

Abstract
The NIH LINCS program is generating extensive multidimensional datasets including biochemical-, genome-wide transcriptional-, and phenotypic cellular response signatures to a variety of small molecule and genetic perturbations with the goal to create a sustainable, widely applicable and readily accessible systems biology knowledge resource. Integration and analysis of diverse LINCS datasets depends on the availability of sufficient metadata to describe the assays and screening results, and on their syntactic, structural, and semantic consistency. Here we report metadata specifications for the most important molecular and cellular components and recommend them for adoption beyond the LINCS project. We focus on the minimum required information to model LINCS assays and results based on a number of use cases and we recommend controlled terminologies and ontologies to annotate assays with syntactic consistency and semantic integrity. We also report specifications for a Simple Annotation Format (SAF) to describe assays and screening results based on our metadata specifications with explicit controlled vocabularies. SAF specifically serves to programmatically access and exchange LINCS data as a prerequisite for a distributed information management infrastructure. We applied the metadata specifications to annotate large numbers of LINCS cell lines, proteins, and small molecules. The resources generated and presented here are freely available.
Introduction
Modern high-throughput screening technologies based on miniaturized assay technologies have enabled the production of vast datasets in the life sciences including genomics, proteomics, transcriptomics, and chemical biology. During the last decade, both the number of publically funded data production projects and the size of datasets have been rising dramatically, providing access to unprecedented amounts and diversity of data in the public domain. Examples of such projects funded by the National Institutes of Health (NIH) include The Cancer Genome Atlas (TCGA)1, the Encyclopedia Of DNA Elements (ENCODE) project2, Cancer Target Discovery and Development (CTD2) Network3, and the Molecular Libraries Probe Center Network (MLPCN)4.
Here we focus on a more recent NIH-funded project, the Library of Integrated Network-based Cellular Signatures (LINCS) program5. The LINCS project aims to generate an extensive reference set of cellular response data to a variety of small molecule and genetic perturbations with the goal to improve our understanding of complex human diseases such as cancer. Common patterns from these data (signatures) include information about gene transcription, protein binding, cell proliferation, cell signaling and other cellular phenotypes. LINCS assays span a variety of technologies, model systems, readouts and perturbations. To produce an integrated view across the diverse LINCS data resources requires i) defining which biological entities and concepts, experimental parameters, and results must be included in such an integrated view; ii) uniquely identifying the entities of interest, such as small molecule compounds, proteins, cells, siRNAs, etc. so that they can be unambiguously associated with the assays and the screening results; and iii) standardized data formats in which datasets can be exchanged or queried. A fundamental requirement of useful metadata standards for LINCS, and other projects, is their free and open accessibility and well-defined relationships with other standards.
Types of standards that are relevant for reporting biological screening experiments and results include i) minimum information checklists, ii) controlled vocabularies and ontologies, and iii) data format specifications. Various minimum information specifications have been developed to facilitate reproducibility and critical evaluation and interpretation of biological experiments and their results by others. Such standards relevant to LINCS include Minimum Information About a Cellular Assay (MIACA)6, Minimum Information About an RNAi Experiment (MIARE)7, Minimum Information About a Protein Affinity Reagent (MIAPAR)8, and Minimum Information About a Bioactive Entity (MIABE)6. These are available via the Minimum Information for Biological and Biomedical Investigations (MIBBI) project9. MIBBI checklists are now part of the larger BioSharing effort10, which also catalogs other standards (such as terminologies) and databases that use such standards. The ISA framework, including the ISA-Tab file format and software tools, enables the use of such standards; ISA refers to the specific metadata categories ‘Investigation’, ‘Study’ and ‘Assay’. Among many projects it has also been used at LINCS11.
Many controlled vocabularies and biomedical ontologies exist and several have become widely used as standards, such as medical subject headings (MeSH)12 and the Gene Ontology (GO)13. However, existing vocabularies and ontologies are still far from comprehensive; and in many cases ontologies have been developed for specific purposes and are not mapped to one another, thus complicating unique identification of biological entities across domains14. To address this challenge in the domain of chemical biology high-throughput screening, we have recently developed BioAssay Ontology (BAO) and demonstrated its utility in classification and analysis of screening experiments and results15–17. We leveraged BAO and several other ontologies to develop the metadata terminologies required to integrate, interpret and analyze LINCS data.
Because of the scale and diversity of data generated, the LINCS consortium does not maintain a central repository containing all data. Towards building a distributed federated LINCS information infrastructure, we have developed data format specifications to facilitate exchange and integrated access of LINCS data across the consortium via web services.
In this paper we describe the metadata standards developed in the LINCS consortium with the goal to generate an integrated view across the diverse LINCS data resources as described above. The metadata standards and annotated datasets including cell lines, proteins and small molecules are freely available for download at the LINCS5-, LINCS Information FramEwork (LIFE)18-, Harvard Medical School (HMS) LINCS19 websites.
Data and Methods
A). LINCS assays and data
Data generated in the LINCS project are described on the LINCS website5 and links to individual LINCS Center websites therein. Briefly, data considered for the current version of metadata standards include transcript expression data, and biochemical and cell phenotypic responses obtained with a variety of assay technologies. Landmark gene (L1000) expression signatures were generated by using multiplex ligation-mediated amplification with the Luminex FlexMAP optically addressed and barcoded microsphere and a flow cytometric detection system20. The LINCS (L1000) along with original Connectivity Map (v1) data are available via The LINCS Connectivity Map Project (LINCS cloud)21. Kinase biochemical profiles are generated using the DiscoveRx KINOMEscan22 technology based on a competition binding assay and phage tag PCR amplification, or the KiNativ23 proteomics assay based on labeling active kinase Lys sites with biotinylated ATP or ADP probes and mass spectrometry detection. Cell-based assays are read out via imaging or bulk fluorescence measurement to quantify phenotypic responses. These data are available via the HMS LINCS Explorer24. LINCS data across the consortium can be queried and explored via the LIFE search engine25.
B). Metadata standards development
LINCS metadata standards were developed in the LINCS Data Working Group (DWG). We set up a DWG private Google website/wiki and used Google spreadsheets linked to the website to enable convenient sharing and collaborative authoring of the metadata standards with change control. The site and documents have 180 registered users, so a relatively large group has access to the DWG activities and provides input. The DWG documented various use cases related to research and tools development goals of the LINCS consortium. We prioritized an initial list of use cases that were relevant to guide the development of the herein reported metadata standards (see results). For each use case, the relevant LINCS assays (and result types) were listed and required parameters and annotations for screening result sets were determined as the basis for formalizing relevant and important metadata. We first focused on assay reagents (molecular entities and model systems) used to carry out LINCS assays, specifically: cells (primary cells and cell lines), proteins, small molecules, siRNA/shRNA, antibodies, and “other” reagents that do not fit any of the previous categories. We reviewed and summarized applicable elements from various minimum information standards9 including MIAME, MIACA, MIAPAR, MIAPE-MSI, MIAPE-MS, MIABE, MIQE, MIFlowCyt, MIARE. For each of the reagent categories, we created a Google shared spreadsheet that lists all metadata entities describing reagents of that category (compare Tables 1 and 2). Each metadata descriptor is captured in a separate row and includes several parameters including: ID, Name, how the descriptor relates to a specific (material) instance of the reagent (invariant canonical or batch-specific representation), description, importance (three levels: essential, recommended, optional), ontologies or other references to be considered for controlled vocabulary, URL of considered reference resources, and comments/additional notes (for development purposes). To determine suitable vocabularies for metadata entities where controlled terms are required, we reviewed available thesauri, taxonomies, and ontologies; we followed a similar approach as previously described in comparing domain coverage of ontologies17. In many cases, comprehensive vocabularies that cover important entities relevant to LINCS assays were not available, thus requiring an ongoing effort to curate this information from various sources and to build the controlled vocabularies within the LINCS project; these include cell line names/symbols, and unique labels for established small molecules such as approved drugs or probes developed in the NIH Molecular Libraries Program (MLP) and screened at LINCS. Each spreadsheet was developed iteratively to allow input from the DWG. Once the primary contributors agreed on the content, the document was released to the entire LINCS DWG for review and refinement. Once approved by the DWG, this version was frozen at the DWG site and publicly released at the LINCS website5, both in a structured format (Excel) and as Adobe PDF. It should be noted that the development of these standards is an ongoing process to accommodate new use cases and new LINCS data types. With the public release of a standards document we cloned a new editable document at the DWG site for the LINCS consortium to evolve and improve the standards, which, following the same process, will be released in the future.
