

Abstract
Motivation
Short bioactive peptides encoded by small open reading frames (sORFs) play important roles in eukaryotes. Bioinformatics prediction of ORFs is an early step in a genome sequence analysis, but sORFs encoding short peptides, often using non-AUG initiation codons, are not easily discriminated from false ORFs occurring by chance.
Results
AnABlast is a computational tool designed to highlight putative protein-coding regions in genomic DNA sequences. This protein-coding finder is independent of ORF length and reading frame shifts, thus making of AnABlast a potentially useful tool to predict sORFs. Using this algorithm, here, we report the identification of 82 putative new intergenic sORFs in the Caenorhabditis elegans genome. Sequence similarity, motif presence, expression data and RNA interference experiments support that the underlined sORFs likely encode functional peptides, encouraging the use of AnABlast as a new approach for the accurate prediction of intergenic sORFs in annotated eukaryotic genomes.
Availability and implementation
AnABlast is freely available at http://www.bioinfocabd.upo.es/ab/. The C.elegans genome browser with AnABlast results, annotated genes and all data used in this study is available at http://www.bioinfocabd.upo.es/celegans.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Obtaining the complete inventory of protein-coding genes in sequenced genomes is a main goal in the current genomic age (Kersey et al., 2016). In silico prediction of protein-coding regions during genome annotation initially relied on start/stop codon position and ORF length to differentiate between a protein-encoding ORF from an ORF arising from chance occurrence. But such methods are generally not sufficiently robust for finding small exons or small protein-coding genes that were precluded from automatic annotation protocols (Chugunova et al., 2018; Li et al., 2000; Samayoa et al., 2011). It is becoming increasingly obvious that the diversity of short biologically active peptides has been underestimated. In fact, experimental approaches such as ribosome profiling and mass spectrometry, have revealed an increasing number of small proteins and peptides that elude in silico identification even in model organisms (Aspden et al., 2014; Calviello et al., 2016; Ingolia et al., 2009; Nesvizhskii, 2014; Raj et al., 2016). These peptides, unlike classical peptide hormones and neuropeptides which are translated as larger precursor proteins followed by proteolytic processing, are encoded directly from small open reading frames (sORFs) (Couso et al., 2017; Slavoff et al., 2013). The small size of sORFs (encoding proteins less than 100 amino acids in length), joined to the fact that some of these peptides start with a non-AUG initiation codon, make their in silico prediction even more complicated in eukaryotic, but also in prokaryotic genomes (Cao et al., 2020; Orr et al., 2020; Ruiz-Orera et al., 2019).
The need to discern functional sORFs from the predominant majority occurring by chance in the genome has propitiated the development of a number of bioinformatic tools to meet growing needs for accurate and reliable sORFs prediction (Dinger et al., 2008). Many of these computational approaches rely on common general principles including sequence or domain conservation, or codon and amino-acid preference metrics (reviewed in Chugunova et al., 2018, Dinger et al., 2008). However, the in silico identification of functional sORFs, even when combining these approaches to increase prediction accuracy, remains challenging (Pueyo et al., 2016).
The computational tool AnABlast has been developed as a reliable new approach for locating protein-coding regions in genomic DNA sequences of both, prokaryotes and eukaryotes (Jimenez et al., 2015; Rubio et al., 2019). This algorithm identifies putative protein-coding sequences independently of the presence of start–stop codons, and efficiently highlights very small protein-coding regions in genomic sequences that, at present, can only be uncovered in silico by this strategy (Casimiro-Soriguer et al., 2020; Jimenez et al., 2015). Thus, AnABlast could provide a different approach to predict sORFs encoding bioactive peptides. Here, we use this algorithm to scan the Caenorhabditis elegans genome in the search for new sORFs. This nematode has been established as a multicellular eukaryote model for the study of genetics and developmental biology, and its genome has been exhaustively annotated. Initial analysis of the complete genome sequence of C.elegans by the WormBase consortium revealed over 19 000 coding genes, but this number has been continuously increasing as a consequence of both, new experimental data and improved protein-coding gene prediction algorithms (Yoshimura et al., 2019). At present, the C.elegans genome sequence (WS228) available in the WormBase database predicts 24 610 coding genes (Dubaj Price et al., 2019), but the identification of sORFs still represents a difficult task. By analysing the entire C.elegans genome, here, we show that AnABlast is highly efficient in locating yet unknown intergenic sORFs, as well as new small exons of known genes in this model organism.
