

Abstract
Knowledge of the underpinnings of cancer initiation, progression and metastasis has increased exponentially in recent years. Advanced “omics” coupled with machine learning and artificial intelligence (deep learning) methods have helped elucidate targets and pathways critical to those processes that may be amenable to pharmacologic modulation. However, the current anti-cancer therapeutic armamentarium continues to lag behind. As the cost of developing a new drug remains prohibitively expensive, repurposing of existing approved and investigational drugs is sought after given known safety profiles and reduction in the cost barrier. Notably, successes in oncologic drug repurposing have been infrequent. Computational in-silico strategies have been developed to aid in modeling biological processes to find new disease-relevant targets and discovering novel drug-target and drug-phenotype associations. Machine and deep learning methods have especially enabled leaps in those successes. This review will discuss these methods as they pertain to cancer biology as well as immunomodulation for drug repurposing opportunities in oncologic diseases.
Keywords: drug repurposing, drug discovery, machine learning, deep learning, artificial intelligence
Introduction
In past years, medical oncology has witnessed an unprecedented explosion in the understanding of cancer pathophysiology and pathogenesis. With the advancement of next generation sequencing technologies such as single-cell RNA sequencing, we are better equipped to explore and model complex phenomen such as cancer heterogeneity, resistance and etiologies at a granular level. Moreover, the establishment and expansion of multi-institutional and large-scale biospecimen collection and bioinformatics initiatives, such as the Cancer Genome Atlas (TCGA), have allowed for the aggregation, curation and analysis of an unprecedented amount patient-derived data that has led to the identification of novel therapeutic targets as well as the implication of well-known targets in new disease areas.
In spite of this extraordinary growth in the cancer biology field, progress in the area of drug discovery remains stagnant, still plagued with long development time to market and exorbitantly high costs despite the systematic implementation of high-throughput screening technologies. To date, bringing a drug to market takes about a decade with research and development (R&D) costs reaching approximately US $2.8 billion (1). Candidate drugs may fail at many points along the drug development pipeline due to numerous reasons such as poor pharmacokinetics, toxicity or lack of clinical efficacy.
A promising solution to the considerable drug development challenges of novel compounds is the use of existing drugs for the treatment of new diseases. Approved drugs have undergone all phases of clinical trials in order to reach the market and thus have a known and accepted safety profile. If a new clinical indication for an approved drug is suggested, that drug may re-enter the clinical trial process at Phase II thus substantially reducing the R&D risk, time and cost (2).
In the past few years, drug repurposing research has benefited greatly by the systematic adoption of computational strategies spanning a large area of unique methodologies and approaches. Molecular modeling of therapeutic protein targets allows for the understanding of the structural biology as well as high throughput “virtual screenings” to identify novel drug candidates. Advances in machine learning and artificial intelligence(in particular deep learning) provide new insights into how drugs bind to targets and how their physicochemical properties relate to phenotypic changes. In addition, the utilization of such methods also help identify novel anti-cancer targets from the large-scale cancer datasets already collected by multiple initiatives. As the amount of chemical and bioactivity data grows due to the adoption of high-throughput and multi-omics drug profiling assays, these methods are poised to make substantial contributions in cancer drug discovery. Furthermore, the increased accessibility of these public dataset collections further facilitates the potential of computational approaches. These approaches need not be limited in using experimental and biological datasets. The utilization of clinical datasets such as EHR (electronic health records) has also shown promising results. In this review, we discuss the current computational strategies used for drug repurposing in oncology with an emphasis on machine and deep learning.
Structure-Based Machine Learning
Structure-based virtual screening methods have augmented the drug discovery process since the inception of in-silico molecular modeling (Figure 1). In the most general sense, structure-based virtual screening aims to prioritize small molecules potentially targeting a protein target of interest, subsequently leading to the reduction of experimental costs and increase of successful outcomes. The typical approach includes the prediction of the binding affinity (or related affinity scores) between small molecules and the target of interest. This binding affinity/score is then used to rank-order compounds for prioritization, and the general rule is that the greater the predicted affinity the greater the likelihood of a true small molecule-target interaction.
Figure 1. Overview of structure-based machine learning strategies for identification of drug-target interactions.
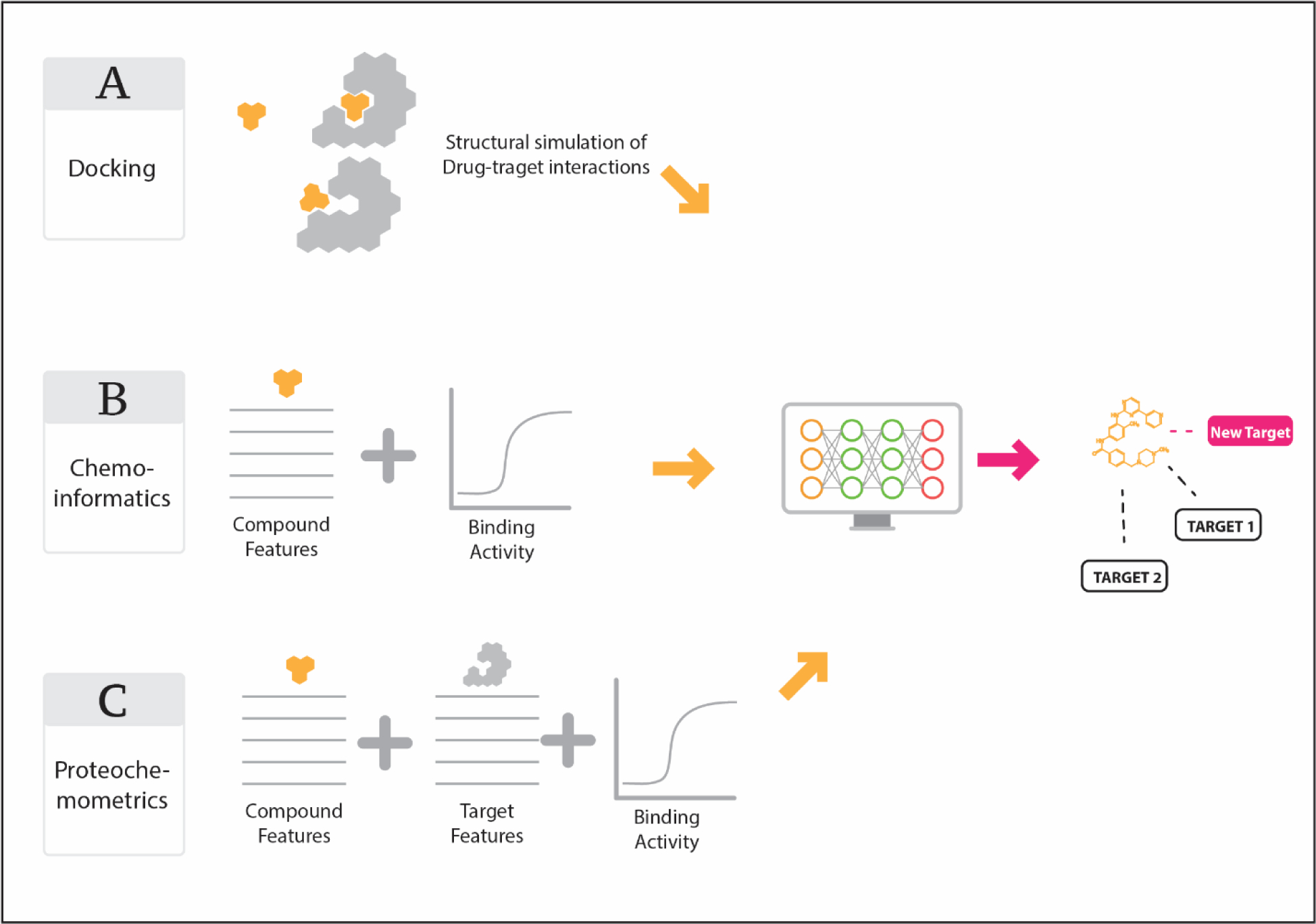
(A) Docking of small molecules into binding sites of three-dimensional protein target models. (B) Cheminformatics methods (i.e. QSAR) where chemical descriptors of small molecules are leveraged to create models relating descriptors to quantifiable target binding biological endpoints (i.e. binding vs non-binding, IC50, etc.). (C) Proteochemometrics similar to (B) but with additional descriptors derived from the protein target such as amino acid sequence.
Molecular docking is one of the most widely used structure-based techniques for predicting and prioritizing drug-target interactions. Briefly, the interactions between a drug and a target’s binding sites are being assessed and quantified calculating the free energy of binding (ΔG), where lower or more negative ΔG values correspond to greater binding affinity. A big advantage of molecular docking methods is the interoperability of the predicted results as ΔG values are standardized and can be compared across millions of drugs that can be virtually screened. To calculate the ΔG, docking programs utilize scoring functions, which are mathematical formulas that incorporate physicochemical parameters of the drug and protein. Multiple docking programs exist that use a variety of scoring functions such as Glide (4) and AutoDock (5). The differences in scoring functions arise due to which parameters are used and how they are related to binding affinity. Overall, docking has proved to be a very effective drug repurposing tool for many diseases such as HIV (7), Dengue virus (8) and multiple cancer types (9). One such recent example is the study from Hafeez et al. where they used docking approaches to reveal how ormeloxifene is able to modulate the epithelial to mesenchymal transition (EMT) process implicated in cancer development and progression (10). They showed that ormeloxifene, an anti-contraceptive selective estrogen receptor modulator, to reduce prostate cancer growth through inhibition of beta-catenin signaling. Using docking, they revealed it could bind to beta-catenin and GSK3beta, thus explaining its unique effect on the EMT process, thus rendering it a promising drug repurposing pursuit for prostate cancer especially given its favorable safety profile.