Table 1.
Metadata categories required for the development of the metadata specifications for the LINCS assays.
| Metadata categories╲LINCSAssays | Apoptosis assay | Cue Signal Response assay | Cell viability assay | L1000 assay | KINOMEscan assay | Cell cycle state assay | KiNativ assay |
|---|---|---|---|---|---|---|---|
| Cell line | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | |
| Primary cell | ☑ | ☑ | |||||
| Protein | ☑ | ☑ | ☑ | ||||
| Antibody | ☑ | ☑ | |||||
| Small molecule | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ | ☑ |
| siRNA/shRNA | ☑ |
Table 2.
Selected metadata standards fields for LINCS reagent categories cell line, protein reagent, small molecule, siRNA/shRNA, and antibody.
| Annotation descriptor | Related to | Importance | Terminology / ontology | |
|---|---|---|---|---|
| Cell line metadata | ||||
| Identification | Cell line name | Canonical | 1 | Cell line ontology / LINCS database |
| Cell line ID | Canonical | 1 | ||
| Provider | Batch | 1 | ||
| Provider ID | Batch | 1 | ||
| Description | Organism | Canonical | 1 | NCBITaxon |
| Organ | Canonical | 1 | Uber Anatomy ontology | |
| Tissue | Canonical | 1 | ||
| Cell type | Canonical | 1 | Cell type ontology | |
| Growth property | Canonical | 1 | provider database | |
| Disease | Canonical | 1 | Human disease ontology | |
| Mutation | Canonical | 1 | COSMIC | |
| Genetic modification | Canonical | 1 | ||
| Recommended culture condition | Canonical | 2 | ||
| Verification profile Patch | Batch | 1 | ATCC; NIST; CLO | |
| Protein reagent metadata | ||||
| Identification | Protein name | Canonical | 1 | UniProt |
| Protein ID | Canonical | 1 | ||
| Gene symbol | Canonical | 2 | NCBI Geneρ | |
| Gene ID | Canonical | 2 | ||
| Provider | Batch | 1 | ||
| Provider ID | Batch | 1 | ||
| Description | Source (isolation, purification, synthesis) | Batch | 1 | |
| Source organism | Batch | 2 | NCBITaxon | |
| Modification (form) | Batch | 2 | ||
| Isoform detail | Canonical | 2 | UniProt | |
| Protein complex (Subunit information) | Canonical | 1 | Protein ontology | |
| Protein type | Canonical | 3 | UniProt | |
| Purity | Batch | 2 | ||
| Protein sequence | Canonical | 2 | UniProt / NCBI Protein | |
| Small molecule metadata | ||||
| Identification | Small molecule name | Canonical | 1 | DrugBank, PubChem, ChEMBL |
| Small molecule LINCS ID | Canonical | 1 | LlNCS / LIFE | |
| Provider patch | Batch | 1 | ||
| Provider ID | Batch | 1 | ||
| PubChem CID | Batch | 1 | pubChem | |
| ChEBI ID | Canonical | 2 | ChEBI | |
| InChI key | Canonical | 2 | ||
| SMILES | Canonical | 1 | ||
| Description | Target information | Canonical | 2 | UniProt |
| Molecular mass | Canonical | 1 | ||
| Molecular formula | Canonical | 2 | ||
| Salt information | Batch | 1 | ||
| Purity | Batch | 3 | ||
| Solubility patch | Batch | 3 | ||
| Purification method patch | Batch | 3 | ||
| siRNA/shRNA metadata | ||||
| Identification | Probe name | Canonical | 1 | NCBI Probe |
| Probe ID | Canonical | 1 | ||
| Probe type | Canonical | 1 | ||
| Provider | Batch | 1 | ||
| Provider ID | Batch | 1 | ||
| Description | Construct information | Canonical | 2 | |
| Target gene symbol | Canonical | 1 | NCBI Gene | |
| Target gene ID | Canonical | 1 | ||
| siRNA/shRNA sequence | Canonical | 2 | NCBI Probe | |
| Validation information | Batch | 2 | ||
| Antibody reagent metadata | ||||
| Identification | Antibody name | Canonical | 1 | NIF antibody registry |
| Antibody ID | Canonical | 1 | ||
| Provider | Batch | 1 | ||
| Provider ID | Batch | 1 | ||
| Description | Target protein name | Canonical | 1 | UniProt |
| Target protein ID | Canonical | 1 | ||
| Target gene symbol | Canonical | 2 | NCBI Gene | |
| Target gene ID | Canonical | 2 | ||
| Target organism | Canonical | 1 | NCBI Taxon | |
| Immunogen information | Canonical | 2 | provider database | |
| Antibody clonality | Canonical | 1 | NIF antibody registry | |
| Antibody isotype | Canonical | 1 | ||
| Source organism | Canonical | 1 | NCBI Taxon | |
| Antibody purity | Batch | 2 | ||
| Antibody specificity | Canonical | 3 | NIF antibody registry | |
| Antibody engineering | Canonical | 1 | ||
| Antibody type (primary or secondary) | Batch | 1 | ||
| Antibody labeling | Canonical | 1 | ||
C). Assay Simple Annotation Format (SAF)
We first developed the requirements of the data format to encode the annotation for LINCS assays and screening results. The primary purpose of SAF is to facilitate programmatic data exchange. We defined specific requirements: the data format must work seamlessly with Javascript and web services in particular Representational State Transfer (REST) Application Programing Interface (API); it should support a wide variety of applications; it must be easy to process and to write applications; it should be reasonably simple and human readable. JSON, a lightweight data-interchange format26, fits these requirements well and thus is a straight-forward choice (compared to XML for example). For each assay we worked out the fields, data types and content required to exchange the information and how they are linked to the assay metadata standards and controlled vocabularies. Specifically, BAO version 2.0 classes and the LINCS metadata standards were used to annotate specific assay types from HMS LINCS DB (http://lincs.hms.harvard.edu/db/) and SAF annotations were developed for each assay types by mapping HMS LINCS DB fieldnames to specific SAF elements, which rely on classes from the BAO and corresponding LINCS metadata representations. SAF includes separate sections for the assay annotations and the result sets, which are encoded as tag – value pairs (see results). SAF files thus represent a portable database-independent means of exchanging these annotations. Full SAF specifications are available at http://lifekb.org/index.php/dcc/SAF. We have made SAF-annotated screening results available through the HMS LINCS DB web services API; instructions and documentation are available at http://lincs.hms.harvard.edu/resources/software/hms-lincs-database/. SAF-annotated screening results are pulled from this service to upload results into the LIFE software system developed at the University of Miami25.