2 Materials and methods
2.1 AnABlast search strategy
AnABlast search for putative protein-coding sequences was used following described methods (Rubio et al., 2019) but analysing the complete genome. Due to the long length of the C.elegans chromosomes, they were used as the reference database in a similarity search using TBLASTN and the millions of protein sequences of non-redundant UniRef50 database (one entry per cluster searched) (2016_02 version) as query sequences (Altschul, 1997). Optimal parameters to identify protein-coding sequences were previously established (Casimiro-Soriguer et al., 2020). Briefly, to get non-restricted alignments, a threshold bit-score of 30 was used. Then, AnABlast takes the positions from the acquired hits and counts the number of alignments (belonging to different non-redundant proteins) that matches each genomic position. The similarity hits including low-scored alignments (short stretches of similarity, named protomotifs) are usually accumulated in coding sequences but rarely in non-coding sequences (Pérez et al., 2004; Thode et al., 1996). Thus, profile of accumulated AnABlast protomotifs yields peaks that accurately marks putative protein-coding regions even in the presence of sequencing errors, or in highly divergent/degenerated evolutionary sequences. To validate AnABlast accuracy for predicting sORFs in C.elegans, genomic sequences from ribosome profiling sORFs (Olexiouk et al. 2018) and curated small genes from UniProtKB entries were analysed in http://www.bioinfocabd.upo.es/ab/. In this validation, the number of accumulated alignments matching the assessed protein-coding sequence was used to estimate accuracy of prediction. sORFs as short as 9 amino acids were identified with a peak height threshold of 15 (Supplementary Table S1). However, in the genome-wide search of new putative intergenic sORFs, a restrictive peak height threshold of 70 was applied to minimize the risk of false positives (Casimiro-Soriguer et al., 2020). Under these conditions, curated proteins as short as 31 amino acids (the smallest peptide entry reported in UniProtKB for C.elegans) were identified.
2.2 In silico analysis of the selected sequences
For in silico analysis of the selected sequences, C.elegans annotations in the genomic regions of the candidates were downloaded from WormBase database at February 1, 2018. Annotations were gathered from the tracks of WormBase browser, including gene coordinates, RNA expression, proteomics and similarity sequences. Expression evidence is associated to an AnABlast candidate peak when reported RNA sequences extend at least 20% along the candidate sequence. To evaluate the uniqueness of AnABlast in searching sORFs, the new protein-coding sequences underlined by this program were subjected to other available gene finders (Alioto, 2012; Goodswen et al., 2012, Nachtweide et al., 2019), and those predicted also by any other gene finder tool (about 60%) were discarded. The amino-acid sequence delimited by each selected AnABlast peak was further studied by using BLAST (Altschul, 1997), Pfam for domain sequences (El-Gebali et al., 2019) and Sma3s for functional annotation (Casimiro-Soriguer et al., 2017).
2.3 Knock-down RNAi assay by feeding
In knock-down RNAi assay by feeding, the pL4440 plasmid was used. This plasmid is an E.coli vector that contains two T7 promoters surrounding the multicloning site. E.coli expressing the T7 polymerase generates double-stranded RNA of the DNA fragment contained in the polylinker. C.elegans feeding on E.coli producing this double-stranded RNA induces the degradation of the endogenous RNA. The RNAi clones used in this study were obtained from the selected DNA sequences underlined by AnABlast as putative protein-coding sequences. DNA fragments ranging between 0.2 and 0.4 kb were PCR amplified and cloned into a pL4440 vector by ligation after digestion with restriction enzymes. Oligonucleotides for each clone were designed to specifically target only the corresponding putative sORF in the C.elegans genome. All RNAi clones were verified by DNA sequencing. C.elegans strains were cultured and maintained using standard procedures (Stiernagle, 2006). Two different fragments of the unc-22 gene were used as positive control, one of 445 bp (including the exon 20) and the other of 362 bp (including the exon 23). L1 of N2 were synchronized in M9 buffer for 16 h at 20°C and seeded in plates with E.coli strains that carry either the empty vector pL4440 (control) or the AnABlast DNA sequence-targeting as previously described in the study by Kamath et al. (2003). Plates were incubated at 20°C and young adults were counted at 47–48 h each h for 6–7 h (N > 150). Normality of data was assessed prior to performing the t-test. The images were taken at 50 h in Olympus SZX16 stereoscope equipped with a PLAPO 1× lens and an Olympus DP73 camera.
3 Results
AnABlast uncovers protein-coding sequences through a new computational approach. This algorithm searches for protein-coding sequences by the significant abundance in databases of short stretches of amino acid sequences (protomotifs) found in virtually translated query DNA sequences (Jimenez et al., 2015; Rubio et al., 2019). Interestingly, AnABlast search is independent of protein-coding sequence length, of the reading frame and of start–stop codon signals, suggesting that this tool could be of particular use in the identification of sORF-encoded peptides.
3.1 AnABlast validation for the search of sORFs
To use this approach in the search of new sORFs, we first evaluated and verified accuracy of AnABlast in identifying intergenic sORFs using two independent sets of small protein-coding sequences reported in the nematode. In first place, a repository of small ORFs identified by ribosome profiling was used (Olexiouk et al., 2018, available at http://www.sorfs.org/database). Ribosome profiling identify ribosome-protected RNA fragments, thus identifying genomic regions with sORFs that have the potential to be translated (Ingolia et al., 2009). Most of them are found in different locations relative to protein-coding genes, including regulatory 5′- and 3′-UTRs mRNAs regions or overlapping main ORFs (Chugunova et al., 2018). At present, this repository stores 86 758 C.elegans entries, most of them (83 942) located at annotated genes. Intergenic sORF-encoding peptides are more resistant to identification. Intergenic sORFs account for only 120 entries. After removing redundant entries (identifying the same genomic interval), we obtained a set of putative 28 sORF-encoded peptides derived from ribosome profiling, which likely represent new intergenic sORFs in the nematode. As shown in Supplementary Table S1, a basic search for conserved motifs or similar proteins in databases indicates that 13 out of these 28 putative sORFs are evolutionary related to other known proteins. Except for one (arnold_n2_2014:505973), reported RNA sequence data also provide evidences that they likely identify functional sORFs. At present, three of them (arnold_c14_2014:229152, nedialkova_2015:204387 and nedialkova_2015:27) are annotated as curated genes in WormBase (see remarks in Supplementary Table S1). Importantly, with the exception of sORF arnold_n2_2014:505973, AnABlast highlighted all these sORFs as well, representing 92% (12/13) of sORFs entries encoding peptides evolutionary related to others in databases.
