sites and metal sites from “closest” to “nanoscale”. The catalytic performance of these catalysts was compared to physical mixtures of zeolite and Pt/γ-Al2O3 powders, which provide a “microscale” distance between sites. Several beneficial effects on catalytic activity and selectivity for n-heptane hydroisomerization were observed when Pt nanoparticles are located on the γ-Al2O3 binder in nanoscale proximity with zeolite acid sites, as opposed to Pt nanoparticles located inside zeolite crystals. On ZSM-5-based catalysts, mostly monobranched isomers were produced, and the isomer selectivity of these catalysts was almost unaffected with an intimacy ranging from closest to microscale, which can be attributed to the high diffusional barriers of branched isomers within ZSM-5 micropores. For composite catalysts based on large-pore zeolites (zeolite Beta and zeolite Y), the activity and selectivity benefitted from the nanoscale intimacy with Pt, compared to both the closest and microscale intimacies. Intracrystalline gradients of heptenes as reaction intermediates are likely contributors to differences in activity and selectivity. This paper aims to provide insights into the influence of the metal–acid intimacy in bifunctional catalysts based on zeolites with different framework topologies.
Keywords: bifunctional catalysts, intimacy, diffusion, zeolites, alkane hydroisomerization, molecular modeling
1. Introduction
Solid bifunctional catalysts, that combine two independent catalytic functions within one catalyst particle, are a popular class of catalysts, which typically have unique properties that cannot be obtained by the use of monofunctional catalysts.1−5 For example, bifunctional metal–acid catalysts find application as highly stable catalysts in large-scale hydrocracking and hydroisomerization processes in oil refineries for the production of transportation fuels and chemicals.6 Bifunctional catalysts are also investigated for the hydro-deoxygenation and subsequent hydrocracking of biomass-derived feedstock toward hydrocarbons.7,8 Other investigated applications are in the hydrogenation of CO or CO2 where beneficial effects on product selectivity toward lower olefins (C2–C4) and aromatics are reported for bifunctional catalysts.2−4,9
Bifunctional catalysts, used for the conversion of hydrocarbons, consist of a metal function catalyzing (de)hydrogenation, which can be highly active noble metals such as platinum or palladium, or more abundant but less active metal sulfides such as nickel and nickel-molybdenum sulfide or cobalt-molybdenum sulfide. The acid function for industrial catalysts is commonly obtained using halogenated alumina, zeolites, or amorphous silica-alumina and is responsible for alkene protonation and subsequent isomerization and cracking.10 Catalysts for industrial hydroisomerization processes are based on either Pt supported on chlorinated Al2O3 (6–7 wt % Cl) or Pt supported on Mordenite zeolite. The latter catalyst operates at higher temperature that is unfavorable in view of the thermodynamic equilibrium of the isomerization reaction,11 but with the advantage of being less sensitive to sulfur and water.
Zeolites are crystalline aluminosilicate materials possessing a regular structure of micropores (<2 nm in diameter) that are of a similar size as hydrocarbon molecules, and when the kinetic diameter of a molecule approaches the size of a micropore, its diffusivity may vary greatly.12,13 Hampered intracrystalline diffusion in micropores may lead to concentration gradients within zeolites crystals, compromising catalytic activity and/or selectivity, for example, by promoting undesired secondary cracking reactions.14−16 Several studies report methods to overcome intracrystalline diffusion limitations in zeolites, for example, by shortening the average micropore length using nanocrystalline zeolites17 or using postsynthesis treatments to create mesopores in zeolite crystals that act as highways of diffusion.18−21
A zeolite possesses shape-selective properties when the fitting of a hydrocarbon molecule in a zeolite pore directly influences its conversion.22 For example, in hydroisomerization of n-hexane or n-heptane over medium-pore ZSM-5-based catalysts, the terminally monobranched isomers (e.g., 2-methylpentane) are preferentially formed over the internally monobranched isomers, which is attributed to the faster diffusion in ZSM-5 micropores of the former (i.e., product shape selectivity).23 Bulky dibranched hexanes and heptanes are formed to a limited extent over these catalysts because of steric constraints and fast cracking of these molecules in ZSM-5 micropores.23,24 Large-pore (12-membered ring) zeolites such as zeolite Beta and zeolite Y impose much less steric constraints on branched isomers of hexane and heptane and are therefore often considered to be “nonshape-selective” zeolites for these processes, with an isomer product distribution that is close to thermodynamic equilibrium.24,25
Hydrocarbon adsorption and diffusion in zeolites have been studied by a variety of experimental techniques: pulsed field gradient nuclear magnetic resonance,26 uptake rate measurements (e.g., combined with a tapered element oscillating microbalance),27,28 and also single-molecule fluorescence microscopy has been employed to determine the diffusion coefficient of a fluorescent hydrocarbon probe in a zeolite matrix.29 Theoretical studies have also often focused on hydrocarbon diffusion, either by establishment of a microkinetic model of a catalytic process14,23 or by molecular modeling using (dynamically corrected) transition state theory,30 Monte Carlo simulations,24,31,32 or molecular dynamics.33
For bifunctional metal–acid catalysts, the diffusivity of the alkene intermediates between metal and acid sites determines the maximum distance between these sites before the activity or selectivity is affected by diffusion limitations.34,35 Microporous all-silica materials have been used to study alkene adsorption and diffusion, where the alkene properties are very similar to their alkane equivalents.36 However, the strong adsorption and high reactivity of alkenes on protonic sites hinder such studies in H-zeolites, often used for catalysis, but a significantly lower diffusivity of alkenes versus the alkane equivalent is to be expected.37 Another factor that may contribute to this difference in diffusivity is the dipole–dipole interaction between a polar aluminum-rich zeolite and the C=C bond of alkenes, causing the alkene to interact more strongly than the alkane equivalent.38,39
Noh et al. have recently studied different ZSM-5 crystal sizes in n-C7 hydroisomerization, whereby the metal function was provided by physical mixing with Pt/SiO2 and was thus located outside the zeolite crystals.15 For a range of differently sized ZSM-5 crystals, a constant turnover frequency per acid site was obtained, but the use of the largest ZSM-5 crystals led to a lower turnover frequency, which was attributed to the limited diffusion of heptenes between extracrystalline regions and the intracrystalline zeolite acid sites. A number of examples have also indicated that for large-pore zeolites, hampered diffusion of alkenes may also impact selectivity, as was inferred by Zečević et al. using a zeolite Y/γ-Al2O3 composite (50/50 wt) as a support for Pt nanoparticles, which were either located in zeolite Y or on the γ-Al2O3 binder, closely outside (i.e., within nanoscale proximity) the zeolite Y crystals.40 In the conversion of n-C10 and n-C19, both catalysts had a similar catalytic activity, indicating that the rate of the reaction was not limited by intracrystalline diffusion limitations of linear alkenes, but favorable effects on isomer selectivity were reported for the catalyst with Pt nanoparticles located on the γ-Al2O3 binder. It was suggested that the formation of branched alkene intermediates, formed on Pt nanoparticles inside the zeolite crystals, led to higher concentrations in the zeolite, resulting in enhanced cracking. Whenever Pt nanoparticles are located on the γ-Al2O3 binder, alkene intermediates coming from “outside” may react in the outer layers of the zeolite crystals and branched alkenes may quickly diffuse out. Similar favorable effects on selectivity were reported in the conversion of n-heptane, a hydrocarbon with relatively high diffusivity, when Pt nanoparticles were located closely outside zeolite Beta crystals on a zeolite Beta/Al2O3 composite,41 and for one-dimensional ZSM-22 and mordenite zeolites,42 but not for ZSM-5 over a range of crystallite sizes.17
A number of recent studies have reported innovative approaches to prepare bifunctional catalysts with varying levels of metal–acid intimacy at the nanometer length scale, such as preferential adsorption of a Pt complex on a composite support40−44 or impregnation of previously synthesized Pt colloids to locate the metal function outside zeolite crystals.17 Easy-to-prepare physical mixtures of a supported metal catalyst and zeolite are a more “traditional” method to vary the metal–acid intimacy at the micrometer or millimeter length scale, which are being applied in the form of fine powders, grains, or stacked beds.15,16,45−47 This approach is only suitable to obtain catalysts with a metal–acid intimacy at the micrometer length scale and often leads to suboptimal catalytic performance.45,46 Locating the metal function in the zeolite crystallite17,40,48,49 provides a very close metal–acid intimacy, but for catalysts with high metal loadings, this could affect the acid function of the zeolite, which compromises a direct comparison with catalysts with the metal function outside the zeolite.50,51
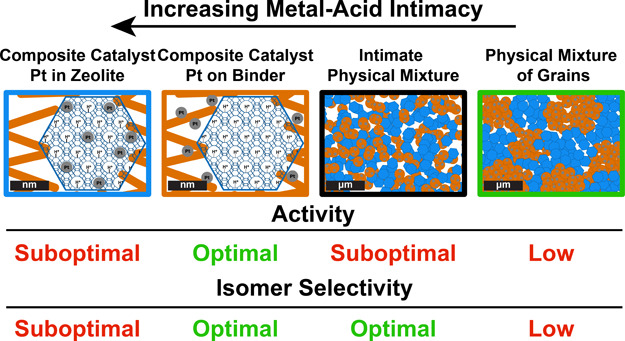
In this study, we evaluated the effects of intimacy between metal and acid sites for the zeolite-based catalyst in n-heptane hydroisomerization, using commercially available zeolites (ZSM-5, zeolite Beta, and zeolite Y) that have three-dimensional pore structures and different micropore sizes. Pt/γ-Al2O3/zeolite composite catalysts with a 50/50 wt ratio between the zeolite and γ-Al2O3 component were used, while the location of Pt nanoparticles was varied from inside the zeolite crystals (closest intimacy) to on the γ-Al2O3 binder and thereby providing an intimacy in the range of 5–500 nm (nanoscale). These catalysts were compared to physical mixtures with similar bulk composition: an intimate physical mixture of Pt/γ-Al2O3 and zeolite prepared by mixing in a mortar followed by pelletizing (pressure: 650 kg cm–2) or a physical mixture of grains (75–212 μm) (Figure 1). Furthermore, using molecular simulations, diffusion coefficients of mono- and dibranched heptane isomers in the straight channels of the ZSM-5 zeolite were computed, which shows to be a determining factor in the isomer product distribution of this zeolite. The results of this study provide new insights into factors, viz., zeolite acidity, micropore size, shape selectivity, and metal–acid intimacy, that determine the catalyst performance and are of great relevance for the preparation of zeolite-based metal–acid catalysts.
