

Abstract
Purpose:
We hypothesized that the combination of a local stimulus for activating tumor-specific T cells and an anti-immunosuppressant would improve treatment of gliomas. Virally encoded IL15Rα-IL15 as the T-cell activating stimulus and a prostaglandin synthesis inhibitor as the anti-immunosuppressant were combined with adoptive transfer of tumor-specific T cells.
Experimental Design:
Two oncolytic poxviruses, vvDD vaccinia virus and myxoma virus, were each engineered to express the fusion protein IL15Rα-IL15 and a fluorescent protein. Viral gene expression (YFP or tdTomato Red) was confirmed in the murine glioma GL261 in vitro and in vivo. GL261 tumors in immunocompetent C57BL/6J mice were treated with vvDD-IL15Rα-YFP vaccinia virus or vMyx-IL15Rα-tdTr combined with other treatments, including vaccination with GARC-1 peptide (a neoantigen for GL261), rapamycin, celecoxib, and adoptive T-cell therapy.
Results:
vvDD-IL15Rα-YFP and vMyx-IL15Rα-tdTr each infected and killed GL261 cells in vitro. In vivo, NK cells and CD8+ T cells were increased in the tumor due to the expression of IL15Rα-IL15. Each component of a combination treatment contributed to prolonging survival: an oncolytic virus, the IL15Rα-IL15 expressed by the virus, a source of T cells (whether by prevaccination or adoptive transfer), and prostaglandin inhibition all synergized to produce elimination of gliomas in a majority of mice. vvDD-IL15Rα-YFP occasionally caused ventriculitis-meningitis, but vMyx-IL15Rα-tdTr was safe and effective, causing a strong infiltration of tumor-specific T cells and eliminating gliomas in 83% of treated mice.
Conclusions:
IL15Rα-IL15-armed oncolytic poxviruses provide potent antitumor effects against brain tumors when combined with adoptive T-cell therapy, rapamycin, and celecoxib.
Introduction
The requirements for successful immunotherapy of solid tumors are still being delineated. In preclinical models, single modalities are sometimes effective. Adoptive transfer of engineered neoantigen-specific T cells can be effective as a single treatment modality for flank-located, established solid tumors (1). Likewise, the presence of IL15 in the local tumor microenvironment can, as a single treatment modality modality, eradicate solid flank fibrosarcomas (2, 3). The requirements for treating tumors within the brain are likely to be more stringent. The immunologic characteristics of brain tumors are different from other cancers, even if they originate as metastatic tumors from other tissues. For example, tumors within the brain have deficient dendritic cell (DC) function, increased regulatory T cells (Treg), and increased TGFβ compared with tumors located in the periphery (4). A comparison of primary lung cancer and brain metastases within the same patients found more PD-L1 expression and fewer CD8+ T cells in the brain metastases compared with primary lung tumors (5). With mouse B16-OVA cells placed in the brain or in the flank, adoptively transferred tumor-infiltrating lymphocytes (TIL) isolated from the brain tumors reduced lytic activity compared to TILs isolated from flank tumors (6). The presence of a tumor within the brain can cause a redistribution of adoptively transferred T cells, sequestering T cells in the bone marrow (7).
We hypothesize that an immunotherapy for gliomas might be more successful if it includes (i) expansion of T cells that recognize tumor neoantigens, either by adoptive transfer or vaccination; (ii) attraction of T cells to the tumor and creation of inflammation to allow extravasation; (iii) attraction of natural killer (NK) cells complementary to T cells; and (iv) reversal of factors that suppress antigen-presenting cells, T cells, and NK cells. We have already observed that these factors have a small impact on survival of glioma-bearing mice when used individually. We hypothesize that when these attributes are combined, they will synergize and eradicate tumors.
The GL261 glioma model has a tumor-specific neoantigen, GARC-1 (a mutation in the IMP3 gene), a peptide that can be presented by mouse MHCH2-Db and recognized by T cells in C57BL/6J mice (8,9). This antigen has previously been targeted by a conventional vaccine strategy, which, when combined with an anti-TGFb antibody, resulted in prolonged survival (10). To attract and activate T cells and NK cells, we chose viral delivery of an IL15Rα-IL15 superagonist fusion protein to the tumor. IL15 has long been recognized as a promising cytokine for cancer treatment (11). However, systemically administered IL15 has had considerable toxicity in both animal studies and clinical trials (12). Local expression of the cytokine mediated by an oncolytic virus could be safer and more effective than systemic delivery. To reverse immunosuppression, we targeted prostaglandin E2 using celecoxib, a cyclooxygenase-2 (COX-2) inhibitor. Prostaglandin E2 (PGE-2), the product of COX-2 activity, has multiple immunosuppressive actions. It (i) causes dendritic cells to induce Tregs (13); (ii) induces and maintains myeloid-derived suppressor cells (MDSC; ref. 14); and (iii) directly inhibits the ability of antigen-specific T cells to kill their tumor targets (15). Tumors induce COX-2 expression in infiltrating myeloid cells, shifting them to M2 macrophages and MDSCs (16). The COX-2 inhibitor celecoxib has little or no effect on gliomas as a single treatment (17), but it enhances immunotherapies against gliomas (17-22). To increase the infection efficiency of tumor cells by either vaccinia virus or myxoma virus, we included rapamycin treatment (23-27). Rapamycin is a complex drug (28): under certain conditions, it can be immunosuppressive (29), but in other cases, it can enhance antigen presentation by DCs (30) and promote survival of antigen-specific CD8+ T cells (31).
In this study, we combined adoptive cell transfer (ACT) of T cells with one of two oncolytic poxviruses: double-deleted (vvDD) vaccinia virus or myxoma virus (vMyx). vvDD is an attenuated oncolytic poxvirus with enhanced tumor selectivity due to deletion of thymidine kinase and vaccinia growth factor (32). vvDD-IL15Rα-YFP effectively treats colon cancer in mice when combined with anti-PD-1 (33). In humans, it was found to be safe when delivered either intravenously or intratumorally in phase I clinical trials (34, 35). However, the safety of intracranial delivery of vvDD is still uncertain. In rats, no deaths or neurologic symptoms were noted when non-tumor-bearing rats were treated with vvDD-EGFP, whereas vvDD-EGFP was extremely toxic to tumor-bearing rats (24). This finding indicates the importance of further evaluation of the safety profile of vvDD in the brain.
Myxoma virus (vMyx) is another promising oncolytic poxvirus, which is restricted to lagomorphs in nature (36). Despite its narrow host range, vMyx can productively infect and kill cancer cells from several species including human (26, 37) and mouse (23, 25, 27), making vMyx a potential candidate for oncolytic virotherapy. The safety of vMyx has been demonstrated in both immunocompromised and immunocompetent murine preclinical trials with different tumors including glioma (38), medulloblastoma (39), melanoma (25, 27), pancreatic cancer (40), and myeloma (41). We engineered both vvDD and myxoma virus to express an IL15Rα-IL15 fusion protein. Co-expression of IL15 with its receptor α (IL15 superagonist) generates a more potent ligand compared with IL15 alone (42, 43).
Materials and Methods
Generation of vvDD-IL15Rα-YFP
The generation of vvDD-IL15Rα-YFP was described previously (33). Briefly, the murine IL15Rα-IL15 gene, which was from plasmid pBS-IL15Rα-IL15-tdTomatoRed, was cloned into pCMS-IRES to form a new shuttle vector pCMS-IL15-Rα. CV-1 cells were infected by vSC20, a Western Reserve (WR) strain vaccinia virus with a deletion in vaccinia growth factor (vgf), followed by transfection of shuttle vector pCMS-IL15-Rα which contains YFP gene (33). Homologous recombination allows genes of IL15Rα-IL15 and YFP from the shuttle vector to be inserted into the thymidine kinase (TK) gene within the vSC20 genome, leading to the generation of the double deletion (vgf-, TK-) vvDD-IL15Rα-YFP vaccinia virus. The new virus was selected by expression of YFP. Control viruses expressed the chemokine CXCL11 or red fluorescent protein (RFP).
Generation of vMyx-IL15Rα-tdTr
Generation of vMyx-IL15Rα-tdTr was described previously (44). Briefly, the recombinant virus expressing the IL15Rα-IL15 fusion protein (vMyx-IL15Rα-tdTr) was created by homologous recombination in RK-13 cells infected with wild type (WT) vMyx-Lau followed by transfection with the engineered recombination vector pBS-IL15Rα-IL15-tdTomatoRed. After the recombinant virus was plaque-purified away from parental virus, recombinant virus expressing IL15Rα-IL15 fusion protein was propagated and titrated by focus formation on RK-13 cells. Fluorescent virus foci were harvested, repropagated, and titrated on RK-13 cells. This process was repeated three times to isolate a purified virus which contains two exogenous genes, IL15Rα-IL15 fusion protein and tdTomato. Genomic structure of the recombinant virus was confirmed by PCR sequencing. Control virus expressed tdTomato but not IL15Rα-IL15.
Cell culture
Glioma 261 (GL261) is a murine glioma cell line. GL261 WT cells were purchased from the National Cancer Institute Division of Cancer Treatment and Diagnosis Repository (NCI-DCTD) and tested negative for mycoplasma. Cells were cultured in DMEM (10013CV, Corning Incorporated) containing 5 mmol/L HEPES, 1.3 mmol/L L-glutamine, 50 μmol/L 2-ME, penicillin, streptomycin, and 10% FBS at 37°C and 5% CO2. GL261 neurosphere (GL261 NS) cells, which are cancer stem-like nonadherent cells, were cultured in untreated cell culture flasks (08-757-501, Corning Incorporated) with DMEM/F12+GlutaMAX (10565018, Life Technologies) culture medium containing penicillin/streptomycin (17-602E, Lonza), B27 with vitamin A (17504044, Life Technologies), 20 ng/mL recombinant human epidermal growth factor (EGF; 236-EG-200, R&D Systems), 20 ng/mL recombinant human fibroblast growth factor (FGF; 233-FB-025/CF, R&D Systems), and 5 mg/mL heparin (H3149100KU, Sigma-Aldrich) in an incubator at 37°C with 5% CO2 (45).