No similar sequences or motifs were found in the remaining 15 sORFs. AnABlast accumulates alignments in the proper DNA-coding strand and reading frame in 5 of these 15 sORFs (arnold_c14_2014:151752, hendriks_2014, hendriks_2014:658572, hendriks_2014:824117 and stadler_2012:54). Independent RNA-Seq data also provide evidences that all these five genomic regions encode sORFs (arnold_c14_2014:151752 and hendriks_2014:824117 are now curated genes in WormBase) (Supplementary Table S1). Albeit only ‘intergenic’ C.elegans sORFs were selected from the repository, 4 of the 11 sequences with no AnABlast peaks were located at the 5′-UTR (nedialkova_2015:412435, stadler_2012:256915), the 3′-UTR (stadler_2012:639775) and an intron (stadler_2012:136758). Thus, these sequences were removed from our analysis. Overall, AnABlast identifies around 45% (5/11) of putative intergenic sORFs with no significant sequence similarity or recognized sequence signatures.
In a second validation assay, we analysed the set of small proteins reported in the UniProtKB. This database accounts for 117 entries of C.elegans proteins with less than 100 residues, 73 of which are characterized proteins encoded by curated annotated genes in the nematode genome. As shown in Supplementary Table S2, near 92% (67/73) of the DNA regions coding for these small proteins accumulated AnABlast protomotifs in the proper coding strand and reading frame, a proportion of true positives similar to that found in canonical genes (Casimiro-Soriguer et al., 2020). Ribosome profiling sORF entries were also found in 59% (43/73) of the small protein-coding sequences, largely coincident with positive AnABlast identifications. Overall, 54% (40/73) were highlighted by both AnABlast and sORFs entries, 37% (27/73) by AnABlast only, 4% (3/73) by reported sORFs only and another 4% (3/73) lack both AnABlast peaks and reported sORFs.
Therefore, according to our validation results using the set of intergenic sORFs reported from ribosome profiling and the set of curated genes encoding small proteins, we conclude that AnABlast may be particularly helpful to underlie intergenic sORFs in silico.
3.2 Discovery of new protein-coding regions in the C.elegans genome
To underlie putative new sORFs in the C.elegans genome, AnABlast profiles were generated for the entire genome of this nematode. The complete set of AnABlast results from the C.elegans genome analysis, annotated genes, available expression data, the repository of sORFs entries and predictions from established gene-finder algorithms can be accessed at the genome browser http://www.bioinfocabd.upo.es/celegans.
As expected, the vast majority of AnABlast peaks were related to already annotated genes (see in the genomic browser), but a number of non-annotated AnABlast sequences, hopefully underlying putative new sORFs located at intergenic regions, were also found. Among them, sequences matching intergenic sORFs from the ribosome profiling repository and those also predicted by established gene finders were discarded, to show the AnABlast unique capability. Finally, to focus on the search of sORF sequences, only AnABlast peaks identifying sequences coding for less than 200 amino acids were selected for further analysis. Overall, 92 AnABlast regions were selected as new putative sORFs. Among them, 82 were distantly located to any annotated gene (arbitrarily, at more than 500 nucleotide), suggesting that these sequences could identify new intergenic sORFs (Supplementary Table S3), while the remaining 10 peaks were adjacent (less than 500 nucleotides) to annotated genes, which therefore could well be new small exons of known genes (Supplementary Table S4).
3.3 Characterization of putative novel C.elegans sORFs
Different approaches can be used to support predicted AnABlast sequences as functional sORFs. In a first approach, DNA sequences highlighted by AnABlast (proper strand and reading frame) were virtually translated, and conventional searches for motifs and similarities to reported proteins were performed. Since sORFs may initiate with start codons other than AUG (Cao et al., 2020; Hellens et al., 2016, Orr et al., 2020), predicted protein sequences lacking an initiation methionine were equally considered. AnABlast predicts coding sequences including those with non-AUG initiation codons, and displays the putative protein-coding sequence delimited by the peak, but unfortunately, it does not unequivocally identify the initiation codon. As shown in Supplementary Table S3, some of the identified sequences share stretches of significant similarity to proteins in reference databases, and/or match recognized motif signatures (El-Gebali et al., 2019; The UniProt Consortium, 2017). AnABlast peaks G71 and G75 are representative examples of predicted sORFs with both, motifs and significant homologous proteins found in related Caenorhabditis species (Fig. 1).
Fig. 1.