Figure 1.

Schematic representation of the investigated samples. Pt/γ-Al2O3/zeolite composite catalysts with Pt nanoparticles located in the zeolite provide the “closest” intimacy between metal sites, while catalysts with Pt nanoparticles located on the γ-Al2O3 binder provide a “nanoscale” intimacy. In this case, by placing Pt nanoparticles closely outside zeolite crystals, an intimacy between metal and acid sites between 5–500 nm is created. As comparison, an intimate physical mixture and a physical mixture of grains are prepared, which provide a “microscale” metal–acid intimacy. The alumina component is indicated in orange, zeolite is indicated in blue, and Pt nanoparticles are indicated in gray.
2. Experimental Methods
2.1. Composite Preparation
H-ZSM-5/γ-Al2O3 and H-zeolite Beta/γ-Al2O3 composites were prepared by adding 50 wt % ZSM-5 zeolite powder (Zeolyst CBV3024E, Si/Al = 15 at/at, particle size ∼20–200 nm) or zeolite Beta powder (Zeolyst CBV814E, Si/Al = 12.5 at/at, particle size ∼20–50 nm) with 50 wt % pseudo-boehmite (HMPA, Shell) in a mortar. Additional properties of the zeolites are reported in Table S1. Then, 2 wt % (based on the total mass of the solid components) acetic acid (Alfa Aesar, glacial, 99+%) and distilled water were added and mixed for ∼5 min, resulting in a homogenous and viscous paste. Acetic acid was added as a peptizing agent for the boehmite particles, to aid in mixing and adhesion with the zeolite particles.52 The mixture was dried overnight at 120 °C in static air followed by calcination at 550 °C for 2 h with a ramp of 5 °C min–1. Afterward, the product was crushed and sieved to obtain grains of 212–500 μm diameter. The zeolite Y/γ-Al2O3 composite (based on Zeolyst CBV760, Si/Al = 30 at/at, particle size ∼200–1000) was obtained from Shell Technology Center in the form of extrudates. The extrudates were crushed and sieved toward a fraction of 212–500 μm and used as such for Pt deposition.
2.2. Catalyst Synthesis
2.2.1. Pt/γ-Al2O3/Zeolite Catalyst with Pt Nanoparticles Located inside Zeolite Crystals
Zeolite/γ-Al2O3 particles (1 g) were suspended in 300 mL of Milli-Q water at room temperature and stirred for 1 h (∼500 rpm). Aqueous solution (40 mL) containing 10.2 mg of Pt(NH3)4(NO3)2 (Sigma-Aldrich, 99.995% purity) was added dropwise to the suspension (intake 0.5 wt % Pt). After 3 h of stirring, the suspension was filtered and washed with 150 mL of Milli-Q water and dried in static air overnight at 120 °C. The dried catalyst was calcined in a flow of 20% O2/N2 (GHSV ∼ 8500 h–1) at 350 °C with a ramp of 0.2 C min–1, a procedure based on Graaf et al. for the preparation of highly dispersed Pt nanoparticles.53 Afterward, the catalyst was reduced in a flow of H2 (GHSV ∼ 3300 h–1) for 3 h at 600 °C, using a ramp of 5 °C min–1.
2.2.2. Pt/γ-Al2O3/Zeolite Catalyst with Pt Nanoparticles Located on the γ-Al2O3 Binder
Zeolite/γ-Al2O3 particles (1 g) were suspended in 300 mL of Milli-Q water at room temperature and stirred for 1 h (∼500 rpm). Aqueous solution (40 mL) containing 12.5 mg of H2PtCl6·6H2O (Merck, ∼40 wt % Pt) was added dropwise to the suspension (intake 0.5 wt % Pt). After 3 h of stirring, the suspension was filtered and washed with 150 mL of Milli-Q water and dried in static air overnight at 120 °C. The dried catalyst was reduced in a flow of H2 (GHSV ∼ 3300 h–1) for 3 h at 600 °C with a ramp of 5 °C min–1. When preparing smaller amounts of catalysts, the volume of Milli-Q water in the suspension was proportionally decreased, while using similar GHSV’s.
2.2.3. Pt/γ-Al2O3 + Zeolite Physical Mixtures
Physical mixtures were prepared using γ-Al2O3 in powder form as a support using the Pt deposition procedure as described above for locating Pt nanoparticles on γ-Al2O3. After Pt deposition, the support was pressed and sieved into a sieve fraction of 212–500 μm in diameter and reduced in H2 (GHSV ∼ 3300 h–1) for 3 h at 500 °C, with a ramp of 5 °C min–1. To produce H-ZSM-5 and H-Beta for physical mixtures, the parent zeolites in ammonium form were calcined in static air at 550 °C for 2 h. Intimate physical mixtures were prepared by mixing the Pt/γ-Al2O3 powder (0.8 wt % Pt) with the zeolites in a 50/50 wt ratio in a mortar, resulting in an overall Pt loading of 0.4 wt %. The resulting mixture was then pelletized (pressure: 650 kg cm–2) and crushed and sieved into a fraction of 75–212 μm to be used for catalytic testing. For a physical mixture of grains, Pt/γ-Al2O3 powder and zeolite powder were separately pressed and sieved into a fraction of 75–212 μm and were carefully mixed with a spatula before loading the reactors.
2.3. Characterization
Elemental analysis of Pt was performed at Kolbe Mikroanalytisches Laboratorium using an inductively coupled plasma-optical emission spectrometer (ICP-OES, PerkinElmer) after sample dissolution according to standard in-house procedures. N2 physisorption measurements of the supports were conducted on a Micromeritics TriStar 3000 at liquid nitrogen temperature. The samples were dried overnight at 300 °C in N2 flow before measurements. Ammonia temperature-programmed desorption (NH3-TPD) was performed on a Micromeritics AutoChem II equipped with a thermal conductivity detector (TCD) calibrated for ammonia. The catalyst (80–100 mg) was dried in a He flow for 1 h at 600 °C (10 °C·min–1). The temperature was then decreased to 100 °C, and ammonia (10 vol % in He) was adsorbed in a pulsewise manner until oversaturation. The physisorbed ammonia was removed by flowing He for 2 h at 100 °C, after which ammonia desorption was monitored up to 600 °C with a ramp of 10 °C·min–1. Quantification of ammonia desorption was performed by deconvolution of the TCD signals with three Gaussian functions. HAADF-STEM imaging was performed on an FEI Talos F200X transmission electron microscope, equipped with a high-brightness field emission gun (X-FEG). For these analyses, catalysts were embedded in Epofix resin, left to cure in air overnight at 60 °C, and cut to 70 nm sections using a Reichert-Jung Ultracut E ultramicrotome with a Diatome Ultra 35° diamond knife. Sections were deposited on carbon/Formvar-coated copper TEM grids.
2.4. n-C7 Hydroisomerization
Catalytic experiments were conducted in an Avantium Flowrence 16 parallel fixed-bed reactor setup. Stainless-steel reactors (internal diameter = 2 mm) were loaded with 25 mg of the catalyst in a sieve fraction of 75–212 μm. The product stream coming from the reactors was analyzed using an online GC (Agilent 7890A or Agilent 7890B) where the hydrocarbon products were analyzed on an Agilent J&W PoraBOND Q or HP-PONA column, respectively, connected to an FID. Before catalytic tests, catalysts were reduced at 300 °C (2 h; 5 °C min–1) in a 25% H2/He flow. n-C7 hydroisomerization tests were performed with the following conditions: a molH2 moln-C7–1 ratio of 9, a feed rate of 2.6 gn-C7·gcat–1·h–1, and a total pressure of 10 bar. n-Heptane was obtained from Acros Organics (99+%, pure). He 5.0, N2 5.0, and H2 6.0 gases were obtained from Linde gas. Results from catalytic tests were obtained by taking the average of two GC measurements performed under identical conditions. No catalyst deactivation was observed in the described experiments. The definitions of n-C7 conversion, product yield, and selectivity are provided in Supporting Information 1.
2.5. Molecular Modeling
The free energy profiles and diffusion coefficients of different heptane isomers along the straight channels of ZSM-5 zeolite at zero loading are computed from Monte Carlo simulations in the canonical ensemble. The RASPA software package is used for the simulations.33,54 Heptane isomers are modeled using the TraPPE force field. Interactions between heptane isomers and the zeolites are described by Lennard-Jones interactions, which are truncated and shifted at 12 Å. No tail corrections are applied. The simulation box consists of 2 × 2 × 2 unit cells with periodic boundary conditions. The zeolite structure was taken from the IZA database.55 The zeolite is modeled as a rigid structure. Simulations are performed at 207, 255, 294, and 387 °C. The length of the channels is divided into 1000 bins, and the probability that a heptane isomer is in each of these slices is calculated. To compute the free energy profiles along a channel, the possible positions for the molecule are restricted to a single channel, and trial moves attempting to move the molecule outside the channel are automatically rejected. The free energy of the molecule at each slice is given by
where T is the absolute temperature, kB is the Boltzmann factor, and Fi and pi are the Landau free energy at slice i and the probability of a molecule being in slice i, respectively. Transition state theory is used to calculate the upper bound for the diffusion coefficient of each of the heptane isomers. A molecule at the top of the free energy barrier is in equilibrium with the surrounding; therefore, the velocity distribution is according to the Maxwell distribution in this temperature. It is assumed that half of the molecules that reach the top of the barrier pass. Consequently, the hopping rate is given by
where m is the mass of the molecule, p(q*) is the probability of being on the top of the free energy barrier, and q* is the reaction coordinate at the top of the free energy barrier. Upper estimates for diffusion coefficients follow then from
where λ is the distance between two free energy barriers. More details regarding the simulation techniques can be found elsewhere.30,56
3. Results and Discussion
3.1. Acidity and Pt Nanoparticle Location of Zeolite Composite Catalysts
The prepared Pt/γ-Al2O3/zeolite catalysts (survey including designations shown in Table 1) were based on three commercially available zeolites whose properties (product code, Si/Al ratio, ring size, pore diameter and pore tortuosity, and particle size) are indicated in Table S1 and the structural properties obtained from N2 physisorption are provided in Figure S1 and Table S2. The zeolite Beta and Y used in this study possessed intercrystalline and intracrystalline mesoporosity, respectively, as evident from the hysteresis loop and pore size distribution, whereas the ZSM-5 crystals were almost exclusively microporous. When the parent zeolites were transformed into zeolite/γ-Al2O3 composites, a bimodal porosity was obtained in all cases, displaying both zeolite microporosity and mesoporosity, which is mostly attributed to the γ-Al2O3 binder.
