

Abstract
Background
Procalcitonin (PCT) is an acute phase response protein, which can be used as an indicator for early diagnosis of infection. At present, the main detection methods for PCT are electrochemiluminescence and enzyme-linked immunofluorescence. We aimed to explore the accuracy of PCT determination in a domestic chemiluminescence detection system and its correlation with other systems.
Methods
Clinical specimens were collected, and the precision, linearity, biological reference interval, contamination rate, Clinical reportable scope, and methodological comparison of the determination of PCT in a Chinese chemiluminescence detection system were evaluated and preliminarily verified by referring to Clinical and Laboratory Standards Institute (CLSI) documents or industry standards.
Results
The results of precision verification showed that the coefficient of variation (CV) values of the variation coefficient of precision in the samples of low and high values were 2.07% and 0.83% respectively, while the CV values of the total variation coefficient of precision were 3.05% and 1.81% respectively; these findings all met the experimental requirements. The results of linear verification test showed that the linear range was 0.006–96.96 ng/mL, and the linear relationship was well within the detection range (R2 =0.9891). The biological reference interval and the carrying contamination rate were also verified. The clinical reportable range was 0.02–369.585 ng/mL. The results showed that the correlation coefficient between the Mindray CL900I and the Roche E602 was 0.9986, and that between the Mindray CL900I and the Snibe 2000 was 0.983. Meanwhile, when the PCT was higher than 0.1 ng/mL, the correlation coefficient was 100%.
Conclusions
The domestic chemiluminescence detection system has a good performance in the determination of calcitonin, as indicated by the measures of precision, linearity, biological reference interval, carrying contamination rate, and Clinical reportable scope, and can thus be used for clinical specimen detection. The results of methodological comparison showed that the correlation coefficient between the Mindray CL900I and Roche E602 was 0.9986, while the correlation coefficient between the Mindray CL900I and the Snibe 2000 was 0.983. The test results were consistent with the experimental requirements.
Keywords: Procalcitonin (PCT), domestic chemiluminescence detection system, performance evaluation
Introduction
Procalcitonin (PCT) is a hormonally inactive precursor of calcitonin composed of 116 amino acids, which is mainly produced and secreted by thyroid follicle cells. Under the action of special proteases, PCT is broken down into amino-terminal peptide, active calcitonin, and calcitonin (1). As an acute phase response protein, PCT has a half-life of 20–24 h in humans and is stable (2). Recent studies have confirmed that PCT can be used as an early diagnostic indicator of infection and its level in serum is positively correlated with the severity of infection. PCT increase in local infections, viral infections, chronic non-specific inflammation, cancer fever, graft-host rejection or autoimmune diseases is insignificant. However, it is significantly increased in severe bacterial, fungal, and parasitic infections, as well as sepsis and multiple organ failure. Therefore, PCT has been widely used in the diagnosis of bacterial infections, differential diagnosis, guiding antimicrobial drug administration, and mortality assessment (3). At present, the clinical detection methods of PCT mainly include electrochemiluminescence and enzyme-linked immunofluorescence. In recent years, chemiluminescence immunoassay has developed extremely fast. It combines the high sensitivity of chemiluminescence and the high specificity of antibody technology. With the characteristics of high sensitivity, good separation effect, short determination time, small inter-batch error, and no radioactive contamination, it has entered the routine application of clinical medicine from the laboratory. There are several chemiluminescence detection systems on the market in China, therefore making it necessary to explore their accuracy and compare them with imported systems. In this paper, we will discuss the accuracy of the domestic chemiluminescence detection system for PCT and its correlation with different imported systems.
We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/atm-20-7047).
Methods
Source of specimen
Specimens
Serum was collected from May to July 2019 from inpatients and health check-ups at the Concord Hospital affiliated to Tongji Medical College of Huazhong University of Science and Technology. Collection included 40 inpatients, comprising 20 males and 20 females, and 20 health check-ups, comprising 10 males and 10 females. All samples were collected with the informed consent of the patients, and approval was received from the medical ethics committee of Union Hospital Affiliated with Tongji Medical College of Huazhong University of Science and Technology. All procedures performed in this study involving human participants were in accordance with the Declaration of Helsinki (as revised in 2013).