Viral killing and infection in vitro
A total of 2 × 105 GL261 WT cells were cultured per well of a 24-well plate, then infected with PBS, vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP (DMEM culture medium with 2% FBS) at multiplicity of infection (MOI) 5 for 1 hour, then switched to DMEM culture medium with 10% FBS. At 24, 48, and 72 hours postinfection, viable cells were counted using a hemacytometer (Hausser Scientific), followed by flow cytometry analysis of tdTomato Red (vMyx-tdTr and vMyx-IL15Rα-tdTr), RFP (vvDD-RFP), and YFP (vvDD-IL15Rα-YFP) to identify the infection of different viruses using BD LSR Fortessa Flow Cytometry Analyzer (BD Biosciences). Brightfield and fluorescent images of GL261 WT cells were taken 48 hours after virus infection with an Olympus CKX41 microscope (Olympus Life Science).
ELISA analysis of murine IL15Rα-IL15 fusion protein
A total of 2 × 105 GL261WT cells were cultured per well of a 24-well plate, then infected with PBS, vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP (DMEM culture medium with 2% FBS) at MOI 5 for 1 hour, then switched to DMEM culture medium with 10% FBS. At 48 hours postinfection, supernatants of cells from each treatment were collected, followed by brief centrifugation to remove cellular debris. Supernatants were diluted 1:100, 1:1,000, and 1:10,000 followed by the measurement of murine IL15Rα-IL15 fusion protein with Mouse IL15/IL15R Complex Uncoated ELISA kit (88721522, Invitrogen), according to the manufacturer's instructions.
Rapamycin effect on viral infection
A total of 2 × 105 GL261WT cells were cultured per well of a 24-well plate for 2 hours before virus inoculation in the DMEM culture medium containing 10% FBS with or without 100 nmol/L rapamycin (LC Laboratories). Cells were then inoculated with vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP at MOI 5 in DMEM medium containing 2% FBS with or without 100 nmol/L rapamycin for 1 hour. Then, cells were cultured with fresh DMEM (10% FBS) medium with or without 100 nmol/L rapamycin for 2 days at 37°C with 5% CO2. Cells were fixed and analyzed for tdTomato red (vMyx-IL15Rα-tdTr) or YFP (vvDD-IL15Rα-YFP) expression by flow cytometry using BD LSR Fortessa Flow Cytometry Analyzer (BD Biosciences).
Animals
C57BL/6J mice were purchased from The Jackson Laboratory and maintained as colonies in the animal facilities at the University of Illinois Urbana-Champaign (Urbana, IL). Mice of both sexes were used in experiments when they were 2 to 3 months old. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Illinois Urbana-Champaign.
Intracranial tumor establishment
GL261 NS cells were harvested, washed twice with Hanks' Balanced Salt Solution (HBSS; 21023CV, Corning Incorporated) and stereotaxically infused into the brains of mice anesthetized with isoflurane (59399-106-01, Akorn). A total of 5 × 104 GL261 NS cells in 0.5 μL HBSS were infused into ventral striatum (0.5 mm rostral; 2.25 mm lateral; 3.3 mm ventral). Mice were euthanized at 75% of baseline body weight or when they exhibited symptoms of neurologic impairment, lethargy, or pain, in accordance with IACUC guidelines.
Intratumoral virus injection
Tumor-bearing mice were anesthetized and stereotaxically injected intratumorally with approximately 2 × 106 plaque-forming unit (pfu) vvDD-IL15Rα-YFP, vvDD-CXCL11, vvDD-RFP vaccinia virus, vMyx-tdTr, vMyx-IL15Rα-tdTr, or PBS control in 1 μL PBS.
Emulsion vaccination
The emulsion vaccine for each mouse contained 50 μg GARC-1 peptide (AALLNKLYA, GenScript) and 50 μg CpG ODN (tlrl-1826-1, InvivoGen) in 50 μL PBS mixed with same volume of Incomplete Freund's adjuvant (IFA; F5506–10 mL, Sigma-Aldrich). Reagents were sonicated intermittently and emulsified through a double needle. One hundred microliters of emulsified vaccine was injected subcutaneously.
TriVax vaccination
TriVax vaccination was used to generate GARC-1–specific T cells for adoptive transfer, and in separate experiments for therapeutic effect. The TriVax vaccine consists of 200 μg peptide GARC-1, 50 μg anti-mouse CD40 monoclonal antibody (Clone FGK4.5/FGK45; BE0016-2, Bio X Cell), and 50 μg poly-IC (vac-pic, InvivoGen). This vaccine was injected retro-orbitally to each mouse in 200 μL PBS. For the booster immunization, a quarter of the same dose was injected retro-orbitally to each mouse in 50 μL PBS.
Adoptive CD8+ T-cell transfer
CD8+ T-cell donor mice were injected with 200 μL TriVax retro-orbitally followed by 50 μL TriVax 10 days later as a booster. Splenocytes from donor mice spleens were prepared by mechanical tissue dissociation through nylon mesh followed by ammonium chloride potassium (ACK) buffer lysis of erythrocytes. CD8+ T cells, isolated from these splenocytes by Dynabeads Untouched Mouse CD8 Cells Kit (11417D, Invitrogen), were resuspended with HBSS. An average of 5 × 106 CD8+ T cells in 200 μL HBSS was injected retro-orbitally to recipient mice.
Oral drug delivery
Cookie powder was made by pulverizing cookies (Oreo cookies without the cream) using a mortar and pestle. Cookie powder with medication was made by combining 16 g of plain cookie powder with 40 mg of rapamycin (LC Laboratories) with or without 100 mg of celecoxib (0093-7306-06, TEVA Pharmaceuticals). Each tablet was made by compressing 100 mg of powder in a manual tablet press. The daily dose was 10 mg/kg rapamycin and 25 mg/kg celecoxib. Mice were singly housed and bedding was inspected to ensure that the cookies were eaten. Blood levels of celecoxib were assayed by HPLC-MS.
Tissue sectioning and histology
After mice were euthanized, brains were snap-frozen in OCT embedding medium (23-730-571, Fisher Healthcare) for cryosectioning. Five-micron cryosections were fixed in cold 95% ethanol for 20 minutes and blocked with Superblock (37515, Thermo Fisher Scientific) for 30 minutes, followed by overnight incubation with primary antibodies, which were diluted in 5% Superblock in 20% glycerol PBS. The slides were then washed with PBS and incubated with secondary antibodies for 2 hours. Slides which were incubated with a biotinylated secondary antibody were washed with PBS and incubated with streptavidin-fluorophore for 1 hour. Finally, slides were stained with 4′,6-diamidino-2-phenylindole (DAPI; D1306, Life Technologies). Control slides omitted primary antibody.
Antibodies for different markers are listed below. Primary antibodies: rabbit anti-M-T7 (Myxoma; generated in the lab of Dr. Grant McFadden), rabbit anti-vaccinia (Vaccinia; YVS8101, Accurate Chemical & Scientific Corporation), rat anti-mouse Ly-49G2 (NK cell; 555314, BD Pharmingen), rat anti-mouse CD8a (CD8+ T cells; 14080882, Invitrogen), rat anti-mouse FOXP3 (Treg; 14477180, Invitrogen), biotinylated anti-mouse IL15 (IL15Rα-IL15; BAF447, R&D Systems). Secondary antibodies: donkey anti-rabbit-Alexa Fluor 647 (Myxoma, Vaccinia; 711606152, Jackson ImmunoResearch), streptavidin-Alexa Fluor 647 (IL15Rα-IL15; 016600084, Jackson ImmunoResearch), biotinylated rabbit anti-rat antibody (NK cells, CD8+ T cells, Treg; BA4001, Vector). Tertiary reagent: streptavidin-Alexa Fluor 647 (NK cells, CD8+ T cells, Treg; 016600084, Jackson ImmunoResearch).
At least six sections were assessed and the section with the largest extent of tumor was evaluated by manually counting stained cells by an investigator blind to treatment condition. As a check of the reliability of using a single section, pairs of sections 100 μm apart from 9 mice were stained and NK cells counted; values obtained on paired sections were highly correlated (r = 0.97, P < 0.05). Some 5-μm cryosections were fixed in cold 95% ethanol overnight and stained with hematoxylin and eosin.
Analysis of tumor-infiltrating lymphocytes
Mice with GL261 NS tumors were injected intratumorally with 2 × 106pfus of vMyx-IL15Rα-tdTr or PBS control 11 days after tumor infusion. An additional control group had no brain tumor. Virus treated tumor-bearing mice also received rapamycin and celecoxib daily, and two days after the virus treatment, they received adoptive transfer of T cells from donor mice vaccinated with TriVax GARC-1 (GARC-1 peptide, anti-CD40, and Poly-IC) 7 days before harvest. The transferred cells included approximately 5 × 105 GARC-1–specific CD8+ T cells (8% of the transferred CD8+ cells), determined by staining with GARC-1-Db dimers (DimerX; 551323, BD Biosciences). Treated mice received a TriVax GARC-1 injection one day following the adoptive transfer. Six days after virus injection and four days after adoptive transfer, mice were euthanized; the right striatum was dissected and mechanically dissociated with a Biomasher II homogenizer (199623, Research Products International Corp) in 500 μL OPTI-MEM media. Following ACK treatment to lyse RBCs, lymphocytes were enriched on a Percoll step gradient (70%, 37%, and 30% Percoll, collecting cells at the 37/70 interface). Cells were analyzed by flow cytometry for anti-CD8 and GARC-1-Db DimerX binding.
