

INTRODUCTION
Lung cancer represents 11.6% of all cancers globally, with more than 2 million new cases worldwide in 2018.1 Across all disease stages, non–small-cell lung cancer (NSCLC) has an overall 5-year relative survival rate of 23%; for metastatic NSCLC, the same rate is 6% in the United States.2 Various subgroups of metastatic NSCLC, such as KRAS-mutated NSCLC, are typically associated with poorer survival.3
CONTEXT
Key Objective
To provide an overview of the epidemiology and prognostic and predictive value of KRAS mutations in non–small-cell lung cancer (NSCLC), summarizing current treatment approaches and highlighting unmet needs.
Knowledge Generated
Specific RAS mutations can render RAS proteins constitutively active, resulting in uncontrolled cell proliferation. KRAS-mutated NSCLC is heterogeneous. KRAS G12C is the most prevalent of the KRAS mutations. KRAS mutations, particularly KRAS G12C, are indicative of poor overall survival, with comutational status also affecting prognosis. Therapies targeting KRAS are beginning to show clinical potential, most notably with KRASG12C inhibitors. Strategies for KRASG12C inhibition are discussed.
Relevance
In Western populations, KRAS mutations are one of the most common oncogenic drivers in NSCLC, with KRAS G12C mutation representing nearly half of KRAS mutations. KRAS inhibition offers potential for targeted treatment of NSCLC; KRASG12C inhibitors are under clinical development and may represent an effective therapeutic option for patients with NSCLC.
Advances in the understanding of NSCLC oncogenic drivers have led to the development of targeted therapies for several molecular subsets of lung adenocarcinoma, contributing to an improvement in patient outcomes and survival.4-6 Although KRAS mutations are observed in approximately one-third of patients with lung adenocarcinoma in Western populations,7 no approved targeted therapies exist for this important molecular subset.
OVERVIEW OF KRAS IN NSCLC
Introduction and Prevalence of KRAS Mutations
