

Abstract
Summary
Proteomics is a powerful tool for protein expression analysis and is becoming more readily available to researchers through core facilities or specialized collaborations. However, one major bottleneck for routine implementation and accessibility of this technology to the wider scientific community is the complexity of data analysis. To this end, we have created ProVision, a free open-source web-based analytics platform that allows users to analyze data from two common proteomics relative quantification workflows, namely label-free and tandem mass tag-based experiments. Furthermore, ProVision allows the freedom to interface with the data analysis pipeline while maintaining a user-friendly environment and providing default parameters for fast statistical and exploratory data analysis. Finally, multiple customizable quality control, differential expression plots as well as enrichments and protein–protein interaction prediction can be generated online in one platform.
Availability and implementation
Quick start and step-by-step tutorials as well as tutorial data are fully incorporated in the web application. This application is available online at https://provision.shinyapps.io/provision/ for free use. The source code is available at https://github.com/JamesGallant/ProVision under the GPL version 3.0 license.
1 Introduction
Mass spectrometry-based shotgun proteomics is a powerful tool that allows researchers a means to investigate the proteome of an organism in an unbiased manner. However, the data analysis associated with proteomics often has a steep learning curve and thus presents a barrier for first-time users.
To address this, tools such as Perseus (Tyanova and Cox, 2018; Tyanova et al., 2016), LFQ-analyst (Shah et al., 2020) and various R packages (Choi et al., 2014; Gatto and Lilley, 2012; Gatto et al., 2015; Gierlinski et al., 2018) have been created. Perseus is currently a widely used companion tool for analysing data from the popular MaxQuant proteomics analysis platform (Cox and Mann, 2008). Perseus provides a wealth of functionality to interface with various label-free and label-based proteomics experiments. However, the proteomics data analysis pipeline can be daunting to newcomers and requires a significant time investment as it is not immediately evident which steps are required. Alternative R-based tools, such as Proteus, LFQ-analyst and MSstats (Choi et al., 2014) provide the usability of powerful open source tools from the R-language with a focus on allowing users to analyze data in an automated approach. However, either knowledge of the R-language is required or only data that incorporates the maxLFQ (Cox et al., 2014) algorithm is supported. Furthermore, automated data analysis provides an attractive option when data are routinely analyzed, but may not be beneficial on a per-use basis where altering key parameters can result in various statistical outcomes.
Here, we have created ProVision, a web-based and user-friendly proteomics data analysis platform for downstream analysis of MaxQuant output. The platform currently supports label-free data with and without the maxLFQ algorithm as well as tandem mass tag (TMT) data. Importantly, ProVision has been created to complement the reactive nature of the R-shiny web framework. Therefore, users can interact with important filtering and statistical parameters and view the effects in real time as the changes propagate through the platform. Default parameters are provided to guide unfamiliar users through the analytical steps, thereby addressing a potential learning curve for new users. In addition, ProVision aims to consolidate the proteomics data analysis workflow in one platform by providing built-in functionality to perform hypothesis tests, pathway and gene ontology enrichment using Webgestalt as well as protein–protein interactions using STRING (Liao et al., 2019; Szklarczyk et al., 2019; Wang et al., 2017). Using this platform, the time spent learning and performing proteomics data analysis is reduced and biologically relevant conclusions can be reached within one platform. ProVision thus provides an exploratory data analysis platform where users can gain insight into their data and tweak parameters when needed, reach biologically relevant conclusions through hypothesis testing and enrichment analysis as well as create custom figures with a high degree of control within the browser. This has the potential to dramatically decrease the turnaround time for proteomics experiments, resulting in faster conclusions and accelerated discovery.
2 Results
ProVision was created using the R-shiny web platform and styled with shinydashboard, shinyjs and shinywidgets as well as custom HTML/CSS and Javascript to create a user-friendly experience.
The platform requires the proteingroups.txt file from MaxQuant and processes data by extracting relevant columns based on the experiment type, while removing identifications flagged as contaminants. From this point, the data is fully reactive, and each filtering parameter has a default value that can be changed based on user preference. With this feature, it is possible to experiment with critical parameters that will affect the outcome, while simultaneously retaining statistical rigour. Thereby providing end-users with a dynamic data analysis pipeline for specific use cases. Multiple quality control plots, such as Q–Q plots (Fig. 1A), histograms (Fig. 1B), scatterplots (Fig. 1C), correlation heat maps (Fig. 1D) and principal component analysis (Fig. 1E) can be created.
Fig. 1.