Results
A). LINCS use cases
One of the central goals of the LINCS project is to evolve more comprehensive systems-level views of normal and diseased states of cellular systems that can be applied for the development of new biomarkers and therapeutics. Towards that goal the LINCS consortium is cataloging, integrating and analyzing changes in gene expression and other cellular processes that occur as a response to different types of perturbations. Various LINCS consortium use cases were documented at the DWG site to coordinate the development of LINCS tools, including data integration and analysis, new algorithms, end user software tools and user interfaces. Simple use cases to assure LINCS datasets could be annotated to facilitate these LINCS goals and that were relevant to guide the development of the herein reported metadata standards include: i) identify screening model systems related to a specific disease or a disease group of interest or a particular tissue or organ of interest; ii) identify small molecule compounds active against a specific kinase target of interest; iii) query a broad kinase binding profile for a kinase inhibitor of interest; iv) identify small molecule compounds that inhibit cell growth in cell lines associated with a disease of interest; v) identify small molecule compounds with a protein target that corresponds to the gene target of a reference siRNA / shRNA; v) query gene expression signatures for a small molecule of interest (for example one that inhibits a kinase of interest).
Following the approach described in the methods section, we first reviewed the data types, detection technologies and assay formats currently used in LINCS assays. We then developed lists of metadata terms required to annotate LINCS assays and screening results, including recommended terminologies (vocabularies). We started with the following LINCS assay types: apoptosis assay, cell cycle state assay, small molecule binding assay (KINOMEscan and KiNativ), cell viability assay, and L1000 transcriptional response profiling assay. Table 1 shows the required metadata categories to be associated with these assay types. Figure 1 summarizes how the proposed reagent metadata standards relate to selected LINCS assays (and results) and other important concepts, such as protein and gene that are related to the mechanism of action of how a particular phenotypic response is mediated. Note that the same entity (e.g. protein) can have multiple distinct roles and these need to be separated in the metadata scheme. For example, protein kinases are specific, biochemically purified protein reagents in the KINOMEscan binding assay. In the broader context of all LINCS assays (most of which are cell-based) and datasets, protein kinases are conceptual targets of small molecules or antibodies. Thus, in our metadata standards, each protein reagent is directly related to a “parent” conceptual protein.
Figure 1.
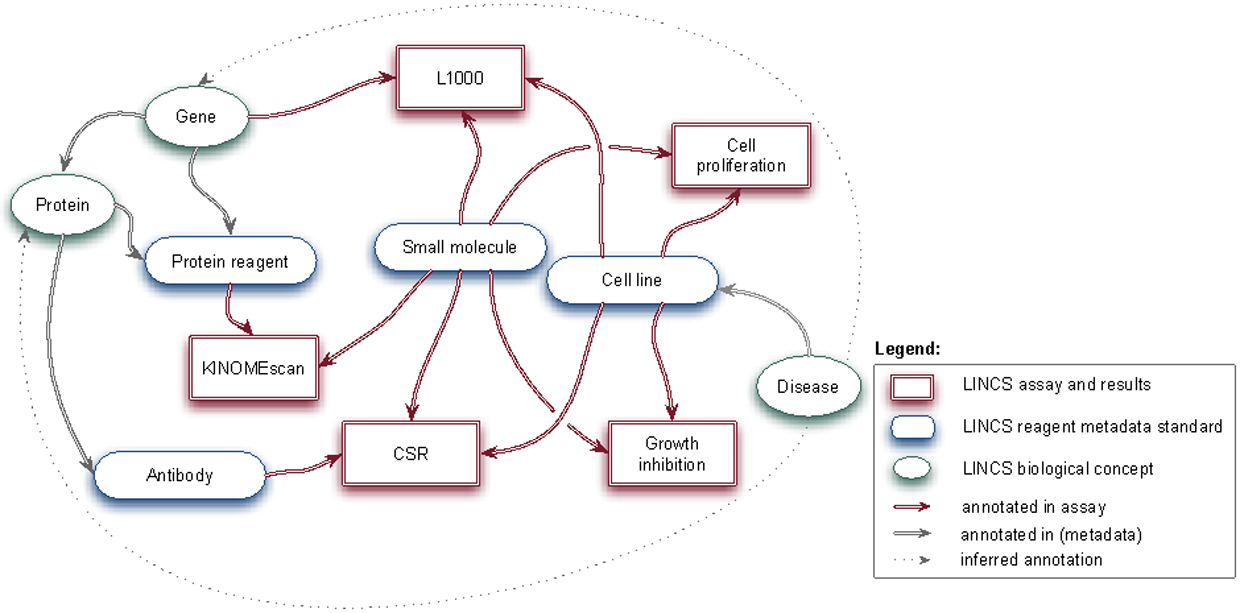
Illustration of how LINCS metadata standards relate to LINCS assays (and results) and biological entities.
B). Model vs. confounder metadata
In our approach, we have made a clear distinction between “model” metadata and “confounder” metadata. Model metadata are those required to understand, interpret, and meaningfully integrate experimental results. These include global identifiers for experimental reagents (e.g. key information about cell lines and small molecule perturbations) and critical experimental parameters (e.g. tested perturbagen concentrations and time points studied). Model metadata should be queryable in software tools and are often shown in published figures that illustrate important conclusions drawn from the data. Confounder metadata, on the other hand, include other details required to reproduce experiments, but that are less important for interpreting experimental results. Examples of confounder metadata include specific batch numbers for reagents, detailed descriptions of the experimental equipment used (model of a centrifuge used in a particular step in an assay protocol), etc. To describe LINCS assay protocols, for the most part, we make model metadata explicit, while other experimental details (confounder metadata) are captured as free text in standard operating procedures (SOP) implemented by the LINCS data production centers that describe how the assays are run. The specific parameters that are included in the model metadata are determined by use cases. This approach leaves the option to make additional metadata explicit at a later time (by curating the experimental procedures), should they be required for new use cases. Model metadata fields are required in our LINCS metadata standards. Confounder metadata (with some exceptions such as batch-specific identification of reagents) are considered optional.
