

Abstract
Background
Diabetic kidney disease (DKD) is the primary cause of end-stage renal disease. However, the pathogenesis of DKD remains unclarified, and there is an urgent need for improved treatments. Recently, many crucial genes closely linked to the molecular mechanism underlying various diseases were discovered using weighted gene co-expression network analysis.
Methods
We used a gene expression omnibus series dataset GSE104948 with 12 renal glomerular DKD tissue samples and 18 control samples obtained from the gene expression omnibus database and performed weighted gene co-expression network analysis. After obtaining the trait-related modules, gene ontology and Kyoto encyclopedia of genes and genomes enrichment analyses of the modules were conducted and the key gene associated with DKD was selected from the top two most significant gene ontology terms using the maximal clique centrality method. Finally, we verified the key gene using protein-protein interaction analysis, additional datasets, and explored the relationship between the key gene and DKD renal function using the Nephroseq v5 online database.
Results
Among the 10 gene co-expression modules identified, the darkorange2 and red modules were highly related to DKD and the normal biological process, respectively. Majority of the genes in the darkorange2 module were related to immune and inflammatory responses, and potentially related to the progression of DKD due to their abnormal up-regulation. After performing sub-network analysis of the genes extracted from the top two most significant gene ontology terms and calculating the maximal clique centrality values of each gene, FCER1G, located at the center of the protein-protein interaction network, was identified as a key gene related to DKD. Furthermore, gene expression omnibus validation in additional datasets also showed that FCER1G was overexpressed in DKD compared with normal tissues. Finally, Pearson’s correlation analysis between the expression of FCER1G and DKD renal function revealed that the abnormal upregulation of FCER1G was related to diabetic glomerular lesions.
Conclusions
Our study demonstrated for the first time that FCER1G is a crucial gene associated with the pathogenesis of DKD.
Keywords: Diabetic kidney disease (DKD), pathogenesis, weighted gene co-expression network analysis (WGCNA), key gene, FCER1G
Introduction
Diabetic kidney disease (DKD), as a common diabetic microangiopathy, manifests as proteinuria or impaired renal function clinically (1). If untreated, the end-stage renal disease caused by DKD is fatal (2). Furthermore, kidney disease can also lead to macrovascular complications (3), and diabetic patients with renal complications have a higher mortality (4). Currently, the treatment for DKD is limited and further exploration of the underlying pathogenesis is urgently needed. Studies have shown that the molecular mechanisms underlying DKD are complicated, involving metabolic factors, oxidative stress, renal hemodynamic changes, as well as growth factors and cytokines (5,6). Furthermore, there is extensive evidence that supports that the genetic background affects the progress of nephropathy in patients with diabetes (7,8) as only a subgroup of these patients progress to nephropathy, whereas the rate of progression varies between patients (9). The expression of genes or proteins, distinguished from the relatively static DNA sequences, represents the dynamics of this disease. In recent years, high-throughput technologies and bioinformatics have developed rapidly and research on gene expression profiling has been widely used to detect key genes related to the pathogenesis of diseases. Weighted gene co-expression network analysis (WGCNA) is an effective data mining method widely used to screen out key genes in various diseases, such as tumors (10-13), chronic diseases (14,15), immune (16-18), and mental diseases (19,20), and kidney diseases (21-23). Based on the detected gene correlations, WGCNA constructs a network and divides the genes into several co-expression modules, each of which contains genes that may have common biological regulatory functions. The modules that are the most relevant to a certain disease are worthy of further study, whereas the most central genes in a crucial module are regarded as key genes. Thus, WGCNA is applied to identify biologically relevant key genes, which may be utilized as therapeutic targets.
In the present study, we used WGCNA to explore the key genes associated with DKD with the aim to provide a new insight into DKD biomarker discovery.
We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/atm-20-1087).
Methods
Microarray data information
We searched the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and obtained GSE104948 matrix format files, containing 12 DKD renal glomerular tissue samples, and 18 normal samples, from patients with DKD and healthy living donors, respectively.
Data pre-processing
We conducted probe annotation to map probes to gene symbols using Affymetrix platforms in a Perl environment, with log2-transformed expression values. All the microarray probes that matched multiple genes were removed. In the case where a gene matched several probes, we calculated the average value of the different probes as the final gene expression value.