Flow cytometry
For flow cytometry analysis of CD8 and IFNγ protein levels, 1 × 106 cells/well were transferred to a 96-well round-bottom plate (3799, Corning Incorporated) and restimulated with GARC-1 peptide for one hour. Then Brefeldin A (BFA; 420601, BioLegend) was added to each well with the final concentration of 5 μg/mL followed by 3-hour incubation. Anti-mouse CD8a Alexa 488 antibody (100723, BioLegend) was added to each well after cells were washed with cell staining buffer (420201, BioLegend) followed by a 0.5-hour incubation on ice. Fixation buffer (420801, BioLegend) was added to each well after another round of cell wash followed by a 20-minute incubation at room temperature. Then, cells were washed and resuspended with intracellular staining permeabilization wash buffer (421002, BioLegend) before undergoing an overnight incubation with anti-mouse IFNγ Alexa 647 antibody (505814, BioLegend) at 4°C. On the following day, cells were washed with intracellular staining permeabilization wash buffer and resuspended in cell staining buffer before analysis of Alexa 488 and 647 on the Accuri C6 Flow Cytometry Analyzer (BD Biosciences).
For flow cytometry analysis of GARC-1–specific CD8+ T cells, an immunofluorescence staining protocol of DimerX (551323, BD Biosciences) was followed. Briefly, GARC-1 peptide was loaded to H-2Db: Ig protein followed by overnight incubation at 37°C. On the following day, PE Rat Anti-mouse IgG1 (550083, BD Biosciences) was incubated with the mixture for 1 hour at room temperature in the dark. Then Purified Mouse IgG1 λ Isotype Control was added, followed by a 1-hour incubation at room temperature in the dark to form the staining cocktail. At the same time, cells were incubated with Mouse BD Fc Block (553142, BD Biosciences) and anti-mouse CD8a Alexa 647 (100724, Biolegend) for 10 minutes on ice in the dark. Then GARC-1-DimerX staining cocktail was added to each sample followed by 1-hour incubation on ice in the dark. Afterwards, cells were washed and resuspended with 0.5% PBS/BSA before analysis of PE and Alexa 647 on the Accuri C6 Flow Cytometry Analyzer (BD Biosciences).
Statistical analysis
GraphPad Prism software was used for all statistical analyses and graph presentation. Numbers of NK cells, CD8+ T cells, and viable GL261 cells were analyzed by one-way ANOVA, using the Sidak correction for multiple comparisons. The percentage of GARC-1–specific CD8+ T cells was analyzed by Student t test. Survival data were recorded from the time of the tumor cell implantation until euthanasia and were plotted using a Kaplan–Meier curve. Survival treatment groups were compared with a log-rank (Mantel–Cox) test. Significance was considered P < 0.05.
Results
GL261 cells can be infected and lysed by vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP and express virally encoded protein in vitro
To verify the susceptibility of GL261 cells to infection and lysis by vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP, GL261 WT cells were incubated with each virus at an MOI of 5 in vitro. At 24, 48, and 72 hours postinfection, viable cells (Trypan blue excluding) were counted using a hemacytometer; infected cells were analyzed for fluorescent protein expression as an indicator of viral infection. Brightfield and fluorescent images of infected cells were taken at 48 hours postinfection. Results showed that GL261 WT cells were killed by all four types of viruses (Fig. 1A and C). vvDD-RFP or vvDD-IL15Rα-YFP most effectively killed GL261 WT cells. In addition, GL261 WT cells expressed virally encoded fluorescent protein after virus inoculation (Fig. 1B and C): tdTomato Red (vMyx-tdTr, vMyx-IL15Rα-tdTr), RFP (vvDD-RFP), and YFP (vvDD-IL15Rα-YFP). These data showed that GL261 cells can be infected, express virally-encoded protein, and are lysed by vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP in vitro. IL15Rα-IL15 is virally encoded in vMyx-IL15Rα-tdTr and vvDD-IL15Rα-YFP. To determine whether infected GL261 WT cells can express and secrete IL15Rα-IL15 in vitro, supernatants of GL261 WT cells infected by the four different viruses (MOI 5, 48 hours postinfection) were collected and analyzed by ELISA against IL15Rα-IL15. Supernatant from cells infected by vMyx-tdTr or vvDD-RFP did not show detectable IL15Rα-IL15, whereas there was a significant increase in IL15Rα-IL15 production in the supernatant from cells infected by vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP (Fig. 1D). The production of IL15Rα-IL15 from vvDD-IL15Rα-YFP–treated cells was 20-fold more than the production from vMyx-IL15Rα-tdTr–treated cells, which further indicates that GL261 WT cells are more susceptible to vvDD-IL15Rα-YFP than vMyx-IL15Rα-tdTr. Interestingly, the production of IL15Rα-IL15 may decrease the oncolytic effect and slow virus infection rate for vMyx-IL15Rα-tdTr compared with vMyx-tdTr. However, vvDD was not affected by expression of IL15Rα-IL15. The rationale for this phenomenon needs further exploration.
Figure 1.
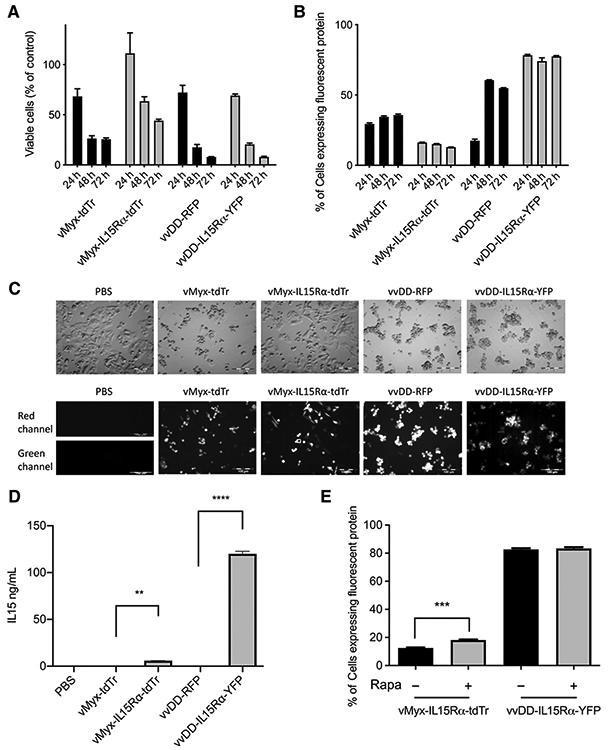
In vitro characterization of vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, and vvDD-IL15Rα-YFP. A total of 2 × 105 GL261WT cells were cultured per well of a 24-well plate, then infected with PBS, vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP at MOI 5 for 24, 48, or 72 hours. A, Viable cell counts. B, Flow cytometry analysis of tdTomato Red (vMyx-tdTr and vMyx-IL15Rα-tdTr), RFP (vvDD-RFP), and YFP (vvDD-IL15Rα-YFP). Mean values and SEM are shown. C, Brightfield and fluorescence images of GL261 WT cells were taken 48 hours after virus infection. Red channel: tdTomato Red (vMyx-tdTr and vMyx-IL15Rα-tdTr), RFP (vvDD-RFP); green channel: YFP (vvDD-IL15Rα-YFP). Scale bar, 100 μm. D, Supernatants of cells from each treatment were collected 48 hours after virus infection. The production of IL15Rα-IL15 was measured by ELISA. Mean ELISA values and SEM are shown. There was a significant increase in IL15Rα-IL15 expression in the supernatants of vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP–treated cells compared with vMyx-tdTr or vvDD-RFP–treated cells. P values: **, < 0.01; ****, < 0.0001. E, GL261 WT cells were treated with or without 100 nmol/L rapamycin 2 hours prior to the 1-hour inoculation of vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP at MOI 5. Cells were fixed and analyzed for tdTomato red (vMyx-IL15Rα-tdTr) or YFP (vvDD-IL15Rα-YFP) expression by flow cytometry at 48 hours p.i. Mean ± SEM, n = 3, P value: ***, < 0.001.
Rapamycin has the potential to enhance infection of vMyx and vvDD virus on GL261 model
Previous work has shown that rapamycin enhances the infection of various types of human and murine cancer cells by myxoma virus and vaccinia virus (23-27, 39, 46). We therefore assessed the effect of rapamycin on infection of GL261 by vMyx or vvDD. GL261 WT cells were treated in the presence or absence of 100 nmol/L rapamycin, added 2 hours prior to the 1-hour inoculation of vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP at an MOI of 5. Cells were fixed and analyzed for tdTomato red (vMyx-IL15Rα-tdTr) or YFP (vvDD-IL15Rα-YFP) by flow cytometry analysis at 48 hours postinfection (p.i.). The significant increase of percentage of tdTomato red positive cells (Fig. 1E) indicates that rapamycin enhances the infection of vMyx-IL15Rα-tdTr on GL261 cells, whereas under these conditions rapamycin did not show any effect on vvDD-IL15Rα-YFP in GL261 cells. However, Lun and colleagues (24) found that rapamycin enhanced vvDD infection of rat F98 and RG2 glioma cell lines at lower MOIs. On the basis of these findings, we included rapamycin in the following survival experiments with both vMyx and vvDD vaccinia virus.
GL261 NS tumors can be infected and express virally-encoded protein in vivo and IL15Rα-IL15 superagonist carried by vMyx or vvDD enhances immune cell infiltration
We examined whether gliomas initiated from GL261 NS can be infected and express virally encoded protein in vivo and whether the IL15Rα-IL15 superagonist can enhance immune responses by attracting NK cells and CD8+ T cells to the tumor location. Glioma tumors were established by direct intracranial injection of 5 × 104 GL261 NS cells/mouse into the right striatum. After 10 days, mice received intratumoral injection of PBS, vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFP (2 × 106 pfu/mouse). Tissue slides of brains harvested 5 days after intratumoral injection were sectioned and stained with antibodies anti-M-T7 (a vMyx-encoded protein), YVS8101 anti-vaccinia, anti-Ly-49G2 (an NK-cell marker), anti-CD8, or anti-IL15. Compared with PBS-injected tumor, the M-T7 positive signal shown in vMyx-tdTr or vMyx-IL15Rα-tdTr–treated tumor indicates that vMyx infects GL261 NS cells in vivo and infected cells express vMyx-encoded protein (Fig. 2A). Compared with PBS-injected tumor, the vaccinia positive signal (Fig. 2A) shown in vvDD-RFP or vvDD-IL15Rα-YFP–treated tumor and the IL15-positive signal (Supplementary Fig. S1) shown in vvDD-IL15Rα-YFP–injected tumor, indicate that vvDD infects GL261 NS cells in vivo and the infected cells express vvDD-encoded protein.