Molecular Docking using Machine Learning
While docking has potential for finding new targets for drugs, the major inherent limitation at this time is the accuracy of the scoring functions used. As previously stated, existing classical scoring functions are numerous and based on a presumptive functional relationship between binding affinity and the structure of drug-target complexes. This may lead to a considerably high false positive rate (11) and thus, utilizing new approaches, such as machine learning, are essential for improving the docking accuracy. By leveraging machine learning, scoring functions can be substantially improved in an unbiased way since drug-target relationships are inferred directly from the training data and are not influenced by presumptive relationships. Moreover, the predictive models, can be used to identify the key features of the datasets that contribute the most to the binding affinity and therefore provide a better understanding of the forces that drive the drug-target interactions.
The adoption and use of deep learning methods for molecular docking has been extremely successful mainly due to the accessibility to deep learning computational frameworks such as Tensorflow. Typically, these methods are applied to post-docking drug-target complexes in order to more accurately predict binding poses and drug activity. Such methods play a pivotal role in drug repurposing, as existing drugs can be virtually screened quickly across a large number of new clinically relevant targets. In such a study, Wallach et al. utilized Convoluted Neural Networks (CNN) to develop AtomNet (12). AtomNet utilizes a three-dimensional (3D) grid representation placed on drug-target complex to extract structural features such as atom types and structural protein-ligand interaction fingerprints (SPLIFs), which are then unfolded into a one-dimension floating point vector. The training set was obtained from the scPDB database (13), which includes annotated 3D crystal structures of true drug-target interactions. AtomNet performed significantly better than commonly used docking algorithms such as Surflex-Dock (14) and DOCK (15) in prioritizing true active compounds from a set of decoys. Similarly, Ragoza et al. employed a CNN to 3D representations of drug-target interactions (16). Their model was able to outperform AutoDock Vina (5) in discriminating between correct and incorrect binding poses as well as between active and inactive compounds. The aforementioned CNN-driven deep learning models were tuned to a dichotomous outcome (i.e. active versus inactive) and, therefore, are inherently limited in that they are unable to provide information about the strength of a drug-target interaction. More recently, Stepniewska-Dziubinska et al. developed Pafnucy, also a CNN based on 3D convolution, to estimate binding affinities of drug-target complexes. Pafnucy had outperformed all state-of-the-art functions when evaluated against the CASF-2013 “scoring power” benchmark (17). Nguyen et al. were also successful in applying deep learning to enhance docking scoring functions for the prediction of binding poses, binding affinities and free energies of binding for a variety of cancer-related targets (18).
Ligand-Based Cheminformatics Modeling Using Machine Learning
A second approach for elucidating new drug-target interactions is that of ligand-based cheminformatics methods. In contrast to molecular docking methods, ligand-based cheminformatics methods do not require target protein-derived information. Rather, features generated from the chemical structure graph or any other suitable description of a drug are related to known target binding data (i.e. binding affinity) or phenotypic changes (i.e. high-throughput cancer cell viability assays) (Figure 1). These features, also termed descriptors, can include physical, chemical, topological or other types of quantifiable information, typically derived from a drug’s chemical structure. Other examples include atom types, electronic parameters, hydrophobicity, shape and many others. The collection of these values make up a chemical fingerprint for a drug and can be used for downstream analysis.
These structure-derived fingerprints are then related to target binding or phenotype data where the underlying assumption is that drugs with similar structures/properties exhibit similar bioactivity (19). Similarity between two drugs, via their chemical fingerprints, is quantified by metrics such as the Tanimoto coefficient. Numerous drug-target associations have been elucidated using the chemical similarity technique (20). While chemical similarity has been successful, there are instances where small chemical modifications (i.e. minor functional group changes) can result in large changes in activity, also known as “activity cliffs”, leading to false-positive associations (21).
In order to circumvent the above limitations, machine learning has been incorporated to better correlate drug features with bioactivity. This is often referred to as quantitative structure-activity relationship (QSAR). Generally speaking, machine learning is used either for categorical classification of drugs with respect to a target (i.e. inhibitor versus non-inhibitor) or predicting values along a continuous variable (i.e. binding affinity). Binding data used to produce machine learning models can be obtained from numerous sources including primary literature and online databases. These databases include BindingDB (22), PubChem (23), PharmGKB (24), ChEMBL (25), among others. Machine learning applied to these rich bioassay datasets has resulted in the successful identification of novel drug-target associations in multiple studies and has paved the way for a systematic drug repurposing framework where features from existing drugs can be leveraged.
In one such study, Deshmukh et al. applied Support Vector Machine (SVM) and Random Forest (RF) machine learning algorithms to PubChem bioassay data to create classification models of inhibitors/non-inhibitors of flap endonuclease 1 (FEN1), an enzyme critical for DNA repair/replication and whose inhibition has been shown to increase cancer cell sensitivity to chemotherapy (26). The SVM models outperformed the RF model and it was further used to screen a database of >50,000 compounds. The top five-ranked compounds were selected for in-vitro screening of which one (JFD00950) was identified as a novel FEN1 inhibitor with cytotoxic activity against the DLD-1 colon cancer cell line. In another study, Algamal et al. used adjusted adaptive least absolute shrinkage and selection operator (AALASSO) to predict the anti-cancer potency (IC50) of imidazo[4,5‐b]pyridine derivatives using 4885 chemical descriptors (27). A three-dimensional (3D)-QSAR model of maslinic acid and its derivatives for their activity against the MCF-7 breast cancer cell line was developed by Alam and Khan using topological molecular features such as shape, electrostatic pattern and hydrophobic regions (28). Using this model, they virtually screened the complete ZINC database (29) of millions of compounds and identified 593 potential hits. After applying ADMET filters, they found P-902 as a novel lead candidate with the potential to bind the NR3C1 glucocorticoid receptor using in-silico docking. Similarly, Taha et al. performed 3D-QSAR modeling on inhibitors of phosphoinositide 3-kinase gamma (PI3Kγ) and screened the National Cancer Institute (NCI) list of compounds to identify 19 compounds with nanomolar inhibition of PI3Kγ (30).
Ligand-based deep learning approaches have also been of benefit. In one such model, Allen et al. created a multi-task deep neural network (MTDNN) models to predict kinase activity of small molecules across the human kinome (31). Briefly, comprehensive kinase bioactivity data was obtained from ChEMBL and the commercial database Kinase Knowledgebase KKB (32). The total aggregated data included 668,920 measurements distributed across 342 targets and 317,228 unique compounds. In comparison to other machine learning methods, MTDNN had superior performance and was -+most generalizable. In another study, Rifaioglu et al, leveraged two-dimensional structural compound representations as input in order to predict drug-target interactions using their convolutional neural network-driven method, DEEPscreen (33). Apart from kinase bioactivity data, deep learning models such as MTDNN and DEEPscreen would enable large scale virtual screening of large compound databases for predictive activity profiling against kinases important for multiple cancer types and thus facilitate the quick and cost-effective repurposing of existing drugs.
In addition to inhibiting relevant protein cancer targets, small molecule modulation of microRNAs (miRNAs) is also of significant interest in cancer therapeutics. In general, MicroRNAs can alter oncogene function post-transcriptionally through their binding to the 3’ non-translated region of transcripts (34) and therefore their inhibition could prove to be of great therapeutic benefit. Jamal et al. were the first to create a predictive model of small molecule modulators of miRNAs (35). They integrated bioassay datasets found in PubChem and 179 molecular descriptors into machine learning algorithms and identified chemical scaffolds that may be critical for miRNA binding. Similarly, Wang et al. employed a random forest machine learning method using small molecule-miRNA binding data from the SM2miR database (36) and were able to predict the experimentally validated miRNA associations for commonly used small molecules 5-fluorouracil, 17β-estradiol, and 5-aza-2’-deoxycytidine (37). In another study, Qu et al. similarly created a machine learning framework called HeteSim that effectively predicts miRNA - small molecule associations (38). A detailed overview of computational methods used for the accurate prediction of small molecule-miRNA binding pairs can be found in the review by Chen et al. (39).
Proteochemometrics Modeling Using Machine Learning
As a logical continuation of the QSAR methodology, the combination of features extracted from both the drug and the target protein has been particularly beneficial for developing predictive models of drug activity. These combinatorial approaches are known as proteochemometrics (PCM) and can uncover target features relevant to binding such as important amino acids. PCM has been used to find numerous novel inhibitors especially for adenosine receptors. Adenosine G protein-coupled receptors have always been a prominent target for cancer research as their upregulation/activation has been implicated in various tumor processes (40). For example, activation of the adenosine A2A receptor suppresses regulatory T cell function and inhibits cytotoxic NK cell activity, thus attenuating anti-tumor immunity (41). Van Westen et al. developed a PCM model from bioactivity data of all adenosine receptor subtypes (humans and rats) that was used to screen a database of >100,000 compounds from which six were experimentally confirmed to be high-affinity adenosine receptor binders (42). Similarly, Cortes-Ciriano et al. used PCM to predict inhibitor potency against cyclooxygenase (COX), proteins implicated in prostaglandin production, which contribute to tumorigenesis and metastasis by promoting inflammation and angiogenesis (43, 44). Their PCM model incorporated 11 mammalian COX amino acid sequences and chemical descriptors for over 3,200 compounds. They implemented an ensemble machine learning model to predict inhibitor potency using IC50 values derived from ChEMBL with good accuracy and were further able to identify sub-structure chemical moieties implicated in potency and selectivity. Kundu et al. applied Random Forest PCM machine learning to predict binding affinity using data from the PDBbind Database (45) with input features from proteins (including sequence and secondary structure information) and ligands (including physicochemical properties) (46).