RAS genes encode small membrane-bound guanine nucleotide-binding proteins (G proteins), including NRAS, HRAS, and KRAS.8,9 These spherical proteins have a relatively smooth molecular surface without readily accessible binding pockets and have high affinity for guanosine diphosphate (GDP) and guanosine triphosphate (GTP).10,11 RAS proteins have a slow dissociation rate from the GDP-bound state. Guanine nucleotide exchange factors (GEFs), such as son-of-sevenless (SOS), promote conversion from the inactive GDP-bound state to the active GTP-bound state (Fig 1).10,12 Similarly, GTPase-activating proteins accelerate conversion back to the inactive state when they are recruited to the plasma membrane in direct proximity to RAS.10
FIG 1.
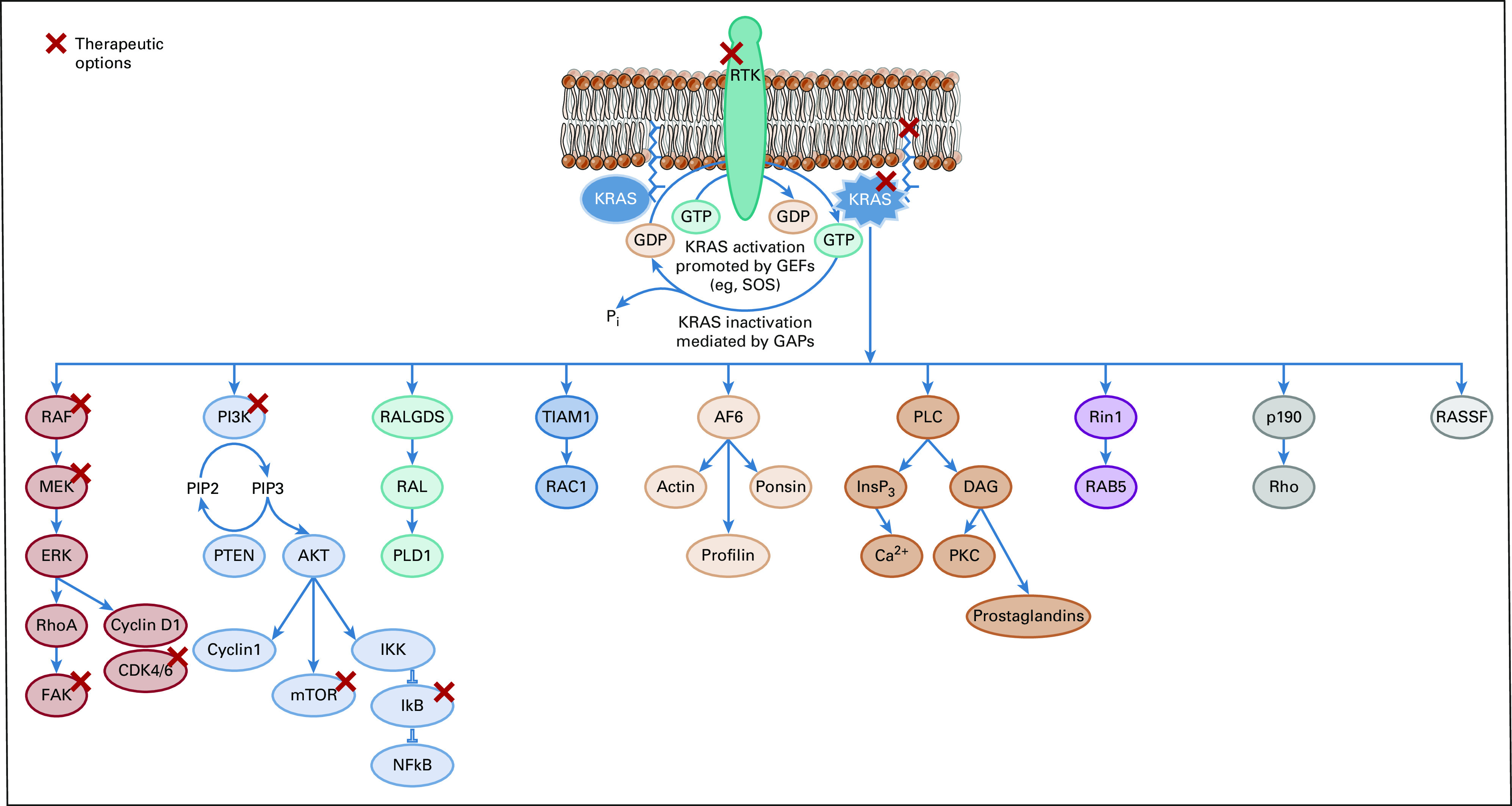
Overview of the process of phosphorylation of the KRAS protein and the downstream effector pathways and potential therapeutic targets for inhibitors of KRAS activity. GDP, guanosine diphosphate; GEF, guanine nucleotide exchange factors; GTP, guanosine triphosphate; SOS, son-of-sevenless.
KRAS proteins are highly homologous (90%) with other RAS proteins in the G or catalytic domain sequence.13 The G domain binds guanine nucleotides, activating signal transduction. It includes four main regions in the inactive GDP-bound (Fig 2A) and the active GTP-bound protein (Fig 2B): amino acid residues 10-17 (the phosphate-binding loop), 30-40 (switch I), 60-76 (switch II), and 116-120 and 145-147 (the base-binding loops).8,9,13 In contrast, the C-terminus, which incorporates farnesyl or prenyl groups, is a hypervariable region. The sequence divergence between different RAS proteins contributes to differential post-translational processing, subcellular trafficking, cellular localization, and subsequent response to therapy.13 There is approximately 8% sequence homology in the C-terminus between KRAS and the other RAS proteins.8,13 In common with the other RAS proteins, KRAS proteins are binary switches, cycling between active GTP-bound and inactive GDP-bound states in normal cells during signal transduction to regulate downstream signaling pathways.9,10 This results in conformational changes in the switch I and II regions (Fig 2), enabling GTP-bound KRAS to interact with and activate proteins regulating effector pathways.9,13,14 In the active state, KRAS relays upstream signals from cell surface receptors to a number of downstream pathways, including RAF/MEK/ERK, PI3K/AKT/mTOR, RALGDS/RAL, and TIAM1/RAC, that control normal cell function and proliferation (Fig 1).8,13,15
FIG 2.

Structure of the KRAS protein in (A) guanosine diphosphate (GDP)–bound state and (B) guanosine triphosphate (GTP)–bound state, with the four main regions highlighted.