Figure 2.
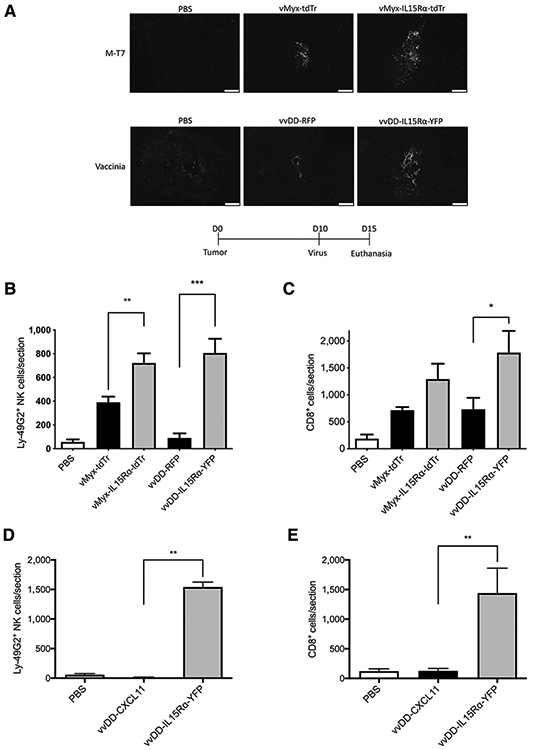
In vivo characterization of vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, and vvDD-IL15Rα-YFP. C57BL/6J mice were implanted with GL261 NS cells intracranially followed 10 days later by injectionwith PBS, vMyx-tdTr, vMyx-IL15Rα-tdTr, vvDD-RFP, or vvDD-IL15Rα-YFPfor A-C, and PBS, vvDD-CXCL11, or vvDD-IL15Rα-YFP for D and E (n = 3;2 × 106 pfu intratumoral). Mice were euthanized 5 days after the virus treatment and tumor sections were analyzed for presence of the viruses, NKcells, and CD8+ T cells by immunostaining for M-T7 (a myxoma-encoded protein), vaccinia virus, Ly-49G2 (4D11 antibody) for NK cells and CD8, respectively. Representative tumor sections are shown. A, Staining for vMyx or vvDDvirus in tumors. Scale bar, 200 μm. B, Numberof Ly-49G2+ NKcells per tumor section for each condition, mean values and SEM are shown. One-way ANOVA showed significant increase in NK cell accumulation in vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP–treated tumors compared with vMyx-tdTr or vvDD-RFP treatments. P values: **, < 0.01; ***, < 0.001. C, Number of CD8+ cells per tumor section for each condition. Mean values and SEM are shown. One-way ANOVA showed significant increase in CD8+ cell accumulation in vvDD-IL15Rα-YFP–treated tumors compared with vvDD-RFP treatment. P value: *, < 0.05. D, Number of Ly-49G2+ NK cells per tumor section for each condition. Mean values and SEM are shown. One-way ANOVA showed significant increase in NK-cell accumulation in vvDD-IL15Rα-YFP–treated tumors compared with both vvDD-CXCL11 and PBS treatments. P value: **, < 0.01. E, Number of CD8+ cells per tumor section for each condition. Mean values and SEM are shown. One-way ANOVA showed significant increase in CD8+ cell accumulation in vvDD-IL15Rα-YFP–treated tumors compared with both vvDD-CXCL11 and PBS treatments. P value: **, < 0.01.
To explore the physiologic function of IL15Rα-IL15 superagonist expressed in tumors in vivo, mouse brain sections from different treatment groups were stained with anti–Ly-49G2 or anti-CD8 antibodies. There was a significant increase of tumor-infiltrated Ly-49G2+ NK cells if the tumor was treated with vMyx-IL15Rα-tdTr or vvDD-IL15Rα-YFP, compared with tumor treated with viruses without IL15Rα-IL15 (vMyx-tdTr or vvDD-RFP; Fig. 2B). There was also a significant effect of vvDD-IL15Rα-YFP treatment on tumor-infiltrating CD8+ T cells compared with vvDD-RFP treatment (Fig. 2C). The same trend was evident comparing vMyx-IL15Rα-tdTr and vMyx-tdTr but was short of significance (P < 0.10). This might be due to small sample size (n = 3) or to the 20-fold higher IL15Rα-IL15 production when tumor cells were treated with vvDD-IL15Rα-YFP compared with vMyx-IL15Rα-tdTr (Fig. 1D).
In a separate experiment, we compared the ability of vvDD-IL15Rα-YFP and vvDD-CXCL11 to attract NK cells and CD8+ T cells into the brain, because we had previously found vvDD-CXCL11 attracts CD8+ T cells to murine mesothelioma tumors (47). vvDD-IL15Rα-YFP again produced a dramatic infiltration of NK and T cells into the tumor (Fig. 2D and E; Supplementary Fig. S1). Surprisingly, with vvDD-CXCL11 treatment, there were fewer T cells in the brain than after treatment with the vvDD expressing only RFP (Fig. 2C compared to Fig. 2E), and mostly as perivascular cuffs (Supplementary Fig. S1). While confirming the efficacy of the IL15Rα-IL15 superagonist, this result reflects the complexity of the role of the CXCL11/CXCR3 axis in modulating T-cell infiltration into the brain, as described in studies of experimental autoimmune encephalomyelitis (48).
vvDD-IL15Rα-YFP as a single treatment shows limited ability to prolong survival of glioma-bearing mice
We tested the hypothesis that the attraction of NK cells and CD8+ T cells by IL15Rα-IL15 could lead to a significant survival benefit using vvDD-IL15Rα-YFP. Glioma-bearing mice received intratumoral administration of vvDD-IL15Rα-YFP 7 days after tumor implantation. Results showed a prolonged median survival for the vvDD-IL15Rα-YFP treatment group compared to the control group, 42 days compared with 32 days, respectively (Supplementary Fig. S2). This difference was short of statistical significance (P = 0.055), indicating the limited efficacy of oncolytic vvDD-IL15Rα-YFP as a single treatment.
Tumor-specific CD8+ T cells are generated by vaccination
One possible explanation for the limited efficacy is that a large portion of the CD8+ T cells in the tumor may not be tumor specific; instead, they may be naïve CD8+ T cells attracted to inflammation without the ability to kill tumor cells. Thus, we hypothesized that generating more tumor-specific CD8+ T cells might improve the treatment with oncolytic vMyx and vvDD.
One way to generate more tumor-specific CD8+ T cells is to vaccinate animals with a tumor-specific antigen. To test this idea, a vaccine combining GARC-1 peptide (a GL261 neoantigen), CpG oligodeoxynucleotide1826 (CpG ODN) and Incomplete Freund′s adjuvant (IFA) was emulsified and injected subcutaneously in left flank followed by a booster injection 16 days later. Five days after the booster injection, cells from axillary and brachial lymph nodes on the left side and spleens were harvested and exposed to GARC-1 peptide. The significantly increased number of IFNγ-releasing CD8+ T cells in the lymph nodes or the spleen (Fig. 3A and B) produced in response to GARC-1 exposure indicates that tumor-specific CD8+ T cells were successfully generated by vaccination.
Figure 3.
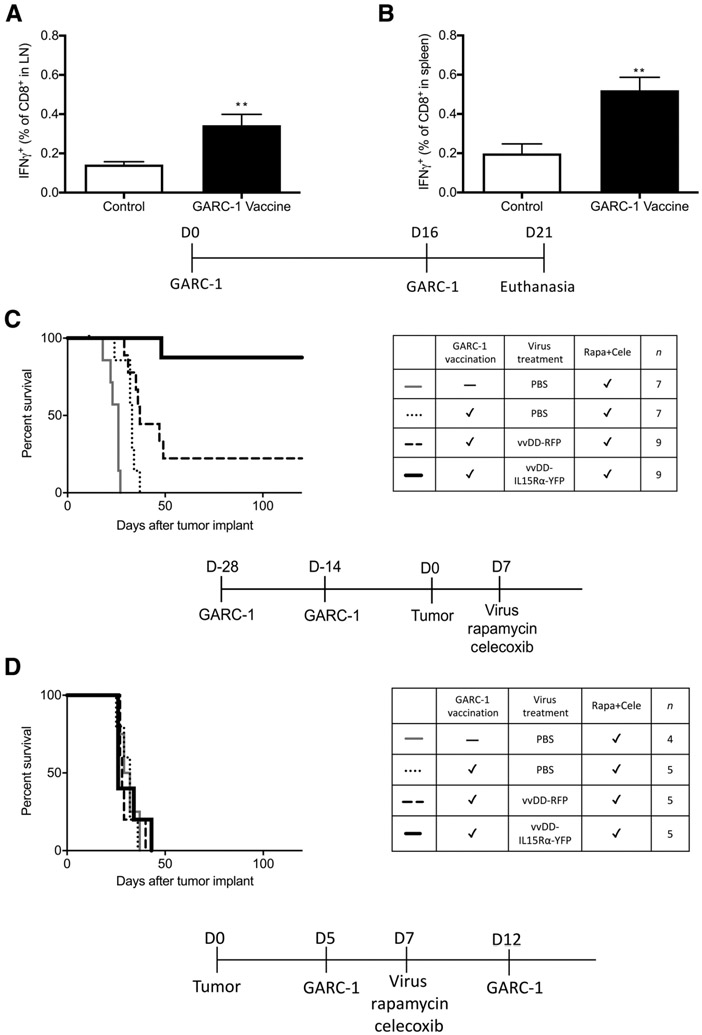
Tumor antigen-specific CD8+ T cells are generated by emulsion vaccination and have the potential to prolong survival of C57BL/6J mice bearing intracranial GL261 NS tumors treated with intratumoral vvDD-IL15Rα-YFP virus, rapamycin, and celecoxib. The vaccine combining 50 μg GARC-1 peptide, 50 μg CpG ODN in 50 μL PBS, mixed with 50 μL IFA for each mouse was emulsified by sonication and injected subcutaneously in left flank (100 μL) followed by a booster injection 16 days later. Five days after the booster injection, cells from axillary and brachial lymph nodes on the left side and spleens were harvested and exposed to GARC-1 peptide followed by flow cytometry analysis of CD8 and IFNγ. A, Lymph node percentage of GARC-1–specific CD8+ T cells among all CD8+ T cells. Mean and SEM from 5 mice per group are shown. **, P < 0.01 for vaccinated group versus control group. B, Spleen percentage of GARC-1–specific CD8+ T cells among all CD8+ T cells. Mean and SEM from 5 mice per group are shown. **, P < 0.01 for vaccinated group versus control group. C, Survival of tumor-bearing mice treated with prevaccination of GARC-1, intratumoral vvDD-IL15Rα-YFP, rapamycin, and celecoxib. Mice received GARC-1 emulsion vaccine on day −28 and −14; tumor on day 0; and 1 μL virus (2 × 106 pfu) or PBS injection, rapamycin, and celecoxib on day 7 (medication treatment continued until day 73). Mice that received the combination treatment lived significantly longer than other groups. P < 0.01 for vvDD-IL15Rα-YFP–treated group compared with each of the other groups. Prevaccinated vvDD-RFP and prevaccinated PBS were also different from unvaccinated PBS-treated mice, P < 0.05. Prevaccinated vvDD-RFP differed from prevaccinated PBS, P < 0.05. D, Survival of tumor-bearing mice treated with postvaccination with emulsion vaccine of GARC-1, intratumoral vvDD-IL15Rα-YFP, rapamycin, and celecoxib. Mice received tumor implantation on day 0; GARC-1 emulsion vaccine on day 5 and 12; and 1 μL virus (2 × 106 pfu) or PBS injection, rapamycin, and celecoxib on day 7 (medication treatment continued until day 73). There was no significant difference between any treatment groups.