Table 1. Properties of the Prepared Composite Catalysts with Their Sample Designationa.
| sample designation | Pt nanoparticle location (TEM) | Pt nanoparticle size (nm) | Pt weight loading (wt %) | total acidity (mmolNH3·gcat–1) |
|---|---|---|---|---|
| Pt-Z/A | ZSM-5 zeolite | 1.8 ± 0.8 | 0.4 | 0.76 |
| Pt-A/Z | γ-Al2O3 binder | 1.1 ± 0.2 | 0.4 | 0.80 |
| Pt-B/A | zeolite Beta | 1.4 ± 0.3 | 0.4 | 0.73 |
| Pt-A/B | γ-Al2O3 binder | 1.0 ± 0.3 | 0.4 | 0.75 |
| Pt-Y/A | Zeolite Y | 1.2 ± 0.2 | 0.2 | 0.51 |
| Pt-A/Y | γ-Al2O3 binder | 1.1 ± 0.2 | 0.2 | 0.54 |
Location and size of Pt nanoparticles were determined by HAADF-STEM analysis, while the Pt weight loading was determined by ICP after sample destruction. Total acidity was determined by NH3-TPD in the range of 100–600 °C.
After the preparation of the composite supports, the location of Pt nanoparticles could be controlled using a previously reported methodology.40 Pt was selectively deposited inside zeolite crystals by ion exchange of [Pt(NH3)4]2+ (aq) with zeolite protons (pH ∼ 5), while strong electrostatic adsorption at pH ∼ 3 of the [PtCl6]2– (aq) complex was used to selectively deposit Pt on the γ-Al2O3 binder. Considering the range in zeolite particle sizes (Table S1), locating Pt nanoparticles on the γ-Al2O3 binder results in a bifunctional intimacy between 5 and 500 nm. Although, in earlier work,57 it has been suggested that Pt particles may end up at the external surface of zeolite crystals when [Pt(NH3)4]2+ (aq) is used as a precursor, we have taken great care during ion exchange and thermal pretreatments in combination with extensive characterization of the location of the Pt nanoparticles58 to assure that they predominantly reside inside the zeolite crystals of the zeolite/γ-Al2O3 composites. To prevent overlap of the zeolite and γ-Al2O3 components in HAADF-STEM images, the Pt/γ-Al2O3/zeolite catalyst was ultramicrotomed into 70 nm sections before analysis. In Figure 2, the HAADF-STEM images of the composite catalyst are provided and point out that Pt nanoparticles were predominantly located in the zeolite component for the catalyst prepared with [Pt(NH3)4]2+ (aq) or on the γ-Al2O3 binder for the catalyst prepared with [PtCl6]2– (aq). The relatively large white areas present in the γ-Al2O3 component of Figure 2e are caused by overlapping γ-Al2O3 needles that result in a strong contrast with respect to the dark background.
Figure 2.

HAADF-STEM images of 70 nm-thick microtome cut sections of the Pt/γ-Al2O3/zeolite composite catalysts. Catalysts with the Pt nanoparticles located in the zeolite: Pt-Z/A, ZSM-5 (a); Pt–B/A, zeolite Beta (c); and Pt–Y/A, zeolite Y (e). Catalysts with Pt nanoparticles located on the γ-Al2O3 binder: Pt-A/Z, ZSM-5 (b); Pt-A/B, zeolite Beta (d); and Pt-A/Y, zeolite Y (f). Pt nanoparticles appear as bright white dots, while γ-Al2O3 has a needle-like structure, and zeolite crystals have a more uniform contrast. The ZSM-5 crystals were relatively large with a rectangular shape (a,b), while zeolite Beta crystals were smaller and spherical (c,d). Zeolite Y had large mesopores (>5 nm) that are clearly visible within the zeolite Y component in images (e,f).
The prepared catalyst exhibited a Pt nanoparticle size of 1–2 nm with narrow size distributions (Table 1), while previous studies have shown that such HAADF-STEM Pt particle sizes show good agreement with bulk techniques such as EXAFS.53,59 The Pt loadings were between 0.2 and 0.4 wt % Pt (Table 1), and discrepancies in Pt weight loading between the composite catalysts can be attributed to variations of the uptake of the Pt precursor during the catalyst preparation. Despite differences in Pt weight loading between catalysts based on different zeolites, earlier research on the effect of Pt to the acid site ratio on Al-rich zeolites (Si/Al = 3 at/at) has pointed out that with a Pt loading of ≥0.2 wt % Pt, the rate of the reaction on the metal sites is not rate limiting and the performance is not affected.60,61 Furthermore, the high hydrogen pressures applied in the catalytic experiments will further advance establishment of a quasi equilibrium between paraffins and olefins.62 In Table 1, the total acidity of the catalysts is provided, which was obtained from NH3-TPD over a temperature range of 100–600 °C. In the Supporting Information, the desorption profiles of the Pt/γ-Al2O3/zeolite catalyst and composite supports are provided, which were however too ill-defined to distinguish individual contributions (Figure S2, Table S3). Besides the composites, the single components (parent zeolites and γ-Al2O3) were also studied with NH3-TPD, and the desorption profiles of the zeolites could be deconvoluted, revealing the presence of low- and high-temperature bands (Figure S3, Table S4). The high temperature band (300–400 °C) is commonly assigned to ammonia desorption from Brønsted acid sites and shows a correlation with the Si/Al ratio.63 The band maximum of the high-temperature NH3 desorption is at a higher temperature for ZSM-5 (Figure S3a) than for zeolite Beta and zeolite Y (Figure S3b,c), which is indicative of slower transport of ammonia out of the smaller micropores of the relatively large ZSM-5 particles (Table S1). The desorption of ammonia at lower temperatures can be attributed to weakly adsorbed ammonia on Lewis acid sites or weakly acidic hydroxyl groups such as those observed on the γ-Al2O3 binder (Figure S3d,e). More detailed studies on the acidities of the different components of composite catalysts can be found in the literature.64,65
When comparing the total acidities of the Pt/γ-Al2O3/zeolite composite catalysts, prepared with either [PtCl6]2– (aq) or [Pt(NH3)4]2+ (aq), a slight but systematic higher total acidity is observed for the former (Table 1). The use of [Pt(NH3)4]2+ (aq) yields catalysts with Pt nanoparticles inside the zeolite crystals, but the three-dimensional pore structures of the zeolites combined with low Pt weight loadings (<0.4 wt % Pt) cannot explain alterations in the zeolite acid function, for example, pore blockage by Pt nanoparticles. Rather, the higher NH3 uptake for the catalyst prepared with [PtCl6]2– (aq) is pointing toward a partial chlorination of the surface of the γ-Al2O3 binder enhancing its acidity. In a recent paper on Pt/γ-Al2O3/zeolite composite catalysts, the effect of residual chlorine from the Pt precursor was studied in detail using (pyridine) FTIR and NH3-TPD, revealing that acidity from residual chlorine had no consequences for catalysis.66
For ZSM-5, zeolite Beta, and zeolite Y, numerous acidity studies are available in the literature, using NMR67 or FTIR with basic probe molecules (e.g., CO, pyridines, and acetonitrile)68,69 sometimes in conjunction with catalytic tests,70 to describe the various acidic sites of these zeolites. There is general agreement that the strong Brønsted acid sites, resulting from a bridging hydroxyl group of charge-balancing tetrahedrally coordinated aluminum atoms (Al3+) with a silicon atom (Al–OH–Si), are the active sites for isomerization and cracking reactions of hydrocarbons. Recent computational studies have suggested that the acid strength of these sites on high-silica zeolites (Si/Al > 10 at/at), such as those used in this study, is independent of framework or the location of the acid site within the framework.71−73
3.2. Catalytic Activity and Isomer Vs Cracking Selectivity in n-Heptane Isomerization
The n-heptane hydroisomerization experiments of the Pt/γ-Al2O3/zeolite composite catalysts and physical mixtures of the parent zeolites and Pt/γ-Al2O3 were performed at a feed rate of 2.6 gn-C7·gcat–1·h–1, a total pressure of 10 bar, and 9 molH2/moln-C7.
As is apparent from Figure 3a, the catalytic activity of composite catalysts followed a trend of ZSM-5-based catalysts as the most active, followed by zeolite Beta and zeolite Y. As stated in the Introduction, the activity of zeolite-based catalysts is highly dependent on its accessibility which is determined by, for example, the porosity and zeolite crystal size. The lower relative activity of the zeolite Y-based catalysts can be attributed to the lower number of acid sites on this zeolite because of the relatively high Si/Al ratio. However, for the hydroisomerization of light n-alkanes, a high relative activity for ZSM-5 with smaller micropores, followed by zeolite Beta and zeolite Y, is more often observed, and the origin has been debated.11,15,77 Noh et al. have attributed the higher reactivity for zeolites with smaller micropores to stronger van der Waals interactions between the carbocation and the zeolite framework, leading to a more stable transition state.15 In other studies, the trend in activity is attributed to the stronger adsorption (a more negative enthalpy of adsorption) of n-hydrocarbons on zeolites with smaller micropores, which lowers the net activation energy.76,77 Denayer et al. have reported adsorption enthalpies of n-heptane at zero coverage: −79.6 kJ mol–1 for ZSM-5, −72.6 kJ mol–1 for zeolite Beta, and −50.1 kJ mol–1 for USY zeolite.78,79 However, from the apparent activation energies that were calculated from the catalytic data (Figure S4, Table S5), we do not find that the adsorption enthalpy only can explain the trends in activity of the zeolites.77,80 Furthermore, composite catalysts with Pt nanoparticles located on the γ-Al2O3 binder displayed, in all cases, a higher catalytic activity than the catalysts with Pt nanoparticles located inside the zeolite crystals (Figure 3a), while apparent activation energies were similar between the composite catalysts based on the same zeolite (Table S5). We postulate that these differences in catalytic activity are caused by intracrystalline concentration gradients in heptenes, as a result of the different locations of Pt nanoparticles which will be discussed in more detail in Section 4.