KRAS mutations greatly diminish GTP hydrolysis, thereby leading to increased GTP-bound mutant KRAS in the active state. Constitutively active KRAS then initiates downstream signaling and leads to uncontrolled cell proliferation and survival.16 As a result, KRAS mutations are oncogenic drivers associated with numerous human cancers, including lung cancer.10 The mechanisms through which distinct KRAS mutations lead to RAS activation are not thoroughly understood and might differ by allelotype.12 This has made targeting RAS and KRAS extremely challenging in the clinic.9,11,17
Across all cancers, the most common RAS mutations are KRAS mutations (85%), with NRAS (12%) and HRAS (3%) much less frequent. Globally, KRAS mutations are associated with approximately 1 million cancer-related deaths annually.10 Oncogenic KRAS mutations occur at codons 12 and 13 (ie, within the phosphate-binding loop) and at codon 61 (ie, within switch II) of the G domain. The most common mutation is at codon 12 and accounts for 80% of all KRAS mutations.9,18 Single-base substitution mutations at position 12 lead to stabilization of KRAS-GTP binding, resulting in structural changes in KRAS and consequently constitutively activated KRAS (GTP-bound state).9,19 For example, the KRAS G12C mutation is a single point mutation with a glycine-to-cysteine substitution at codon 12. The cysteine 12 residue is located close to the nucleotide-binding pocket and switches I and II. The glycine-to-cysteine substitution has an effect on GEF binding and downstream signaling, leading to cell proliferation and survival.13
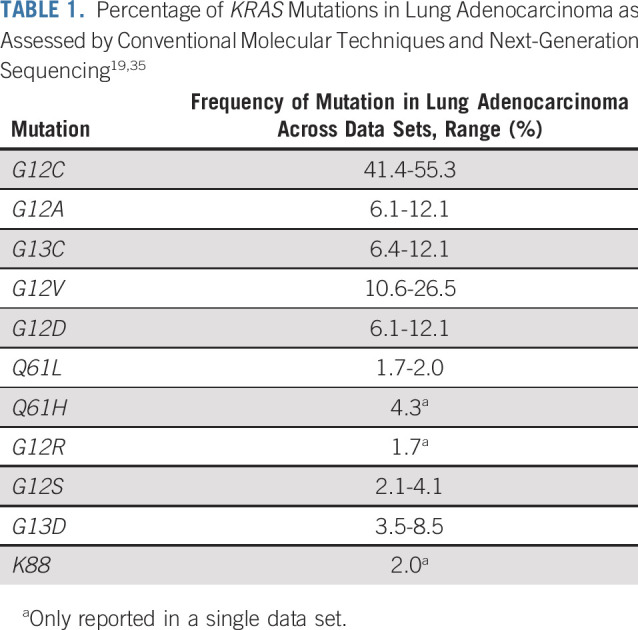
In NSCLC, KRAS is one of the most frequently mutated oncogenes. According to a large genomic study of human lung tumors, KRAS mutations are more commonly observed in the subtype of adenocarcinoma than in squamous cell carcinoma or large cell carcinoma, with a mutation frequency of 32.6% in adenocarcinoma.20 The incidence of KRAS mutations is between 8% and 24% in large clinical trials21 and between 27% and 32% in a series of patients with adenocarcinoma.7,22 The majority of KRAS mutations are found in current and former smokers23; however, KRAS G12D is also found in never-smokers.24 In Western populations, approximately one-quarter to one-third of patients with NSCLC have a KRAS mutation.25-27 The overall incidence of KRAS mutation is lower (approximately 5%-10%) in Indian,28,29 Chinese,30-32 and Iranian populations.33 Across Western populations with NSCLC, the prevalence of the KRAS G12C mutation is approximately 12%-14% (40%-50% of all KRAS mutations).8,19,22,25-27,34 KRAS G12V mutations account for approximately 20%-25% of KRAS mutations in NSCLC,19,22 and others include KRAS G12D, KRAS G12A, and KRAS G12S (Table 1).19,35 The proportion of KRAS G12C–mutated subtypes is slightly lower (32%-34%) across large databases of Asian populations.31,36 KRAS G12C and KRAS G12D mutations are approximately equally distributed across disease stage in patients with NSCLC.27 Of note, in KRAS G12C–mutant tumors, KRAS G12C was a truncal mutation in the majority of patients with NSCLC, suggesting that it is likely a driver mutation in most of these patients.35
TABLE 1.
Percentage of KRAS Mutations in Lung Adenocarcinoma as Assessed by Conventional Molecular Techniques and Next-Generation Sequencing19,35
The clinicopathological features of NSCLC with KRAS mutations are discussed in Appendix 1 (online only).
Relationship Between KRAS Mutation and Other Oncogenic Mutations, PD-L1 Expression, and Tumor Mutational Burden
Concurrent alterations in the expression of at least one other gene (tumor suppressor genes: CDKN2A, KEAP1, STK11, TP53; KRAS downstream effector genes: PIK3CA, BRAF, AKT1) exist in approximately half of patients with KRAS mutations (Appendix 2, online only, includes a discussion of additional coalterations). The most common comutations are TP53 (approximately 40%) and STK11 (12%-20%).26,27,37 Furthermore, approximately one-quarter of patients with KRAS mutations have more than one concurrent genetic alteration,26 which depends on the underlying KRAS mutation. For example, KRAS G12C mutations tend to co-occur with ERBB2 amplifications and ERBB4 mutations, KRAS G12V mutations co-occur with PTEN mutations, and KRAS G12D mutations co-occur with PDGFRA mutations.26 The presence of comutations tends to indicate a highly aggressive subgroup (Table 2); comutations may also have negative overall impact on response to treatment (Appendix 2). In contrast, KRAS mutations are generally mutually exclusive with other driver oncogenes in NSCLC, such as EGFR, ALK, and ROS1 mutations in the treatment-naïve setting, and occur relatively infrequently in combination with BRAF mutations.7,22,27
TABLE 2.