Instruments and reagents
The equipment used for the experimental group was a fully automated chemiluminescence immunoassay analyzer (Mindray CL900I, Shenzhen Mindray Bio-Medical Electronics Co, Ltd, Shenzhen, China). Matching reagents included a Mindray calcitonin (PCT) quantitative chemiluminescence assay kit (Mindray, no. 20190101), and a Mindray calcitonin calibrator (PCT) (Mindray, no. 20190101).
The equipment used for the control group was an automated electrochemiluminescence immunoassay analyzer (Roche, Mannheim, Germany). The reagents used were a Roche electrochemiluminescence calcitonin (PCT) quantitation kit (Roche, no. 40830901) (abbreviated as chemiluminescence 1); The other equipment used for the control group was MAGLUMI 2000 (Snibe, Shenzhen, China). The matching reagent was a New Industries calcitonin PCT chemiluminescence quantitation test kit (Snibe, no. 0681800101) (abbreviated as chemiluminescence 2).
Research methodology
Precision evaluation
Total precision
According to Clinical and Laboratory Standards Institute (CLSI) EP15-A2, two levels of quality control sera were used. One batch was used per day for each level, and each batch was repeated three times for a total of five days. The experiment also needed to include daily quality control, such that if there was a quality control or operational error within a batch, the results of that batch could be rejected and another batch of results could be made. The mean, standard deviation, and coefficient of variation (CV) for each level to obtain intrabatch precision and total precision were calculated. According to the Clinical Laboratory Improvement Amendments of 1988 (CLIA '88), the in-lot precision was determined as ≤1/4 of the allowable total error (TEa). The total precision was ≤1/3 TEa or, and if not specified in the CLIA '88, the precision data described in the reagent instructions were used or verified in accordance with the requirements of the inter-room quality assessment of the Clinical Laboratory Center of the Ministry of Health of China.
In-batch precision
According to the In Vitro Diagnostic Reagents Analytical Performance Assessment Series Guidelines and current domestic industry standards, the CV of the validation material (mixed fresh serum or quality control products) was calculated for 20 consecutive tests in one batch, and the CV within the batch was less than that declared in the instruction manual.
Linear range determination
According to CLSI EP6-A (4), in the case of collecting low value and high value serum specimens (concentration as far as possible to cover the linear range of the reagent statement), the closer the high value specimen is to the highest value declared by the manufacturer, the better, and the closer the low value specimen to the lowest value declared by the manufacturer, the better. As to related items of reagents and calibrators, it should maintain the same lot number during the evaluation process. A series of diluted serum concentrations were formed by using low concentration (L) and high concentration (H) sera in proportion to each other (5L, 4L+1H, 3L+2H, 2L+3H, 1L+4H, and 5H). Each experimental sample was tested twice on the assay system. The mean value of the recorded results was calculated, and the measured value and the expected value were compared using regression statistical analysis. Regression and correlation coefficients were calculated, and the average slope method was used to determine the expected value (X) of the diluted samples in the series. The mean value of repeated measurements for all samples was the measured value (Y). According to industry-recognized standards, different manufacturers’ detection systems have different linear ranges, requiring (I) linear results with a linear distribution within the claimed linear range and (II) a correlation coefficient of R>0.99.
Validation of biological reference intervals
With reference to The National Committee for Clinical Laboratory Standards (NCCLS) C28-A2 (5) (How Clinical Laboratories Determine and Establish Biological Reference Intervals) document, 20 specimens of healthy medical examiners who had passed a physical examination were selected and tested. The assay was performed on the system, and the results were tallied and validated against the reference intervals provided in the reagent instructions or established by the laboratory. The raw data were retained. If the results of 20 specimens were within the reference interval provided in the reagent instructions or established by the laboratory, or if only 2 specimens exceeded the reference interval, the validation was passed. Otherwise, a reference interval had to be established.
Carrying contamination rate experiments
According to the CLSI’s Defining, Establishing, and Verifying Cntervals in the Clinical Laboratory guidelines, one specimen each of high concentration H and very low concentration L were taken, and the high concentration sample was divided into 10 cups while the low concentration sample was divided into 11 cups. The total of 21 samples were continuously measured in the following sequence: L1, L2, L3, H1, H2, L4, H3, H4, L5, L6, L7, L8, H5, H6, L9, H7, H8, L10, H9, H10, and L11. High represents the concentration value of high-concentration samples, Low represents the concentration value of low-concentration samples. The carry contamination was calculated as “(mean of High-Low) − (mean of Low-Low)”. Carry contamination <3 times (Low-Low) standard deviation (SD) was verified.