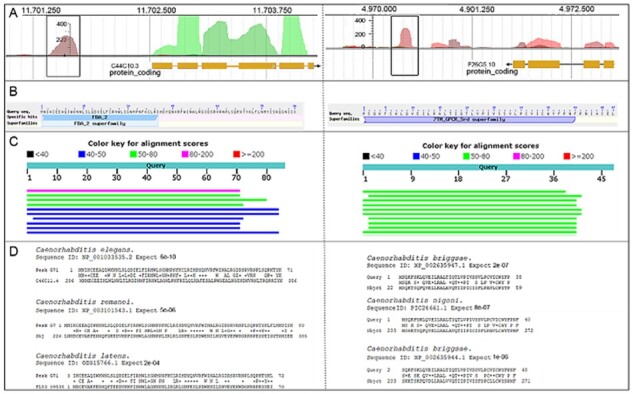
Putative new sORFs identified by AnABlast in peaks G71 (X:11701412.11701730) and G75 (V:4970301.4970493). (A) Peaks G71 (left) and G75 (right) (square boxes). Annotated exons of their respective adjacent genes C44C10.3 and F26G5.10 are shown (yellow boxes). (B) F-box associated FBA2-motif signature and serpentine-type 7TM GPCR chemoreceptor motif underlined in peaks G71 (left) and G75 (right), respectively. (C) Top 10 BLASTp hits of G71 (left) and G75 (right) protein sequences. (D) G71 (left) and G75 (right) protein sequence alignments to proteins found in the indicated Caenorhabditis sp. Sequence ID and alignment significance (expected E-value) are indicated
The WormBase database provides genome-wide data of Ribo-seq, RNA-Seq and proteomic experimental results. Therefore, we also analysed the existence of reported RNA and peptide sequences in the selected genomic regions to support the accuracy of AnABlast in searching for functional sORFs. Reported RNA-Seq data provide evidences of transcription in 18 of the proposed new sORFs. In one of them, evidences of translation from polysome data was also observed (see Supplementary Table S3 and the corresponding genomic intervals at the genome browser). Some of them showed either significant similarity to other proteins (G30, G45 and G54), harboured significant motifs (G26, G58), or both (G31) (Fig. 2).
Fig. 2.
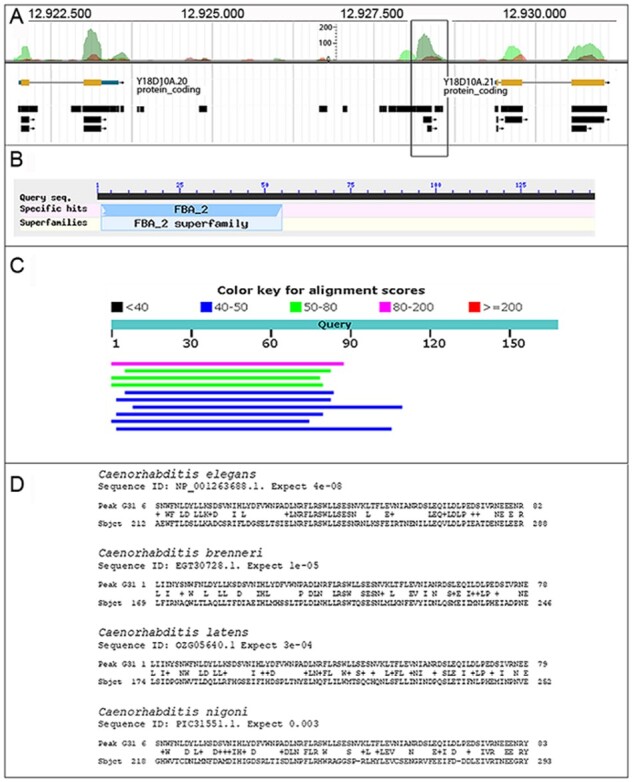
Putative new sORFs identified by AnABlast in peak G31 (I:12928143.12928563). (A) AnABlast profile of peak G31 (square) and annotated exons (yellow boxes) of the flanking genes Y18D10A.20 andY18D10A. RNA-Seq data (black boxes) available in WormBase are shown. (B) FBA2-Motif signature identified in predicted G31 protein sequence. (C) Top 10 BLASTp hits of G31 protein sequence. (D) G31 protein sequence alignments to proteins in the indicated Caenorhabditis sp. Sequence ID and alignment significance (expected E-value) are indicated
Evidence of expression itself (RNA-Seq and/or polysome data) is a useful clue for the identification of functional sORFs (Andrews et al., 2014). Remarkably, 12 out of 18 AnABlast-predicted coding regions showing significant evidences of transcription (RNA-Seq data) lack any detectable similarity or motif signature (Supplementary Table S3), reinforcing the potential use of AnABlast in searching for putative sORFs that escape to conventional in silico strategies.
3.4 Characterization of putative novel small exons of known genes
In the identified putative protein-coding sequences, 10 AnABlast peaks were located at less than 500 bp to the 5′ or to the 3′ end of known genes, suggesting that these coding regions could be small exons of the adjacent genes rather than intergenic sORFs (Supplementary Table S4). To explore this possibility, amino-acid sequences predicted by AnABlast and that of the adjacent gene were concatenated, and the resulting sequence used to BLAST search for homologous proteins in non-redundant protein data bases. When the new predicted exon belongs to the adjacent gene, homologous proteins expanding significant similarity along the added exon may be identified into the data bases. This approach suggested that at least E6, E13, E26 and E28 encode putative new exons of annotated genes (Fig. 3).