Construction of the weighted gene co-expression network
Consensus clustering was applied to recognize the types of samples and remove outlier samples before performing WGCNA.
We conducted WGCNA according to the protocol included in the WGCNA package using the R platform (version 3.5.3) (24).
Initially, Pearson’s correlation was used to evaluate the correlation between two genes. Subsequently, the pairwise correlations of all genes formed a correlation matrix.
Next, the scale-free fit index and average connectivity for soft-thresholding powers (β) in the range of 1 to 30 were calculated to select the most proper β value and translate the correlation matrix to a scale-free network. When the scale-free fit index reached 0.85, the β value with the maximum average connectivity was selected as the most appropriate for scale-free network translation.
Finally, the scale-free network was transformed to a topological overlap matrix (TOM) to understand the indirect relationship between genes. Based on the TOM, we conducted average linkage hierarchical clustering and grouped all the genes into several co-expression modules.
In a given module, the module eigengenes (MEs) were the chief components of the module, which represented the expression pattern of a certain module (24). The correlations between the MEs of different modules were determined, and highly correlated modules were merged into one (Pearson’s correlation ≥0.75).
Further, the adjacency of eigengenes in modules and the correlation among randomly picked genes were calculated to evaluate the reliability of the constructed modules. The results were visualized using heatmaps.
Identification of trait-related modules
The modules that were positively correlated with DKD were involved in the pathogenesis of DKD, while modules that were positively correlated with normal trait were important in maintaining the normal biological function. After obtaining the gene co-expression network and modules using WGCNA, the correlation between the modules and traits can be analyzed, and the module with the highest correlation with a trait can be selected as a trait-related module for further analyses. In our study, DKD and normal are the sample traits, while MEs represent the expression pattern of a given module. We used Pearson’s test to calculate the correlation coefficient between the MEs and clinical traits to identify the trait-related modules.
Gene significance (GS) represents the relationship between individual genes and clinical traits, while module membership (MM) indicates the associations between the gene expression value and the MEs in a given module. When a certain module had a high correlation between GS and MM, it indicated that the genes in that module contributed greatly to this module and the corresponding traits (25). We calculated the GS and MM of trait-related modules, and then visualized the results using a scatter plot diagram.
Enrichment analyses and differentially expressed genes (DEGs) analyses of the trait-related modules
We performed gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analyses of the trait-related modules using clusterProfiler based on Hypergeometric test to better understand the biological functions (26). For the enrichment analyses, a P value of <0.01 (P<0.05 after Benjamin-Hochberg correction) was considered statistically significant. The top 10 most significant terms were selected for visualization. Furthermore, the DEGs based on Empirical Bayes test were analyzed to compare the differences between the DKD and normal samples using the limma package (27), and the cut-off criteria for DEGs were set as |log FC| >1 and P<0.05.
Identification of key genes in trait-related modules
The key genes located at the center of the co-expression network had a relatively strong correlation with the other genes. Therefore, the key genes of the trait-related modules may be important in the pathogenesis of DKD. After identifying the most significant trait-related GO terms using GO enrichment analysis, we extracted the sub-networks of the gene clusters of the top two most significant terms from the whole WGCNA network. Following this, we imported the two gene clusters and their weighted correlations of the sub-WGCNA networks into the Cytoscape software to analyze the relationships among genes (28). We ran cytohubba (a plugin of Cytoscape) to calculate the maximal clique centrality (MCC) value of each gene in the sub-networks (29), and screened the key genes, which were defined as the genes possessing the top 10 MCC values.
Protein-protein interaction (PPI) network creation
Since most proteins function by interacting with other proteins, investigating their interactions and functions as an integrated system may help us to better understand their function. The STRING2 database (30) contains information about the interactions between proteins, derived from experimental studies. To explore the biological relationships among genes in the trait-related module, we identified the PPI pairs among them with a confidence score of 0.700 and built the PPI network using the STRING online analysis tool (http://www.string-db.org). Subsequently, the exported data were imported into Cytoscape for visualization.
GEO validation
To verify the robustness of our results in external datasets, we compared the expression level of the key genes associated with DKD and normal controls in three additional datasets downloaded from GEO.