Clinical reportable range (CRR test methods)
The concentration of the sample near 90% of the upper limit of high value was collected. After the sample was diluted, it was measured three times in parallel. The mean value of the three replicates and the deviation between the diluted concentration and the theoretical concentration were calculated. A deviation of less than the maximum allowable error (Tea, 30%) was the widest requirement, and the best requirement was less than 1/2 of Tea; if the deviation between the expected value and the theoretical value was ≤1/2 of Tea, the concentration level selected for the test multiplied by the number of dilutions was considered the upper limit of the clinical reportable range.
Methodological comparisons
Forty fresh patient samples that met the required sample size for the test method were retained (samples were evenly distributed within the linear range) to ensure that the samples were of the common type used in routine tests and that the analyses were stable and reliable prior to measurement. The selected samples were tested with the CL900I (Mindray) analytical system and corresponding reagent kits according to CLSI EP15-A2 (6). Also, they were compared with the reference system (Roche E602 testing system) for methodological comparison. The results was “Y = aX + b”, with R2 >0.97 and a relative bias of less than 1/2 TEA compared with reference system (0.5 ng/mL). According to the EP9-A2 document, if the index cannot be met, you can expand the sample range or partition bias analysis; linear fitting is recommended to eliminate outliers and calculate the average of the difference between the two methods of measurement results (Y-X). When it exceeds 4 times the average, the sample is judged to be an outlier.
Statistical treatment
The experimental results were analyzed by one-dimensional linear regression analysis. The bias was determined using the Bland-Altman method, with the mean of the two methods of detection as the x-axis and the difference as the y-axis.
Results
Precision analysis results
The within-batch precision CVs of the Mindray automated chemiluminescence immunoassay systems were 2.07% and 0.83% at level1, 0.50 ng/mL and level2, 10.00 ng/mL, respectively. The total precision CVs were 3.05% and 1.81%, respectively. The intrabatch precision and total precision CV values of the Mindray automatic chemiluminescence immunoassay system were both <5% (Table 1).
Table 1. In-batch precision and total precision of Mindray’s fully automated chemiluminescent immunoassay system.
| Sample | Mean (ng/mL) | Within-day precision SD | Within-batch precision | Total precision | |||
|---|---|---|---|---|---|---|---|
| SD | CV (%) | SD | CV (%) | ||||
| Level 1 | 0.50 | 0.013 | 0.010 | 2.07 | 0.015 | 3.05 | |
| Level 2 | 9.42 | 0.158 | 0.079 | 0.83 | 0.171 | 1.81 | |
SD, standard deviation; CV, coefficient of variation.
Results of linear range determination
The low concentration sample (0.006 ng/mL, abbreviated as L) and the high concentration sample (97.960 ng/mL, abbreviated as H) were mixed in a certain ratio to make a series of six different levels of diluted serum, which were used for linear experiments (Table 2). The linear regression analysis is shown in Figure 1.
Table 2. Theoretical concentrations and results of linear analysis.
| Dilution ratio (L:H) | Theoretical concentration (ng/mL) | Measured value (ng/mL) | Average value (ng/mL) | |
|---|---|---|---|---|
| 1st | 2nd | |||
| Level 1 | 0.50 | 0.013 | 0.010 | 2.07 |
| Level 2 | 9.42 | 0.158 | 0.079 | 0.83 |
| Level 1 | 0.50 | 0.013 | 0.010 | 2.07 |
| Level 2 | 9.42 | 0.158 | 0.079 | 0.83 |
| Level 1 | 0.50 | 0.013 | 0.010 | 2.07 |
| Level 2 | 9.42 | 0.158 | 0.079 | 0.83 |
L, low concentration sample; H, high concentration sample.
Figure 1.

Linear regression analysis plot.
Results of validation of biological reference intervals
Twenty cases of normal human blood samples, both male and female, aged from 20 to 70 years old, were randomly selected. They were calibrated and quality-controlled with Mindray’s original reagents; the quality control data qualified this batch of samples. The results of 19 cases were in the range of 0–0.05 ng/mL, and the result of 1 case was 0.054 ng/mL, which met the requirement that there should not be 2 samples out of the range. Thus, the reference range passed the verification.