Deep learning approaches incorporating protein and ligand chemical features for PCM modeling have also recently emerged. DeepDTA is a CNN-based drug-target affinity prediction model that learns from just sequence information of proteins and compounds (amino acids and SMILES, respectively) (47). This is notable in that DeepDTA, using only raw sequences as input, performed as well as other established machine learning methods that implement multiple tools and algorithms. Cui et al. established DeepCSeqSite based on deep convolutional neural networks for predicting protein-ligand binding residues using the BioLip database (48) for model training (49). PCM deep learning also has utility for enriching docking results through refinement of scoring functions and increasing ligand pose prediction accuracy. For example, Jimenez et al. employed 3D-convolutional neural networks for predicting binding affinity from three-dimensional ligand-protein structures obtained from PDBbind (50).
Machine Learning on Cell Phenotype Data
Although associating a target’s inhibition with a desired clinical outcome has been successful in multiple cases, our limited knowledge of the complete interactome prohibits any systematic target-phenotype extrapolations. This, however, can be partially overcome by studying the direct phenotypic effects of drugs on cells and complex tissues with dose-response, cell death and apoptosis assays. Moreover, the ability to easily scale such assays into a high-throughput format allows the generation of large-scale datasets and enables the utilization machine and deep learning approaches for the prediction of drug-phenotype associations (Figure 2).
Figure 2. Schematic of machine learning applied to (A) phenotypic and (B) transcriptomic data.
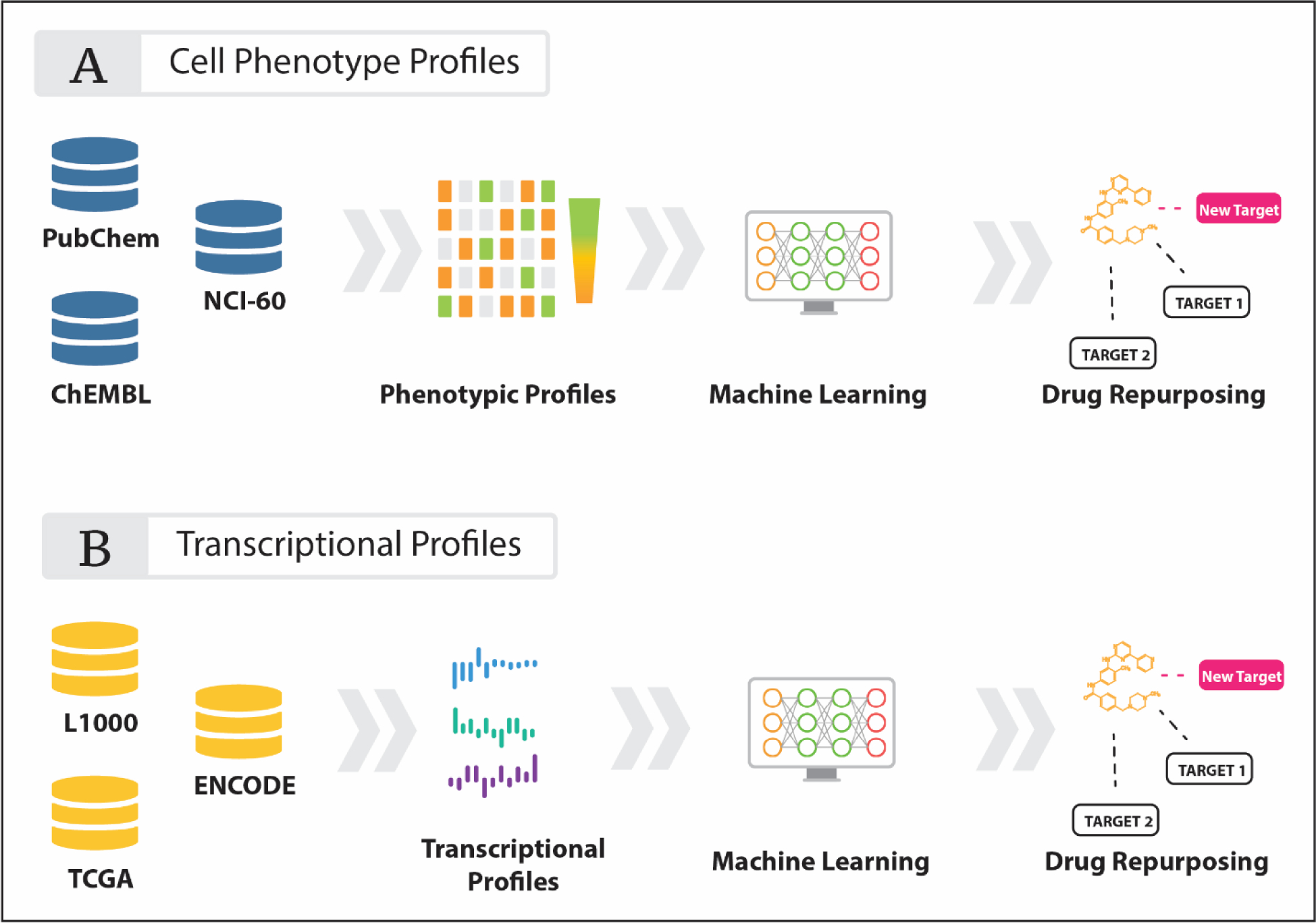
Bioassay data can be obtained from numerous publicly available databases such as PubChem, ChEMBL, TCGA and L1000 and implemented into machine learning systems.
These high-throughput phenotypic datasets are in most cases publicly accessible through dedicated data repositories and portals that enable the researchers to quickly access, explore and analyze them. One of the largest and widely used repositories is the PubChem bioassay repository (51). As of 2017, PubChem contained over 1.2 million assays comprising ~2.2 million chemicals, ~65,000 protein/gene targets and over 3,000 species comprising ~230 million bioactivity outcome data points. ChEMBL (25) is also another large-scale chemical bioactivity repository with. Publicly available tools such as DPubChem (52) have also been developed to allow for streamlining machine learning applications for investigators.
Developing machine learning models based on the above large-scale datasets has resulted in interesting new drug repurposing opportunities. One such example is the study from Kadurin et al. in which they applied adversarial autoencoders to full dose response data from the NCI-60 cell line assay to develop a deep learning model (53). They then pipelined the PubChem database of over 72 million compounds and were able to validate their model by finding molecules with already proven anti-cancer activity as well as revealed new molecules with potential anti-cancer activity, but had not yet been studied for that indication.
High throughput imaging assays also serve as rich sources of phenotypic data in drug screenings. For example, morphological changes of cells exposed to drugs may be captured via cell staining microscopy and morphology information can be extracted using software such as CellProfiler (54) and transformed into image-based compound fingerprints amenable to machine learning for the prediction of compound activity (55). In a recent study, Simm et al. created a high-dimensional image-based fingerprint for compound-treated cells in a three-channel screen for glucocorticoid receptor translocation (56). 524,371 compounds were screened against H4 brain neuroglioma cells and 842-dimensional vectors for each compound were extracted to form fingerprints. These fingerprints, combined with known drug bioactivity data from orthogonal assays (~1,200 bioassays across all screened compounds), were used as input for different machine and deep learning protocols.
Machine learning applied to chemical phenotypic screens may also conversely help to identify new disease-relevant targets - a process known as “target deconvolution”. This is possible through exploitation of drug polypharmacology. Using diverse libraries of drugs with known target interaction profiles (i.e. inhibitory and binding constant values), one can screen a library to see effects on a particular phenotype (i.e. cell growth) and further apply machine learning to find important targets. Gujral et al. utilized this approach to identify cancer cell type-specific kinases relevant to cell migration (57). They first created an optimized library of 32 kinase inhibitors that cover a significant portion of the kinome and screened those molecules against six different cancer cell types in real time wound closure assays using 96-well plates. EC50 (50% reduction in cell migration) values at 12-hour time points were obtained. They then employed elastic net regularization on the EC50 values and drug-target interaction map to narrow down the list of candidate kinases to less than 30 for each cancer cell type. To further distinguish which kinases are causal to the cell migration phenotype from non-causal or even non-expressed kinases, they subsequently used mRNA expression profiling to further prune the list of kinases and performed gene knockdown experiments for validation. Al-Ali et al. similarly used support vector machines (SVMs) to relate kinase inhibition data to neurite growth - a process critical for neural regeneration in central nervous system injury (58). Using SVMs, they were not only able to identify kinase targets whose inhibition promoted neurite growth, but also found kinase “anti-targets” whose inhibition blocked growth. They further found growth-promoting synergy among a 2-drug combination that collectively covers the whole span of robust targets. Their SVM model, termed idTRAX, was recently applied to triple negative breast cancer cell lines to identify cell line-specific kinases critical for cell survival (i.e. inhibition of FGFR2 specifically kills MFM-223 cells) (59). Coupling target deconvolution methods for cancer cell-specific phenotypes with the fast growing datasets of drug-target interaction profiles will further enable novel precision drug repurposing opportunities.
Transcriptomics-Based Machine Learning
The advent of Next Generation Sequencing (NGS) technologies have enabled the cost-effective, reliable and large-scale generation of transcriptional profiles that provide considerable insights into the complex and aberrant regulatory network in different cancers. The Cancer Genome Atlas (TCGA) has been at the forefront of this NGS data generation effort, providing the largest uniformly processed multi-omic cancer dataset from 33 tumor types (60). Gene expression profiles obtained from thousands of patient samples deposited in the TCGA, as well as other public databases such as the NCBI Gene Expression Omnibus (61), have enabled the adoption of comprehensive systems biology approaches that together with machine learning approaches can elucidate new disease target and next generation treatments.