Prevaccination with GARC-1 peptide together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib provides a survival benefit to mice with intracranial tumors
After observing that tumor-specific CD8+ T cells can be generated by vaccination, we hypothesized that these tumor-specific CD8+ T cells together with vvDD-IL15Rα-YFP virus, rapamycin, and celecoxib treatment could provide a survival benefit for glioma-bearing mice. To test this hypothesis, vaccination was completed twice before a tumor was established. Virus was injected into the tumor a week after tumor implantation, and a tablet containing rapamycin and celecoxib was fed to the mice. Tablets were administered for 66 days starting from the day of virus injection. Blood celecoxib concentration was 21 ± 3 μmol/L at 2 hours after feeding (n = 9). The survival results (Fig. 3C) show that prevaccination of GARC-1 together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib treatment cured 87% of mice with brain glioma. Data from Fig. 3C also imply that GARC-1 prevaccination and vvDD infection without IL15Rα-IL15 are not sufficient by themselves; IL15Rα-IL15 is an important element of the treatment.
To mimic the clinical situation where patients are treated after they are diagnosed with a tumor, the vaccination of GARC-1 was conducted after tumor implantation (postvaccination) followed by the same combination treatment. Results of postvaccination of GARC-1 peptide showed that there was no significant survival benefit (Fig. 3D). One possible reason for this result is that the emulsion vaccine may require a longer time for mice to generate sufficient numbers of tumor-specific CD8+ T cells; by the time the immune system has the ability to respond against the emulsion vaccine, the tumor has become too advanced to destroy. If this is the case, then finding a way to increase the number of GARC-1–specific CD8+ T cells within a short period of time would be crucial to the success of this combination treatment.
TriVax vaccination of GARC-1 together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib provides a survival benefit to mice with intracranial tumors
TriVax vaccination, which consists of peptide, poly-IC, and anti-CD40 antibody, was reported to yield a high amount of peptide-specific CD8+ T cells within a short period of time (49). We injected the TriVax vaccine with GARC-1 peptide retro-orbitally and blood was taken to perform flow cytometry analysis in order to detect GARC-1–specific CD8+ T cells using a DimerX Db probe (Fig. 4). TriVax and emulsion experiments shown in Fig. 4A and B were conducted on different days, so they were not compared statistically, but it is apparent that vaccination with TriVax produces a higher percentage of tumor-specific CD8+ T cells than the emulsion vaccine.
Figure 4.
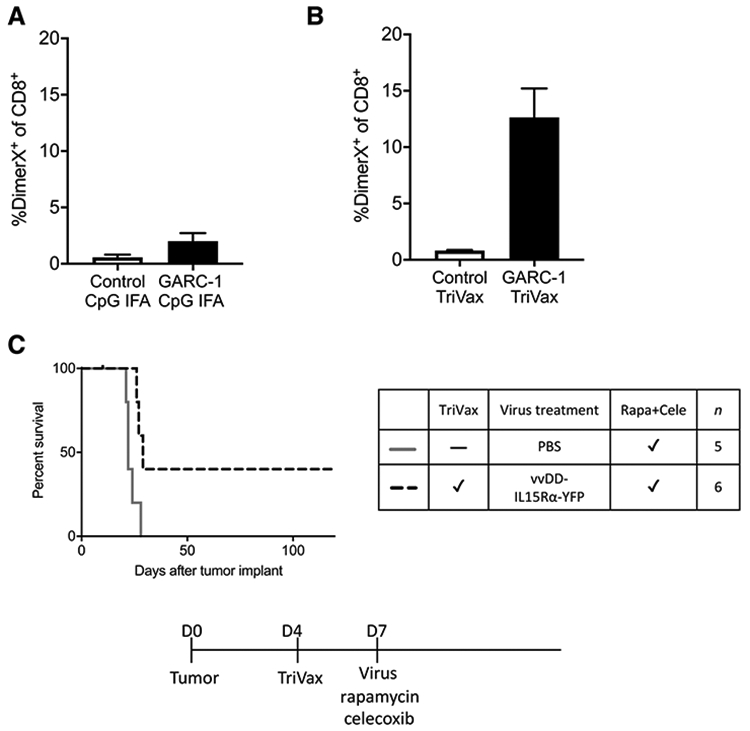
Improved response to TriVax vaccination. Mice were vaccinated by emulsion vaccine subcutaneously or TriVax vaccine retro-orbitally. Blood was collected and lysed by ACK, then cells from blood were stained with DimerX Db-GARC-1 (recognizing GARC-1–specific CD8+ T cells) and anti-CD8 antibody followed by flow cytometry analysis. Mean and SEM, n = 3. A, The percentage of GARC-1–specific CD8+ T cells generated by emulsion vaccine among all CD8+ T cells in the blood. B, The percentage of GARC-1–specific CD8+ T cells generated by TriVax vaccine among all CD8+ T cells in the blood. C, Survival of tumor-bearing mice treated with TriVax vaccination, intratumoral vvDD-IL15Rα-YFP, rapamycin, and celecoxib. Mice received tumor on day 0; TriVax vaccination on day 4; and 1 μL virus (2 × 106 pfu) or PBS injection, rapamycin, and celecoxib on day 7 (medication treatment continued until day 73). Mice that received the combination treatment lived significantly longer than the control group, P < 0.001.
To test whether TriVax postvaccination combined with vvDD-IL15Rα-YFP, rapamycin, and celecoxib provides a survival benefit to glioma-bearing mice, TriVax vaccine was injected 4 days after tumor implantation followed at day 7 by vvDD-IL15Rα-YFP virus injection, rapamycin, and celecoxib treatment. The survival curve (Fig. 4C) shows that in contrast to the results from previous emulsion postvaccination (Fig. 3D), TriVax postvaccination together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib did provide a moderate survival benefit to tumor-bearing mice. This result also suggests that the key to the success of this combination treatment is the amount of tumor-specific CD8+ T cells generated and the time to generate them.
Adoptive transfer of tumor-specific CD8+ T cells together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib provides a survival benefit to mice with intracranial tumors
Adoptive transfer of tumor-specific CD8+ T cells to recipient mice allows control of the timing and quantity of tumor-specific CD8+ T cells received by tumor-bearing mice. CD8+ T cells were isolated from splenocytes of donor mice which received TriVax vaccination twice. Eight percent to 12% of CD8+ T cells harvested from donor mice were GARC-1 tumor-specific CD8+ T cells by peptide-DimerX staining. A survival experiment was conducted to determine effects of adoptive T-cell therapy combined with oncolytic virotherapy. vvDD-IL15Rα-YFP virus, rapamycin, and celecoxib treatment was given to mice a week after tumor implantation. Two days later, 5 × 106 CD8+ T cells that were isolated from donor mice were injected retro-orbitally into each of the tumor-bearing mice followed by a single TriVax booster injection the next day as a way to keep transferred CD8+ T cells functional in recipient mice (50). The 100% survival in Fig. 5A strongly indicates that adoptive transfer of GARC-1 tumor-specific CD8+ T cells together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib is highly effective in mice with glioma. In addition, the results illustrate the synergy of IL15Rα-IL15 and adoptive transfer of T cells, evident by the limited efficacy of vvDD-IL15Rα-YFP, rapamycin, and celecoxib without ACT, and limited efficacy of ACT with rapamycin, celecoxib, and a vvDD-RFP virus that does not express IL15Rα-IL15 (Fig. 5A).
Figure 5.