Clinical validation
We used the Nephroseq v5 online database (http://v5.nephroseq.org), an integrated data-mining platform for gene expression data sets of kidney diseases, to validate the correlation between the key genes and clinical traits of DKD using Pearson’s correlation analysis. A P value of <0.05 was considered statistically significant.
Results
Evaluation and preparation of data
Figure 1 shows the flowchart of our study. We obtained the expression profiles of 11,884 genes from 30 renal glomerular samples and all genes were used for further analysis. After performing cluster analysis for all the samples, one outlier DKD sample was removed and the final results are shown in Figure 2A. We found that the samples were divided into two clusters: one cluster contained 11 DKD samples, while the other contained 18 normal samples, which indicated a high consistency between samples of the same type.
Figure 1.

Study flowchart.
Figure 2.

Gene cluster analysis. (A) Sample dendrogram and trait heatmap. The branches of the dendrogram correspond to clustered samples. (B) Construction of the scale-free network with a suitable soft-thresholding power (β). The red line represents the value of the scale-free fit index (0.85).
Construction of the weighted gene co-expression network
We selected β=19 as the soft-threshold to construct a scale-free network (Figure 2B). The co-expression modules were confirmed using Dynamic Tree Cutting and represented by different colors. Ultimately, modules with diverse sizes and colors were generated (Figure 3A), whereas the number of genes in the modules ranged from 99 to 4,156.
Figure 3.

Construction of the weighted gene co-expression network. (A) Cluster dendrogram. Each branch represents a module. The original and merged modules are respectively shown in the two colored bars below. (B) Eigengene adjacency heatmap. The colors of the squares from red to blue indicate the adjacency of the corresponding modules from high to low. (C) Heatmap plot of the selected genes. The colors from light to deep red represent a low to high interaction. The left and top of the figure show the gene dendrogram and module allocation.
The eigengenes adjacency of the modules was analyzed to further evaluate the adjacency of all modules (Figure 3B). Most modules had a low adjacency to other modules, which meant the clustering was independent and precise.
The interaction relationships of 1,000 randomly selected genes are presented in the network heatmap (Figure 3C). The results revealed that the genes in the same module were highly correlated, while they were weakly correlated to those in other modules. Thus, the reliability of the modules was verified.
Selection of trait-related modules
Figure 4A shows the correlations between modules and traits. The darkorange2 module (1,429 genes) was the most positively related to DKD (r=0.88, P=3e–10), while the association between the red module (367 genes) and normal trait was higher than that of other modules (r=0.8, P=2e–07). A scatterplot of the GS vs. MM in the darkorange2 module was plotted (Figure 4B), in which the GS and MM had a highly significant correlation (cor =0.8, P<1e−200). Most genes were distributed in the upper right corner, with the GS ranging from 0.5 to 1 and the MM from 0.7 to 1. The red module also had a similar significant association between the GS and MM (cor =0.58, P=2.3e–34) (Figure 4C). Hence, we identified the darkorange2 and red modules as the most valuable modules for subsequent analysis.
Figure 4.

Selection of trait-related modules. (A) Module-trait relationships heatmap. Rows correspond to modules, while columns correspond to traits. The correlation and P values are shown in the cells. Colors from green to red correspond to the correlation between the module and the trait form low to high. (B) The relationship between the module membership and gene significance in the darkorange2 module. (C) The relationship between the module membership and gene significance in the red module.
Enrichment analyses and DEGs analyses of the trait-related modules
We conducted GO and KEGG enrichment analyses of the genes in the darkorange2 and red modules. The top 10 GO items in the darkorange2 module are shown in Figure 5.
Figure 5.

The top 10 most statistically significant terms of the enrichment analyses of the darkorange2 module: (A) biological process (BP) terms; (B) cellular component (CC) terms; (C) molecular function (MF) terms; (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The length of the strip represents the gene count and the color scale corresponds to the significance of the enrichment.
Regarding their biological process (BP) (Figure 5A), the enriched genes were primarily related to immunity, inflammation and extracellular matrix (ECM) organization. The top four significant GO terms were the following: leukocyte migration (102 genes, P=1.57e–22), T cell activation (96 genes, P=8.55e–20), ECM organization (77 genes, P=2.56e–18) and extracellular structure organization (83 genes, P=1.26e–17).