Fig. 3.
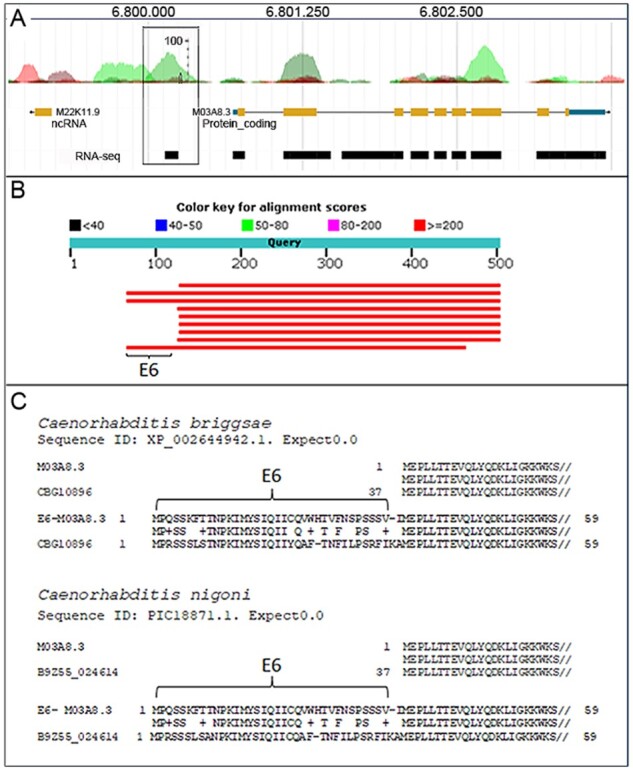
Putative new exon identified by AnABlast peak E6 (X:6799982.6800363). (A) AnABlast profiles showing peak E6 (square box) and the adjacent peaks matching exons encoding the M03A8.3 protein. RNA-Seq data (black boxes) available in WormBase are shown. (B) Top 10 hits in BLASTp search of concatenated E6-M03A8.3 sequences. E6 sequence is indicated. (C) Protein sequence alignment of the annotated M03A8.3 protein sequence (upper) and the concatenated E6-M03A8.3 protein sequence (lower) to protein CBG10896 (Sequence ID: XP_002644942.1) of C. briggsae and protein B9Z55_024614 (Sequence ID: PIC18871.1) of C.nigonias indicated. Alignment significance (expected E-value), and E6 amino-acid sequences are indicated
AnABlast E6 DNA region is located at the 5′ end of the gene encoding the hypothetical protein M03A8.3, a protein containing a PH-like domain. Remarkably, BLAST search of concatenated E6-M03A8.3 protein sequences indicates that a stretch of E6 expands the amino-terminal end of M03A8.3 protein homologs identified in C.briggsae and C.nigoni (Fig. 3). According to RNA sequences available in WormBase, DNA regions coding for both E6 and M03A8.3 are likely transcribed at similar levels in C.elegans (Fig. 3). Thus, we suggest that genomic DNA predicted by AnABlast to encode E6 is likely a new non-annotated exon of the gene encoding M03A8.3 in the C.elegans genome.
AnABlast-coding sequence E13 locates at the 5′ end of the gene encoding two protein isoforms, F20G2.3a and F20G2.3b. While F20G2.3a homologs containing E13 sequences were not found, the concatenated E13-F20G2.3b sequence was similar to protein F28H7.8 found in the parasitic nematode Toxocara canis, suggesting that E13 could be a putative exon of the F20G2.3b isoform (Fig. 4). Some weak intronic signatures can be predicted in the DNA sequence encoding E13. However, while RNA-Seq data provide evidences that both F20G2.3a and F20G2.3b DNA regions are transcribed, expression is not observed in the E13 genomic interval (neither RNA transcripts nor translated peptides). Thus, the E13 DNA sequence may represent a pseudo exon, rather than an actual exon of this gene.
Fig. 4.
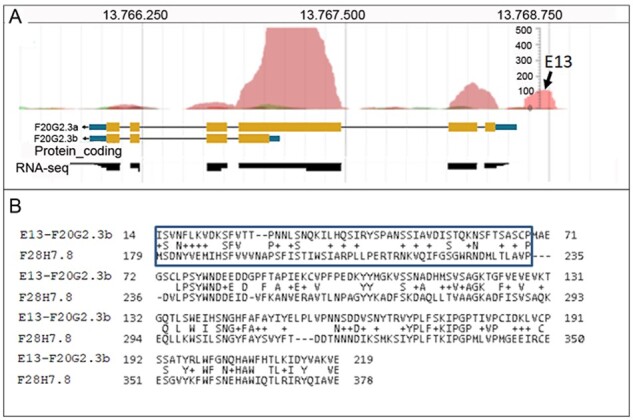
Putative new exon underlined by AnABlast peak E13 (V:13768591.13768795). (A) AnABlast profiles showing the peak E13 and adjacent peaks matching exons encoding F20G2.3a and F20G2.3b isoforms of the adjacent gene. RNA-Seq data (black boxes) available in WormBase are shown. (B) Sequence alignment of concatenated E13-F20G2.3b protein sequence to protein F28H7.8 (ID: KHN82554.1) (E-value: 1e-31) of the nematode Toxocara canis. E13 amino-acid sequence is indicated (square box)
Peaks E23 and E28 can also be identified in homologous proteins when concatenated to predicted protein sequences coded by their respective adjacent genes (Supplementary Fig. S1), suggesting that both are putative new exons of these hypothetical genes. Overall, these results suggest that AnABlast accurately identifies small protein-coding regions, either new sORFs or new small exons of known genes.