Figure 3.

Catalytic activity of the Pt/γ-Al2O3/zeolite composite catalysts (a), n-heptane conversion as a function of temperature, at a feed rate of 2.6 gn-C7·gcat–1·h–1, a total pressure of 10 bar, and 9 molH2 moln-C7–1. Weight-based product yields toward monobranched isomers (closed symbols), dibranched isomers (asterisks), and C3 and C4 cracking products (open symbols) as a function of conversion for the ZSM-5-based catalyst (b), zeolite Beta-based catalyst (c), and zeolite Y-based catalyst (d). Catalysts with Pt nanoparticles located in the zeolite are indicated in blue, while catalysts with Pt nanoparticles located on the γ-Al2O3 binder are indicated in orange. The differences in temperature of samples based on the same zeolite are within a margin of 5–10 °C, while differences between catalysts based on ZSM-5 and zeolite Y are ∼50 °C.
In Figure 3b–d, the yields toward monobranched and dibranched isomers and cracking products (propane and butanes) are plotted as a function of conversion for the composite catalysts, which were observed to be the main reaction products for all catalysts, indicative of the predominance of acid-catalyzed isomerization and cracking (Figure S5).10,35 Earlier research has pointed out that for hydroisomerization of n-alkanes on zeolite-based bifunctional catalysts, such isomer/cracking yield curves are unique functions of conversion over a broad range of reaction temperatures, because the overall isomerization and cracking rates increase simultaneously upon an increase in temperature.62,81 From Figure 3b and Table 2, it can be observed that both ZSM-5-based catalysts had a virtually identical selectivity toward monobranched isomers and cracked products, while only a limited amount of dibranched isomers were formed.
Table 2. Maximum Yield (%) of Mono- and Dibranched Heptane Isomers Obtained over Pt/γ-Al2O3/Zeolite Catalysts at a Feed Rate of 2.6 Gn-C7·gcat–1·h–1, a Total Pressure of 10 bar, and 9 molH2/moln-C7.
| Pt-“zeolite”/A |
Pt-A/“zeolite” |
|||
|---|---|---|---|---|
| mono (%) | di (%) | mono (%) | di (%) | |
| ZSM-5 | 28.5 | 0.4 | 28.9 | 0.4 |
| zeolite Beta | 53.9 | 9.4 | 59.4 | 11.1 |
| zeolite Y | 52.7 | 9.6 | 58.7 | 11.1 |
Similar findings for ZSM-5 have been reported before in the literature; more specifically, Kim et al. have observed that the location of Pt nanoparticles for ZSM-5-based catalysts (inside or outside zeolite crystals) did not impact the isomer selectivity below a zeolite crystallite size of 300 nm (this study: 20–200 nm), while the zeolite porosity, Si/Al ratio, and zeolite particle size did show to influence the total isomer yield that could be obtained.17,42 For the large-pore zeolite Beta and zeolite Y (Figure 3c,d), locating Pt nanoparticles on the γ-Al2O3 binder had a beneficial effect on isomer selectivity, both to mono- and dibranched isomers. Considering the maximum isomer yields of these catalysts (Table 2), zeolite Beta and zeolite Y perform similarly, despite the significant structural differences such as micropore size, zeolite particle size, porosity, and Si/Al ratio between the zeolites. Earlier research has pointed out that there are no great differences in diffusivity of skeletal heptane isomers in the micropores of these 12-membered ring zeolites.24 Zeolite Y-based catalysts were studied before using an n-C19 feed, resulting in a larger enhancement in isomer selectivity (∼50%) by locating Pt nanoparticles outside zeolite Y than those observed here for n-C7.40 The relatively high diffusivity of the branched heptane isomers allows relatively quick diffusion of the zeolite, and therefore, catalysts with Pt nanoparticles located in the zeolite also have a high isomer yield.
3.3. Isomer Product Distribution of Composite ZSM-5 Zeolite-Based Catalysts
The formation of isomers of heptene is commonly attributed by the protonated cyclopropyl mechanism (PCP), which consists of relatively fast reaction steps for the formation and shift of methyl groups.35 In large-pore zeolites, the reaction is nonselective with respect to the isomer formed, and therefore, in principle, the distribution between either monobranched or dibranched isomers is dictated by the thermodynamic equilibrium.24 The isomer product distribution on medium-pore ZSM-5 catalysts is subjected to shape selectivity, which has been studied in various previous studies.23,24 The isomer product distribution for Pt-Z/A and Pt-A/Z, catalysts with the Pt nanoparticles in ZSM-5 or on the γ-Al2O3 binder, respectively, is provided in Figure 4, showing that the catalysts had a high selectivity toward 2- and 3-methylhexanes and minute amounts of 2,3- and 2,4-dimethylpentane were formed. Meanwhile, on zeolite Beta and zeolite Y catalysts, mono-, di-, and tribranched isomers were formed (Figure S6), while the ratio between the 2-methylhexane and 3-methylhexane was ∼0.7, in good agreement with the ratio predicted by the thermodynamic equilibrium (Figure S7). The results of the ZSM-5 catalysts show clear evidence of shape selectivity in n-heptane hydroisomerization, resulting in a very low selectivity to di- and tribranched isomers and a 2-methylhexane/3-methylhexane ratio that is deviating from the equilibrium value.23 Previous research has pointed out the that the isomer product distribution of ZSM-5 is mainly governed by the free energy barriers for diffusion of different heptane isomers within micropores, that is, product shape selectivity.24 The isomers formed within the zeolite with a high free energy barrier for diffusion are more likely to undergo cracking and leave the zeolite as fast diffusing propane and i-butane. The extent to which the slower diffusing isomers are formed with respect to the faster diffusing isomers is known to change as a function of zeolite crystallite size.14,17,24 Based on the free energy barriers, upper limits of the diffusion coefficients for different heptane isomers were computed at different temperatures and are provided in Table 3. One methyl side group in heptane caused the diffusion coefficient in the straight channels of ZSM-5 to drop by approximately 2–3 orders of magnitude. The data confirm that the internally branched 3-methylhexane is slower diffusing than the terminally branched 2-methylhexane, leading to a deviation in the ratio between the two, which changes as a function of crystallite size.14 Dibranched isomers with geminal methyl groups (2,2- and 3,3-dimethylpentane) have very low diffusion coefficients. For isomers with vicinal methyl groups (2,3-dimethylpentane), the diffusion coefficient is higher and increases further for the isomer with more isolated methyl groups (2,4-dimethylpentane). These trends are reflected by the isomer selectivities of the ZSM-5 catalysts, in Figure 4, showing that small amounts of the 2,3 and 2,4-dimethylpentane were formed, whereas no 2,2- and 3,3-dimethylpentane were observed. Furthermore, the 2-methylhexane/3-methylhexane ratio does not differ between Pt-Z/A and Pt-A/Z catalysts (Figure S7). These observations are therefore indicative of the large diffusional differences between skeletal isomers of heptane in ZSM-5 that determine the isomer product distribution.14,23,24 The results also show that the product distribution is not affected by the location of Pt nanoparticles in composite supports.82
Figure 4.

Isomer product distribution for the Pt/γ-Al2O3/ZSM-5 catalyst: Pt-Z/A (a) and Pt-A/Z (b). The Y axis denotes the selectivity, that is, the fraction of the total products formed at a certain conversion level. Data between brackets are the total isomer yield and total n-heptane conversion at the indicated temperature.
Table 3. Upper Bounds of Diffusion Coefficients of the Heptane Isomers in the Straight Channels of ZSM-5 Zeolite, Computed with Transition State Theory.