Regarding cellular component (CC) (Figure 5B), the enriched genes were related with the ECM (97 genes, P=4.02e–20), receptor complex (66 genes, P=3.14e–11), and cytoplasmic vesicle lumen (57 genes, P=3.98e–09).
Regarding their molecular function (MF) (Figure 5C), the enriched genes were mainly correlated with cell adhesion molecule binding (72 genes, P=4.47e–08), glycosaminoglycan binding (46 genes, P=1.1e–10), and cytokine activity (42 genes, P=2.49e–8).
Figure 5D shows the top 10 terms of the KEGG enrichment analysis in darkorange2 where most of the genes were related to immune and inflammatory responses, for instance cytokine-cytokine receptor interaction (62 genes, P=1.78e–09) and the chemokine signaling pathway (43 genes, P=7.37e–08). Furthermore, most genes were also related to ECM organization, like ECM-receptor interaction (21 genes, P=2.37e–05).
Figure 6 presents the GO and KEGG enrichment analyses of the red module, in which most of the genes were linked to the metabolic process. For instance, the top four GO-BP terms were “small molecule catabolic process” (P=6.51e−34), “organic acid catabolic process” (P=6.14e−32), “carboxylic acid catabolic process” (P=6.14e−32), “cellular amino acid metabolic process” (P=1.54e−27) and the top 4 KEGG terms were “fatty acid degradation” (P=4.06e−13), “glycine, serine and threonine metabolism” (P=3.17e−11), “valine, leucine and isoleucine degradation” (P=4.16e−10), “carbon metabolism” (P=4.42e−10).
Figure 6.

The top 10 most statistically significant terms of enrichment analyses of the red module: (A) biological process (BP) terms (B) cellular component (CC) terms; (C) molecular function (MF) terms; (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
We performed DEGs analysis in the darkorange2 and red modules. A total of 246 DEGs were confirmed in the darkorange2 module, including 231 upregulated and 15 downregulated genes in DKD compared with the control (Figure 7A). In the red module, 70 DEGs were identified, including 68 downregulated and 2 upregulated genes in DKD compared with the control (Figure 7B). These results indicated that most genes in DKD were abnormally activated or upregulated compared with their levels in normal tissues.
Figure 7.

Differentially expressed genes (DEGs) analysis. (A) Heatmap of DEGs in the darkorange2 module. Columns correspond to samples, and rows correspond to the gene expression level. Colors from green to red correspond to the gene expression level from low to high. (B) Heatmap of DEGs in the red module.
Identification of the key gene associated with DKD
The top-ranking clusters of the GO-BP enrichment analysis were “leukocyte migration” and “T cell activation”. Thus, we extracted the genes of these two clusters from the darkorange2 module for sub-network analysis. The top 150 genes with weighted correlations in the sub-network analysis were imported into Cytoscape. Then, we calculated the MCC values of each gene using the Cytohubba plug-in and screened the key genes with the top 10 MCC values (Figure 8A,B). Finally, FCER1G, the gene with the largest MCC value in both selected sub-networks, was identified as the key gene potentially associated with DKD.
Figure 8.

Identification of the key gene associated with diabetic kidney disease (DKD). (A) Weighted gene co-expression network analysis (WGCNA) sub-network analysis of the leukocyte migration cluster in the darkorange2 module. Each node corresponds to a gene. Colors from yellow to red correspond to the top 10 maximal clique centrality (MCC) values from low to high. (B) WGCNA sub-network analysis of the T cell activation cluster in the darkorange2 module. The MCC value of FCER1G with red color was the largest in both sub-networks.
PPI network creation
We interrogated the STRING database for constructing PPI networks to determine the biological connectivity of genes in darkorange2. A total of 657 interaction pairs were identified among those genes, and the proteins with their interaction scores were then imported into Cytoscape for visualization (Figure 9). FCER1G, which was centrally located in the network, was one of the top 9 central genes in the PPI network with more than 20 interaction pairs.
Figure 9.

Protein-protein interaction (PPI) network creation. A PPI network with 167 nodes and 657 edges was created in the darkorange2 module.
GEO validation
We analyzed the expression of the key gene in three additional datasets (GSE47183, GSE99339, GSE99340), and found that FCER1G was overexpressed in DKD compared with the levels in normal tissues in each dataset, thus verifying our finding (Figure 10).