3.5 Functional analysis of putative new sORFs by RNA interference
AnABLast is better suited for situations where the predictions are tested by experimental validation. To support our intergenic sORFs search strategy, we carried out experimental RNAi interference to knock-down expression in all 82 identified intergenic sORF sequences (See Section 2 and Supplementary Table S5).
The nematode C.elegans is a genetically tractable model system that has been widely used to investigate the molecular mechanisms of aging and longevity, and the development of RNA interference (RNAi) technology has provided a powerful tool for performing large-scale genetic screens in this organism (Kamath et al., 2003). RNAi is an endogenous cellular mechanism triggered by double-stranded RNA (dsRNA), which leads to the degradation of homologous RNAs in the cytoplasm. RNAi is relatively easy to use since feeding worms with bacteria expressing dsRNA is enough to knockdown gene expression (Kamath et al., 2003). To gain insight into the function of the candidate sORFs provided by AnABlast, DNA sequences were designed to specifically target only the corresponding putative sORF in the C.elegans genome. Expression of all the selected DNA sequences were knocked down with RNAi, and developmental-associated phenotypes were examined. Importantly, three of the selected sequences (peaks G71, G98 and G107) yielded RNAi-dependent phenotypic defects, suggesting that at least these three DNA sequences likely identify functional intergenic sORFs.
Peak G71 encodes a predicted 86 amino acids peptide, with neither significant homologs in databases nor reported RNA expression (Supplementary Table S3). When this DNA sequence was knocked down with RNAi, a significant delay in the L4 to adult transition was observed (Fig. 5 and Supplementary Table S6). Detailed in silico analysis indicated that the 86 amino acid predicted sequence contains an F-box domain (Pfam: PF00646). About 326 C.elegans annotated genes contain known F-box sequences, indicating that it is a frequent domain in the worm genome. F-box bearing proteins usually bind SCF complexes, which in turn, function in the ubiquitination of cell cycle regulatory proteins (Kipreos et al., 2000). In C.elegans, however, the F-box domain is also found in FOG-2 proteins triggering spermatogenesis during development (Hu et al., 2019). According to the observed phenotype in peak G71 knocked down worms (Fig. 5), the proposed F-box-containing peptide could play a role in exit of the L4 state during the nematode development.
Fig. 5.
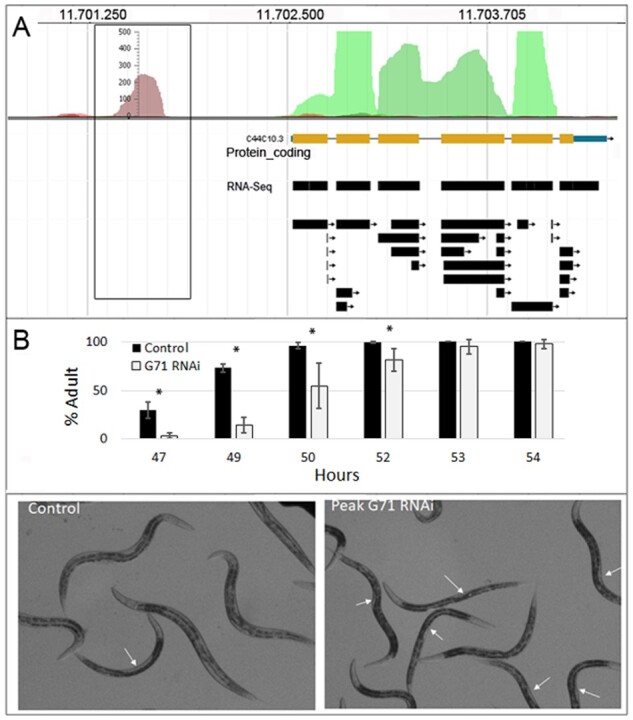
Analysis of putative sORF encoded by AnABlast peak G71 (X:11701412.11701730). (A) AnABlast profiles showing peak G71 (square box) and the adjacent peaks matching exons encoding protein C44C10.3. RNA-Seq data (black boxes) available in WormBase are shown. (B) Average worms (%) that reaches adulthood from L1 (time 0 h) in worms subject to E71 RNAi (grey) with respect to the control (black). Standard deviation bars are indicated. * means P ≤ 0,05. Photographs of the phenotype caused by G71 RNAi with respect to control at 49 h are shown (lower panels). Arrows indicate the typical non-mature vulva of L4 animals
Peak G98 encodes a putative 35 amino acids peptide without reported RNA expression, as well as without significant homologs (Supplementary Table S3). RNAi knockdown experiments of this sequence yielded a slight delay in development that can be visualized in the transition of the last developmental stage (L4 larval stage to adult) (Supplementary Fig. S2A and Table S6). The sequence of this putative peptide harbours a BEACH domain (Pfam: PF02138), a highly conserved motif which functions in lysosomal protein trafficking, but also endomembrane signalling during development (Khodosh et al., 2006). Thus, the putative BEACH-containing peptide encoded by AnABlast peak G98 could also affect different developmental steps.