| diffusion
coefficients (m2·s–1) |
||||
|---|---|---|---|---|
| C7 isomer | 207 °C | 255 °C | 294 °C | 387 °C |
| n-heptane | 4.3 × 10–8 | 4.2 × 10–8 | 4.2 × 10–8 | 4.1 × 10–8 |
| 2-methylhexane | 1.1 × 10–10 | 2.0 × 10–10 | 3.1 × 10–10 | 6.1 × 10–10 |
| 3-methylhexane | 1.8 × 10–11 | 3.0 × 10–11 | 4.9 × 10–11 | 9.7 × 10–11 |
| 2,2-dimethylpentane | 1.1 × 10–17 | 9.4 × 10–17 | 4.5 × 10–16 | 3.9 × 10–15 |
| 2,3-dimethylpentane | 1.3 × 10–14 | 4.1 × 10–14 | 8.2 × 10–14 | 3.3 × 10–13 |
| 2,4-dimethylpentane | 5.0 × 10–12 | 1.0 × 10–11 | 1.9 × 10–11 | 4.4 × 10–11 |
| 3,3-dimethylpentane | 3.5 × 10–18 | 1.8 × 10–17 | 9.8 × 10–17 | 9.5 × 10–16 |
3.4. Cracking Product Distribution of Composite Zeolite-Based Catalysts
Because of their high octane number, dibranched heptane isomers are more desired products than monobranched isomers for heptane hydroisomerization. However, neighboring methyl groups enable energetically favorable (β-scission, type B) cracking reactions,10 and their slow micropore diffusivity further increases the probability of cracking,15,24 and consequently, they are only formed to a limited extent. To get more insight into the cracking behavior of the composite catalysts, the dimethylpentanes/i-butane ratio is plotted, which scales the amount of dimethylpentanes that are able to leave the zeolite micropores relative to those being cracked on acid sites, toward propane and i-butane (Figure 5a). For ZSM-5-based catalysts, almost no dibranched products were observed (Section 3.3), and therefore, the ratio is close to zero for the entire conversion range. For the large-pore (Beta, Y) zeolites, placing Pt nanoparticles on the γ-Al2O3 binder significantly suppresses the cracking of dibranched isomers, that is, by a factor of 2–3, over a broad conversion range. Furthermore, the plot shows that below a conversion level of ∼80%, for both zeolite Y-based catalysts, less dibranched isomers are cracked than on the zeolite Beta catalyst. Poursaeidesfahani et al. have shown that such a difference in isomerization versus cracking tendency depends on the height of the free energy barriers of diffusion and/or the micropore length as determined by zeolite crystal size.24 Here, both zeolite Beta and Y possess significant mesoporosity, while zeolite Beta consists of smaller zeolite particles than zeolite Y that, in principle, should lead to a lower cracking tendency.17 The higher cracking tendency of zeolite Beta with respect to zeolite Y is therefore expected to be caused by a combination of micropore size and higher acid site density. In Figure 5b, the ratio between n-butane and i-butane is plotted as a function of cracking conversion, that is, the amount of C7 that is cracked toward propane and butanes. This ratio can be used as an indication of the cracking of monobranched versus dibranched isomers: i-butane and propane are formed from cracking (β-scission, type B) of all dibranched isomers, except 2,3-dimethylpentane, while n-butane is either formed by the cracking of monobranched isomers or 2,3-dimethylpentane (β-scission, type C) or hydrogenolysis (metal-catalyzed cracking).83 At a cracking conversion of >10%, a ratio of 10 or higher is observed for all studied catalysts, indicative of the predominance of acid-catalyzed cracking, which is further confirmed by the cracking product distribution patterns in Figure S6, showing negligible methane and ethane formation from hydrogenolysis. The higher relative i-butane production for the ZSM-5 catalysts compared to zeolite Beta and zeolite Y may be explained by the preferential formation of geminal dibranched isomers, especially 2,2-dimethylpentane, which is unique for the ZSM-5 framework.24,82 Because of the high free energy barrier of diffusion of geminal dibranched isomers in ZSM-5, they are subsequently cracked toward i-butane and propane. The large-pore zeolite Beta and Y do not possess such shape selectivity, which might therefore result in slightly more contributions of Type C β-scission reactions of 2,3-dimethylpentane, yielding n-butane. For all studied catalysts with Pt nanoparticles located on the γ-Al2O3 binder, it is apparent that more i-butanes are formed over n-butanes compared to catalysts with Pt nanoparticles in the zeolite. A similar difference has been observed for ZSM-22 catalysts and shows to be more subtle for the zeolites with a three-dimensional pore structure and larger micropores.42 These observations point out that when Pt nanoparticles are located in the zeolite, the acid cracking reactions are prone to more pronounced confinement effects (shape selectivity) within the zeolite micropores, leading to the formation of n-butane. To assess if the isomerization of the i- and n-butanes influences the i-butane/n-butane ratio, the ratios from Figure 5b were compared to the equilibrium ratio (Figure S8), which should be approached upon secondary isomerization. Previous studies have confirmed that shape selectivity is absent for butanes in ZSM-5, zeolite Beta, or zeolite Y because of the relatively small size of the C4 hydrocarbons.84 The i-butane/n-butane ratios obtained in the catalytic experiments were significantly higher than the equilibrium value, while the slight downward trend at higher cracking conversions might be attributed to conversion of i-butane toward n-butane. Nevertheless, the systematically high i-butane/n-butane ratio over a broad cracking conversion range confirms the relative inertness of butanes under the conditions of n-C7 hydroisomerization. Furthermore, from the product distributions, it is apparent that to a limited extent, the so-called bimolecular reactions took place for zeolite Beta- and Y-based catalysts (Figures S6, S8), resulting in the formation of 3-ethylpentane and C5–C6 cracking products, while hardly any of these product were observed for ZSM-5.83,85
Figure 5.

Ratio of dimethylheptanes over i-butane as function of n-C7 conversion (a) and the ratio between i-butane and n-butane as a function of C7 cracking conversion (b). The composite catalyst with Pt nanoparticles located in the zeolite is indicated in blue, the catalyst with Pt nanoparticles located on the γ-Al2O3 binder is indicated in orange, ZSM-5-based catalysts are indicated as triangles, zeolite Beta-based catalysts are indicated as circles, and zeolite Y-based catalysts are indicated as squares. Different n-C7 conversion or C7 cracking conversion levels were obtained by changing the temperature of the reactor between 200 and 350 °C. Differences in temperature in between catalysts based on the same zeolite are within 10 °C difference at isoconversion.
3.5. Catalytic Performance of Composite Catalysts and Physical Mixtures
In Figure 6, trends in activity (a) and maximum heptane isomer selectivity (b) are shown for the different catalysts over a broad intimacy range, and in the Supporting Information, the cracking product distribution (Figure S6) and more detailed catalytic data (Figure S9) are provided. Comparing catalytic activity of the physical mixtures to the composite catalysts, it is apparent that a close metal–acid intimacy is crucial for all catalysts, and catalysts with Pt nanoparticles on the γ-Al2O3 binder, with a metal–acid intimacy at the nanoscale, were optimal for catalytic activity. Interestingly, the catalytic activity for the intimate physical mixture of Pt/γ-Al2O3 and ZSM-5 was higher than that for the ZSM-5 composite catalyst with Pt nanoparticles located inside the zeolite crystal, while for the other zeolites, a worse performance of the physical mixtures was observed. The apparent activation energies for the intimate physical mixtures with respect to the composite catalysts were similar (ZSM-5, zeolite Beta) or slightly higher for the zeolite Y-based physical mixtures (Figure S5, Table S5). The physical mixtures of grains had a significantly lower catalytic activity than the composite catalysts and intimate physical mixtures, and all displayed a lower apparent activation energy that can be attributed to diffusion limitations as a result of the large distance between metal and acid sites.34 The intimate physical mixtures appeared to be in a transition zone between the composite catalysts and the physical mixtures of grains: the apparent activation energies were not directly affected by diffusion limitations, but the greater average distance between metal and acid sites compared to the composite catalysts negatively affects the catalytic activity. The maxima in isomer yield for the composite catalysts and physical mixtures are provided in Figure 6b and show that the selectivity of the ZSM-5-based catalyst was rather constant over the intimacy range from Pt nanoparticles in ZSM-5 zeolites to intimate physical mixtures of Pt/γ-Al2O3 and ZSM-5 zeolite. For zeolite Beta and zeolite Y, the intimate physical mixtures have similar selectivity toward isomers as catalysts with Pt nanoparticles located on the γ-Al2O3 binder, although higher temperatures are required to obtain the optimum isomer yield. The isomer yield for the physical mixtures of grains was worse for all zeolites. Considering both catalytic activity and isomer selectivity, catalysts with Pt nanoparticles located outside the zeolite, but in close (nanoscale) vicinity to zeolite acid sites, are optimal for catalytic performance.
Figure 6.

Conversion for each zeolite at a fixed temperature (ZSM-5, 230 °C; zeolite Beta, 250 °C; and zeolite Y, 270 °C) (a) and maximum heptane isomer yield (b) with metal–acid intimacy ranging from closest to microscale. ZSM-5-based catalysts are indicated as blue triangles, the zeolite Beta-based catalyst is indicated as orange circles, and zeolite Y-based catalysts are indicated as black squares.
4. General Discussion
In the foregoing sections of the paper, we have discussed the effect of the metal–acid intimacy in zeolite-based bifunctional catalysts (composite catalysts and physical mixtures based on ZSM-5, zeolite Beta, and zeolite Y) on differences in the performance in n-heptane hydroisomerization. We observed that the metal–acid intimacy, altered by varying the location of Pt nanoparticles, for zeolite-based catalysts plays its role for catalyst activity and selectivity. Of all catalysts based on the same zeolite, the composite catalysts with Pt nanoparticles on the γ-Al2O3 binder had the highest catalytic activity and optimal isomer selectivity. For large-pore zeolite Beta and zeolite Y, locating Pt nanoparticles in the zeolite was detrimental for isomer yield, whereas for the ZSM-5-based catalysts, the isomer yield was rather constant ranging from the closest intimacy to the microscale of the intimate physical mixture. Finally, for all composite catalysts, locating Pt nanoparticles in the zeolite increased the amount of n-butanes formed over i-butanes.
To explain the enhancement in activity and isomer selectivity, we argue that the average reactant diffusion pathway and related reaction events on the composite catalysts with Pt nanoparticles on the γ-Al2O3 binder are different with respect to catalysts with Pt nanoparticles located in the zeolite. The heptenes formed on Pt nanoparticles outside the zeolite upon entering the zeolite may adsorb and react on the first Brønsted acid site they encounter, while still being close to the zeolite outer surface. After conversion to a methyl branched heptene, we propose that it is “easier” for the isomer to diffuse out than to diffuse into the zeolite crystal, that is, there is an anisotropy in the diffusion coefficient based on direction, as was earlier proposed for Pt/γ-Al2O3/ZSM-22 catalysts.42 This implies that the heptene isomers in the outer surface layer should “remember” the zeolite pore entrance to rapidly diffuse out and subsequently be hydrogenated on Pt nanoparticles on the γ-Al2O3 binder. This mechanism effectively decreases the average residence time in the zeolite micropores of heptenes formed outside the zeolite, with respect to heptenes formed on Pt nanoparticles inside the zeolite, which has shown to be a determining factor in the catalyst selectivity.15 We anticipate that, on average, heptenes formed inside the zeolite are located deeper in the zeolite with lower diffusion coefficients, resulting in cracking reactions and producing quickly diffusing butenes and propene. This reaction mechanism is beneficial for the catalyst activity, for the large-pore zeolite Beta and zeolite Y, and also for the selectivity toward branched isomers.