C). Metadata specifications
The full LINCS metadata specifications are publically available at the LINCS project and LIFE websites (http://www.lincsproject.org/data/data-standards/, http://lifekb.org/index.php/data-standards). In the following we briefly describe these standards. Table 2 lists the most important metadata descriptors of each LINCS reagent category including the descriptor name, how it relates to a specific (material) instance of the reagent (invariant canonical or batch-specific representation), its importance level (1-essential; 2-recommended / if available; 3-optional), and – for controlled vocabulary – the recommended reference terminology / ontology. Resources for controlled vocabulary that are applied in the metadata standards are listed below. Metadata for each LINCS reagent category in Table 2 are separated into two sections: identification of the reagent and reagent-specific descriptors. For all details for each of the reagent categories, we refer to the full specifications.
i). Cell lines and primary cells
LINCS assays interrogate a variety of disease models. Cell lines are immortalized cells, while primary cells are mortal and generally undergo a finite number of cell divisions after which they reach senescence. To describe cell lines and primary cells, we incorporated some of the elements proposed in MIACA. The underlying theme among all cell types is their association with a tissue or organ from which the cells were derived. In many cases (especially with cell lines), the cells are also associated with a disease. We proposed explicit fields to describe the source (vendor or laboratory), origin (organism, organ and tissue), cell type (epithelial, neuronal stem cell, etc.), associated disease / disease model (e.g. type of cancer), growth properties (adherent or suspension), genetic modifications (transfection, transduction), inherent mutations (mutations in receptors, oncogenes, tumor suppressors), and culture conditions (culture medium and the medium components such as serum, growth factors). Cell line source and culture conditions are batch-specific information, while the others are canonical (do not change between batches). In addition, permanent cell lines require reporting of cell line authentication, such as short tandem repeat (STR) profiling, while primary cells require the passage number, donor details, such as the age, ethnicity, gender, etc.
ii). Small molecules
Small molecules are used as perturbagens in LINCS experiments. Some of the minimal information standards proposed in MIABE were included in our specifications, such as compound name and ID (PubChem CID, ChEBI ID), canonical structure representation (SMILES, InChI key), software used to generate a canonical structure representation, important molecular descriptors, chemical salt, etc. Known biological targets of small molecules should be annotated if known using standard symbols; this is in particular important for approved drugs or clinical compounds with a known mechanism as suggested in MIABE. Small molecule metadata also include substance-specific batch information, such as compound provider, salt form, molecular mass, purity, aqueous solubility. For FDA-approved drugs, we proposed to report additional information, such as drug indication and mechanism of action. If available, protein data bank (PDB) identifiers of corresponding target-small molecule co-crystal structures should also be reported.
iii). Protein reagents
Standardized description of protein reagents is critical to link results of different LINCS assay types. Protein reagents need to be identified in a manner that enables screening results associated with a specific protein reagent (e.g. KINOMEscan) to be linked with data obtained by other assays in which that protein participates as a (material) component, e.g. in a cell-based assay read out via the L1000 transcript profiling method (see Figure 1). Although this is a fairly obvious requirement, it is not trivial to implement, because a protein reagent expressed recombinantly is typically not the exact same entity or in the same state as its corresponding assay participant in a living cell (e.g. kinase domain binding assay vs. corresponding kinase occurring in a specific cell line used for a growth inhibition assay). In this first version of metadata standards we take a rudimentary approach. We use the UniProt accession and approved Gene symbol (NCBI Gene) and accession number to identify and reference proteins and their coding genes, respectively. Although we recognize limitations, for the purpose of our current simple use cases, this is sufficient. Linking protein and gene identifiers in addition is relevant to integrate RNAi reagent gene targets (see below). The recommended explicit fields for proteins include a standardized name, both for the protein and the gene that encodes it, source of protein (e.g., chemically synthesized, purified from natural source, recombinantly expressed), protein modifications (e.g., mutations, post-translational modification), protein purity, subunit information for components of a protein complex, isoform information (derived from either alternative promoter usage, alternative splicing, alternative translation initiation, frameshifting). We are currently working on a formal description of proteins that will allow ambiguity (more or less specific definition of proteins), because in some cases the exact entity and state of a protein reagent or model system participant is not definitively known (full length, functional domain, exact sequence, mutation, phosphorylation state, etc.).
iv). Inhibitory RNAs (siRNA, shRNA)
RNA interference is a standard methodology to transiently knockdown gene expression in living cells. This can be achieved using different types of small RNA molecules, including siRNA, shRNA and miRNA. Information that is relevant to identify and describe these perturbations include probe ID, name, source / provider, target gene symbol and accession number, sequence of the probe, and modifications to the probe (e.g., chemical modification) if any are specified.
v). Antibody reagents
Antibodies are extremely useful because of their high target specificity in detection of proteins, capture of proteins for isolation, purification and quantification, and selective inhibition of protein function (e.g. membrane receptor). Important metadata to be reported include a standardized name and ID of the antibody, identity of the target protein, target organism, information on the immunogen (name, source, modification of the protein/peptide), antibody clonality, antibody isotype, antibody purity, antibody specificity, and whether it was used as a primary or secondary antibody in an assay.
vi). Other reagents
This category serves to generically describe reagents that fall outside of any of the previously listed specific categories. An example is lipopolysaccharide, a component of the outer membrane of gram-negative bacteria that triggers an immune response similar to that initiated by a bacterial infection. Information that is relevant to be reported about these reagents include a standardized name and ID, provider information, purity, and source.
D). Resources and controlled vocabularies used in the metadata standards
BAO was initially developed to describe high-throughput assays and therefore already includes many terms and definitions for assay-related entities and concepts17. The Ontology for Biomedical Investigations (OBI)27 is an important mid-level ontology to integrate various domain-specific experimental ontologies. One of the main objectives of BAO was to describe screening outcomes (endpoints) and to enable classification and aggregation of these results by categories that relate to the biology (e.g. target) of the assay, the detection method, the assay design (how a signal is generated), and the model system. In contrast, OBI has a more operational focus (how is an investigation performed, how are the samples processed, etc.). However, the ontologies are not incompatible and we plan to align BAO with OBI to facilitate future integration with other biomedical investigations. We recently extended BAO to enable more flexible modeling of profile endpoints and signatures that are generated in LINCS assays (manuscript submitted). BAO is specifically used as a reference to the SAF (see below). We formally defined the LINCS assays in BAO; these include the KINOMEscan, KiNativ, cell viability, transcriptional response profiling, apoptosis, and cue signal response (CSR) assays; as such BAO serves as an important reference to the metadata standards, directly or via imported ontologies. To facilitate the unique identification of reagents and assay annotations, we recommend several other ontologies (Supporting table S1). Disease should be captured using standardized terminology from the Human Disease Ontology28. Organism names should be obtained from NCBI Organismal classification29; organ and tissue names from Uber Anatomy ontology30; cell type information from Cell type ontology31; cell line nomenclature from Cell Line Ontology (CLO)32 and cell line repositories. However, not all LINCS cell lines are in CLO and we are therefore developing a LINCS cell line database with links to CLO as applicable. Gene mutations inherent in cell lines can be obtained from Catalogue Of Somatic Mutations In Cancer (COSMIC) database from Sanger33; cell line authentication using short tandem repeat (STR) profiling from the cell line repositories, e.g., American Type Culture Collection (ATCC); information on subcellular components, molecular functions, and biological processes from GO13; protein name and ID from UniProt34; siRNA name, ID and sequence information from the NCBI Probe database35; and antibody information from the Neuroscience information framework (NIF) antibody registry (if available)36 and vendor catalogs.