In addition to finding pharmacologically relevant cancer targets, large-scale transcriptomics datasets can be used to delineate drug mechanisms of action and uncover drug repurposing opportunities (Figure 2). The Connectivity Map (CMap) approach is among the most well-known approaches where drugs that induce discordant gene expression profiles to a disease-specific gene expression profile are likely to be therapeutic candidates for that disease (63). Over the past years, connectivity mapping has been successful in illuminating unforeseen cancer drug repurposing opportunities. Sirota et al. predicted and validated cimetidine, an off-patent approved anti-ulcer drug with favorable safety profile, to be indicated for lung adenocarcinoma (64). This effect was replicated recently in xenograft model of A549 lung adenocarcinoma cells (65). Jahchan et al. also identified tricyclic antidepressants to inhibit small cell lung cancer as well as neuroendocrine tumors (66). Previous success of CMap in cancer (67) and other disease states (68–71) led to a significant expansion of the reference drug perturbation gene expression dataset to increase the likelihood of uncovering new drug indications. A major effort was established by the NIH LINCS Consortium (72, 73) to establish a 1,000-fold scale-up of the original CMap using a novel gene expression profiling method called the L1000 that is of low cost and high throughput (74). Using the L1000 assay, over 42,000 unannotated perturbagens (including drugs, gene knockdowns and gene overexpressions) were screened against 77 cell lines to create over a million gene expression perturbation signatures. These transcriptional response signatures have helped describe and elucidate the effects of drug treatments on the transcriptional level and have been instrumental in the cancer drug repurposing opportunities, such as emetine, a pro-emetic, for atypical meningiomas (75). Furthermore, apart from single drug predictions, the use of the L1000 data has also aided in the development of methods for predicting synergistic drug combinations such as the SynergySeq (76) and SynGeNet (77) platforms.
In addition to the typical signature anti-correlated matching, the large amount of data generated from L1000 is ideal for machine learning approaches to recapitulate and find novel drug-target interactions. Xie et al. applied a supervised deep neural network (DNN) to L1000 gene expression data and a drug-target interaction activity dataset from DrugBank (78) and accurately predicted established drug-target pairs using external validation sets (79). Filzen et al. similarly utilized the Pylearn2 deep learning framework for model building from the L1000 along with a different drug-target annotation dataset obtained from Merck (CHEMGENIE) (80). Chiu et al. used DNN combining inputs from TCGA mutation linked to cancer-specific gene expression data and relating them to drug response (81). Their DNN model, called DeepDR, predicted IC50 values for 265 drugs and found a novel use for the chemotheraputic drug vinorelbine in TTN-mutated tumors. Zhang et al. integrated TCGA copy number data in addition to mutation and gene expression data to develop their deep learning model, AuDNNsynergy, to predict synergistic drug combinations (82). Zhou et al. designed a gene expression-weighted cosine method called EMUDRA from Connectivity Map and LINCS data and experimentally validated the repurposing of the antibiotic rifabutin as an inhibitor of triple negative breast cancer cell growth (83). Combining drug perturbation gene expression with that of cancer cell lines and pathway activation data, Aliper et al. used DNNs and predict the potassium channel-blocking vasodilator drug pinacidil as a central nervous system (CNS) agent (84) with the potential to be repurposed for poor-prognosis CNS cancers such as glioblastoma (86).
EHR-Based Machine Learning
In addition to the utilization of cheminformatic, phenotypic and transcriptomic datasets, electronic health records (EHRs) also contain a wealth of information that can be mined for drug discovery, repurposing and adverse drug event (ADE) predictions. Recent advances in natural language processing (NLP) has enabled investigators to take the largely unstructured data sources (i.e. clinic notes, web forum posts) and translate them into a machine-interpretable format, thus enabling the application of machine and deep learning analytics. For example, in an effort to improve upon the underreporting of ADEs through current means such as the FDA Adverse Events Reporting System, Li et al. employed a deep learning model on EHR notes to more accurately detect ADEs than the most current state-of-the-art (87).
An important drug repurposing opportunity comes from off-label use data of approved drugs. Both EHR and social media data have been successfully used to determine off-label uses. In one such study, Jung et al. built a training set of known drug usage using the Medi-Span Drug Indications Database and used it to train a support vector machine (SVM) classifier that was able to discriminate between known drug use indications and off-label use from annotated clinical text, thus producing 10,765 potential novel drug-indication pairs that could be used for EHR-based drug repurposing (88). In another EHR-based study, Xu et al. developed a machine learning framework that led to the association between metformin, an anti-diabetic medication, and improved cancer survival rates (89). By integrating two EHRdatabases from the Mayo Clinic and Vanderbilt University Medical Center to their tumor registries, they generated two independent cohorts totaling over 100,000 patients between 1995–2010. While controlling for other covariates such as smoking, metformin was found to be associated with decreased mortality after a cancer diagnosis. The repurposing potential for metformin as an anti-cancer therapeutic has also been confirmed in several preclinical and epidemiological studies for breast cancer (90).
As NLP and deep learning algorithms increase in sophistication with a growing body of clinical data available from EHR and online websites, we imagine that other cancer drug repurposing instances will arise from similar endeavors. There has also been an increased involvement of large private entities such as Google and Amazon that have developed their own deep learning platforms (Google AutoML and Amazon Comprehend Medical) and application programming interface (APIs) (91). With involvement of these entities and their supercomputing capabilities, identification of new drug repurposing opportunities through NLP/deep learning will undoubtedly be accelerated.
Drug Repurposing and Immunotherapy
A major property of poor prognosis malignancies is their ability to evade detection and killing by the patient’s immune system (92). The tumor’s intrinsic biology and its immune microenvironment both play critical roles in tumor immune evasion. The relative proportions and absolute amounts of different immune cell types within the tumor microenvironment determine a tumors ability for immune evasion. For example, dominant presence of regulatory T cells confers an immunotolerant phenotype. A complex interplay of immune cell types, expressed antigens and secreted factors alters the ability of the immune system to eradicate tumor cells. A detailed discussion of the mechanisms of immune evasion can be found in the following reference (94).
Much attention has been placed on immunomodulation of the tumor microenvironment for therapy. CD4+ FoxP3+ T regulatory cells (Tregs), which provide a suppressive effect on effector T cells implicated in tumor surveillance, have been shown to represent a large proportion of tumor-infiltrating lymphocytes (TILs) in melanoma and breast cancer immune checkpoint inhibitor (ICI) non-responders (102). Thus, the attenuation or abolition of tumor-infiltrating Treg function has been a subject of great interest in immuno-oncology therapeutics (103). However, a major concern of Treg depletion is increased immune-related adverse events such as induction of autoimmune disorders (105). In lieu of depletion, biologic- and small molecule-driven alteration of Treg function is under intense study (106). The long-established chemotherapeutic cyclophosphamide was recently found to inhibit Tregs at a low dose thus posing an effective drug repurposing opportunity immunotherapy (107). Wang et al. found Treg function in tumor infiltrates to be dependent on the enhancer of zeste homolog 2 (EZH2) protein was also identified (108). Pharmacological inhibition of EZH2 resulted in Tregs acquiring a pro-inflammatory function that enhanced anti-cancer immunity. Thus, application of machine and deep learning methods to find novel inhibitors of EZH2 may be a promising strategy for repurposing drugs for Treg-driven immunomodulation.
In addition to direct regulation of Tregs, other immune or tumor cell protein targets implicated in immunomodulation are of pharmaceutical interest. Indoleamine 2,3-dioxygenase (IDO) is a target with a multifaceted effect on tumor microenvironment immunosuppression. IDO catalyzes the degradation of tryptophan along the kynurenine pathway (109). Depletion of tryptophan in the tumor microenvironment consequently activates the GCN2 kinase pathway in neighboring cells, thus biasing naive CD4+ T cells to differentiate into Treg cells (110, 111). In addition, tryptophan metabolites (e.g. kynurenine) generated from IDO catalysis bind aryl hydrocarbon receptor (AHR) to induce Treg cells and a tolerogenic phenotype of dendritic cells (112). Apoptotic tumor cells also stimulate IDO expression in neighboring cells and can contextually alter the balance between pro-inflammatory and immunosuppressive states in the microenvironment (113). In addition to IDO, the adenosine A2A receptor is yet another target of interest for immunomodulation (114). Elevated adenosine levels have been recognized in the tumor microenvironment resulting in an immunosuppressive effect on NK and T cells (115, 116). Antagonism of the A2A receptor is thus an additional viable strategy for immune checkpoint blockade (117).
Application of virtual screenings and machine learning has helped identify novel inhibitors of IDO and A2AR. Zhang et al. took a library of known IDO inhibitors and developed naive Bayesian and recursive partitioning models that identified three new IDO inhibitors with low-micromolar inhibitory concentrations and activity in cell-based assays (118). Interestingly, these inhibitors were of the tanishone family with molecular scaffolds derived from the Chinese herb Danshen, which is widely used for cardiovascular and cerebrovascular diseases. Zhou et al. employed a ligand-based 3D-QSAR using pharmacophore models derived from known IDO inhibitors to find novel backbones for next generation IDO inhibitors (119). SVMs applied to the repertoire of A2AR inhibitors helped identify novel pyrazolo-triazolo-pyrimidine derivatives with A2AR activity (120). SVMs were also used by Shao et al. to identify novel compounds with the ability to simultaneously inhibit A2AR and dopamine D2 receptor at low-micromolar concentrations (121). Liu et al. applied recurrent neural networks (RNN) to design a library of novel A2AR inhibitors that exhibited large diversity in their coverage of the chemical space (122). Tian et al. performed docking virtual screening of over 2.65 million compounds against A2AR crystal structures to find a novel A2AR inhibitor with nanomolar binding affinity (123).
The vascular endothelial growth factor (VEGF)-VEGFR2 axis is also of great pharmacologic interest in cancer therapy. Inhibition of this pro-angiogenic pathway through VEGFR2 kinase inhibition has been efficacious for numerous cancers (124) by reducing immunosuppression by preventing recruitment of immature dendritic cells, myeloid-derived suppressor cells and Tregs (125, 126). Anti-angiogenic agents targeting this axis promote a pro-inflammatory microenvironment (127). Drug repurposing efforts to inhibit VEGFR2 have been underway. Our group previously employed proteochemometrics to identify the approved anti-helmenthic drug mebendazole as a VEGFR2 inhibitor and anti-angiogenic (128). Kang et al. developed a naïve Bayesian machine learning model using a training set of 2,598 inhibitors and 7,764 decoys and screened 1,841 FDA-approved drugs (129). They discovered flubendazole, rilpivirine and papaverine to inhibit VEGFR2 with sub- to low-micromolar IC50 values.