For ZSM-5, the 10-membered ring pores of the zeolite impose such strict confinement on heptene intermediates that the yield and product distribution of isomers remain unaffected for the catalysts with a different Pt nanoparticle location (inside or outside the zeolite). This behavior can be attributed to the presence of a three-dimensional micropore network and intersections that facilitate isomerization and cracking and erase the “memory effect” mentioned above and is therefore fundamentally different from one-dimensional 10-membered ring zeolites such as ZSM-22 that have generally high selectivities toward branched isomers.16,42 Other evidence of the impact of concentration gradients in heptenes in the zeolites being affected by Pt location is the higher i-butane/n-butane ratio when Pt nanoparticles are placed outside the zeolite, with respect to inside, which was observed for all composite catalysts.42
For the physical mixtures, the greater distance between metal and acid sites may lead to concentration gradients of heptenes.46 Nevertheless, optimal isomer selectivity could also be obtained using intimate physical mixtures, which can be attributed to a similar transport of olefins with the zeolite as for the composite catalysts with Pt nanoparticles on the γ-Al2O3 binder. The large metal–acid distances present in the physical mixtures of grains negatively affected both the activity and isomer selectivity for all catalysts.
The results of this study are therefore in general agreement with previously reported results where beneficial effects have been reported when Pt nanoparticles located are outside zeolite crystals and in nanoscale intimacy with zeolite acid sites.40−42
5. Conclusions
In this study, beneficial effects on activity for heptane hydroisomerization were observed when Pt nanoparticles are placed on the γ-Al2O3 binder of zeolite/γ-Al2O3 composite supports as opposed to Pt located inside zeolite crystals. The activity of the ZSM-5-based catalyst appeared to be rather unsensitive for Pt nanoparticle location within the nanoscale to microscale range, while the catalytic activity of large-pore zeolites benefitted from a nanoscale metal–acid intimacy. For both zeolite Beta- and zeolite Y-based catalyst, beneficial effects on isomer selectivity were observed while cracking was suppressed when Pt nanoparticles were placed on the γ-Al2O3 binder. The enhanced selectivity of large-pore zeolites toward isomers is ascribed to the shorter residence time of intermediates in zeolite micropores as a consequence of concentration gradients of heptenes. For ZSM-5, no significant differences in selectivity were observed between catalysts over a range of metal–acid intimacies, which can be explained by the large diffusional barriers of heptane/heptene isomers in the ZSM-5 zeolite that dictates the product distribution. In general, it is concluded that the catalytic performance of the zeolite-based bifunctional catalyst for the conversion of hydrocarbons is affected by pore-mouth mechanisms and intracrystalline diffusion of reaction intermediates in zeolite crystals.
Acknowledgments
We acknowledge the European Research Council EU FP7 ERC Advanced Grant no. 338846 for support. T.J.H.V. acknowledges NWO-CW for a VICI grant. Remco Dalebout, Laura Barberis, and Nikos Nikolopoulos (UU) are acknowledged for performing N2 physisorption measurements. Shell Projects and Technology is acknowledged for supplying the zeolite Y/γ-Al2O3 extrudates and HMPA pseudo-boehmite.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c03138.
Calculations for catalytic parameters, additional properties of commercial zeolites, characterization by N2 physisorption, NH3-TPD, results from thermodynamic calculations, and catalytic results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Coonradt H. L.; Garwood W. E. Mechanism of Hydrocracking. Reactions of Paraffins and Olefins. Ind. Eng. Chem. Process Des. Dev. 1964, 3, 38–45. 10.1021/i260009a010. [DOI] [Google Scholar]
- Weber J. L.; Dugulan I.; de Jongh P. E.; de Jong K. P. Bifunctional Catalysis for the Conversion of Synthesis Gas to Olefins and Aromatics. ChemCatChem 2018, 10, 1107–1112. 10.1002/cctc.201701667. [DOI] [Google Scholar]
- Cheng K.; Gu B.; Liu X.; Kang J.; Zhang Q.; Wang Y. Direct and Highly Selective Conversion of Synthesis Gas into Lower Olefins: Design of a Bifunctional Catalyst Combining Methanol Synthesis and Carbon-Carbon Coupling. Angew. Chem., Int. Ed. 2016, 55, 4725–4728. 10.1002/anie.201601208. [DOI] [PubMed] [Google Scholar]
- Jiao F.; Li J.; Pan X.; Xiao J.; Li H.; Ma H.; Wei M.; Pan Y.; Zhou Z.; Li M.; Miao S.; Li J.; Zhu Y.; Xiao D.; He T.; Yang J.; Qi F.; Fu Q.; Bao X. Selective Conversion of Syngas to Light Olefins. Science 2016, 351, 1065–1068. 10.1126/science.aaf1835. [DOI] [PubMed] [Google Scholar]
- Sartipi S.; Parashar K.; Makkee M.; Gascon J.; Kapteijn F. Breaking the Fischer-Tropsch Synthesis Selectivity: Direct Conversion of Syngas to Gasoline over Hierarchical Co/H-ZSM-5 Catalysts. Catal. Sci. Technol. 2013, 3, 572–575. 10.1039/c2cy20744c. [DOI] [Google Scholar]
- Thybaut J. W.; Marin G. B.. Multiscale Aspects in Hydrocracking. Advances in Catalysis; Elsevier Inc., 2016; Vol. 59, pp 109–238. [Google Scholar]
- Peng B.; Yao Y.; Zhao C.; Lercher J. A. Towards Quantitative Conversion of Microalgae Oil to Diesel-Range Alkanes with Bifunctional Catalysts. Angew. Chem., Int. Ed. 2012, 51, 2072–2075. 10.1002/anie.201106243. [DOI] [PubMed] [Google Scholar]
- Martens J. A.; Verboekend D.; Thomas K.; Vanbutsele G.; Gilson J.-P.; Pérez-Ramírez J. Hydroisomerization of Emerging Renewable Hydrocarbons Using Hierarchical Pt/H-ZSM-22 Catalyst. ChemSusChem 2013, 6, 421–425. 10.1002/cssc.201200888. [DOI] [PubMed] [Google Scholar]
- Gao P.; Li S.; Bu X.; Dang S.; Liu Z.; Wang H.; Zhong L.; Qiu M.; Yang C.; Cai J.; Wei W.; Sun Y. Direct Conversion of CO2 into Liquid Fuels with High Selectivity over a Bifunctional Catalyst. Nat. Chem. 2017, 9, 1019–1024. 10.1038/nchem.2794. [DOI] [PubMed] [Google Scholar]
- Weitkamp J. Catalytic Hydrocracking-Mechanisms and Versatility of the Process. ChemCatChem 2012, 4, 292–306. 10.1002/cctc.201100315. [DOI] [Google Scholar]
- Chica A.; Corma A. Hydroisomerization of Pentane, Hexane, and Heptane for Improving the Octane Number of Gasoline. J. Catal. 1999, 187, 167–176. 10.1006/jcat.1999.2601. [DOI] [Google Scholar]
- Ruthven D. M.; Post M. F. M.. Diffusion in Zeolite Molecular Sieves. In Studies in Surface Science and Catalysis; van Bekkum H., Jansen J. C., Jacobs P. A., Flanigen E. M., Eds.; Elsevier Science B.V., 2001; Vol. 137, pp 525–577. [Google Scholar]
- Kärger J.; Ruthven D. M.; Theodorou D. N.. Medium-Pore (Ten-Ring) Zeolites. Diffusion in Nanoporous Materials; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp 653–728. [Google Scholar]
- Vandegehuchte B. D.; Choudhury I. R.; Thybaut J. W.; Martens J. A.; Marin G. B. Integrated Stefan-Maxwell, Mean Field, and Single-Event Microkinetic Methodology for Simultaneous Diffusion and Reaction inside Microporous Materials. J. Phys. Chem. C 2014, 118, 22053–22068. 10.1021/jp506056r. [DOI] [Google Scholar]
- Noh G.; Shi Z.; Zones S. I.; Iglesia E. Isomerization and β-Scission Reactions of Alkanes on Bifunctional Metal-Acid Catalysts: Consequences of Confinement and Diffusional Constraints on Reactivity and Selectivity. J. Catal. 2018, 368, 389–410. 10.1016/j.jcat.2018.03.033. [DOI] [Google Scholar]
- Noh G.; Zones S. I.; Iglesia E. Isomer Sieving and the Selective Formation of Terminal Methyl Isomers in Reactions of Linear Alkanes on One-Dimensional Zeolites. J. Catal. 2019, 377, 255–270. 10.1016/j.jcat.2019.07.022. [DOI] [Google Scholar]
- Kim J.; Kim W.; Seo Y.; Kim J.-C.; Ryoo R. N-Heptane Hydroisomerization over Pt/MFI Zeolite Nanosheets: Effects of Zeolite Crystal Thickness and Platinum Location. J. Catal. 2013, 301, 187–197. 10.1016/j.jcat.2013.02.015. [DOI] [Google Scholar]
- Tromp M.; van Bokhoven J. A.; Garriga Oostenbrink M. T.; Bitter J. H.; de Jong K. P.; Koningsberger D. C. Influence of the Generation of Mesopores on the Hydroisomerization Activity and Selectivity of N-Hexane over Pt/Mordenite. J. Catal. 2000, 190, 209–214. 10.1006/jcat.1999.2778. [DOI] [Google Scholar]
- de Jong K. P.; Zečević J.; Friedrich H.; de Jongh P. E.; Bulut M.; van Donk S.; Kenmogne R.; Finiels A.; Hulea V.; Fajula F. Zeolite Y Crystals with Trimodal Porosity as Ideal Hydrocracking Catalysts. Angew. Chem., Int. Ed. 2010, 49, 10074–10078. 10.1002/anie.201004360. [DOI] [PubMed] [Google Scholar]
- Ban S.; Van Laak A. N. C.; Landers J.; Neimark A. V.; De Jongh P. E.; De Jong K. P.; Vlugt T. J. H. Insight into the Effect of Dealumination on Mordenite Using Experimentally Validated Simulations. J. Phys. Chem. C 2010, 114, 2056–2065. 10.1021/jp909900a. [DOI] [Google Scholar]