Figure 10.

Gene expression omnibus (GEO) validation: the expression of FCER1G in diabetic kidney disease (DKD) compared with normal tissues in the GSE47183, GSE99339, GSE99340 datasets.
Clinical validation
We used the Nephroseq v5 online database to explore the correlation between the expression of FCER1G and clinical traits of DKD. As shown in Figure 11, there was a negative correlation between the expression of FCER1G in DKD glomeruli and the glomerular filtration rate (GFR) (r=−0.61, P=0.003), indicating that the abnormal upregulation of FCER1G was associated with impaired renal function, which may aggravate the development of DKD.
Figure 11.
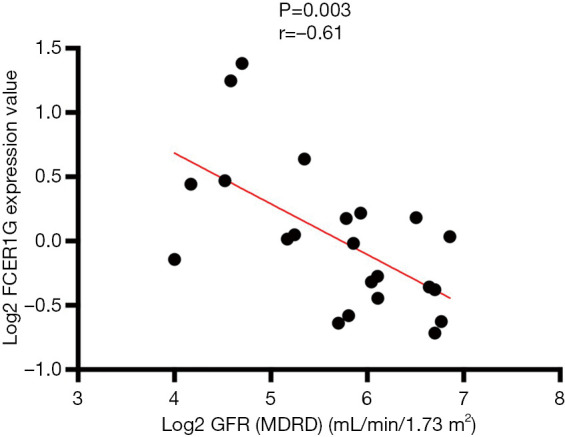
Correlation between the expression of FCER1G in diabetic kidney disease (DKD) glomeruli and the glomerular filtration rate (GFR), a P value of <0.05 was statistically significant.
Discussion
We used WGCNA to identify the key gene related to the molecular mechanisms underlying DKD. We concluded that the darkorange2 module had the most significant association with DKD and we investigated it further to understand more regarding the underlying pathogenesis. The results of the subsequent GO enrichment analyses showed that leukocyte migration and T cell activation were chiefly enriched. Furthermore, KEGG enrichment analysis identified genes primarily related to cytokine-cytokine receptor interaction and chemokine signaling pathways, revealing that the majority of genes were related to immune and inflammatory responses. DEGs analysis found that major genes in the darkorange2 module were upregulated in DKD, indicating an abnormal activation of inflammatory and immune responses. Based on the importance of the darkorange2 module in the DKD trait, we selected the top two most significant GO-BP terms for WGCNA sub-network analysis, which revealed that FCER1G is the key gene associated with DKD. Moreover, PPI network analysis indicated that FCER1G was one of the top nine central genes with more than 20 interaction pairs. Validation of external datasets from the GEO database showed that FCER1G was overexpressed in DKD compared with its levels in normal tissues. Furthermore, Pearson’s correlation analysis between the expression of FCER1G and renal function in DKD revealed that the abnormal up-regulation of FCER1G was associated with diabetic glomerular lesions, which may be important in the progress of DKD.
Regarding the red module, it was highly correlated with normal traits. Enrichment analyses revealed that major genes in this module participated in various metabolic processes, which were involved in maintaining normal biological functions. The results also indirectly reflected that DKD is possibly a metabolic disorder. As previously reported, metabolic abnormalities, such as amino acid and lipid abnormalities, were associated with impaired kidney function in DKD (31-35).
Gene FCER1G is located on chromosome 1q23 and encodes the γ subunit of the fragment crystallizable (Fc) region (Fc R) of immunoglobulin E (IgE). Fc R γ is a signal-transducing subunit that plays an essential role in chronic inflammatory programs (36-38). The binding between the Fc of immunoglobulins and the Fc R of immune cells activates cellular effector functions via the antigen-antibody binding reaction. In the normal immune system state, Fc:Fc R recognize and eliminate non-self antigens, while the same combination may trigger destructive inflammation, immune cell activation, phagocytosis, oxidative burst, and cytokine release in a pathological immune state (39).