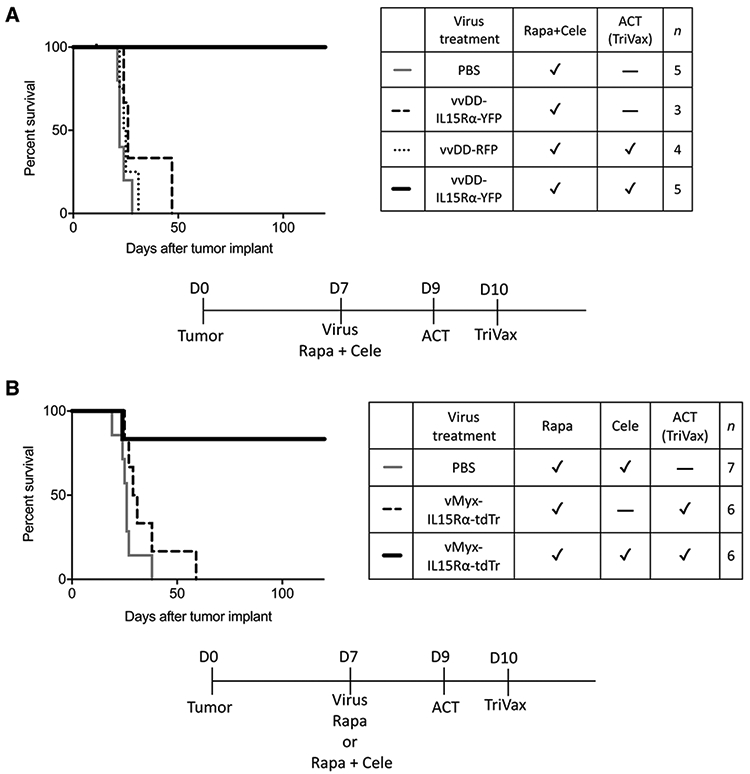
The therapeutic effect of the full combination treatment using vvDD-IL15Rα-YFP or vMyx-IL15Rα-tdTr. A, Survival of tumor-bearing mice treated with intratumoral vvDD-IL15Rα-YFP virus injection, rapamycin and celecoxib, adoptive transfer of CD8+ T cells, and TriVax booster. Mice received tumor on day 0; 1 μL vvDD-IL15Rα-YFP virus (2 × 106 pfu) or PBS injection, rapamycin, and celecoxib on day 7 (medication treatment continued until day 73; adoptive transfer of CD8+ T cells on day 9; and TriVax booster on day 10. Mice that received the full combination treatment survived longer compared with other groups, indicating the requirement for ACT. P < 0.01 for each comparison. B, Survival of tumor-bearing mice treated with intratumoral vMyx-IL15Rα-tdTr virus injection, rapamycin and celecoxib, adoptive transfer of CD8+ T cells, and TriVax booster. Mice received tumor on day 0; 1 μL vMyx-IL15Rα-tdTr virus (2 × 106 pfu) or PBS injection, rapamycin with or without celecoxib on day 7 (medication treatment continued until day 73; adoptive transfer of CD8+ T cells on day 9; and TriVax booster on day 10. Mice that received the full combination treatment survived longer compared with other groups, indicating the requirement for celecoxib and the insufficiency of ACT (TriVax). P < 0.05 for each comparison.
Comparison of the safety profile of vvDD-IL15Rα-YFP and vMyx-IL15Rα-tdTr
Several articles report that vvDD vaccinia virus is safe in animals, and it has been tested in phase I clinical trials (46, 51-54). We also found that mice subjected to direct injection of vvDD-IL15Rα-YFP to the parenchyma of the brain showed only minor weight loss. On the basis of these findings, we believed that there was no safety issue for vvDD vaccinia virus. However, we observed that there was one mouse in each cohort that died within 4 days of vvDD-IL15Rα-YFP injection in survival experiments (3/56 mice treated with vvDD overall). These mice were censored in the survival curves, because their deaths were not attributed to tumor growth. Brains from these mice were harvested and examined by IHC for vaccinia virus protein. Results showed that ependymal cells lining the lateral ventricles of these brains were extensively infected by vaccinia virus. Direct injection of vvDD-IL15Rα-YFP or vvDD-RFP into the lateral cerebral ventricles was uniformly fatal (55).
Therefore, although vvDD-IL15Rα-YFP is safe for non-brain cancers (33), a safer virus expressing IL15Rα-IL15 could be used to replace vvDD. We chose myxoma virus, whose tropism is restricted to the European rabbit, but still has the ability to infect and kill tumor cells from mice and humans. A previous study from our lab found that direct injection of vMyx-tdTr myxoma virus to the lateral ventricle of the mouse brain had no toxic effects, and the virus was cleared by 11 days post-injection (56). We recently observed that intracerebroventricular injection of vMyx-IL15Rα-tdTr did not show any toxic effect on mice either (55), indicating that vMyx-IL15Rα-tdTr myxoma virus is safe to use in the C57BL/ 6J mouse model.
Adoptive transfer of tumor-specific CD8+ T cells together with vMyx-IL15Rα-tdTr, rapamycin, and celecoxib provides a survival benefit to mice with intracranial tumors
Because the vMyx-IL15Rα-tdTr virus was confirmed to be safe to use in mice, we tested whether vMyx-IL15Rα-tdTr can replace vvDD-IL15Rα-YFP in this combination treatment. In this experiment, we also tested the contribution of celecoxib to the efficacy of the combination treatment. vMyx-IL15Rα-tdTr virus and rapamycin with or without celecoxib treatment was given to mice a week after tumor implantation. Two days later, 5 × 106 CD8+ T cells isolated from donor mice were injected retro-orbitally into each of the tumor-bearing mice followed by a single TriVax booster injection as a way to keep transferred CD8+ T cells functional in recipient mice (50). Results showed that the combination treatment using vMyx-IL15Rα-tdTr virus cured 83% of mice (Fig. 5B), which was similar to the efficacy of the combination treatment using vvDD-IL15Rα-YFP. This finding strongly suggests that adoptive transfer of GARC-1 tumor-specific CD8+ T cells together with vMyx-IL15Rα-tdTr, rapamycin, and celecoxib has the potential to cure glioma-bearing mice with reduced toxicity.
The results of Fig. 5B also suggest a synergistic effect of celecoxib in the combination treatment, because the omission of celecoxib produced a treatment with greatly reduced efficacy.
To assess possible mechanisms of the contribution of celecoxib to the vMyx-IL15Rα-tdTr combination treatment, C57BL/6J mice were implanted with GL261 NS cells intracranially on day 0, received 1 μL vMyx-IL15Rα-tdTr virus (2 × 106 pfu) injection, rapamycin with or without celecoxib on day 7 (medication treatment continued until day 12), adoptive transfer of CD8+ T cells on day 9, and TriVax booster on day 10. Mice were euthanized on day 12 and tumor sections were analyzed for presence of NK cells, CD8+ T cells, and Tregs by immunostaining for Ly-49G2 (4D11 antibody), CD8, and Foxp3, respectively. There was a significant increase of infiltrated CD8+ T cells and a significant decrease of infiltrated Foxp3+ Treg in the celecoxib-treated group (Fig. 6A). The ratio of infiltrated CD8+ T cells to Tregs is significantly higher in the celecoxib-treated group, compared to the no celecoxib group (Fig. 6B). These results indicate that celecoxib plays an essential role in promoting the infiltration of CD8+ T cells and preventing the infiltration of Tregs in the combination treatment with vMyx-IL15Rα-tdTr, which may explain the impressive benefit celecoxib brings to the success of the combination treatment (Fig. 5B).
Figure 6.
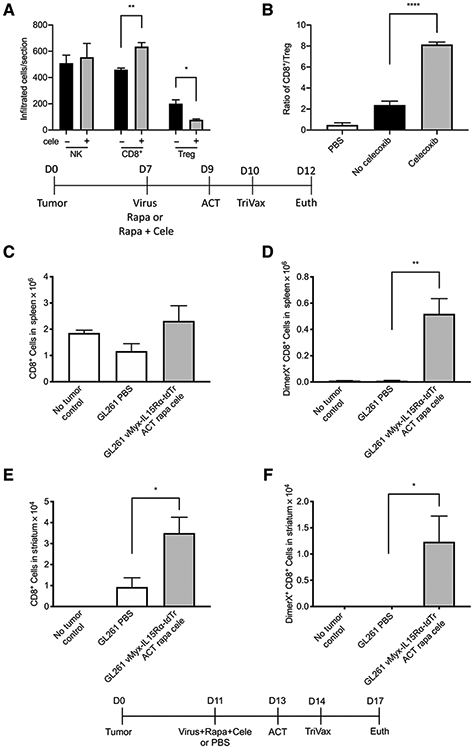
Potential mechanisms of the full combination treatment using vMyx-IL15Rα-tdTr. The effect of celecoxib on the tumor-infiltrated NK, CD8+ T cells, and Tregs when treated with vMyx-IL15Rα-tdTr virus. C57BL/6J mice were implanted with GL261 NS cells intracranially on day 0, then received 1 μL vMyx-IL15Rα-tdTr virus (2 × 106 pfu) injection and rapamycin with or without celecoxib on day 7 (medication treatment continued until day 12), adoptive transfer of CD8+ T cells on day 9, and TriVax booster on day 10. Mice were euthanized on day 12 and tumor sections were analyzed for presence of NK cells, CD8+ T cells, and Tregs by immunostaining for Ly-49G2 (4D11 antibody), CD8, and Foxp3, respectively. A, Number of infiltrated Ly-49G2+ NK cells, CD8+ T cells, and Foxp3+ Tregs per tumor section for each condition are shown (mean and SEM). There was a significant increase of infiltrated CD8+ T cells and significant decrease of infiltrated Tregs when celecoxib was included in the treatment. *, P < 0.05; **, P < 0.01. B, The ratio of tumor-infiltrated CD8+ T cells over tumor-infiltrated Tregs for each condition. There was a significant increase of the ratio when GL261 NS tumor was treated with vMyx-IL15Rα-tdTr and celecoxib. ****, P < 0.0001. C-F, Infiltration of gliomas by tumor-specific T cells following treatment of mice with oncolytic vMyx-IL15Rα-tdTr, rapamycin, celecoxib, and adoptive transfer of tumor-specific CD8+ T cells. Mice bearing GL261 gliomas were treated with a combination oncolytic virus and immunotherapy and euthanized four days after adoptive T-cell therapy. The right striatum and spleen were dissociated, and cells were analyzed by flow cytometry for CD8 and binding of a GARC-1-Db DimerX to detect tumor-specific T cells. Controls received only a PBS infusion or were untreated non-tumor-bearing mice. C, Total number of CD8+ T cells in spleen. D, Total number of GARC-1–specific CD8+ T cells in spleen. **, P < 0.01. E, Total number of CD8+ T cells in gliomas. *, P < 0.05. F, Total number of GARC-1–specific CD8+ T cells in gliomas. *, P < 0.05.