Association Between KRAS Mutations and KRAS G12C Mutations and Survival

KRAS-mutated NSCLC tumor cells are associated with higher PD-L1 expression levels than wild-type cells.38,39 It is possible that the increased PD-L1 expression is due to concurrent TP53 mutation40 or smoking status.41 The upregulation of PD-L1 appears to be via activation of phosphorylated extracellular signal–regulated kinase (ERK) signaling.42 The extent of PD-L1 expression appears to be related to the KRAS mutation subtype.38 KRAS G12D, KRAS G12V, and KRAS G13C mutations are associated with a higher proportion of PD-L1‒positive versus KRAS G12A and KRAS G12C mutations, which are associated with a higher proportion of PD-L1‒negative tumors.43 However, others have reported that KRAS G12C is associated with PD-L1 positivity, yet at low levels of expression.27 Preliminary data also suggest that the distribution of PD-L1 levels is higher in patients with KRAS mutations versus those with wild-type disease.44
Finally, patients with KRAS-mutated NSCLC have a higher tumor mutational burden (TMB) than those with wild-type disease.39,44 Even when adjusted for smoking status, the relationship between KRAS-mutant disease and TMB was maintained; these factors, taken together, may contribute to the positive response to immune checkpoint inhibitors targeting PD-L1 in patients with KRAS mutations.39
Current Treatment Options for Patients With KRAS Mutations: Clinical Outcomes
KRAS status and response to chemotherapy.
KRAS mutations appear to be predictive for treatment outcome and can be associated with chemotherapy resistance.45-49 For example, in vitro studies demonstrated that different KRAS mutations led to differences in sensitivity to cytotoxic chemotherapeutic agents,50 highlighting the need to consider KRAS mutations at the subgroup level. When compared with wild-type cells, KRAS G12C mutation was associated with reduced cisplatin sensitivity in vitro and in vivo.48,50 However, in patients with early-stage NSCLC from the Lung Adjuvant Cisplatin Evaluation (LACE)–Bio database, few biomarkers appeared to have prognostic or predictive effect, with the possible exception of a subset of KRAS mutations.51 Patients with codon 12 KRAS mutations and those with wild-type disease appeared to derive no benefit from adjuvant chemotherapy.52 In contrast, those with codon 13 KRAS mutations had significantly worse overall and disease-free survival.52 More recent data have demonstrated a similar overall response to chemotherapy (approximately 20%) in patients with KRAS G12C tumors and those with wild-type disease in the locally advanced or metastatic setting.44
KRAS status and response to targeted therapy.
A meta-analysis from 2008 identified an association between KRAS mutations and lack of response to EGFR–tyrosine kinase inhibitors (TKIs; predominantly gefitinib).53 This association remained when potentially confounding factors such as ethnicity, investigational treatment (erlotinib v gefitinib), and previous treatment were taken into account.53 This finding results from the mutual exclusivity of EGFR mutations with KRAS mutations in NSCLC (Appendix 3, online only). In a single-center analysis, no patient with KRAS-mutated disease responded to gefitinib or erlotinib compared with 35.7% of patients with KRAS wild-type disease achieving a complete or partial response.54 Similarly, in a pooled analysis of four trials comparing EGFR-TKI with placebo and including 968 patients with advanced disease, KRAS mutation was associated with an extremely low overall response rate (1.3%).55
KRAS status and response to immunotherapy.