IL-6 is another immunomodulatory target shown to suppress T cell-mediated anti-tumor immunity (130). IL-6 blockade, in combination with other treatment modalities, provides a beneficial anti-tumor response (131). As IL-6 binds GP130, Chen et al. employed a novel combinatorial approach to target GP130 and found the FDA-approved drug bazedoxifene, indicated for osteoporosis, to block that interaction and inhibit the increased proliferation of triple-negative breast cancer cells induced by IL-6 (132). Han et al. also found the molecule CGP-60474, a CDK inhibitor, to reduce IL-6 levels in murine macrophage cell lines (133). They applied Connectivity Mapping on sepsis-related microarray gene expression studies using the L1000 perturbation set and found CGP-60474 to reverse the sepsis gene signature. Subsequent analysis of the anti-inflammatory effect of CGP-60474 revealed it to inhibit IL-6 levels.
Macrophage polarization has also been implicated in tumor biology. M2/repair-type macrophages promote tumor growth whereas M1/kill-type proinflammatory macrophages halt growth (134). High M1/M2 macrophage ratio decreases cancer susceptibility, and modulation of M2 to M1 phenotype (or alteration of the M1/M2 ratio) seems to be a viable immunotherapy modality. Some targetable proteins involved in the modulation of macrophage polarization, such as the TRPV4 ion channel (135) and cannabinoid receptor 2 (136), have already been elucidated. Structure based approaches noted above could be leveraged to identify drug repurposing opportunities against those targets. In addition, some drugs have also been shown to alter macrophage polarization such as praziquantel (137) and capsaicin (138). Machine and deep learning methods applied to these drugs in conjunction with their quantifiable phenotypic effect on macrophages could further help uncover unrelated drugs with similar capabilities.
Considerations in Machine Learning- and AI-Driven Methods for Drug Repurposing
The accuracy of any proposed machine learning- or AI-driven model is contingent upon the quality, type and amount of data in the training set as well as the validation methods of those models. With respect to the former, the training set dictates the applicability domain of the model the query molecule space from which the model can make accurate predictions (139). For example, if a model is created from a small set of inhibitors for a target containing the same chemical backbone, then the model would be successful in predicting new active compounds with similar backbones but not actives with diverse structures. This narrowness in a model’s applicability domain is also known as overfitting.
Deep learning approaches are particularly prone to overfitting, especially as a network’s complexity increases. Numerous methods help to reduce overfitting including: (1) making the network smaller (removing layers), (2) neuron dropout regularization (ignoring neuron units during the training phase at random), and (3) early stopping (termination of training at the junction where validation performance starts to deteriorate) (140, 141). Rifaioglu et al. implemented neuron dropout regularization in their model training of DEEPScreen, validated using Monte Carlo Cross-validation, to predict drug-target interactions from two-dimensional structural images of drugs (33). Chiu et al. utilized the early stopping method in the development of their highly complex deep neural network that integrates drug, TCGA tumor mutational data and CCLE tumor gene expression data that predicts tumor response to drugs (81).
Type and number of input descriptors also affect model performance. Typically, protein and ligand structural, chemical and physical features are represented as “fingerprints” - vectors of varying sizes containing 0’s or 1’s that numerically represent the absence or presence of a feature, respectively. Fingerprints can be used to represent both two-dimensional (e.g. atom pairs, atom types, linear atom connectivity) and three-dimensional features (e.g. neighboring covalently bound and unbound atoms and atom functional group categories in 3D space). Thousands of fingerprints exist and have been extensively used in cheminformatics, and their relatedness and performance has been evaluated (142). In addition to fingerprints, integers and floating numbers can be used to represent properties such as molecular weight or solvent-accessible surface area. Other potential descriptor inputs include biological data such as mutations, gene expression and others. Thus, a variety of descriptor inputs exists for model generation.
The choice of input descriptor depends on the prediction problem. For example, pharmacophore fingerprints may work better for discovering new chemotypes. Pharmacophores are vectorized 3D representations of chemical functional groups (e.g. hydrophobic centers, hydrogen bond donors and acceptors, etc.) and the distances between them necessary to establish specific ligand-target interactions. As pharmacophores are more abstract representations of atom types, they can be used to screen for structurally diverse ligands with the ability to recapitulate similar interactions to the same target binding site. Use of pharmacophores in machine learning-based predictions is exemplified by Taha et al. (30). They generated pharmacophore models and other 2D descriptor fingerprints from a known set of PI3Kgamma inhibitors and created a QSAR model of the optimal combination of pharmacophores/2D descriptors using IC50 values as the biological endpoint. The resulting QSAR model was then used to screen the National Cancer Institute list of >200,000 compounds and identified 9 compounds with diverse chemotypes and sub-micromolar IC50 values against PI3Kgamma. Alam et al. also generated pharmacophore models of maslinic acid analogs and subsequently subjected them to QSAR modeling using their IC50 inhibitor concentrations against the MCF-7 breast cancer cell line (28). The ZINC database was then screened using the developed QSAR model resulting in the identification of compound P-902 with potential anticancer activity against MCF-7 breast cancer cells. Zhou et al. similarly employed QSAR modeling of IDO1 inhibitor pharmacophores to discover novel backbones for next generation IDO inhibitors (119).
In addition to pharmacophore fingerprints, circular fingerprints (e.g. ECFP) also offer a good input choice that can exhibit good performance. ECFPs are fingerprints that are iteratively generated by first assigning a feature to a selected atom in a molecule and then radially expanding to neighboring atoms (143). These features may represent atom types, bond connectivity, atomic weight, atomic charge, number of attached hydrogens, pharmacophoric property, among many others. The number of iterations is up to the discretion of the user. Structural protein-ligand interaction fingerprints are also widely used in proteochemometrics as they numerically represent three-dimensional structural information derived from biologically relevant protein-ligand interactions (144, 145). Interaction fingerprints could be considered as protein residues within a certain distance of each ligand atom or the center of the entire ligand obtained from crystallography or structural molecular modeling. Like ECFPs, interaction fingerprints are successful at predicting new ligands for a target. For example, van Westen et al. implemented both ligand-based circular and interaction fingerprints to predict novel adenosine receptor ligands (42). They found six novel high-affinity ligands, two of which resemble structures of known adenosine receptor binders while others exhibited diverse chemistry. Interestingly, the combination of circular and interaction fingerprints was able to uncover new chemotypes while also prioritizing compounds with similar chemotypes. Another descriptor used by Rifaioglu et al. is the actual two-dimensional structural image of drugs as input for their CNN (33). Focused on gene expression-driven inputs, Aliper et al. utilized transcriptional drug perturbation responses across different cell types as inputs for their deep neural network (84). They found that pathway-level information performed significantly better than gene level data due to reduction of dimensionality while keeping biological relevance. Choice and applicability of data types and fingerprints used in machine learning- and AI-driven methods are highly nuanced to the problem posed. There usually is an iterative process in model development where different descriptors/combinations are tried such that performance is maximized, which changes across desired biological endpoints and targets.
Dataset size is also a crucial aspect of in machine learning. In general, more data incorporated into the training set generally results in better model performance (less overfitting), especially for deep neural networks. For example, Allen et al. found that for predicting drug-kinase activity using deep learning, model performance was significantly less when the number of active compounds for a kinase was less than 500 and further degraded when there was less than 50 (31). This is with respect to single-task learning (i.e. predicting drug activity to a single kinase). Interestingly, they found that their drug-kinase multi-task deep neural network, where they predicted drug-kinase activity across 342 kinases simultaneously using a well-curated aggregate dataset, continued to perform well even with low number of actives. Their model was further validated by 5-fold cross validation.
Data quality is also of critical importance. Generally, data inputted into models are typically generated from high-throughput screens. For example, PubChem (23) contains information from biological assays for drug-target binding events (e.g. binding affinity) or drug-induced changes in gene expression, phosphorylation as well as cell viability. Such assays are performed by various groups under different conditions, and data is being generated at an exponential pace. Thus, in order to more optimally integrate this data into machine learning- and AI-assisted drug discovery efforts, the data needs conform to community standards of findability, accessibility, interoperability and reusability (FAIR standards) (146, 147). Metadata (e.g. data regarding experimental conditions, species studied, tissue obtained, etc.) is of critical importance as it describes the nature of the conditions under which the data was collected and provides further context from which the model can learn. For example, biological phenotypes due to drugs such as estrogen receptor transcriptional response may be different across cell lines due to differences in expressed co-activators, co-repressors, etc. Such information can be captured by well-annotated metadata and help fine-tune models for greater accuracy. Thus, appropriate data management with standards becomes a priority for drug discovery data generation (148). Our group has formally defined the details of bioassays (149–151) and drug targets (152) in an effort to lay the foundations for future data generation and analytics. By upholding community-accepted data standards, poor quality data can easily be filtered out prior to model generation, and data generated in the future will conform to the same standards and thus be easily integrated in various machine-learning and AI-driven drug discovery efforts while minimizing the need for extensive manual curation. As the amount of data continues to exponentially increase along with computing capabilities, we anticipate that future modeling efforts will be exceedingly more accurate and uncover new drug repurposing opportunities for cancer therapy.