- Wang G.; Johannessen E.; Kleijn C. R.; de Leeuw S. W.; Coppens M.-O. Optimizing Transport in Nanostructured Catalysts: A Computational Study. Chem. Eng. Sci. 2007, 62, 5110–5116. 10.1016/j.ces.2007.01.046. [DOI] [Google Scholar]
- Smit B.; Maesen T. L. M. Towards a Molecular Understanding of Shape Selectivity. Nature 2008, 451, 671–678. 10.1038/nature06552. [DOI] [PubMed] [Google Scholar]
- Vandegehuchte B. D.; Thybaut J. W.; Marin G. B. Unraveling Diffusion and Other Shape Selectivity Effects in ZSM5 Using N-Hexane Hydroconversion Single-Event Microkinetics. Ind. Eng. Chem. Res. 2014, 53, 15333–15347. 10.1021/ie500164q. [DOI] [Google Scholar]
- Poursaeidesfahani A.; de Lange M. F.; Khodadadian F.; Dubbeldam D.; Rigutto M.; Nair N.; Vlugt T. J. H. Product Shape Selectivity of MFI-Type, MEL-Type, and BEA-Type Zeolites in the Catalytic Hydroconversion of Heptane. J. Catal. 2017, 353, 54–62. 10.1016/j.jcat.2017.07.005. [DOI] [Google Scholar]
- Thybaut J. W.; Marin G. B.; Baron G. V.; Jacobs P. A.; Martens J. A. Alkene Protonation Enthalpy Determination from Fundamental Kinetic Modeling of Alkane Hydroconversion on Pt/H-(US)Y-Zeolite. J. Catal. 2001, 202, 324–339. 10.1006/jcat.2001.3292. [DOI] [Google Scholar]
- Kärger J.Diffusion Measurements by NMR Techniques. Molecular Sieves—Science and Technology; Springer-Verlag Berlin Heidelberg, 2008; Vol. 7, pp 85–133. [Google Scholar]
- Van Donk S.; Broersma A.; Gijzeman O. L. J.; Van Bokhoven J. A.; Bitter J. H.; De Jong K. P. Combined Diffusion, Adsorption, and Reaction Studies of n-Hexane Hydroisomerization over Pt/H-Mordenite in an Oscillating Microbalance. J. Catal. 2001, 204, 272–280. 10.1006/jcat.2001.3393. [DOI] [Google Scholar]
- Zhu W.; Kapteijn F.; Moulijn J. A. Diffusion of Linear and Branched C6 Alkanes in Silicalite-1 Studied by the Tapered Element Oscillating Microbalance. Microporous Mesoporous Mater. 2001, 47, 157–171. 10.1016/S1387-1811(01)00372-9. [DOI] [Google Scholar]
- Hendriks F. C.; Meirer F.; Kubarev A. V.; Ristanović Z.; Roeffaers M. B. J.; Vogt E. T. C.; Bruijnincx P. C. A.; Weckhuysen B. M. Single-Molecule Fluorescence Microscopy Reveals Local Diffusion Coefficients in the Pore Network of an Individual Catalyst Particle. J. Am. Chem. Soc. 2017, 139, 13632–13635. 10.1021/jacs.7b07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbeldam D.; Beerdsen E.; Vlugt T. J. H.; Smit B. Molecular Simulation of Loading-Dependent Diffusion in Nanoporous Materials Using Extended Dynamically Corrected Transition State Theory. J. Chem. Phys. 2005, 122, 224712. 10.1063/1.1924548. [DOI] [PubMed] [Google Scholar]
- Maesen T. L. M.; Schenk M.; Vlugt T. J. H.; Jonge J. P. d.; Smit B. The Shape Selectivity of Paraffin Hydroconversion on TON-, MTT-, and AEL-Type Sieves. J. Catal. 1999, 188, 403–412. 10.1006/jcat.1999.2673. [DOI] [Google Scholar]
- Dubbeldam D.; Torres-Knoop A.; Walton K. S.. On the Inner Workings of Monte Carlo Codes. Molecular Simulation; Taylor & Francis, 2013; pp 1253–1292. [Google Scholar]
- Dubbeldam D.; Calero S.; Ellis D. E.; Snurr R. Q. RASPA: Molecular Simulation Software for Adsorption and Diffusion in Flexible Nanoporous Materials. Mol. Simul. 2016, 42, 81–101. 10.1080/08927022.2015.1010082. [DOI] [Google Scholar]
- Weisz P. B. Polyfunctional Heterogeneous Catalysis. Adv. Catal. 1962, 13, 137–190. 10.1016/S0360-0564(08)60287-4. [DOI] [Google Scholar]
- Martens J. A.; Jacobs P. A.. Introduction to Acid Catalysis with Zeolites in Hydrocarbon Reactions. Studies in Surface Science and Catalysis, 2001; Vol. 137, pp 633–671. [Google Scholar]
- Dvoyashkina N.; Freude D.; Stepanov A. G.; Böhlmann W.; Krishna R.; Kärger J.; Haase J. Alkane/Alkene Mixture Diffusion in Silicalite-1 Studied by MAS PFG NMR. Microporous Mesoporous Mater. 2018, 257, 128–134. 10.1016/j.micromeso.2017.08.015. [DOI] [Google Scholar]
- Kazansky V. B. Adsorbed Carbocations as Transition States in Heterogeneous Acid Catalyzed Transformations of Hydrocarbons. Catal. Today 1999, 51, 419–434. 10.1016/S0920-5861(99)00031-0. [DOI] [Google Scholar]
- Ruthven D. M.; Reyes S. C. Adsorptive Separation of Light Olefins from Paraffins. Microporous Mesoporous Mater. 2007, 104, 59–66. 10.1016/j.micromeso.2007.01.005. [DOI] [Google Scholar]
- Hartmann M.; Kunz S.; Himsl D.; Tangermann O.; Ernst S.; Wagener A. Adsorptive Separation of Isobutene and Isobutane on Cu3(BTC)2. Langmuir 2008, 24, 8634–8642. 10.1021/la8008656. [DOI] [PubMed] [Google Scholar]
- Zecevic J.; Vanbutsele G.; De Jong K. P.; Martens J. A. Nanoscale Intimacy in Bifunctional Catalysts for Selective Conversion of Hydrocarbons. Nature 2015, 528, 245–248. 10.1038/nature16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Moussa O.; Tinat L.; Jin X.; Baaziz W.; Durupthy O.; Sayag C.; Blanchard J. Heteroaggregation and Selective Deposition for the Fine Design of Nanoarchitectured Bifunctional Catalysts: Application to Hydroisomerization. ACS Catal. 2018, 8, 6071–6078. 10.1021/acscatal.8b01461. [DOI] [Google Scholar]
- Cheng K.; Wal L. I.; Yoshida H.; Oenema J.; Harmel J.; Zhang Z.; Sunley G.; Zečević J.; Jong K. P. Impact of the Spatial Organization of Bifunctional Metal–Zeolite Catalysts on the Hydroisomerization of Light Alkanes. Angew. Chem., Int. Ed. 2020, 59, 3592–3600. 10.1002/anie.201915080. [DOI] [PubMed] [Google Scholar]
- Harmel J.; van der Wal L. I.; Zečević J.; de Jongh P. E.; de Jong K. P. Influence of Intimacy for Metal-Mesoporous Solid Acids Catalysts for n -Alkanes Hydro-Conversion. Catal. Sci. Technol. 2020, 10, 2111–2119. 10.1039/C9CY02510C. [DOI] [Google Scholar]
- Samad J. E.; Blanchard J.; Sayag C.; Louis C.; Regalbuto J. R. The Controlled Synthesis of Metal-Acid Bifunctional Catalysts: Selective Pt Deposition and Nanoparticle Synthesis on Amorphous Aluminosilicates. J. Catal. 2016, 342, 213–225. 10.1016/j.jcat.2016.08.002. [DOI] [Google Scholar]
- Batalha N.; Pinard L.; Pouilloux Y.; Guisnet M. Bifunctional Hydrogenating/Acid Catalysis: Quantification of the Intimacy Criterion. Catal. Lett. 2013, 143, 587–591. 10.1007/s10562-013-1003-9. [DOI] [Google Scholar]
- Chu H. Y.; Rosynek M. P.; Lunsford J. H. Skeletal Isomerization of Hexane over Pt/H-Beta Zeolites: Is the Classical Mechanism Correct?. J. Catal. 1998, 178, 352–362. 10.1006/jcat.1998.2136. [DOI] [Google Scholar]
- Weber J. L.; Krans N. A.; Hofmann J. P.; Hensen E. J. M.; Zecevic J.; de Jongh P. E.; de Jong K. P. Effect of Proximity and Support Material on Deactivation of Bifunctional Catalysts for the Conversion of Synthesis Gas to Olefins and Aromatics. Catal. Today 2020, 342, 161–166. 10.1016/j.cattod.2019.02.002. [DOI] [Google Scholar]
- Creyghton E. J.; Van Duin A. C. T.; Jansen J. C.; Kooyman P. J.; Zandbergen H. W.; Van Bekkum H. Synthesis of Pt Clusters in Zeolite BEA: Effect of Reduction Rate on Cluster Size and Location. J. Chem. Soc., Faraday Trans. 1996, 4637–4642. 10.1039/ft9969204637. [DOI] [Google Scholar]
- Iida T.; Zanchet D.; Ohara K.; Wakihara T.; Román-Leshkov Y. Concerted Bimetallic Nanocluster Synthesis and Encapsulation via Induced Zeolite Framework Demetallation for Shape and Substrate Selective Heterogeneous Catalysis. Angew. Chem., Int. Ed. 2018, 57, 6454–6458. 10.1002/anie.201800557. [DOI] [PubMed] [Google Scholar]
- Camblor M. A.; Corma A.; Martínez A.; Martínez-Soria V.; Valencia S. Mild Hydrocracking of Vacuum Gasoil over NiMo-Beta Zeolite Catalysts: The Role of the Location of the NiMo Phases and the Crystallite Size of the Zeolite. J. Catal. 1998, 179, 537–547. 10.1006/jcat.1998.2249. [DOI] [Google Scholar]
- Francis J.; Guillon E.; Bats N.; Pichon C.; Corma A.; Simon L. J. Design of Improved Hydrocracking Catalysts by Increasing the Proximity between Acid and Metallic Sites. Appl. Catal., A 2011, 409–410, 140–147. 10.1016/j.apcata.2011.09.040. [DOI] [Google Scholar]
- Whiting G. T.; Chung S.-H.; Stosic D.; Chowdhury A. D.; Van Der Wal L. I.; Fu D.; Zecevic J.; Travert A.; Houben K.; Baldus M.; Weckhuysen B. M. Multiscale Mechanistic Insights of Shaped Catalyst Body Formulations and Their Impact on Catalytic Properties. ACS Catal. 2019, 9, 4792–4803. 10.1021/acscatal.9b00151. [DOI] [Google Scholar]
- De Graaf J.; Van Dillen A. J.; De Jong K. P.; Koningsberger D. C. Preparation of Highly Dispersed Pt Particles in Zeolite Y with a Narrow Particle Size Distribution: Characterization by Hydrogen Chemisorption, TEM, EXAFS Spectroscopy, and Particle Modeling. J. Catal. 2001, 203, 307–321. 10.1006/jcat.2001.3337. [DOI] [Google Scholar]
- Dubbeldam D.; Torres-Knoop A.; Walton K. S. On the Inner Workings of Monte Carlo Codes. Mol. Simul. 2013, 39, 1253–1292. 10.1080/08927022.2013.819102. [DOI] [Google Scholar]
- Baerlocher C.; McCusker L. B.. Database of Zeolite Structures. http://www.iza-structure.org/databases/ (accessed Nov 28, 2019).