Currently, the knowledge regarding the function of FCER1G and its relationship with kidney disease is very limited. In a mouse model of glomerulonephritis, deficiency of the Fc R γ chain decreased urinary albumin excretion and alleviated the pathological changes of the glomeruli, such as mesangial thickening, glomerulosclerosis, and infiltration of inflammatory cells (40). Suppression of the Fc R γ chain attenuated renal inflammation in models of immune-mediated glomerulonephritis by reducing inflammation and preventing fibrosis (41). Furthermore, the lack of the Fc R γ chain in a model of lupus prevented severe nephritis, even immune system component deposition in the glomeruli (42,43). Thus, the absence of the Fc R γ chain protected against the inflammatory response and autoimmune glomerulonephritis, which provides another potential pathway for therapeutic intervention (44). Thereby, FCER1G may be important in the immune and inflammatory responses that occur in renal disease.
The relationship between FCER1G and DKD has not been previously reported. However, it has been proposed that immunity and inflammation are tightly linked to DKD. As long ago as 1991, inflammatory cytokines were found to be involved in the molecular mechanisms of DKD (45), and subsequent studies verified their role in the development of DKD (46,47). Renal pathology analysis of patients with DKD indicated that the inflammatory cells deposited in the kidney were related to glomerular sclerosis and interstitial fibrosis (48,49). Although DKD is not considered an “immune-mediated” renal disease, an increasing number of studies demonstrated that immune system components participated in the progression of DKD (50). Clinical research studies have revealed that the activation of T cells (51) and the increasing levels of immune complexes in the circulation (52,53) are related to nephropathy progression in patients with diabetes mellitus. Consistent with our GO enrichment analysis results, several studies in animal models of DKD have also examined the effect of T cells, and T-regulatory cells in DKD (54,55).
Therefore, we propose that FCER1G overexpression regulates immune and inflammatory pathways that are involved in the pathogenesis of DKD.
We identified FCER1G as a key gene related to DKD by constructing a WGCNA network. This is the first study to report that gene FCER1G is a potential biomarker of DKD. Our study provides a new insight into the molecular mechanisms underlying DKD and offers a novel candidate target for the precise treatment of this disease.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was supported by the China National Key R&D Program during the 13th Five-year Plan Period (2018YFC1314003).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/atm-20-1087
Peer Review File: Available at http://dx.doi.org/10.21037/atm-20-1087
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-1087). The authors have no conflicts of interest to declare.
References
- 1.Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol 2013;9:713-23. 10.1038/nrendo.2013.184 [DOI] [PubMed] [Google Scholar]
- 2.Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol 2016;12:73-81. 10.1038/nrneph.2015.173 [DOI] [PubMed] [Google Scholar]
- 3.Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet 2010;375:2073-81. 10.1016/S0140-6736(10)60674-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tancredi M, Rosengren A, Svensson AM, et al. Excess Mortality among Persons with Type 2 Diabetes. N Engl J Med 2015;373:1720-32. 10.1056/NEJMoa1504347 [DOI] [PubMed] [Google Scholar]
- 5.Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nat Rev Dis Primers 2015;1:15018. 10.1038/nrdp.2015.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia 2001;44:1957-72. 10.1007/s001250100000 [DOI] [PubMed] [Google Scholar]
- 7.Qi W, Keenan HA, Li Q, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med 2017;23:753-62. 10.1038/nm.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan Y, Xiao W, Li Z, et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun 2015;6:7841. 10.1038/ncomms8841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jerums G, Panagiotopoulos S, Premaratne E, et al. Integrating albuminuria and GFR in the assessment of diabetic nephropathy. Nat Rev Nephrol 2009;5:397-406. 10.1038/nrneph.2009.91 [DOI] [PubMed] [Google Scholar]