Finally, peak G107 encodes a 38 amino acids peptide, which according to RNAi results could also play a role in development (detected during L4 state progression), with a percentage of animals that did not reach adulthood in the 8 h transition time from L4 to young adult (Supplementary Fig. S2B and Table S6). A BAH domain is found in the amino-acid sequence of this putative peptide (Supplementary Table S3). Proteins containing this domain are usually involved in chromatin remodelling, histone recognition (Yang et al., 2013) and proliferating cell nuclear antigen (PCNA) ubiquitination (Niimi et al., 2015). Therefore, there is a large repertoire of different functions in which the peptide encoded by this proposed new sORF could act during worm development.
RNAi degradation of mRNA is typically not complete, and only a fraction of the functional genes should be expected to develop a distinguishable phenotype using RNAi strategies. In C.elegans, only 19% of the functional protein-coding genes yield observable phenotypes when subjected to RNAi knockdown (Kamath et al., 2003, Lizabeth et al., 2015). In our characterization, this frequency would significantly drop down since our phenotypic assay only covers a limited set of developmental defects. Thus, the observation of RNAi-induced phenotypes in 3 out of 82 sequences assessed suggests that sequences highlighted by AnABlast efficiently uncover genomic regions encoding functional sORFs. Nonetheless, targeting non-coding sequences may occasionally cause phenotypic defects as well (Check, 2007; Xu et al., 2019) and further experimental approaches should be required to unequivocally support that our targeted sORF sequences by RNAi are protein coding.
Perhaps the most fundamental question that can be asked about a DNA sequence is whether or not it encodes protein. Canonical large ORFs are easily uncovered in silico by Gene-Finder algorithms, but finding small ORFs represents an extremely difficult task (Crappé et al., 2013, Kroll et al., 2017). AnABlast identifies sORFs as short as nine amino acids length in validation assays, where lower peak height thresholds can be assessed (Supplementary Table S1). However, in a genome-wide search strategy, improved specificity is required to avoid false positives, with the cost of a diminished sensitivity (Casimiro-Soriguer et al., 2020). Using optimal conditions to this end (see Section 2), AnABlast highlights new putative sOFRs coding for as short as 17 amino acids (Supplementary Table S3), but not below this length. Possibly, a minimal alignment length required for protomotifs accumulation in this strategy limits accuracy to underlie expected sORFs down this length. Nonetheless, our approach has led to the prediction of a set of putative sORFs and small exons in the C.elegans genome which are missed in other strategies.
With the advances in technology, notably ribosome profiling assays and mass spectrometry, the identification of functional small peptides has drastically increased, raising the number of functional sORFs within the eukaryotic genomes (Orr et al., 2020; Slavoff et al., 2013). Based on the remarkable property of AnABlast in highlighting small protein-coding regions, we believe that this computer approach may provide a powerful tool for the identification of elusive intergenic sORFs in sequenced genomes, complementing other in silico approaches (Hanada et al., 2010) as well as ribosome profiling/proteo-genomic methods.
Supplementary Material
Acknowledgements
The authors thank Genetics Group members at the Pablo de Olavide University for their useful comments on the manuscript, and Victor Carranco for technical assistance. They also thank to C3UPO for the HPC support.
Funding
This research was supported by the Ministry of Economy and Competitiveness of the Spanish Government [BFU2016-77297-P].
Conflict of Interest: none declared.
Contributor Information
C S Casimiro-Soriguer, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
M M Rigual, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
A M Brokate-Llanos, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
M J Muñoz, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
A Garzón, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
A J Pérez-Pulido, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
J Jimenez, Centro Andaluz de Biología del Desarrollo (CABD, UPO-CSIC), Universidad Pablo de Olavide, 41013 Sevilla, Spain.