Conclusion
With massive data collection efforts such as the TCGA, our understanding of tumor pathogenesis and survival has evolved significantly. The implementation of newer “omics” technologies such as proteomics and metabolomics lend new insights into cancer cell and immunological processes contributing to disease progression (153–155). The movement toward single cell RNA sequencing has already provided further perspectives into tumor heterogeneity at a highly granular level not previously appreciated from bulk biopsy transcriptomics (156). As these technologies, mature, novel pharmacologically actionable targets will further be uncovered.
At this time, a major limitation in the success of machine and deep learning applications for drug repurposing is the quality and quantity of available data. As the costs of highly automated high-throughput screening platforms continue to decrease, the amount of data produced from drug assay screening is increasing exponentially. Rigorous analytical frameworks for annotating drug discovery data (i.e. ontologies) are also necessary for true standardization and interoperability of data produced and integration into machine learning programs (151, 152, 157, 158). Further, model applicability will be enhanced with more data, better data integration, the true drug-target interaction space is expanding significantly, and the full extent of drug polypharmacology is starting to emerge. Such a large dataset increases its drug domain coverage that could bring equal numbers of classes to the training dataset potentially reducing the class imbalance problem in the machine learning models. The physiological effects of drugs on different cell phenotypes are better understood via increasingly thorough multi-omics profiling and sophisticated systems biology models. Coupled with the advancement of computational drug discovery strategies such as machine learning and artificial intelligence (deep learning), this information can be leveraged to identify new drug-target and drug-cell interactions.
As the development of new molecular entities is risky and suffers from a high cost burden, drug repurposing of approved and investigational drugs has been touted as a strategy with lower barrier to success. The clinical potential of drug repurposing in oncology has yet to be realized at this time. With the ever-increasing amount of biological and pharmacological data, computational methods will certainly provide new perspectives into therapeutic mechanisms and enable drug repurposing within the framework of precision oncology.
Acknowledgements
This work was supported by NIH grants U54HL127624 (BD2K LINCS Data Coordination and Integration Center, DCIC), U24TR002278 (Illuminating the Druggable Genome Resource Dissemination and Outreach Center, IDG-RDOC), and U01LM012630 (BD2K, Enhancing the efficiency and effectiveness of digital curation for biomedical ‘big data’), and the State of Florida Biomedical Research Program, Bankhead Coley grant number 9BC13. The author Sivanesan Dakshanamurthy wishes to acknowledge the support in part by DOD grant CA140882, CCSG grant NIH-P30 CA051008, GUMC Computational Chemistry Shared Resources (CCSR), and GUMC Lombardi Comprehensive Cancer Center.
Abbreviations Used:
- ADE
adverse drug event
- ADMET
absorption, distribution, metabolism, excretion, toxicity
- CNN
convolutional neural network
- DNN
deep neural network
- EHR
electronic health record
- EMT
epithelial-mesenchymal transition
- ICI
immune checkpoint inhibitor
- MDSC
myeloid-derived suppressor cells
- NLP
natural language processing
- PCM
proteochemometric
- QSAR
quantitative structure-activity relationship
- RF
Random Forest
- SVM
Support Vector Machine
- TCGA
The Cancer Genome Atlas
- TIL
tumor immune cell infiltrates
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
REFERENCES
- 1.Ekins S, Puhl AC, Zorn KM, Lane TR, Russo DP, Klein JJ, et al. Exploiting machine learning for end-to-end drug discovery and development. Nat Mater. 2019;18(5):435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nosengo N Can you teach old drugs new tricks? Nature. 2016;534(7607):314–6. [DOI] [PubMed] [Google Scholar]
- 3.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–49. [DOI] [PubMed] [Google Scholar]
- 4.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–49. [DOI] [PubMed] [Google Scholar]
- 5.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain AN. Scoring functions for protein-ligand docking. Curr Protein Pept Sci. 2006;7(5):407–20. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Lin HQ, Wang P, Hu JS, Ip TM, Yang LM, et al. Discovery of a Novel HIV-1 Integrase/p75 Interacting Inhibitor by Docking Screening, Biochemical Assay, and in Vitro Studies. J Chem Inf Model. 2017;57(9):2336–43. [DOI] [PubMed] [Google Scholar]
- 8.Mirza SB, Salmas RE, Fatmi MQ, Durdagi S. Virtual screening of eighteen million compounds against dengue virus: Combined molecular docking and molecular dynamics simulations study. J Mol Graph Model. 2016;66:99–107. [DOI] [PubMed] [Google Scholar]
- 9.Kumar V, Krishna S, Siddiqi MI. Virtual screening strategies: recent advances in the identification and design of anti-cancer agents. Methods. 2015;71:64–70. [DOI] [PubMed] [Google Scholar]
- 10.Hafeez BB, Ganju A, Sikander M, Kashyap VK, Hafeez ZB, Chauhan N, et al. Ormeloxifene Suppresses Prostate Tumor Growth and Metastatic Phenotypes via Inhibition of Oncogenic beta-catenin Signaling and EMT Progression. Mol Cancer Ther. 2017;16(10):2267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YC. Beware of docking! Trends Pharmacol Sci. 2015;36(2):78–95. [DOI] [PubMed] [Google Scholar]
- 12.Wallach I, Dzamba M, Heifets A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv e-prints [Internet]. 2015. October 01, 2015. Available from: https://ui.adsabs.harvard.edu/abs/2015arXiv151002855W. [Google Scholar]
- 13.Desaphy J, Bret G, Rognan D, Kellenberger E. sc-PDB: a 3D-database of ligandable binding sites--10 years on. Nucleic Acids Res. 2015;43(Database issue):D399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spitzer R, Jain AN. Surflex-Dock: Docking benchmarks and real-world application. J Comput Aided Mol Des. 2012;26(6):687–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, et al. DOCK 6: Impact of new features and current docking performance. J Comput Chem. 2015;36(15):1132–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein-Ligand Scoring with Convolutional Neural Networks. J Chem Inf Model. 2017;57(4):942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Han L, Liu Z, Wang R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J Chem Inf Model. 2014;54(6):1717–36. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen DD, Cang Z, Wu K, Wang M, Cao Y, Wei GW. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges. J Comput Aided Mol Des. 2019;33(1):71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maldonado AG, Doucet JP, Petitjean M, Fan BT. Molecular similarity and diversity in chemoinformatics: from theory to applications. Mol Divers. 2006;10(1):39–79. [DOI] [PubMed] [Google Scholar]
- 20.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Stumpfe D, Bajorath J. Advancing the activity cliff concept. F1000Res. 2013;2:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44(D1):D1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–D9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbarino JM, Whirl-Carrillo M, Altman RB, Klein TE. PharmGKB: A worldwide resource for pharmacogenomic information. Wiley Interdiscip Rev Syst Biol Med. 2018;10(4):e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45(D1):D945–D54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deshmukh AL, Chandra S, Singh DK, Siddiqi MI, Banerjee D. Identification of human flap endonuclease 1 (FEN1) inhibitors using a machine learning based consensus virtual screening. Mol Biosyst. 2017;13(8):1630–9. [DOI] [PubMed] [Google Scholar]
- 27.Algamal ZY, Lee MH, Al-Fakih AM, Aziz M. High-dimensional QSAR prediction of anticancer potency of imidazo[4,5-b]pyridine derivatives using adjusted adaptive LASSO. Journal of Chemometrics. 2015;29(10):547–56. [Google Scholar]
- 28.Alam S, Khan F. 3D-QSAR studies on Maslinic acid analogs for Anticancer activity against Breast Cancer cell line MCF-7. Sci Rep. 2017;7(1):6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sterling T, Irwin JJ. ZINC 15--Ligand Discovery for Everyone. J Chem Inf Model. 2015;55(11):2324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taha MO, Al-Sha’er MA, Khanfar MA, Al-Nadaf AH. Discovery of nanomolar phosphoinositide 3-kinase gamma (PI3Kgamma) inhibitors using ligand-based modeling and virtual screening followed by in vitro analysis. Eur J Med Chem. 2014;84:454–65. [DOI] [PubMed] [Google Scholar]
- 31.Allen BK, Ayad NG, Schürer SC. Kinome-wide activity classification of small molecules by deep learning. bioRxiv. 2019:512459. [Google Scholar]
- 32.Schurer SC, Muskal SM. Kinome-wide activity modeling from diverse public high-quality data sets. J Chem Inf Model. 2013;53(1):27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rifaioglu AS, Atalay V, Martin MJ, Cetin-Atalay R, Doğan T. DEEPScreen: High Performance Drug-Target Interaction Prediction with Convolutional Neural Networks Using 2-D Structural Compound Representations. bioRxiv. 2018:491365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho WC. OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer. 2007;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jamal S, Periwal V, Open Source Drug Discovery C, Scaria V. Computational analysis and predictive modeling of small molecule modulators of microRNA. J Cheminform. 2012;4(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Wang S, Meng F, Wang J, Zhang Y, Dai E, et al. SM2miR: a database of the experimentally validated small molecules’ effects on microRNA expression. Bioinformatics. 2013;29(3):409–11. [DOI] [PubMed] [Google Scholar]
- 37.Wang CC, Chen X, Qu J, Sun YZ, Li JQ. RFSMMA: A New Computational Model to Identify and Prioritize Potential Small Molecule-MiRNA Associations. J Chem Inf Model. 2019;59(4):1668–79. [DOI] [PubMed] [Google Scholar]
- 38.Qu J, Chen X, Sun YZ, Zhao Y, Cai SB, Ming Z, et al. In Silico Prediction of Small Molecule-miRNA Associations Based on the HeteSim Algorithm. Mol Ther Nucleic Acids. 2019;14:274–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Guan NN, Sun YZ, Li JQ, Qu J. MicroRNA-small molecule association identification: from experimental results to computational models. Brief Bioinform. 2018. [DOI] [PubMed] [Google Scholar]
- 40.Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA. Adenosine receptors and cancer. Biochim Biophys Acta. 2011;1808(5):1400–12. [DOI] [PubMed] [Google Scholar]