- 10.Xia L, Su X, Shen J, et al. ANLN functions as a key candidate gene in cervical cancer as determined by integrated bioinformatic analysis. Cancer Manag Res 2018;10:663-70. 10.2147/CMAR.S162813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin X, Wang J, Zhang J. Identification of biomarkers of chromophobe renal cell carcinoma by weighted gene co-expression network analysis. Cancer Cell Int 2018;18:206. 10.1186/s12935-018-0703-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Hu AX, Zhao JL, et al. Identification of Key Gene Modules in Human Osteosarcoma by Co-Expression Analysis Weighted Gene Co-Expression Network Analysis (WGCNA). J Cell Biochem 2017;118:3953-9. 10.1002/jcb.26050 [DOI] [PubMed] [Google Scholar]
- 13.Giulietti M, Occhipinti G, Principato G, et al. Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell Oncol (Dordr) 2016;39:379-88. 10.1007/s13402-016-0283-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin J, Yang T, Zeng N, et al. Differential coexpression networks in bronchiolitis and emphysema phenotypes reveal heterogeneous mechanisms of chronic obstructive pulmonary disease. J Cell Mol Med 2019;23:6989-99. 10.1111/jcmm.14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng T, Li K, Zheng P, et al. Weighted Gene Coexpression Network Analysis Identified MicroRNA Coexpression Modules and Related Pathways in Type 2 Diabetes Mellitus. Oxid Med Cell Longev 2019;2019:9567641. 10.1155/2019/9567641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musimbi ZD, Rono MK, Otieno JR, et al. Peripheral blood mononuclear cell transcriptomes reveal an over-representation of down-regulated genes associated with immunity in HIV-exposed uninfected infants. Sci Rep 2019;9:18124. 10.1038/s41598-019-54083-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan S, Wang W, Gao G, et al. Key genes and functional coexpression modules involved in the pathogenesis of systemic lupus erythematosus. J Cell Physiol 2018;233:8815-25. 10.1002/jcp.26795 [DOI] [PubMed] [Google Scholar]
- 18.Sumitomo S, Nagafuchi Y, Tsuchida Y, et al. A gene module associated with dysregulated TCR signaling pathways in CD4(+) T cell subsets in rheumatoid arthritis. J Autoimmun 2018;89:21-9. 10.1016/j.jaut.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 19.MacDonald ML, Ding Y, Newman J, et al. Altered glutamate protein co-expression network topology linked to spine loss in the auditory cortex of schizophrenia. Biol Psychiatry 2015;77:959-68. 10.1016/j.biopsych.2014.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Roy B, Dwivedi Y. Co-expression network modeling identifies key long non-coding RNA and mRNA modules in altering molecular phenotype to develop stress-induced depression in rats. Transl Psychiatry 2019;9:125. 10.1038/s41398-019-0448-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckerman P, Qiu C, Park J, et al. Human Kidney Tubule-Specific Gene Expression Based Dissection of Chronic Kidney Disease Traits. EBioMedicine 2017;24:267-76. 10.1016/j.ebiom.2017.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shved N, Warsow G, Eichinger F, et al. Transcriptome-based network analysis reveals renal cell type-specific dysregulation of hypoxia-associated transcripts. Sci Rep 2017;7:8576. 10.1038/s41598-017-08492-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Yang S, Yan M, et al. Interstitial HIF1A induces an estimated glomerular filtration rate decline through potentiating renal fibrosis in diabetic nephropathy. Life Sci 2020;241:117109. 10.1016/j.lfs.2019.117109 [DOI] [PubMed] [Google Scholar]
- 24.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 2005;4:Article17. [DOI] [PubMed]
- 26.Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 2012;16:284-7. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498-504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014;8 Suppl 4:S11. 10.1186/1752-0509-8-S4-S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447-52. 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niewczas MA, Mathew AV, Croall S, et al. Circulating Modified Metabolites and a Risk of ESRD in Patients With Type 1 Diabetes and Chronic Kidney Disease. Diabetes Care 2017;40:383-90. 10.2337/dc16-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tofte N, Vogelzangs N, Mook-Kanamori D, et al. Plasma Metabolomics Identifies Markers of Impaired Renal Function: A Meta-analysis of 3089 Persons with Type 2 Diabetes. J Clin Endocrinol Metab 2020;105:dgaa173. [DOI] [PubMed]