References
- Alioto T. (2012) Gene prediction. Methods Mol. Biol. (Clifton, N.J.), 855, 175–201. [DOI] [PubMed] [Google Scholar]
- Altschul S.F. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S.J. et al. (2014) Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet., 15, 193–204. [DOI] [PubMed] [Google Scholar]
- Aspden J.L. et al. (2014) Extensive translation of small open reading frames revealed by Poly-Ribo-Seq. Elife, 3, e03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calviello L. et al. (2016) Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods, 13, 165–170. [DOI] [PubMed] [Google Scholar]
- Cao X. et al. (2020) Non-AUG start codons: expanding and regulating the small and alternative ORFeome. Exp. Cell. Res., 391, 111973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro-Soriguer C.S. et al. (2017) Sma3s: a universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics, 17, 1700071. [DOI] [PubMed] [Google Scholar]
- Casimiro-Soriguer C.S. et al. (2020) Ancient evolutionary signals of protein-coding sequences allow the discovery of new genes in the Drosophila melanogaster genome. BMC Genomics, 21, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugunova A. et al. (2018) Mining for small translated ORFs. J. Proteome Res., 17, 1–11. [DOI] [PubMed] [Google Scholar]
- Check E. (2007) RNA interference: hitting the on switch. Nature, 448, 855–858. [DOI] [PubMed] [Google Scholar]
- Couso J.P. et al. (2017) Classification and function of small open reading frames. Nat. Rev. Mol. Cell. Biol., 18, 575–589. [DOI] [PubMed] [Google Scholar]
- Crappé J. et al. (2013) Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC Genomics, 14, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger M.E. et al. (2008) Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput. Biol., 4, e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubaj Price M. et al. (2019) WormBase: a model organism database. Med. Ref. Serv. Q., 38, 70–80. [DOI] [PubMed] [Google Scholar]
- El-Gebali S. et al. (2019) The Pfam protein families database in 2019. Nucleic Acids Res., 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodswen S.J. et al. (2012) Evaluating high-throughput ab initio gene finders to discover proteins encoded in eukaryotic pathogen genomes missed by laboratory techniques. PLoS One, 7, e50609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K. et al. (2010) sORF finder: a program package to identify small open reading frames with high coding potential. Bioinformatics, 26, 399–400. [DOI] [PubMed] [Google Scholar]
- Hellens R.P. et al. (2016) The emerging world of small ORFs. Trends Plant Sci., 21, 317–328. [DOI] [PubMed] [Google Scholar]
- Hu S. et al. (2019) Multi-modal regulation of C. elegans hermaphrodite spermatogenesis by the GLD-1-FOG-2 complex. Dev. Biol., 446, 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T. et al. (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science, 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez J. et al. (2015) AnABlast: a new in silico strategy for the genome-wide search of novel genes and fossil regions. DNA Res., 22, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath R.S. et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature, 421, 231–237. [DOI] [PubMed] [Google Scholar]
- Kersey P.J. et al. (2016) Ensembl genomes 2016: more genomes, more complexity. Nucleic Acids Res., 44, D574–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodosh R. et al. (2006) Bchs, a BEACH domain protein, antagonizes Rab11 in synapse morphogenesis and other developmental events. Development, 133, 4655–4665. [DOI] [PubMed] [Google Scholar]
- Kipreos E.T. et al. (2000) The F-box protein family. Genome Biol., 1, Reviews 3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll J.E. et al. (2017) A tool for integrating genetic and mass spectrometry-based peptide data: proteogenomics viewer: PV: a genome browser-like tool, which includes MS data visualization and peptide identification parameters. Bioessays, 39, 1700015. [DOI] [PubMed] [Google Scholar]
- Li W. et al. (2000) Saturated BLAST: an automated multiple intermediate sequence search used to detect distant homology. Bioinformatics, 16, 1105–1110. [DOI] [PubMed] [Google Scholar]
- Lizabeth A. et al. (2015) The transgenic RNAi project at Harvard Medical School: resources and validation. Genetics, 201, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtweide S. et al. (2019) Multi-genome annotation with AUGUSTUS. Methods Mol. Biol., 1962, 139–160. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A.I. (2014) Proteogenomics: concepts, applications and computational strategies. Nat. Methods, 11, 1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A. et al. (2015) The BAH domain of BAF180 is required for PCNA ubiquitination. Mutat. Res., 779, 16–23. [DOI] [PubMed] [Google Scholar]
- Olexiouk V. et al. (2018) An update on sORFs.org: a repository of small ORFs identified by ribosome profiling. Nucleic Acids Res., 46, D497–D502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr M.W. et al. (2020) Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Res., 48, 1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez A.J. et al. (2004) AnaGram: protein function assignment. Bioinformatics, 20, 291–292. [DOI] [PubMed] [Google Scholar]
- Pueyo J.I. et al. (2016) New peptides under the s(ORF)ace of the genome. Trends Biochem. Sci., 41, 665–678. [DOI] [PubMed] [Google Scholar]
- Raj A. et al. (2016) Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling. Elife, 5, e13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio A. et al. (2019) AnABlast: re-searching for protein-coding sequences in genomic regions. Methods Mol. Biol., 1962, 207–214. [DOI] [PubMed] [Google Scholar]
- Ruiz-Orera J. et al. (2019) Translation of small open reading frames: roles in regulation and evolutionary innovation. Trends Genet., 35, 186–198. [DOI] [PubMed] [Google Scholar]
- Samayoa J. et al. (2011) Identification of prokaryotic small proteins using a comparative genomic approach. Bioinformatics, 27, 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff S.A. et al. (2013) Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol., 9, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. (2006) Maintenance of C. elegans. WormBook, 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res., 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thode G. et al. (1996) Search for ancient patterns in protein sequences. J. Mol. Evol., 42, 224–233. [DOI] [PubMed] [Google Scholar]
- Xu T. et al. (2019) Gene amplification-driven long noncoding RNA SNHG17 regulates cell proliferation and migration in human non-small-cell lung cancer. Mol. Ther. Nucleic Acids, 17, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N. et al. (2013) Structure and function of the BAH domain in chromatin biology. Crit. Rev. Biochem. Mol. Biol., 48, 211–221. [DOI] [PubMed] [Google Scholar]
- Yoshimura J. et al. (2019) Recompleting the Caenorhabditis elegans genoma. Genome Res., 29, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.