- 41.Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. 2016;29:7–16. [DOI] [PubMed] [Google Scholar]
- 42.van Westen GJ, van den Hoven OO, van der Pijl R, Mulder-Krieger T, de Vries H, Wegner JK, et al. Identifying novel adenosine receptor ligands by simultaneous proteochemometric modeling of rat and human bioactivity data. J Med Chem. 2012;55(16):7010–20. [DOI] [PubMed] [Google Scholar]
- 43.Cortes-Ciriano I, Murrell DS, van Westen GJ, Bender A, Malliavin TE. Prediction of the potency of mammalian cyclooxygenase inhibitors with ensemble proteochemometric modeling. J Cheminform. 2015;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider C, Pozzi A. Cyclooxygenases and lipoxygenases in cancer. Cancer Metastasis Rev. 2011;30(3–4):277–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang R, Fang X, Lu Y, Wang S. The PDBbind database: collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J Med Chem. 2004;47(12):2977–80. [DOI] [PubMed] [Google Scholar]
- 46.Kundu I, Paul G, Banerjee R. A machine learning approach towards the prediction of protein–ligand binding affinity based on fundamental molecular properties. RSC Advances. 2018;8(22):12127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozturk H, Ozgur A, Ozkirimli E. DeepDTA: deep drug-target binding affinity prediction. Bioinformatics. 2018;34(17):i821–i9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013;41(Database issue):D1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cui Y, Dong Q, Hong D, Wang X. Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinformatics. 2019;20(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jimenez J, Skalic M, Martinez-Rosell G, De Fabritiis G. KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J Chem Inf Model. 2018;58(2):287–96. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Bryant SH, Cheng T, Wang J, Gindulyte A, Shoemaker BA, et al. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017;45(D1):D955–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soufan O, Ba-Alawi W, Magana-Mora A, Essack M, Bajic VB. DPubChem: a web tool for QSAR modeling and high-throughput virtual screening. Sci Rep. 2018;8(1):9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kadurin A, Aliper A, Kazennov A, Mamoshina P, Vanhaelen Q, Khrabrov K, et al. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget. 2017;8(7):10883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7(10):R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheeder C, Heigwer F, Boutros M. Machine learning and image-based profiling in drug discovery. Curr Opin Syst Biol. 2018;10:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simm J, Klambauer G, Arany A, Steijaert M, Wegner JK, Gustin E, et al. Repurposing High-Throughput Image Assays Enables Biological Activity Prediction for Drug Discovery. Cell Chem Biol. 2018;25(5):611–8 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gujral TS, Peshkin L, Kirschner MW. Exploiting polypharmacology for drug target deconvolution. Proc Natl Acad Sci U S A. 2014;111(13):5048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Ali H, Lee DH, Danzi MC, Nassif H, Gautam P, Wennerberg K, et al. Rational Polypharmacology: Systematically Identifying and Engaging Multiple Drug Targets To Promote Axon Growth. ACS Chem Biol. 2015;10(8):1939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gautam P, Jaiswal A, Aittokallio T, Al-Ali H, Wennerberg K. Phenotypic Screening Combined with Machine Learning for Efficient Identification of Breast Cancer-Selective Therapeutic Targets. Cell Chem Biol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cieślik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nature Reviews Genetics. 2017;19:93. [DOI] [PubMed] [Google Scholar]
- 63.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science. 2006;313(5795):1929–35. [DOI] [PubMed] [Google Scholar]
- 64.Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, et al. Discovery and Preclinical Validation of Drug Indications Using Compendia of Public Gene Expression Data. Science Translational Medicine. 2011;3(96):96ra77–96ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kandela I, Aird F, Reproducibility Project: Cancer B. Replication Study: Discovery and preclinical validation of drug indications using compendia of public gene expression data. eLife. 2017;6:e17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013;3(12):1364–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng HW, Liang YH, Kuo YL, Chuu CP, Lin CY, Lee MH, et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015;6(5):e1753–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brum AM, van de Peppel J, van der Leije CS, Schreuders-Koedam M, Eijken M, van der Eerden BCJ, et al. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(41):12711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishimatsu-Tsuji Y, Soma T, Kishimoto J. Identification of novel hair-growth inducers by means of connectivity mapping. The FASEB Journal. 2010;24(5):1489–96. [DOI] [PubMed] [Google Scholar]
- 70.Vanderstocken G, Dvorkin-Gheva A, Shen P, Brandsma CA, Obeidat M, Bosse Y, et al. Identification of Drug Candidates to Suppress Cigarette Smoke-induced Inflammation via Connectivity Map Analyses. Am J Respir Cell Mol Biol. 2018;58(6):727–35. [DOI] [PubMed] [Google Scholar]
- 71.Brum AM, van de Peppel J, Nguyen L, Aliev A, Schreuders-Koedam M, Gajadien T, et al. Using the Connectivity Map to discover compounds influencing human osteoblast differentiation. Journal of Cellular Physiology. 2018;233(6):4895–906. [DOI] [PubMed] [Google Scholar]
- 72.Koleti A, Terryn R, Stathias V, Chung C, Cooper DJ, Turner JP, et al. Data Portal for the Library of Integrated Network-based Cellular Signatures (LINCS) program: integrated access to diverse large-scale cellular perturbation response data. Nucleic Acids Res. 2018;46(D1):D558–D66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keenan AB, Jenkins SL, Jagodnik KM, Koplev S, He E, Torre D, et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018;6(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171(6):1437–52 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zador Z, King AT, Geifman N. New drug candidates for treatment of atypical meningiomas: An integrated approach using gene expression signatures for drug repurposing. PLoS One. 2018;13(3):e0194701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stathias V, Jermakowicz AM, Maloof ME, Forlin M, Walters W, Suter RK, et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat Commun. 2018;9(1):5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harrold JM, Ramanathan M, Mager DE. Network-based approaches in drug discovery and early development. Clin Pharmacol Ther. 2013;94(6):651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie L, He S, Song X, Bo X, Zhang Z. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genomics. 2018;19(Suppl 7):667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Filzen TM, Kutchukian PS, Hermes JD, Li J, Tudor M. Representing high throughput expression profiles via perturbation barcodes reveals compound targets. PLoS Comput Biol. 2017;13(2):e1005335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chiu YC, Chen HH, Zhang T, Zhang S, Gorthi A, Wang LJ, et al. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med Genomics. 2019;12(Suppl 1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang T, Zhang L, Payne PRO, Li F. Synergistic Drug Combination Prediction by Integrating Multi-omics Data in Deep Learning Models. arXiv e-prints [Internet]. 2018. November 01, 2018. Available from: https://ui.adsabs.harvard.edu/abs/2018arXiv181107054Z. [DOI] [PubMed] [Google Scholar]
- 83.Zhou X, Wang M, Katsyv I, Irie H, Zhang B. EMUDRA: Ensemble of Multiple Drug Repositioning Approaches to improve prediction accuracy. Bioinformatics. 2018;34(18):3151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aliper A, Plis S, Artemov A, Ulloa A, Mamoshina P, Zhavoronkov A. Deep Learning Applications for Predicting Pharmacological Properties of Drugs and Drug Repurposing Using Transcriptomic Data. Mol Pharm. 2016;13(7):2524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang X, Jan LY. Targeting potassium channels in cancer. J Cell Biol. 2014;206(2):151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Molenaar RJ. Ion channels in glioblastoma. ISRN Neurol. 2011;2011:590249-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li F, Liu W, Yu H. Extraction of Information Related to Adverse Drug Events from Electronic Health Record Notes: Design of an End-to-End Model Based on Deep Learning. JMIR Med Inform. 2018;6(4):e12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jung K, Lependu P, Shah N. Automated Detection of Systematic Off-label Drug Use in Free Text of Electronic Medical Records. AMIA Jt Summits Transl Sci Proc. 2013;2013:94–8. [PMC free article] [PubMed] [Google Scholar]
- 89.Xu H, Aldrich MC, Chen Q, Liu H, Peterson NB, Dai Q, et al. Validating drug repurposing signals using electronic health records: a case study of metformin associated with reduced cancer mortality. J Am Med Inform Assoc. 2015;22(1):179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang GH, Satkunam M, Pond GR, Steinberg GR, Blandino G, Schunemann HJ, et al. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis. Cancer Epidemiol Biomarkers Prev. 2018;27(6):627–35. [DOI] [PubMed] [Google Scholar]
- 91.Trivedi HM, Panahiazar M, Liang A, Lituiev D, Chang P, Sohn JH, et al. Large Scale Semi-Automated Labeling of Routine Free-Text Clinical Records for Deep Learning. J Digit Imaging. 2019;32(1):30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. [DOI] [PubMed] [Google Scholar]
- 93.Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35 Suppl:S185–S98. [DOI] [PubMed] [Google Scholar]
- 95.Onyshchenko M The Puzzle of Predicting Response to Immune Checkpoint Blockade. EBioMedicine. 2018;33:18–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen K, Ye H, Lu XJ, Sun B, Liu Q. Towards In Silico Prediction of the Immune-Checkpoint Blockade Response. Trends Pharmacol Sci. 2017;38(12):1041–51. [DOI] [PubMed] [Google Scholar]
- 97.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18(1):248–62. [DOI] [PubMed] [Google Scholar]
- 98.Dawood Z, Coudray N, Kim RH, Nomikou S, Moran U, Weber JS, et al. Prediction of response and toxicity to immune checkpoint inhibitor therapies (ICI) in melanoma using deep neural networks machine learning. Journal of Clinical Oncology. 2018;36(15_suppl):9529-. [Google Scholar]
- 99.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nature Medicine. 2018;24:144. [DOI] [PubMed] [Google Scholar]