E). The Assay Simple Annotation Format (SAF)
We developed the SAF specifications to facilitate data exchange between the HMS LINCS DB and LIFE via a web services API as described in methods. Here we describe the SAF, how it is used and its implementation in a LINCS publication web services API. It is a model that can be extended to the entire LINCS network and potentially beyond.
i). Description of the SAF format and content
The Simple Annotation Format (SAF) is a JSON26-based format for annotating and exchanging assay metadata and results. The chief goal of the SAF is to provide a simple, human readable format for representing and exchanging assay (experiment) data. JSON was chosen for encoding because it is simple to understand, easy for a human to read, ubiquitous and computationally easy to use (Java script, web services, with support in many applications) for data display and storage. Each SAF JSON object can be any subset of results generated by one assay (which is defined by its annotations); in practice it is an operational unit, such as one screening experiment. A SAF file (Supporting figure S1) consists of three logical sections: i) a header (red box); ii) a set of fields describing the scalar elements of the assay (the assay metadata) (blue box); and iii) a set of fields describing the repeating elements (data) of the assay (green box). There is no enforcement on the order of the elements on any of these sections. SAF fields primarily rely on concepts from the BAO (blue text) and LINCS metadata standards (green text), however BAO mappings are not required for all SAF fields. Some fields are used for housekeeping during data exchange (e.g., “endpointFile”, “uri”), while other fields may be outside the scope of the BAO (e.g., “recordedPlate”), but operationally relevant and therefore kept in the SAF. The field names from the HMS LINCS DB were mapped to the SAF elements and to BAO. In parallel, at the HMS LINCS DB, the field names from the SAF – BAO mapping were implemented as display names to achieve a consistent representation of the content across these resources.
Table 3 lists the SAF elements, descriptions and mappings to the HMS database and BAO. Data types include controlled vocabulary, free text, numeric value, and IDs with further differentiation of (LINCS) global and local (center- and / or batch specific) IDs. Table 3 also lists specific example annotations (tags and values) that apply to the KINOMEscan assay. It should be noted that many metadata annotations that refer to the assay are implicitly defined by the name KINOMEscan assay; this means they can be inferred based on the formal definition of the assay in BAO. For example the assay format, assay method, detection technology, etc. do not need to be explicitly annotated, because BAO defines all these details for the (KINOMEscan) assay. That also applies for the semantics of the reported endpoint ‘percent control’. In this particular case, BAO defines the KINOMEscan assay as a competitive binding assay (assay technology described above) that reports ‘percent control’ as the (normalized) percentage of substrate that remains bound to the kinase; 100 percent control thus is formally defined as no binding of the screened compound to the kinase, and vice versa, 0 percent control means 100 percent compound binding. Because compounds bind at the ATP site (competitive with the substrate), this can also be interpreted as 100 percent inhibition of the kinase.
Table 3.
List of the SAF elements, descriptions, mappings and data types with examples that apply to the KINOMEscan assay.
| SAF Element | Element Description | SAF Example | HMS mapping | BAO mapping | Data type |
|---|---|---|---|---|---|
| safVersion | Version of SAF, tightly bound to ontology annotations | “safVersion”:“0.1”, | Numeric value | ||
| bioAssay | Bioassay is defined by the assay (design) method, detection, biology / target, format perturbagen, and reported endpoint | “bioAssay”:“KINOMEscan”, | bioassay | bioassay | controlled vocabulary |
| hmsDatasetiD | Identification of the bioassay | “hmsDatasetiD”:“20020”, | HMS Dataset ID | has bioassay ID | local ID |
| screeningLablinwestigator | Screening facility laboratory investigator | “sereeningLabInvestigator”: “Qingsong Liu”, | Screening Lab Investigator | has screening lab Investigator | free text |
| screeningPrincipalInvestigator | Screening facility principal investigator or head of the laboratory | “screeningPrincipalInvestigator”: “Nathanael Gray”, | Screening Principal Investigator | has screening principal Investigator | free text |
| assayProtocol | Methodology to perform a bioassay | “assayProtocol”: “1 T7 kinase-tagged phage strains are grown in parallel in 24-well or 96 well block in… | Assay Protocol | has assay protocol | free text |
| assayProtocolReference | Reference (publications, urls…) for the assay protocol | “ assayProtocolReference”: “KINOMEscan website: http://kinomescan.com/Techno1ogy/How-it-Works… | Assay Protocol Reference | has PMID | global ID |
| screeningFacility | Screening facility where the assay was performed | “ sereeningFacility”:“HMS”, | research institute | controlled vocabulary | |
| assayDescription | Background information to perform the bioassay | “assayDescription”: “The KINOMEscan assay platform is based on a competition binding assay that is… | Assay Description | has assay narrative | free text |
| assayTitle | Name of a bioassay | “assayTitle”:“Sorafenib KINOMEscan”, | AssayTitle | has assay title | free text |
| smCenterCompoundID | Center specific compound ID, for the parent structure. | smCenterCompoundID:“10008”, | Small Mol HMS LINCS ID | local ID | |
| smSalt | Reference to counter-ions and other addends present in the compound’s formulation | smSalt:“101”, | Salt ID | local ID | |
| smCenterSamplelD | Sample ID of the tested compound, referring to of the tested sample; assigned after local registry of the compound (center specific) | “smCenterSamplelD”:“10008–101-1”, | Small Mol HMS LINCS ID | local ID | |
| smLincsID | Small molecule LINCS ID | smLincsID: “LSM- 1008”, | LINCS ID | has small molecule ID | global ID |
| smName | The primary name for the (parent) compound. | smName: “BAY- 439006”, | SM Name | small molecule | controlled vocabulary |
| ppName | The primary name of the protein. | ppName: “ABL1(E255K) phosphorylated”, | Protein Name | protein | controlled vocabulary |
| ppCenterProteinID | LINCS center-specific protein ID | ppCenterProteinID:“200004”, | HMS Protein ID | has UniProtlD | global ID |
| concUnit | Standardized quantity in which the concentration is expressed/measured | datapointName: “concUnit”, “datapointValue”: “uM” | Concunit | concentration unit | controlled vocabulary |
| assayCompoundConcentration | The concentration of the perturbagen used in the assay to elicit the biological effect or perturbation | datapointName:” assay CompoundConcentration”, “datapointValue”: “10” | Assay compound conc; | has concentration value | numeric value |
| percentControl | Percent control is the response relative to a reference state, typically to a high control | datapointName:“percentControl”,“datapointValue”: “100” | % Control | has percent response value | numeric value |
SAF has also been implemented for LINCS apoptosis, cell cycle state, cell growth inhibition and KiNativ assays.
ii). Implementation of SAF as LINCS Publication Service (LPS)
The SAF provides a mechanism to minimally describe assay and screening result information so that it can be exchanged between screening centers, or accessed programmatically. We have started to use the SAF to annotate LINCS assays so that they can be easily indexed and made searchable by the LIFEwrx KnowledgeBase. The LIFEwrx KnowledgeBase is a searchable repository of LINCS assay data linked to the LIFE ontology and accessible through an easy to use web-based user interface (Figure 2)25. Previously, data were populated in LIFEwrx by an ETL-like process in which data were loaded from the LINCS centers into a staging database where standardization was done. The data were then annotated using the metadata standards, which enriches the information by linking associated concepts (e.g., disease names and categories). All of this information was made searchable and viewable through the search application. Annotating assays using the SAF simplifies this pipeline, because assay information is already in a standard format and linked to ontology concepts (Figure 2). The SAF annotated assays are made available through the HMS LINCS DB web services API, which serves as a LINCS publication service (LPS). Data from the service can be pulled directly by the LPS-driven LIFEwrx ingest pipeline with no special processing (see methods for access and references to SAF and API specifications).