- Vlugt T. J. H.; Krishna R.; Smit B. Molecular Simulations of Adsorption Isotherms for Linear and Branched Alkanes and Their Mixtures in Silicalite. J. Phys. Chem. B 1999, 103, 1102–1118. 10.1021/jp982736c. [DOI] [Google Scholar]
- Cho H. J.; Kim D.; Li J.; Su D.; Xu B. Zeolite-Encapsulated Pt Nanoparticles for Tandem Catalysis. J. Am. Chem. Soc. 2018, 140, 13514–13520. 10.1021/jacs.8b09568. [DOI] [PubMed] [Google Scholar]
- Oenema J.; Hofmann J. P.; Hensen E. J. M.; Zečević J.; Jong K. P. Assessment of the Location of Pt Nanoparticles in Pt/Zeolite Y/γ-Al2O3 Composite Catalysts. ChemCatChem 2020, 12, 615–622. 10.1002/cctc.201901617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zečević J.; Van Der Eerden A. M.; Friedrich H.; De Jongh P. E.; De Jong K. P. Heterogeneities of the Nanostructure of Platinum/Zeolite y Catalysts Revealed by Electron Tomography. ACS Nano 2013, 7, 3698–3705. 10.1021/nn400707p. [DOI] [PubMed] [Google Scholar]
- Guisnet M.; Alvarez F.; Giannetto G.; Perot G. Hydroisomerization and Hydrocracking of N-Heptane on Pth Zeolites. Effect of the Porosity and of the Distribution of Metallic and Acid Sites. Catal. Today 1987, 1, 415–433. 10.1016/0920-5861(87)80007-X. [DOI] [Google Scholar]
- Alvarez F.; Ribeiro F. R.; Perot G.; Thomazeau C.; Guisnet M. Hydroisomerization and Hydrocracking of Alkanes. J. Catal. 1996, 162, 179–189. 10.1006/jcat.1996.0275. [DOI] [Google Scholar]
- Thybaut J. W.; Laxmi Narasimhan C. S.; Denayer J. F.; Baron G. V.; Jacobs P. A.; Martens J. A.; Marin G. B. Acid-Metal Balance of a Hydrocracking Catalyst: Ideal versus Nonideal Behavior. Ind. Eng. Chem. Res. 2005, 44, 5159–5169. 10.1021/ie049375+. [DOI] [Google Scholar]
- Rodríguez-González L.; Hermes F.; Bertmer M.; Rodríguez-Castellón E.; Jiménez-López A.; Simon U. The Acid Properties of H-ZSM-5 as Studied by NH3-TPD and 27Al-MAS-NMR Spectroscopy. Appl. Catal., A 2007, 328, 174–182. 10.1016/j.apcata.2007.06.003. [DOI] [Google Scholar]
- Velthoen M. E. Z.; Lucini Paioni A.; Teune I. E.; Baldus M.; Weckhuysen B. M. Matrix Effects in a Fluid Catalytic Cracking Catalyst Particle: Influence on Structure, Acidity, and Accessibility. Chem.—Eur. J. 2020, 26, 11995–12009. 10.1002/chem.201905867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting G. T.; Chowdhury A. D.; Oord R.; Paalanen P.; Weckhuysen B. M. The Curious Case of Zeolite-Clay/Binder Interactions and Their Consequences for Catalyst Preparation. Faraday Discuss. 2016, 188, 369–386. 10.1039/c5fd00200a. [DOI] [PubMed] [Google Scholar]
- Oenema J.; van Alst R. A.; Meijerink M. J.; Zečević J.; de Jong K. P. The Influence of Residual Chlorine on Pt/Zeolite Y/γ-Al2O3 Composite Catalysts: Acidity and Performance. Appl. Catal., A 2020, 605, 117815. 10.1016/j.apcata.2020.117815. [DOI] [Google Scholar]
- Hunger M.NMR Spectroscopy for the Characterization of Surface Acidity and Basicity. In Handbook of Heterogeneous Catalysis; Ertl G., Knozinger H., Schuth F., Weitkamp J., Eds.; Wiley-VCH Verlag GmbH& Co. KGaA, 2008; pp 1163–1178. [Google Scholar]
- Knozinger H.Infrared Spectroscopy for the Characterization of Surface Acidity and Basicity. In Handbook of Heterogeneous Catalysis; Ertl G., Knozinger H., Schüth F., Weitkamp J., Eds.; Wiley-VCH Verlag GmbH& Co. KGaA, 2008; pp 1135–1163. [Google Scholar]
- Daniell W.; Topsøe N.-Y.; Knözinger H. An FTIR Study of the Surface Acidity of USY Zeolites: Comparison of CO, CD3CN, and C5H5N Probe Molecules. Langmuir 2001, 17, 6233–6239. 10.1021/la010345a. [DOI] [Google Scholar]
- Kotrel S.; Lunsford J. H.; Knözinger H. Characterizing Zeolite Acidity by Spectroscopic and Catalytic Means: A Comparison. J. Phys. Chem. B 2001, 105, 3917–3921. 10.1021/jp002161v. [DOI] [Google Scholar]
- Jones A. J.; Iglesia E. The Strength of Brønsted Acid Sites in Microporous Aluminosilicates. ACS Catal. 2015, 5, 5741–5755. 10.1021/acscatal.5b01133. [DOI] [Google Scholar]
- Rybicki M.; Sauer J. Acid Strength of Zeolitic Brønsted Sites—Dependence on Dielectric Properties. Catal. Today 2019, 323, 86–93. 10.1016/j.cattod.2018.04.031. [DOI] [Google Scholar]
- Boronat M.; Corma A. What Is Measured When Measuring Acidity in Zeolites with Probe Molecules?. ACS Catal. 2019, 9, 1539–1548. 10.1021/acscatal.8b04317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven J. A.; Tromp M.; Koningsberger D. C.; Miller J. T.; Pieterse J. A. Z.; Lercher J. A.; Williams B. A.; Kung H. H. An Explanation for the Enhanced Activity for Light Alkane Conversion in Mildly Steam Dealuminated Mordenite: The Dominant Role of Adsorption. J. Catal. 2001, 202, 129–140. 10.1006/jcat.2001.3265. [DOI] [Google Scholar]
- Van De Runstraat A.; Kamp J. A.; Stobbelaar P. J.; Van Grondelle J.; Krijnen S.; Van Santen R. A. Kinetics of Hydro-Isomerization of n-Hexane over Platinum Containing Zeolites. J. Catal. 1997, 171, 77–84. 10.1006/jcat.1997.1779. [DOI] [Google Scholar]
- Denayer J. F.; Souverijns W.; Jacobs P. A.; Martens J. A.; Baron G. V. High-Temperature Low-Pressure Adsorption of Branched C5-C8 Alkanes on Zeolite Beta, ZSM-5, ZSM-22, Zeolite Y, and Mordenite. J. Phys. Chem. B 1998, 102, 4588–4597. 10.1021/jp980674k. [DOI] [Google Scholar]
- Denayer J. F.; Baron G. V.; Martens J. A.; Jacobs P. A. Chromatographic Study of Adsorption of N-Alkanes on Zeolites at High Temperatures. J. Phys. Chem. B 1998, 102, 3077–3081. 10.1021/jp972328t. [DOI] [Google Scholar]
- Guisnet M.; Fouche V.; Belloum M.; Bournonville J. P.; Travers C. Isomerization of N-Hexane on Platinum Dealuminated Mordenite Catalysts II. Kinetic Study. Appl. Catal. 1991, 71, 295–306. 10.1016/0166-9834(91)85087-C. [DOI] [Google Scholar]
- Froment G. F. Kinetics of the Hydroisomerization and Hydrocracking of Paraffins on a Platinum Containing Bifunctional Y-Zeolite. Catal. Today 1987, 1, 455–473. 10.1016/0920-5861(87)80009-3. [DOI] [Google Scholar]
- Maesen T. L. M.; Schenk M.; Vlugt T. J. H.; Smit B. Differences between MFI- and MEL-Type Zeolites in Paraffin Hydrocracking. J. Catal. 2001, 203, 281–291. 10.1006/jcat.2001.3332. [DOI] [Google Scholar]
- Martens J. A.; Tielen M.; Jacobs P. A. Attempts to Rationalize the Distribution of Hydrocracked Products. III. Mechanistic Aspects of Isomerization and Hydrocracking of Branched Alkanes on Ideal Bifunctional Large-Pore Zeolite Catalysts. Catal. Today 1987, 1, 435–453. 10.1016/0920-5861(87)80008-1. [DOI] [Google Scholar]
- Houžvička J.; Hansildaar S.; Ponec V. The Shape Selectivity in the Skeletal Isomerisation of N-Butene to Isobutene. J. Catal. 1997, 167, 273–278. 10.1006/jcat.1997.1525. [DOI] [Google Scholar]
- Blomsma E.; Martens J. A.; Jacobs P. A. Mechanisms of Heptane Isomerization on Bifunctional Pd/H-Beta Zeolites. J. Catal. 1996, 159, 323–331. 10.1006/jcat.1996.0094. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.