Experimental results on the contamination rate carried
The results of sequentially testing 21 samples are shown in Table 3. High represents the concentration value of high-concentration samples, Low represents the concentration value of low-concentration samples. The carry contamination was calculated as “(mean of High-Low) − (mean of Low-Low)”. Carry contamination <3 times (Low-Low) standard deviation (standard deviation, SD) was verified.
Table 3. Experimental results for the contamination rate.
| Project | Value |
|---|---|
| AVE H-L# | 0.0406 |
| SD H-L# | 0.002701851 |
| AVE L-L# | 0.0396 |
| SD L-L# | 0.002 |
| 3SD L-L# (Permissible error) | 0.005 |
| Carrying contamination | 0.001 |
| Conclusion | Passed |
#, high represents the concentration value of high-concentration samples, low represents the concentration value of low-concentration samples. SD, standard deviation; AVE, average.
Clinical reportable range test results
The results of the Mindray assay range validation test are shown in Table 4, with a clinically reportable range of 0.02–369.585 ng/mL.
Table 4. Results of the reportable range experiment.
| Sample | Diluted 4 times | Original doubling |
|---|---|---|
| Test 1 | 102.625 | 91.376 |
| Test 2 | 101.158 | 93.265 |
| Test 3 | 102.006 | 92.548 |
| Theoretical concentration | 92.396 | |
| Average value | 101.930 | 92.396 |
| CV (%) | 0.936 | 1.032% |
| Bias | 10.318% | |
| Reportable scope | 0.02–369.585 ng/mL | |
CV, coefficient of variation.
Correctness validation results of methodological comparisons
The three-way comparison of Mindray CL900I with the Roche E602 and the as the reference system was completed, with a sample size of 40 cases. The comparison results indicated that the correlation coefficient between the Mindray CL900I and Roche E602 was 0.9986, and the correlation coefficient between the Mindray CL900I and the New Industry Snibe 2000 was 0.983 (Figure 2 and Figure 3). Table 5 shows that the PCT is higher than 0.1 ng. Table 5 shows that when the PCT is higher than 0.1 ng/mL, the result of Mindray CL900I is 100% compatible with the Roche reference system.
Figure 2.
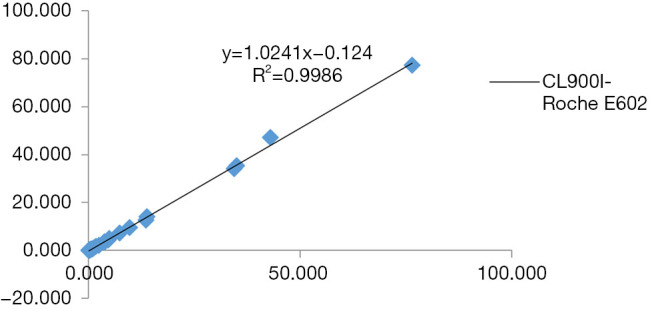
Results of the Mindray CL900I and Roche E602 methodological comparison test.
Figure 3.
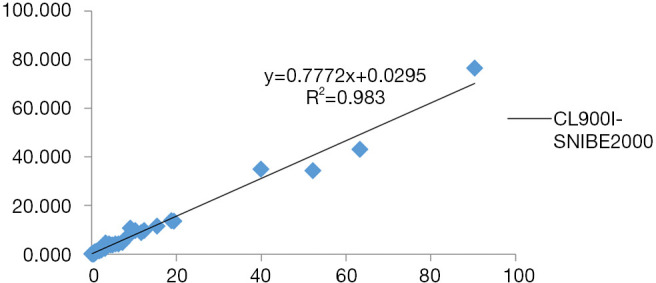
Results of the methodological comparison test between Mindray CL900I and the New Industry Snibe 2000.
Table 5. Results of the comparison of the different systems.
| PCT test results segmented diagnostic reference system compliance rate | |||||
|---|---|---|---|---|---|
| Concentration (ng/mL) | Roche (case) | CL900I-1 (case) | Snibe 2000 | CL900I compliance rate | Snibe 2000 compliance rate |
| <0.1 | 5 | 4 | 0 | 80.00% | 0.00% |
| 0.1–0.25 | 5 | 5 | 2 | 100.00% | 40.00% |
| 0.25–0.5 | 7 | 7 | 1 | 100.00% | 14.29% |
| >0.5 | 23 | 23 | 23 | 100.00% | 100.00% |
Discussion
As a marker of severe bacterial infection, PCT is important for the diagnosis, treatment, and prognosis. Compared to inflammatory factors, such as interleukin-6 (IL-6), IL-8, tumor necrosis factor (TNF), white blood cell count, C -reactive protein, blood sedimentation rate, and other traditional diagnostic indicators, PCT has better accuracy and sensitivity (7-9). Meanwhile, PCT is a distinguishing marker between bacterial and non-bacterial infections, and it can guide the proper use of clinical antibiotics, which is clinically important in shortening or reducing the duration of antibiotic use by assessing the efficacy of antibiotics (8).