Figure 2.
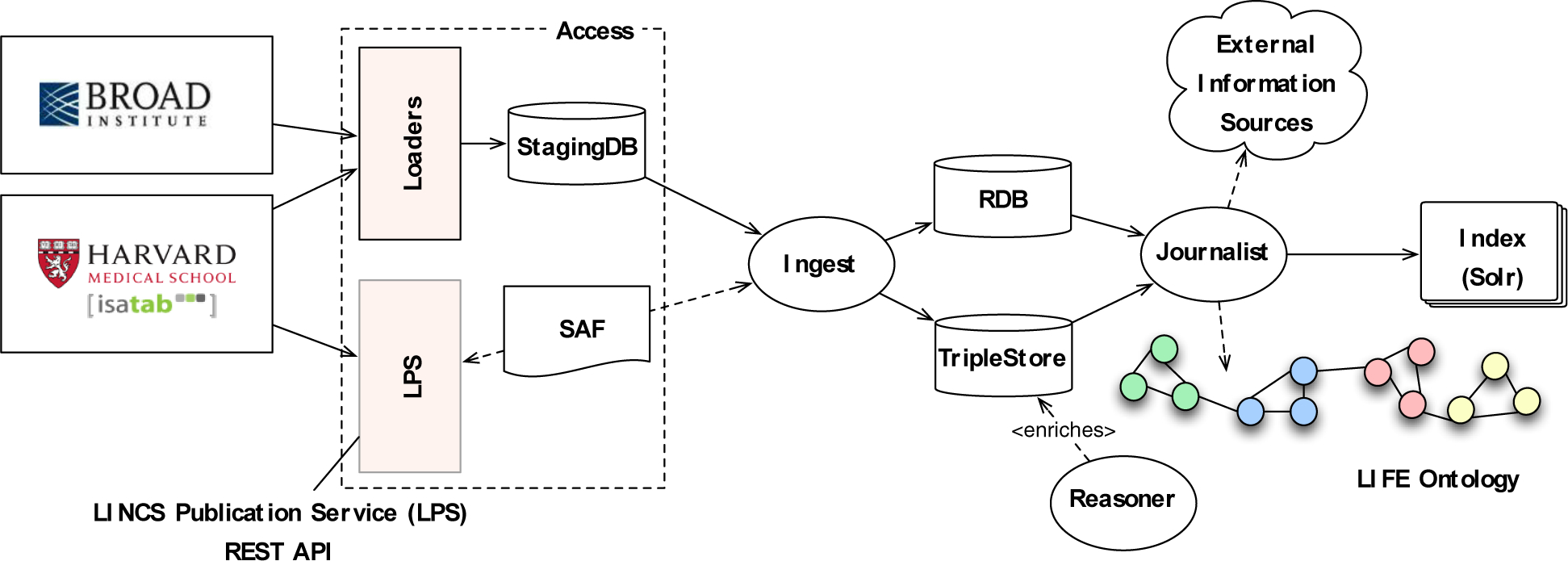
Integration of HMS LINCS data into LIFE via the LINCS Publication Service (LPS) REST API that leverages the SAF. ISA-Tab has been used in a pilot project to annotate some LINCS data at HMS and SAF is used facilitate programmatic access via the LPS.
F). Annotating datasets applying LINCS metadata standards
Applying the metadata standards, we have systematically curated and annotated cell lines, small molecules, and proteins used in LINCS assays. Representative examples for cell lines, proteins and small molecules tested in LINCS assays are shown in Supporting tables S2, S3, and S4, respectively. We describe the currently available resources of annotated cell lines, proteins and small molecules below.
i). Cell line annotation and linkage to disease and tissue
Established cell lines are powerful high-throughput screening disease model systems. This is in particular the case in cancer research; for example the NCI60 screen for effects on viability of multiple cancer-derived cell lines is routinely run on promising lead compounds. To facilitate the integration and analysis of large-scale cell-based screening profiles such as those generated at LINCS, we systematically annotated cell lines with controlled terms identifying associated organs, diseases and mutations leveraging the Human Disease Ontology, the organ Uber Anatomy Ontology; example annotations are shown in Supporting table S2. We initially curated and annotated 567 cell lines. Figures 3 and Supporting figure S2 illustrate the representation of the different types of cancers and their organs of origin among these cell lines. The mutation and disease sub-categorization of different ovarian cancer cell lines tested at LINCS were annotated from COSMIC33 and Human Disease Ontology (Supporting table S5).
Figure 3.
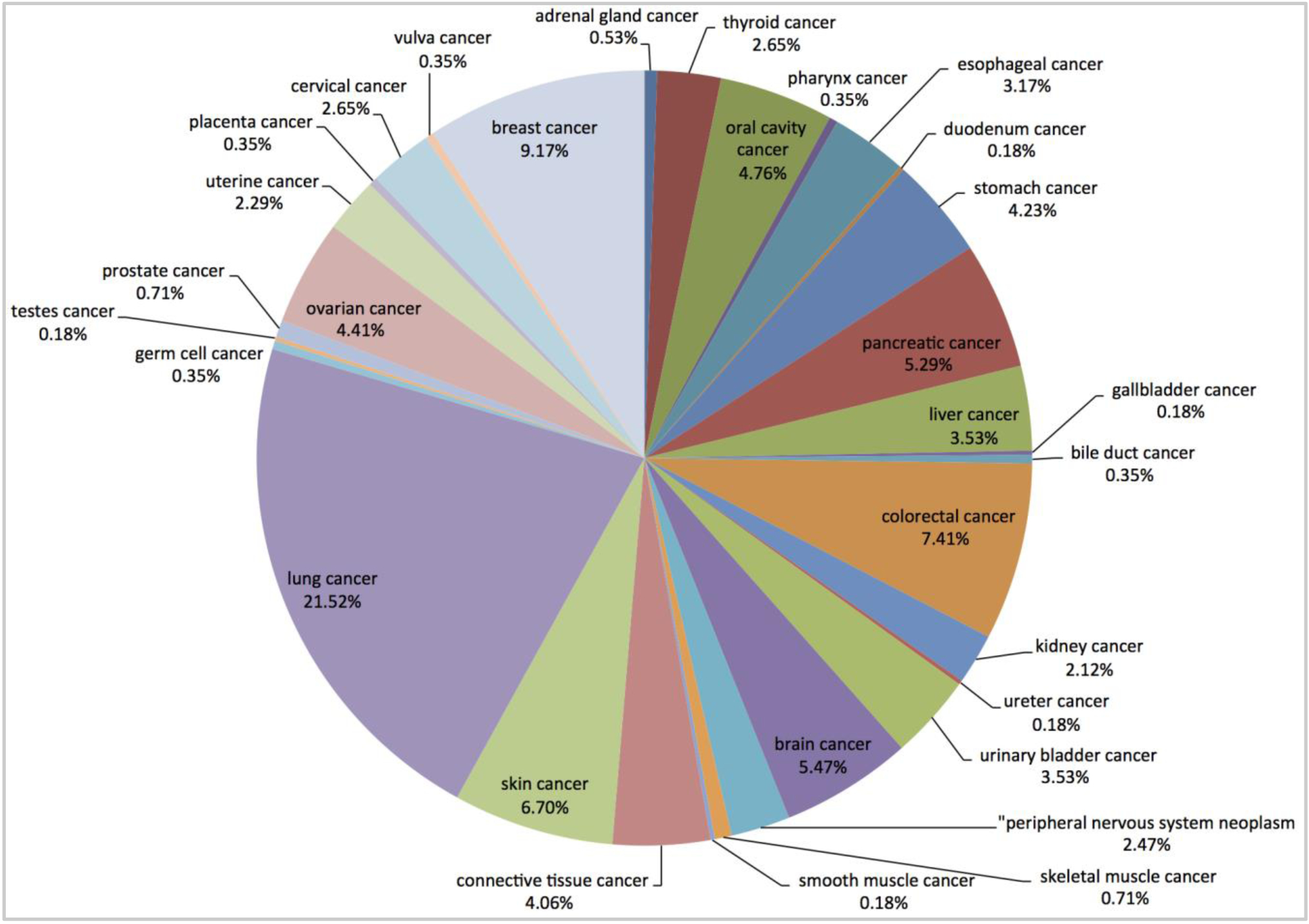
Representation (percentage) of the different types of cancers among cell lines tested in the LINCS assays.