Adoptive transfer of tumor-specific CD8+ T cells together with vMyx-IL15Rα-tdTr, rapamycin, and celecoxib results in tumor-specific T cells infiltrating GL261 gliomas
We investigated whether the combination treatment that resulted in long-term survival of glioma-bearing mice (vMyx-IL15Rα-tdTr, ACT, rapamycin, and celecoxib) produced infiltration of the tumors by tumor-specific T cells. Mice with established GL261 NS tumors were treated with adoptive transfer of GARC-1–specific CD8+ T cells together with vMyx-IL15Rα-tdTr, a TriVax peptide boost, rapamycin, and celecoxib. Six days after virus injection and four days after adoptive transfer, mice were euthanized, and the right striatum from the brain and the spleen were dissected; cells were then dissociated and analyzed by flow cytometry. In the spleen, adoptive transfer of T cells and the TriVax boost did not consistently change the overall number of CD8+ cells (Fig. 6C), but clearly increased the number of GARC-1–specific CD8+ T cells (Fig. 6D), with 24% ± 4% of CD8+ splenocytes stained by the GARC-1-Db reagent. Confirming the histological observations, glioma-infiltrating CD8+ T cells were also increased by the treatment (Fig. 6E), and more than 30% of these cells were specific for the tumor neoantigen GARC-1, resulting in more than 1 × 104 GARC-1–specific CD8+ T cells in the tumors (Fig. 6F). Tumors were also evaluated by standard hematoxylin–eosin staining; characteristic histopathologic features of diffuse glioma including infiltration into the surrounding parenchyma and subpial, perivascular, and perineuronal glioma cell satellitosis were identified in all mice. Numerous apoptotic cells were present in tumors injected with vvDD-IL15Rα-YFP and a moderate number of apoptotic cells were present in tumors injected with vMyx-IL15Rα-tdTr (Supplementary Fig. S4).
Discussion
A modest prolongation of survival for mice with gliomas has been reported for a large number of treatments. However, complete elimination of established gliomas in a majority of animals is not common, and we observed this dramatic response with two different oncolytic poxviruses expressing the IL15Rα-IL15 fusion protein when combined with adoptive transfer of tumor-specific T cells and a prostaglandin synthesis inhibitor. Oncolytic viruses may contribute to an anti-tumor response by multiple mechanisms, such as a direct cytotoxic effect on cancer cells and consequent release of tumor antigens that may generate an immune response. When the virus is engineered to express a cytokine, it becomes a vector for local expression of potent immune-activating agents, while avoiding systemic inflammation that parenteral delivery of the cytokine would produce. We chose IL15 because it activates and maintains function of NK and CD8+ T cells (57) with less vascular leakage potential (58), less activation of Tregs (59), and less activation-induced cell death (AICD) for CD8+ effector T cells (60). We chose the co-expression of IL15 with its receptor α because it greatly enhances the stability and function of the cytokine in vivo (42,61). We confirmed that the murine glioma cell line GL261 can be infected both in vitro and in vivo by the two viruses and that GL261 cells infected with either virus can express functional IL15Rα-IL15 and attract NKand CD8+ T cells. Despite the infiltration of NK cells and CD8+ T cells, the survival benefit was modest, suggesting that either most of the CD8+ T cells were not tumor specific or the tumor microenvironment in the brain is too immunosuppressive for NK and CD8+ T cells to function.
To overcome these problems, we injected GARC-1, a neoantigen for GL261 cells, into mice before tumor implantation, allowing tumor-bearing mice to develop tumor-specific CD8+ T cells ahead of time. We then treated tumors with oncolytic virus and rapamycin and celecoxib starting on the same day as virus treatment. Although rapamycin was originally used as an immunosuppressive drug, it can enhance the spread and replication of oncolytic viruses (24, 26, 27), promote activation of dendritic cells and enhance their antigen-presenting ability (62), and improve the functionality of memory CD8+ T cells (31). We did not directly test its necessity in survival experiments. On the other hand, celecoxib was critical for the effectiveness of the vMyx-IL15Rα-tdTr with ACT treatment. Celecoxib is a COX-2 inhibitor which can selectively inhibit the production of prostaglandins (mainly PGE2; refs. 63, 64) a group of physiologically active lipid compounds with strong immunosuppressive functions in the tumor microenvironment.
We observed that prevaccination of GARC-1 together with vvDD-IL15Rα-YFP, rapamycin, and celecoxib cured 87% of mice with intracranial tumors, whereas postvaccination (mimicking clinical conditions) of GARC-1 with the combination treatment did not work at all. These facts indicate that the generation of enough tumor-specific CD8+ T cells within a short period of time is critical to the success of this treatment.
ACT therapy is another way to generate tumor-specific T cells. For the generation of GARC-1–specific CD8+ T cells, non–tumor-bearing C57BL/6J mice were injected with TriVax (GARC-1, Poly-IC, and anti-CD40 antibody) followed by a booster of TriVax 10 days later. Instead of using traditional lymphodepletion before ACT (65), a single injection of TriVax after ACT was used in this research, based on observations by Celis that a TriVax booster was as effective as lymphodepletion for maintaining survival and expansion of transferred T cells (50). We observed that tumor-bearing mice can be cured by a combination treatment of vvDD-IL15Rα-YFP or vMyx-IL15Rα-tdTr, rapamycin, celecoxib, and adoptive transfer of tumor-specific CD8+ T cells.
Adoptive cell transfer in the clinical setting could be accomplished by engineering T cells to express a tumor-specific T-cell receptor or a chimeric antigen receptor construct. The neoantigen that we targeted in these experiments, GARC-1, was first identified from its immunogenicity (8), but was also independently isolated by a whole-exome screen of mutations (9). The prospects for clinically applying neoantigen targeting are rapidly improving; for example, a phase Ib trial of personalized neoantigen vaccination for glioblastoma has recently been reported (66).
One concern for this combination treatment is the safety of oncolytic vaccinia virus vvDD when the virus is directly injected into the brain. Several preclinical and clinical trials report that it is safe to use vvDD in rodent models (mouse and rat; ref. 46), non-human primates (51), and humans (52-54), but in most cases, the vaccinia virus is injected into the blood or into subcutaneous tumors. One study (46) has shown that there were non-lethal neuropathological changes following the intracerebral (i.c.) injection of vaccinia virus in non–tumor-bearing rats, while another study reported that there were no deaths or symptoms of neurologic impairment when non–tumor-bearing rats were treated with vvDD-EGFP, whereas vvDD-EGFP was extremely toxic to tumor-bearing rats (24), indicating the necessity of further exploration of the safety profile of vvDD in the brain. We directly injected vvDD-IL15Rα-YFP virus into the parenchyma of non–tumor-bearing C57BL/6J mice and confirmed that it is safe in the brain parenchyma, but if the virus reaches the lateral ventricles, it can lead to death (55).
Alternatively, previous studies from our lab showed that another type of oncolytic poxvirus, myxoma virus, is safe to use in mice even when directly injected into the cerebral ventricles (56). On the basis of this safety profile, vMyx-IL15Rα-tdTr virus was combined with rapamycin, celecoxib, and adoptive transfer of tumor-specific CD8+ T cells to treat glioma-bearing mice. This combination treatment produced a result similar to the combination treatment with vvDD-IL15Rα-YFP, indicating that we can replace vaccinia virus with myxoma virus in this treatment to increase safety without greatly impairing efficacy. vvDD was more aggressive than vMyx in infection of GL261 and expression of virally encoded IL15Rα-IL15, and its biological effects were less dependent on celecoxib (Supplementary Fig. S3). However, the risk of ventriculitis and meningitis should preclude the use of vvDD for gliomas.
Analysis of TILs four days following the adoptive transfer of T cells from donor mice showed a pronounced increase in the number of tumor-specific T cells in response to the combination treatment. Future studies will characterize how different components of the effective treatment influence the number and phenotype of tumor-specific T cells to determine the essential features of a treatment that eradicates an established glioma.
Supplementary Material
Translational Relevance.
We developed a complex therapy regimen that included the following elements: tumor-specific T cells, an oncolytic virus that produced local release of IL15Rα-IL15 in the tumor environment, and inhibition of prostaglandin synthesis to minimize immunosuppression. Omission of any one of these resulted in a minimally effective therapy, whereas their combination resulted in eradication of established gliomas. These results encourage clinical trials of combinations of immunotherapy and oncolytic virotherapy, and encourage the use of a prostaglandin inhibitor in conjunction with immunotherapies.