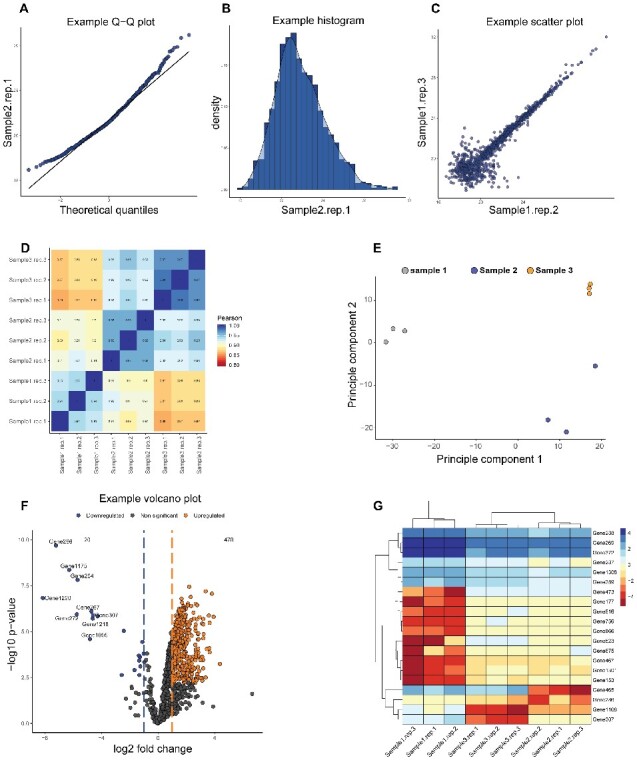
Representation of the various graphs that can be created by Provision using the tutorial data. Quality control plots include (A) Q–Q plots, (B) histograms, (C) scatterplots, (D) correlation heat maps and (E) principle component analysis. Main figures include (F) volcano plots and (G) heat maps. Multiple parameters of every plot can be customized to user preference
Key figures that can be created include volcano plots (Fig. 1F) and heat maps (Fig. 1G) from statistically significant data. Analysis of the protein lists can be further extended by performing gene set enrichments and over-representation analysis using Webgestalt (Liao et al., 2019; Wang et al., 2017). Furthermore, both known and predicted protein–protein interactions can be done within the application using the STRING database (Szklarczyk et al., 2019). These analyses are done directly on the platform and automatically integrates with all upstream data analysis. The plots are created by ggplot2 and thus have multiple customizable options. Furthermore, all plots can be exported to major file formats and can be exported to PDF for vector graphics and downstream processing. The statistical analysis is done using the Limma (Ritchie et al., 2015) package and is fully reactive as well. This allows for any statistical changes made to propagate to the volcano plots, heatmaps, Webgestalt enrichments and STRING networks in real time and update the display. The differential expression as well as the analyzed data can be downloaded in either Excel or text format, with a choice of various delimiters, for downstream analysis. Finally, no uploaded data are stored within the server thus creating a safe environment with the caveat that progress can be lost if the browser window is closed. Both quick start and full tutorials are available online and embedded within the application for users to access. ProVision is under continuous development with source code available to advanced users who would like to contribute or request specific features.
3 Conclusions and outlook
ProVision is an open source web application designed for ease of use and accessibility to newcomers for proteomics data analysis. ProVision aims to assist researchers to reach accurate conclusions based on their unique experimental designs, while providing high-quality customizable graphs and statistics in an intuitive environment. In addition, users can revisit their analysis and change parameters to gain the optimal output if necessary as well identify differentially regulated proteins, pathways and networks. This platform is deployed at https://provision.shinyapps.io/provision/ for general use and a development version is available at https://github.com/JamesGallant/ProVision for advanced users who would like to contribute to its development.
Funding
This work was supported by the NRF-VU Desmond Tutu Doctoral training program for financial assistance to JG. The authors acknowledge the SA MRC Centre for TB Research and DST/NRF Centre of Excellence for Biomedical Tuberculosis Research for financial support for this work. SLS is funded by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation (NRF) of South Africa, award number UID 86539.
Conflict of Interest: none declared.
Contributor Information
James Luke Gallant, Division of Molecular Biology and Human Genetics, Department of Biomedical Science, DST/NRF Centre of Excellence in Biomedical TB Research, SA MRC Centre for Tuberculosis Research, Faculty of Medicine and Health Science, Stellenbosch University, Tygerberg, Cape Town 7505, South Africa; Section Molecular Microbiology, Amsterdam Institute for Molecules, Medicines and Systems, Vrije Universiteit Amsterdam, Amsterdam 1081 HZ, The Netherlands.
Tiaan Heunis, Division of Molecular Biology and Human Genetics, Department of Biomedical Science, DST/NRF Centre of Excellence in Biomedical TB Research, SA MRC Centre for Tuberculosis Research, Faculty of Medicine and Health Science, Stellenbosch University, Tygerberg, Cape Town 7505, South Africa; Biosciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK.
Samantha Leigh Sampson, Division of Molecular Biology and Human Genetics, Department of Biomedical Science, DST/NRF Centre of Excellence in Biomedical TB Research, SA MRC Centre for Tuberculosis Research, Faculty of Medicine and Health Science, Stellenbosch University, Tygerberg, Cape Town 7505, South Africa.
Wilbert Bitter, Section Molecular Microbiology, Amsterdam Institute for Molecules, Medicines and Systems, Vrije Universiteit Amsterdam, Amsterdam 1081 HZ, The Netherlands; Department of Medical Microbiology and Infection Control, Amsterdam UMC, location VUmc, Amsterdam 1081 HZ, The Netherlands.
References
- Choi M. et al. (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics, 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
- Cox J. et al. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Gatto L. et al. (2015) Visualization of proteomics data using R and Bioconductor. Proteomics, 15, 1375–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto L., Lilley K.S. (2012) MSnbase—an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics, 28, 288–289. [DOI] [PubMed] [Google Scholar]
- Gierlinski M. et al. (2018) Proteus: an R package for downstream analysis of MaxQuant output, 10.1101/416511. [DOI]
- Liao Y. et al. (2019) WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res., 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E. et al. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A.D. et al. (2020) LFQ-analyst: an easy-to-use interactive web platform to analyze and visualize label-free proteomics data preprocessed with MaxQuant. J. Proteome Res., 19, 204–211. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D. et al. (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res., 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S. et al. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods, 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Cox J. (2018) Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer research. Methods Mol. Biol. (Clifton, N.J.), 1711, 133–148. [DOI] [PubMed] [Google Scholar]
- Wang J. et al. (2017) WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res., 45, W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]