A list of all (>1,000) annotated cell lines screened at the LINCs consortium is available via the HMS LINCS DB at http://lincs.hms.harvard.edu/db/cells/. Cell lines can also be queried and explored by disease, tissue or assay results via the LIFE software25.
ii). Protein annotations
Deregulation of protein kinases is a hallmark of many diseases, including cancer. LINCS addresses the role of protein kinases using several assay types where activity is either directly measured in biochemical assays (KINOMEScan) or by assessing phenotypes resulting from inhibition in cell-based assays (CSR, apoptosis, cell viability assays, transcriptional response profiling). Protein name, ID, alternate names, posttranslational modification, and mutation status were annotated using standardized terminology from UniProt, NCBI/Protein and Protein Ontology (example shown in Supporting table S3).
A list of proteins reagents (>1,000) is available via the HMS LINCS DB at http://lincs.hms.harvard.edu/db/proteins/ and curation of this list is ongoing. Kinase proteins including phosphorylation status and mutations can also be queried and explored via a kinase domain ontology in the LIFE software25.
iii). Compound annotations
Small molecules tested in the LINCS assays include approved drugs, clinical kinase inhibitors, MLP probes and various other screening compounds. Integration of data from different assays and external resources requires a unique identification of small molecules. We used PubChem CIDs and we annotated the compounds with additional details curated from various sources including DrugBank, PubChem, the NCBI MLP probe reports, the NCATS pharmaceutical collection (NPC), and the Protein Data Bank (PDB). Example records are shown in Supporting table S4.
We made the annotations for LINCS small molecules (> 4,000) available at the LIFE KB website (http://lifekb.org/index.php/data-standards). The list of compounds can also be obtained from LINCS HMS DB (http://lincs.hms.harvard.edu/db/sm/). Compound information can be queried, browsed and downloaded via LIFE25.
Discussion
Formal specifications of metadata are required to make the biological and methodological context of the assays and results explicit. Because of the diversity of methods and data types generated at LINCS, such specifications are critical to generate integrated and interpretable views of diverse LINCS results, and also to link to external resources, such as small molecule activity data in PubChem, ChEMBL, drug information in DrugBank, pathway information, disease data, etc. Here we developed metadata specifications for assays and screening results produced in the LINCS consortium. We focused on the model metadata needed to interpret and link assays and results. Guided by prioritized use cases we determined the required types of biological entities and concepts and the corresponding specifications to uniquely identify each individual entity and to relate them while not impeding human parsing of the data (common names, descriptions, etc.). We reviewed existing minimum information specifications and available established resources for controlled vocabularies. Although these have been a useful starting point, we determined that the LINCS project requires specific metadata standards to fulfill the current and envisioned future use cases. Comprehensive minimum information specifications for the purpose of replicating experiments were not practically applicable given limited data curation resources and the focus on model metadata. Vocabulary resources (including ontologies) to describe many of the important LINCS biological entities and concepts were still lacking. We first developed the required metadata specifications in a smaller core group and then passed them to a larger group at LINCS for review and approval before their public release. We have demonstrated the applicability of these metadata standards by annotating LINCS assays and results. We have made publically available information on over thousand cell lines with detailed annotations including disease and tissue, on over thousand LINCS protein reagents, and on several thousand compounds including many clinical kinase inhibitors and drugs. The various biological entities and concepts and their associated screening assays and results can be queried and browsed based on these metadata in the LIFE software system25. Use cases to develop the LINCS specifications range from relatively simple queries to more complex analyses, and also include the development of software tools and user interfaces to query, explore, and analyze LINCS data. We have already implemented a variety of useful functionality leveraging these metadata standards in the LIFE search engine25.
To facilitate the programmatic exchange of metadata-annotated screening results, we developed specifications for an assay Simple Annotation Format (SAF). The ISA-Tab format was used at HMS to capture important metadata at the time of running assay experiments. Metadata and screening results are deposited to the HMS LINCS DB. SAF is the native format of the LPS REST API, which publishes this information for programmatic access and further processing by other systems such as LIFE (Figure 2). We have described several of the LINCS assays using these SAF specifications and implemented LINCS publication web services to access these data programmatically. This mechanism is also used to upload data into the LIFEwrx knowledgebase. We have shown several examples of curated annotations using the metadata specifications for cell lines, proteins, and compounds, and how an assay is described in SAF; the full lists and details are available at the LINCS5, LIFE18, and HMS LINCS19 websites.
As an example of linking results from different LINCS assays, we illustrate biochemical, cell growth inhibition, cell cycle state (mitosis / apoptosis), and transcriptional responses of a novel Plk-1 inhibitor, BI-2536, that has been shown to inhibit tumor growth in vivo37, has a modest efficacy and favorable safety in relapsed non-small cell lung cancer38 and is also in phase I study in advanced solid tumors39. The presented standards to annotate cell lines and small molecules enable integration of relevant data. In this example, the cell growth inhibition data of a non-small cell lung carcinoma cell line, A549, indicate the cell survival rate of 30% (at the BI-2536 concentration of 0.5 μM) while the KINOMEscan inhibition data confirms its activity in vitro with the Plk-1 inhibition of 81% (at the concentration of 10 μM). Tang et al.40 identified an unexpected bell-shaped dose-response of BI-2536 in the mitosis / apoptosis assay and suggest that low/medium concentrations of the drug inhibit the primary target (Plk1) in its function in promoting progression through mitosis and cells arrest in mitosis and from there move into apoptosis. Meanwhile, medium/higher concentrations of the drug might block mitotic entry altogether, which can protect from cytotoxic effects of antimitotic drugs. At highest concentrations, cytotoxicity due to off-target inhibition of other kinases is seen and the apoptosis/death curve rises again as mitotic index falls. Off targets candidates can readily be identified via the KINOMEscan results for BI-2536. Similarly, gene expression results for BI-2536 in A549 cells and other cell lines can readily be queried and integrated with these results. The utility of the metadata standards is illustrated by their implementation in the LIFE search engine25. For example a simple query of “BI-2536” (LSM-1041) returns various types of LINCS data for this compound, including L1000 transcriptional response, cell cycle state assay, cell growth inhibition, and KINOMEscan results.