Acknowledgments
This research was funded by institutional funds from the University of Illinois Urbana-Champaign. We thank the people of the Biotechnology Center Flow Cytometry facility for cheerful and competent service. B. Tang is supported by the Carle Foundation Hospital and University of Illinois Cancer Scholars for Translational and Applied Research (C*STAR) Graduate Program. We thank Dr. David Kranz for generously sharing his laboratory facilities and expertise for the last 26 years.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
G. McFadden is an employee/paid consultant for, reports receiving commercial research grants from, and holds ownership interest (including patents) in OncoMyx Therapeutics. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Leisegang M, Engels B, Schreiber K, Yew PY, Kiyotani K, Idel C, et al. Eradication of large solid tumors by gene therapy with a T-cell receptor targeting a single cancer-specific point mutation. Clin Cancer Res 2016;22:2734–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu RB, Engels B, Arina A, Schreiber K, Hyjek E, Schietinger A, et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res 2012;72:1964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu RB, Engels B, Schreiber K, Ciszewski C, Schietinger A, Schreiber H, et al. IL-15 in tumor microenvironment causes rejection of large established tumors by T cells in a noncognate T cell receptor-dependent manner. Proc Natl Acad Sci U S A 2013;110:8158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biollaz G, Bernasconi L, Cretton C, Püntener U, Frei K, Fontana A, et al. Site-specific anti-tumor immunity: differences in DC function, TGF-beta production and numbers of intratumoral Foxp3+ Treg. Eur J Immunol 2009;39:1323–33. [DOI] [PubMed] [Google Scholar]
- 5.Zhou J, Gong Z, Jia Q, Wu Y, Yang Z-Z, Zhu B. Programmed death ligand 1 expression and CD8+ tumor-infiltrating lymphocyte density differences between paired primary and brain metastatic lesions in non-small cell lung cancer. Biochem Biophys Res Commun 2018;498:751–7. [DOI] [PubMed] [Google Scholar]
- 6.Jackson CM, Kochel CM, Nirschl CJ, Durham NM, Ruzevick J, Alme A, et al. Systemic tolerance mediated by melanoma brain tumors is reversible by radiotherapy and vaccination. Clin Cancer Res 2016;22:1161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 2018;24:1459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iizuka Y, Kojima H, Kobata T, Kawase T, Kawakami Y, Toda M. Identification of a glioma antigen, GARC-1, using cytotoxic T lymphocytes induced by HSV cancer vaccine. Int J Cancer 2006;118:942–9. [DOI] [PubMed] [Google Scholar]
- 9.Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, et al. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunol Res 2016; 4:1007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G, et al. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res 2009;15:6551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev 2008;222:357–68. [DOI] [PubMed] [Google Scholar]
- 12.Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol 2015;33:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akasaki Y, Liu G, Chung NHC, Ehtesham M, Black KL, Yu JS. Induction of a CD4+ T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J Immunol 2004;173:4352–9. [DOI] [PubMed] [Google Scholar]
- 14.Obermajer N, Wong JL, Edwards RP, Odunsi K, Moysich K, Kalinski P. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest 2012;41:635–57. [DOI] [PubMed] [Google Scholar]
- 15.Göbel C, Breitenbuecher F, Kalkavan H, Hähnel PS, Kasper S, Hoffarth S, et al. Functional expression cloning identifies COX-2 as a suppressor of antigen-specific cancer immunity. Cell Death Dis 2014;5:e1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE2 catabolism in myeloid cells. J Leukoc Biol 2010;88:839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosaka A, Ohkuri T, Okada H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother 2014;63:847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eberstål S, Badn W, Fritzell S, Esbjörnsson M, Darabi A, Visse E, et al. Inhibition of cyclooxygenase-2 enhances immunotherapy against experimental brain tumors. Cancer Immunol Immunother 2012;61:1191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eberstål S, Fritzell S, Sandén E, Visse E, Darabi A, Siesjö P. Immunizations with unmodified tumor cells and simultaneous COX-2 inhibition eradicate malignant rat brain tumors and induce a long-lasting CD8(+) T cell memory. J Neuroimmunol 2014;274:161–7. [DOI] [PubMed] [Google Scholar]
- 20.Eberstål S, Sandén E, Fritzell S, Darabi A, Visse E, Siesjö P. Intratumoral COX-2 inhibition enhances GM-CSF immunotherapy against established mouse GL261 brain tumors. Int J Cancer 2014;134:2748–53. [DOI] [PubMed] [Google Scholar]
- 21.Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res 2011;71:2664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Tian M, Xiu C, Wang Y, Tang G. Enhancement of antitumor activity by combination of tumor lysate-pulsed dendritic cells and celecoxib in a rat glioma model. Oncol Res 2013;20:447–55. [DOI] [PubMed] [Google Scholar]
- 23.Lun X, Alain T, Zemp FJ, Zhou H, Rahman MM, Hamilton MG, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res 2010;70:598–608. [DOI] [PubMed] [Google Scholar]
- 24.Lun XQ, Jang J-H, Tang N, Deng H, Head R, Bell JC, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res 2009;15:2777–88. [DOI] [PubMed] [Google Scholar]
- 25.Stanford MM, Shaban M, Barrett JW, Werden SJ, Gilbert P-A, Bondy-Denomy J, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther 2008;16:52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. Oncolytic virotherapy synergism with signaling inhibitors: rapamycin increases myxoma virus tropism for human tumor cells. J Virol 2007;81:1251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas DL, Doty R, Tosic V, Liu J, Kranz DM, McFadden G, et al. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother 2011;60:1461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrer IR, Araki K, Ford ML. Paradoxical aspects of rapamycin immunobiology in transplantation. Am J Transplant 2011;11:654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cobbold SP. The mTOR pathway and integrating immune regulation. Immunology 2013;140:391–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amiel E, Everts B, Fritz D, Beauchamp S, Ge B, Pearce EL, et al. Mechanistic target of rapamycin inhibition extends cellular lifespan in dendritic cells by preserving mitochondrial function. J Immunol 2014;193:2821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature 2009;460:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lun X, Ruan Y, Jayanthan A, Liu DJ, Singh A, Trippett T, et al. Double-deleted vaccinia virus in virotherapy for refractory and metastatic pediatric solid tumors. Mol Oncol 2013;7:944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther 2018;26:2476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UNM, Ramalingam L, et al. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther 2015;23:202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Downs-Canner S, Guo ZS, Ravindranathan R, Breitbach CJ, O’Malley ME, Jones HL, et al. Phase 1 study of intravenous oncolytic poxvirus (vvDD) in patients with advanced solid cancers. Mol Ther 2016;24:1492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenner F The Florey lecture, 1983. Biological control, as exemplified by smallpox eradication and myxomatosis. Proc R Soc Lond B Biol Sci 1983;218: 259–85. [DOI] [PubMed] [Google Scholar]
- 37.Chan WM, Rahman MM, McFadden G. Oncolytic myxoma virus: the path to clinic. Vaccine 2013;31:4252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lun X, Yang W, Alain T, Shi Z-Q, Muzik H, Barrett JW, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res 2005;65:9982–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lun XQ, Zhou H, Alain T, Sun B, Wang L, Barrett JW, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res 2007;67:8818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther 2012;20:759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartee E, Bartee MY, Bogen B, Yu X-Z. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol Ther Oncolytics 2016;3:16032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Epardaud M, Elpek KG, Rubinstein MP, Yonekura A, Bellemare-Pelletier A, Bronson R, et al. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res 2008;68:2972–83. [DOI] [PubMed] [Google Scholar]
- 43.Bessard A, Sole V, Bouchaud G, Quemener A, Jacques Y. High antitumor activity of RLI, an interleukin-15 (IL-15)-IL-15 receptor alpha fusion protein, in metastatic melanoma and colorectal cancer. Mol Cancer Ther 2009;8:2736–45. [DOI] [PubMed] [Google Scholar]
- 44.Tosic V, Thomas DL, Kranz DM, Liu J, McFadden G, Shisler JL, et al. Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PLoS One 2014;9:e109801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pellegatta S, Finocchiaro G. Dendritic cell vaccines for cancer stem cells. Methods Mol Biol 2009;568:233–47. [DOI] [PubMed] [Google Scholar]
- 46.Lun X, Chan J, Zhou H, Sun B, Kelly JJP, Stechishin OO, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther 2010;18:1927–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology 2016;5:e1091554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Müller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol 2007;179:2774–86. [DOI] [PubMed] [Google Scholar]
- 49.Cho H-I, Celis E. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res 2009;69:9012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho H-I, Reyes-Vargas E, Delgado JC, Celis E. A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Res 2012;72:1986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hodge JW, Schlom J, Donohue SJ, Tomaszewski JE, Wheeler CW, Levine BS, et al. A recombinant vaccinia virus expressing human prostate-specific antigen (PSA): safety and immunogenicity in a non-human primate. Int J Cancer 1995; 63:231–7. [DOI] [PubMed] [Google Scholar]
- 52.Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther 2012;20:749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer 2009;9:64–71. [DOI] [PubMed] [Google Scholar]
- 54.Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin Biol Ther 2011;11:595–608. [DOI] [PubMed] [Google Scholar]
- 55.Tang B, Guo ZS, Bartlett DL, Liu J, McFadden G, Shisler JL, et al. A cautionary note on the selectivity of oncolytic poxviruses. Oncolytic Virother 2019;8:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.France MR, Thomas DL, Liu J, McFadden G, MacNeill AL, Roy EJ. Intraventricular injection of myxoma virus results in transient expression of viral protein in mouse brain ependymal and subventricular cells. J Gen Virol 2011;92(Pt 1):195–9. [DOI] [PubMed] [Google Scholar]
- 57.Jakobisiak M, Golab J, Lasek W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev 2011;22:99–108. [DOI] [PubMed] [Google Scholar]
- 58.Munger W, Dejoy SQ, Jeyaseelan R, Torley LW, Grabstein KH, Eisenmann J, et al. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: comparison with interleukin-2. Cell Immunol 1995; 165:289–93. [DOI] [PubMed] [Google Scholar]
- 59.Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood 2009;114:2417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, et al. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A 2000;97:11445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergamaschi C, Rosati M, Jalah R, Valentin A, Kulkarni V, Alicea C, et al. Intracellular interaction of interleukin-15 with its receptor alpha during production leads to mutual stabilization and increased bioactivity. J Biol Chem 2008;283:4189–99. [DOI] [PubMed] [Google Scholar]
- 62.Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, et al. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol 2012;189:2151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mostofa AGM, Punganuru SR, Madala HR, Al-Obaide M, Srivenugopal KS. The process and regulatory components of inflammation in brain oncogenesis. Biomolecules 2017;7:pii:E34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seibert K, Masferrer JL. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 1994;4:17–23. [PubMed] [Google Scholar]
- 65.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol 2005;26:111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.