Clinically, the quantitative methods commonly used for PCT detection include radioimmunoassay, immunofluorescence, double-antibody sandwich immunochemiluminescence, enzyme immunoassay, and others. In this study, the performance of the domestic chemiluminescence detection system for the determination of PCT was explored through the experimental evaluation of the precision, linear range, biological reference interval, reportable range, carry contamination rate, and correctness validation, and through methodological comparison. The results showed that the domestic chemiluminescence detection system had good intrabatch and interbatch precision within the linear range; the credible maximum dilution factor was 4 times, the clinical reportable range was 0.02–369.585 ng/mL, and it had a good linear gradient relationship within the detection range of the kit. Thus, the methodological comparison test was considered to meet the needs of clinical testing. The three-way comparison of Mindray CL900I, the reference system Roche E602, and the New Industry Snibe 2000 showed good correlation coefficients of 0.9986 and 0.983, respectively. Meanwhile, the Mindray CL900I maintained a high degree of consistency with the Roche E602 in the interpretation of the clinical diagnostic level results. The coincidence rate of Mindray segmented diagnostic test results (exclude bacterial infection concentration (<0.25 ng/mL), recommended antibiotic treatment concentration (0.25–0.5 ng/mL), highly recommended antibiotic treatment concentration (>0.5 ng/mL) with Roche E602 reference system was 100%.
In summary, the Mindray chemiluminescence detection system for PCT detection is efficient but automatic and accurate, and has good correlation with the imported systems.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All samples were collected with the informed consent of the patients, and approval was received from the medical ethics committee of Union Hospital Affiliated with Tongji Medical College of Huazhong University of Science and Technology. All procedures performed in this study involving human participants were in accordance with the Declaration of Helsinki (as revised in 2013).
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/atm-20-7047
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-7047
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-7047). The authors have no conflicts of interest to declare.
References
- 1.Assicot M, Gendrel D, Carsin H, et al. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet 1993;341:515-8. 10.1016/0140-6736(93)90277-N [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacifico L, Osborn JF, Natale F, et al. Procalcitonin in pediatrics. Adv Clin Chem 2013;59:203-63. 10.1016/B978-0-12-405211-6.00007-3 [DOI] [PubMed] [Google Scholar]
- 3.Leng Y, Chen C, Zhang Y, et al. Ability of serum procalcitonin to distinguish focus of infection and pathogen types in patients with bloodstream infection. Ann Transl Med 2019;7:135. 10.21037/atm.2019.03.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clinical and Laboratory Standards Institute. Evaluation of the linearity of quantitative measurement procedures: a statistical approach; Approved Guideline. EP6-A, CLSI, 2003. [Google Scholar]
- 5.Clinical and Laboratory Standards Institute. Defining, establishing, and verifying intervals in the clinical laboratory. C28-A2, CLSI, 2010. [Google Scholar]
- 6.The National Committee for Clinical Laboratory Standards. Method Comparison and Bias Estimation Using Patient Samples; Approved Guideline. NCCLS, EP 9-A, 1995.
- 7.Aloisio E, Dolci A, Panteghini M. Procalcitonin: Between evidence and critical issues. Clin Chim Acta 2019;496:7-12. 10.1016/j.cca.2019.06.010 [DOI] [PubMed] [Google Scholar]
- 8.Covington EW, Roberts MZ, Dong J. Procalcitonin Monitoring as a Guide for Antimicrobial Therapy: A Review of Current Literature. Pharmacotherapy 2018;38:569-81. 10.1002/phar.2112 [DOI] [PubMed] [Google Scholar]
- 9.Bassetti M, Russo A, Righi E, et al. Role of procalcitonin in bacteremic patients and its potential use in predicting infection etiology. Expert Rev Anti Infect Ther 2019;17:99-105. 10.1080/14787210.2019.1562335 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as