During the development of the metadata standards presented here, and in particular when applying them to curate and annotate cell lines, proteins and small molecules, it became apparent that such an effort requires significant resources, which are easy to underestimate. Judged by previous attempts, biocuration and systematic annotation of biological data have not been perceived as high-priority efforts in the community and as a result often appear underresourced41. It is therefore particularly important to optimize and prioritize minimum annotations that enable the scientific use cases and software functionality that involve integrated data views and linking to external information. Here we have developed and applied such minimum annotations in one of the first attempts to describe and make public large diverse datasets reporting biochemical and phenotypic readouts in addition to gene expression data; this is a major goal for the LINCS project. The development of metadata specifications continues to accommodate new use cases, data analysis algorithms, and software tools. It should be noted that the current metadata specifications already enable more complicated use cases that were not originally considered, such as associating kinase targets and genes with diseases. Although causal associations cannot be directly inferred from the LINCS data, the metadata standards in principle include the required details to perform such analyses; for example linking kinase targets (from KINOMEscan) and diseases (linked to cell lines tested in growth inhibition assays) based on the activity of small molecules tested in both assays (compare Figure 1, inferred relations).
In conclusion, the LINCS metadata and SAF specifications facilitate various use cases involving data integration, analysis, development of software tools and programmatic data exchange across a variety of assay types, screening results and external biomedical data. We anticipate that the metadata specifications, the SAF, and annotated cell lines, proteins and small molecules will be useful beyond the LINCS project. All developed resources in this project are freely available.
Supplementary Material
Acknowledgements
This work was funded by the LINCS project grants 1U01HL111561, 3U01HL111561-01S1, and 3U01HL111561-02S1, U54HG006097, U54 HG006093.
References
- 1.The Cancer Genome Atlas (TCGA) http://cancergenome.nih.gov.
- 2.Bernstein BE; Birney E; Dunham I; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Cancer Target Discovery and Development (CTD2) http://ctd2.nci.nih.gov/.
- 4.Roy A; McDonald PR; Sittampalam S; Chaguturu R Open Access High Throughput Drug Discovery in the Public Domain: A Mount Everest in the Making. Curr. Pharm. Biotechnol 2010, 11, 764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Library of Integrated Network-based Cellular Signatures (LINCS) http://lincsproject.org/.
- 6.Orchard S; Al-Lazikani B; Bryant S; et al. Minimum information about a bioactive entity (MIABE). Nat. Rev. Drug Discov 2011, 10, 661–9. [DOI] [PubMed] [Google Scholar]
- 7.Minimum Information About an RNAi Experiment(MIARE) http://miare.sourceforge.net/HomePage.
- 8.Bourbeillon J; Orchard S; Benhar I; et al. Minimum information about a protein affinity reagent (MIAPAR). Nat. Biotechnol 2010, 28, 650–3. [DOI] [PubMed] [Google Scholar]
- 9.Taylor CF; Field D; Sansone SA; et al. Promoting coherent minimum reporting guidelines for biological and biomedical investigations: the MIBBI project. Nat. Biotechnol 2008, 26, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.BioSharing. http://biosharing.org/.
- 11.Sansone S-A; Rocca-Serra P; Field D; et al. Toward interoperable bioscience data. Nat. Genet 2012, 44, 121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medical Subject Headings (MeSH) http://www.nlm.nih.gov/mesh/).
- 13.Gene Ontology Consortium The Gene Ontology (GO) project in 2006. Nucleic Acids Res 2006, 34, D322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harland L; Larminie C; Sansone S-A; et al. Empowering industrial research with shared biomedical vocabularies. Drug Discov. Today 2011, 16, 940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schürer; Vempati U; Smith R; Southern M; et al. BioAssay Ontology Annotations Facilitate Cross-Analysis of Diverse High-Throughput Screening Data Sets. J. Biomol. Screen 2011, 16, 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Visser U; Abeyruwan S; Vempati U; et al. BioAssay Ontology (BAO): a semantic description of bioassays and high-throughput screening results. BMC Bioinformatics 2011, 12, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vempati; Przydzial MJ; Chung C; et al. Formalization, annotation and analysis of diverse drug and probe screening assay datasets using the BioAssay Ontology (BAO). PLoS One 2012, 7, e49198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LINCS Information FramEwork (LIFE) http://lifekb.org/.
- 19.Harvard Medical School LINCS. http://lincs.hms.harvard.edu/.
- 20.Peck D; Crawford ED; Ross KN; et al. A method for high-throughput gene expression signature analysis. Genome Biol. 2006, 7, R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The LINCS Connectivity Map Project. http://lincscloud.org/.
- 22.Fabian MA; Biggs WH 3rd; Treiber DK; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- 23.Patricelli MP; Szardenings AK; Liyanage M; et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- 24.HMS LINCS Explorer. http://lincs.hms.harvard.edu/explore/.
- 25.LINCS Information FramEwork (LIFE) Search Engene. http://life.ccs.miami.edu/.
- 26.JavaScript Object Notation. http://www.json.org/.
- 27.Brinkman RR; Courtot M; Derom D; Fostel JM; He Y; Lord P; Malone J; Parkinson H; Peters B; Rocca-Serra P; Ruttenberg A; Sansone SA; Soldatova LN; Stoeckert CJ Jr.; Turner JA; Zheng J Modeling biomedical experimental processes with OBI. J. Biomed. Semant 2010, 1 Suppl 1, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du P; Feng G; Flatow J; Song J; Holko M; Kibbe WA; Lin SM From disease ontology to disease-ontology lite: statistical methods to adapt a general-purpose ontology for the test of gene-ontology associations. Bioinformatics 2009, 25, i63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The NCBI Taxonomy Homepage. http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/.
- 30.Mungall CJ; Torniai C; Gkoutos GV; Lewis SE; Haendel MA Uberon, an integrative multi-species anatomy ontology. Genome Biol. 2012, 13, R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meehan TF; Masci AM; Abdulla A; Cowell LG; Blake JA; Mungall CJ; Diehl AD Logical development of the cell ontology. BMC Bioinformatics 2011, 12, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarntivijai S; Ade AS; Athey BD; et al. The Cell Line Ontology and its use in tagging cell line names in biomedical text. AMIA Annu. Symp. Proc 2007, 1103. [PubMed] [Google Scholar]
- 33.Catalogue Of Somatic Mutations In Cancer (COSMIC) http://www.sanger.ac.uk/genetics/CGP/cosmic/.
- 34.UniProt. http://www.uniprot.org/.
- 35.NCBI Probe. http://www.ncbi.nlm.nih.gov/probe.
- 36.NIF Antibody Registry. http://antibodyregistry.org/.
- 37.Steegmaier M; Hoffmann M; Baum A; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol 2007, 17, 316–22. [DOI] [PubMed] [Google Scholar]
- 38.Sebastian M; Reck M; Waller CF; et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J. Thorac. Oncol 2010, 5, 1060–7. [DOI] [PubMed] [Google Scholar]
- 39.Frost A; Mross K; Steinbild S; et al. Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr. Oncol 2012, 19, e28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang Y; Xie T; Florian S; Moerke N; et al. Differential determinants of cancer cell insensitivity to antimitotic drugs discriminated by a one-step cell imaging assay. J. Biomol. Screen 2013, 18, 1062–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazumder R; Natale D; Julio J; et al. Community annotation in biology. Biol. Direct 2010, 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.