- 33.Afshinnia F, Nair V, Lin J, et al. Increased lipogenesis and impaired β-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019;4:e130317. 10.1172/jci.insight.130317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang W, Luo Y, Yang S, et al. Ectopic lipid accumulation: potential role in tubular injury and inflammation in diabetic kidney disease. Clin Sci (Lond) 2018;132:2407-22. 10.1042/CS20180702 [DOI] [PubMed] [Google Scholar]
- 35.Eid S, Sas KM, Abcouwer SF, et al. New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia 2019;62:1539-49. 10.1007/s00125-019-4959-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreu P, Johansson M, Affara NI, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell 2010;17:121-34. 10.1016/j.ccr.2009.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang Y, Wang P, Zhao M, et al. Demethylation of the FCER1G promoter leads to FcepsilonRI overexpression on monocytes of patients with atopic dermatitis. Allergy 2012;67:424-30. 10.1111/j.1398-9995.2011.02760.x [DOI] [PubMed] [Google Scholar]
- 38.Kraft S, Kinet JP. New developments in FcepsilonRI regulation, function and inhibition. Nat Rev Immunol 2007;7:365-78. 10.1038/nri2072 [DOI] [PubMed] [Google Scholar]
- 39.Brandsma AM, Hogarth PM, Nimmerjahn F, et al. Clarifying the Confusion between Cytokine and Fc Receptor "Common Gamma Chain". Immunity 2016;45:225-6. 10.1016/j.immuni.2016.07.006 [DOI] [PubMed] [Google Scholar]
- 40.Ravetch JV, Clynes RA. Divergent roles for Fc receptors and complement in vivo. Annu Rev Immunol 1998;16:421-32. 10.1146/annurev.immunol.16.1.421 [DOI] [PubMed] [Google Scholar]
- 41.Kanamaru Y, Pfirsch S, Aloulou M, et al. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol 2008;180:2669-78. 10.4049/jimmunol.180.4.2669 [DOI] [PubMed] [Google Scholar]
- 42.Andrews BS, Eisenberg RA, Theofilopoulos AN, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med 1978;148:1198-215. 10.1084/jem.148.5.1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol 1985;37:269-390. 10.1016/S0065-2776(08)60342-9 [DOI] [PubMed] [Google Scholar]
- 44.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 1998;279:1052-4. 10.1126/science.279.5353.1052 [DOI] [PubMed] [Google Scholar]
- 45.Hasegawa G, Nakano K, Sawada M, et al. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int 1991;40:1007-12. 10.1038/ki.1991.308 [DOI] [PubMed] [Google Scholar]
- 46.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, et al. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol 2011;7:327-40. 10.1038/nrneph.2011.51 [DOI] [PubMed] [Google Scholar]
- 47.Shoukry A, Bdeer Sel A, El-Sokkary RH. Urinary monocyte chemoattractant protein-1 and vitamin D-binding protein as biomarkers for early detection of diabetic nephropathy in type 2 diabetes mellitus. Mol Cell Biochem 2015;408:25-35. 10.1007/s11010-015-2479-y [DOI] [PubMed] [Google Scholar]
- 48.Nguyen D, Ping F, Mu W, et al. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006;11:226-31. 10.1111/j.1440-1797.2006.00576.x [DOI] [PubMed] [Google Scholar]
- 49.Klessens CQF, Zandbergen M, Wolterbeek R, et al. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant 2017;32:1322-9. [DOI] [PubMed] [Google Scholar]
- 50.Pichler R, Afkarian M, Dieter BP, et al. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol 2017;312:F716-31. 10.1152/ajprenal.00314.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bending JJ, Lobo-Yeo A, Vergani D, et al. Proteinuria and activated T-lymphocytes in diabetic nephropathy. Diabetes 1988;37:507-11. 10.2337/diab.37.5.507 [DOI] [PubMed] [Google Scholar]
- 52.Lopes-Virella MF, Carter RE, Baker NL, et al. High levels of oxidized LDL in circulating immune complexes are associated with increased odds of developing abnormal albuminuria in Type 1 diabetes. Nephrol Dial Transplant 2012;27:1416-23. 10.1093/ndt/gfr454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lopes-Virella MF, Hunt KJ, Baker NL, et al. High levels of AGE-LDL, and of IgG antibodies reacting with MDA-lysine epitopes expressed by oxLDL and MDA-LDL in circulating immune complexes predict macroalbuminuria in patients with type 2 diabetes. J Diabetes Complications 2016;30:693-9. 10.1016/j.jdiacomp.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 54.Lim AK, Ma FY, Nikolic-Paterson DJ, et al. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia 2010;53:1772-82. 10.1007/s00125-010-1757-1 [DOI] [PubMed] [Google Scholar]
- 55.Eller K, Kirsch A, Wolf AM, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011;60:2954-62. 10.2337/db11-0358 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as