- 100.Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24(10):1545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wiesweg M, Mairinger F, Reis H, Goetz M, Walter RFH, Hager T, et al. Machine learning-based predictors for immune checkpoint inhibitor therapy of non-small-cell lung cancer. Annals of Oncology. 2019;30(4):655–7. [DOI] [PubMed] [Google Scholar]
- 102.Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. 2017;127(9):3472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci. 2018;1417(1):104–15. [DOI] [PubMed] [Google Scholar]
- 104.Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, et al. Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol. 2009;183(12):7631–4. [DOI] [PubMed] [Google Scholar]
- 105.Liu J, Blake SJ, Harjunpaa H, Fairfax KA, Yong MC, Allen S, et al. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016;76(18):5288–301. [DOI] [PubMed] [Google Scholar]
- 106.Han S, Toker A, Liu ZQ, Ohashi PS. Turning the Tide Against Regulatory T Cells. Front Oncol. 2019;9:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abu Eid R, Razavi GS, Mkrtichyan M, Janik J, Khleif SN. Old-School Chemotherapy in Immunotherapeutic Combination in Cancer, A Low-cost Drug Repurposed. Cancer Immunol Res. 2016;4(5):377–82. [DOI] [PubMed] [Google Scholar]
- 108.Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018;23(11):3262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–42. [DOI] [PubMed] [Google Scholar]
- 111.Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176(11):6752–61. [DOI] [PubMed] [Google Scholar]
- 112.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. Journal of immunology (Baltimore, Md : 1950). 2010;185(6):3190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Johnson TS, McGaha T, Munn DH. Chemo-Immunotherapy: Role of Indoleamine 2,3-Dioxygenase in Defining Immunogenic Versus Tolerogenic Cell Death in the Tumor Microenvironment. Adv Exp Med Biol. 2017;1036:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dhanak D, Edwards JP, Nguyen A, Tummino PJ. Small-Molecule Targets in Immuno-Oncology. Cell Chem Biol. 2017;24(9):1148–60. [DOI] [PubMed] [Google Scholar]
- 115.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103(35):13132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sorrentino R, Pinto A, Morello S. The adenosinergic system in cancer: Key therapeutic target. Oncoimmunology. 2013;2(1):e22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leone RD, Lo YC, Powell JD. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J. 2015;13:265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang H, Liu W, Liu Z, Ju Y, Xu M, Zhang Y, et al. Discovery of indoleamine 2,3-dioxygenase inhibitors using machine learning based virtual screening. Medchemcomm. 2018;9(6):937–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou Y, Peng J, Li P, Du H, Li Y, Li Y, et al. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors by virtual screening. Comput Biol Chem. 2019;78:306–16. [DOI] [PubMed] [Google Scholar]
- 120.Michielan L, Bolcato C, Federico S, Cacciari B, Bacilieri M, Klotz K-N, et al. Combining selectivity and affinity predictions using an integrated Support Vector Machine (SVM) approach: An alternative tool to discriminate between the human adenosine A2A and A3 receptor pyrazolo-triazolo-pyrimidine antagonists binding sites. Bioorganic & Medicinal Chemistry. 2009;17(14):5259–74. [DOI] [PubMed] [Google Scholar]
- 121.Shao Y-M, Ma X, Paira P, Tan A, Herr DR, Lim KL, et al. Discovery of indolylpiperazinylpyrimidines with dual-target profiles at adenosine A2A and dopamine D2 receptors for Parkinson’s disease treatment. PloS one. 2018;13(1):e0188212–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xuhan L, Kai Y, Herman VV, Adriaan PI, Gerard JP Vw. An Exploration Strategy Improves the Diversity of de novo Ligands Using Deep Reinforcement Learning – A Case for the Adenosine A2A Receptor2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tian S, Wang X, Li L, Zhang X, Li Y, Zhu F, et al. Discovery of Novel and Selective Adenosine A2A Receptor Antagonists for Treating Parkinson’s Disease through Comparative Structure-Based Virtual Screening. Journal of Chemical Information and Modeling. 2017;57(6):1474–87. [DOI] [PubMed] [Google Scholar]
- 124.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet. 2016;388(10043):518–29. [DOI] [PubMed] [Google Scholar]
- 125.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, et al. Control of the immune response by pro-angiogenic factors. Front Oncol. 2014;4:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013;73(2):539–49. [DOI] [PubMed] [Google Scholar]
- 128.Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, et al. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012;55(15):6832–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kang D, Pang X, Lian W, Xu L, Wang J, Jia H, et al. Discovery of VEGFR2 inhibitors by integrating naïve Bayesian classification, molecular docking and drug screening approaches. RSC Advances. 2018;8(10):5286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tsukamoto H, Fujieda K, Senju S, Ikeda T, Oshiumi H, Nishimura Y. Immune-suppressive effects of interleukin-6 on T-cell-mediated anti-tumor immunity. Cancer Sci. 2018;109(3):523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen X, Fu S, Tian J, Cao Y, Li C, Lin J. Abstract 864: Repurposing FDA-approved drug bazedoxifene as a novel inhibitor of IL-6 signaling for triple-negative breast cancer. Cancer Research. 2018;78(13 Supplement):864-. [Google Scholar]
- 133.Han H-W, Hahn S, Jeong HY, Jee J-H, Nam M-O, Kim HK, et al. LINCS L1000 dataset-based repositioning of CGP-60474 as a highly potent anti-endotoxemic agent. Scientific Reports. 2018;8(1):14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mills CD, Lenz LL, Harris RA. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 2016;76(3):513–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pairet N, Mang S, Fois G, Keck M, Kuhnbach M, Gindele J, et al. TRPV4 inhibition attenuates stretch-induced inflammatory cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS One. 2018;13(4):e0196055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tomar S, Zumbrun EE, Nagarkatti M, Nagarkatti PS. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J Pharmacol Exp Ther. 2015;353(2):369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kong D, Zhou C, Guo H, Wang W, Qiu J, Liu X, et al. Praziquantel Targets M1 Macrophages and Ameliorates Splenomegaly in Chronic Schistosomiasis. Antimicrob Agents Chemother. 2018;62(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bok E, Chung YC, Kim K-S, Baik HH, Shin W-H, Jin BK. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Experimental & molecular medicine. 2018;50(7):76-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gramatica P Principles of QSAR models validation: internal and external. QSAR & Combinatorial Science. 2007;26(5):694–701. [Google Scholar]
- 140.Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15(1):1929–58. [Google Scholar]
- 141.Angermueller C, Parnamaa T, Parts L, Stegle O. Deep learning for computational biology. Mol Syst Biol. 2016;12(7):878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sastry M, Lowrie JF, Dixon SL, Sherman W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. Journal of Chemical Information and Modeling. 2010;50(5):771–84. [DOI] [PubMed] [Google Scholar]
- 143.Rogers D, Hahn M. Extended-Connectivity Fingerprints. Journal of Chemical Information and Modeling. 2010;50(5):742–54. [DOI] [PubMed] [Google Scholar]
- 144.Deng Z, Chuaqui C, Singh J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein-Ligand Binding Interactions. Journal of Medicinal Chemistry. 2004;47(2):337–44. [DOI] [PubMed] [Google Scholar]
- 145.Da C, Kireev D. Structural Protein–Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. Journal of Chemical Information and Modeling. 2014;54(9):2555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, et al. The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data. 2016;3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sansone S-A, McQuilton P, Rocca-Serra P, Gonzalez-Beltran A, Izzo M, Lister AL, et al. FAIRsharing as a community approach to standards, repositories and policies. Nature Biotechnology. 2019;37(4):358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stathias V, Koleti A, Vidović D, Cooper DJ, Jagodnik KM, Terryn R, et al. Sustainable data and metadata management at the BD2K-LINCS Data Coordination and Integration Center. Scientific Data. 2018;5:180117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Abeyruwan S, Vempati UD, Küçük-McGinty H, Visser U, Koleti A, Mir A, et al. Evolving BioAssay Ontology (BAO): modularization, integration and applications. Journal of Biomedical Semantics. 2014;5(1):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vempati UD, Przydzial MJ, Chung C, Abeyruwan S, Mir A, Sakurai K, et al. Formalization, Annotation and Analysis of Diverse Drug and Probe Screening Assay Datasets Using the BioAssay Ontology (BAO). PLOS ONE. 2012;7(11):e49198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vempati UD, Schurer SC. Development and Applications of the Bioassay Ontology (BAO) to Describe and Categorize High-Throughput Assays In: Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, et al. , editors Assay Guidance Manual. Bethesda (MD)2004. [PubMed] [Google Scholar]
- 152.Lin Y, Mehta S, Kucuk-McGinty H, Turner JP, Vidovic D, Forlin M, et al. Drug target ontology to classify and integrate drug discovery data. J Biomed Semantics. 2017;8(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Edwards NJ, Oberti M, Thangudu RR, Cai S, McGarvey PB, Jacob S, et al. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J Proteome Res. 2015;14(6):2707–13. [DOI] [PubMed] [Google Scholar]
- 154.Armitage EG, Ciborowski M. Applications of Metabolomics in Cancer Studies. Adv Exp Med Biol. 2017;965:209–34. [DOI] [PubMed] [Google Scholar]
- 155.Johnson CH, Spilker ME, Goetz L, Peterson SN, Siuzdak G. Metabolite and Microbiome Interplay in Cancer Immunotherapy. Cancer Res. 2016;76(21):6146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17(9):557–69. [DOI] [PubMed] [Google Scholar]
- 157.Sarntivijai S, Lin Y, Xiang Z, Meehan TF, Diehl AD, Vempati UD, et al. CLO: The cell line ontology. J Biomed Semantics. 2014;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Visser U, Abeyruwan S, Vempati U, Smith RP, Lemmon V, Schurer SC. BioAssay Ontology (BAO): a semantic description of bioassays and high-throughput screening results. BMC Bioinformatics. 2011;12:257. [DOI] [PMC free article] [PubMed] [Google Scholar]