KRAS mutation status tended to be associated with beneficial outcome with immune checkpoint inhibitors such as pembrolizumab, with prolonged progression-free survival (PFS) in KRAS-mutated advanced NSCLC (14.7 months) versus wild-type disease (3.5 months).40 It is possible that comutations with STK11/LKB1 affect the response to immunotherapy.56,57 In a retrospective study, PD-L1 expression seemed to be more relevant for predicting efficacy of immune checkpoint inhibitors in KRAS-mutant NSCLC (approximately 87% current or former smokers) than in patients with other types of NSCLC.43 However, the benefits of immune checkpoint inhibitors may not extend to all KRAS-mutated subgroups. In one study, there was a trend toward improved outcomes after immune checkpoint inhibitors in patients with KRAS G12A or KRAS G12V mutations compared with KRAS G12C mutations; however, this was not statistically significant.43 More recently, in a study enrolling PD-L1–positive patients, pembrolizumab monotherapy did not significantly improve outcomes compared with chemotherapy for patients with wild-type disease.44 However, pembrolizumab monotherapy did improve PFS and overall survival in patients with KRAS-mutant disease, particularly in the KRAS G12C subgroup.44 Median PFS in patients with KRAS G12C tumors was 15 months versus 6 months in those with wild-type disease and 12 months in those with any KRAS mutation; overall survival was not reached in the KRAS G12C subgroup versus 15 months in those with wild-type disease and 12 months in those with any KRAS mutation.44 The addition of chemotherapy to pembrolizumab eliminated the differential effect of KRAS mutations on pembrolizumab activity.58 PFS and overall survival were improved by the combination versus chemotherapy alone in those with wild-type and mutant disease.58
Considering the heterogeneity of responses by KRAS-mutated subtypes of NSCLC to currently available therapies, developing agents that effectively target and inhibit specific KRAS subtypes is key to improving outcomes for patients with KRAS-mutated NSCLC.
STRATEGIES FOR KRAS INHIBITION
Inhibition of KRAS Protein Activity
Early attempts to inhibit RAS proteins focused on decreasing RAS activation by identifying molecules preferentially binding to the RAS-GTP pocket.11,13 As the first RAS protein identified and sequenced, early focus was mostly directed toward HRAS protein inhibition. However, KRAS was subsequently found to be of more clinical relevance and has become the target of renewed research efforts.10 Nonetheless, attempts to directly inhibit formation of the GTP-bound protein have been unsuccessful because of the high binding affinity of RAS proteins for GTP and the lack of accessible binding pockets on RAS proteins large enough to allow small-molecule binding (Table 313,59-66). Furthermore, given the ubiquitous nature of KRAS proteins and their influence on multiple downstream pathways, it was reasoned that arbitrarily inhibiting wild-type and mutant KRAS proteins would likely be associated with unacceptable toxicity.9
TABLE 3.
Overview of Unsuccessful Strategies to Target KRAS Mutations in NSCLC

An alternative approach to the direct inhibition of KRAS proteins involves interfering with the peripheral association of KRAS with the lipid membrane compartments (Fig 1).9 Early research demonstrated that RAS protein isoprenylation was required for peripheral association with plasma and internal membranes and for subsequent oncogenic transformation.67 This led to the development of farnesyl transferase inhibitors, which were found to inhibit tumor growth of HRAS mutant cancers, and subsequently demonstrated activity against KRAS-mutant cancer in vitro68 and in murine models.69 However, clinical trials evaluating farnesyl transferase inhibitors in KRAS-mutated lung cancers have produced disappointing results.8 It appears that the presence of alternative prenylation pathways, via geranylgeranyltransferase type 1, allows for the continued association of KRAS, but not HRAS, with the cell membrane. This difference may account for the different outcomes of farnesyl transferase inhibitors in RAS-mutant cancers.70 Furthermore, it has led to exploration of combined farnesyl transferase and geranylgeranyl transferase type 1 inhibition.13 Other attempts to inhibit the association of KRAS with the cell membrane have been unsuccessful to date. For example, salirasib (trans-farnesylthiosalicyclic acid) was found to have preclinical activity but had no demonstrable activity in the first phase II study in patients with KRAS-mutant lung cancer.59 Salirasib has not been further developed.59 Other research has focused on inhibition of phosphodiesterase delta, a chaperone protein involved in association of KRAS with lipid membrane compartment.71 Despite the interest in targeting membrane association of KRAS, this approach may be relatively nonspecific and is unlikely to discriminate between the association of mutant and wild-type proteins, leading to potential toxicity concerns.13
Preclinical studies found that GEF SOS1 inhibition completely inhibits the RAS/RAF/MEK/ERK pathway in cells with wild-type KRAS and reduces phospho-ERK activity by 50% in KRAS-mutant cell lines regardless of the allelotype.72 Combinations with KRASG12C inhibitors are synergistic; however, clinical efficacy and tolerability have not yet been explored.72
Downstream Inhibition of Key KRAS Pathway Mediators
KRAS proteins have an integral role in regulating numerous downstream effector pathways. Of these, the RAF/MEK/ERK and PI3K/AKT/mTOR pathways are arguably the most important in KRAS-mutated cancers and offer multiple potential opportunities for targeted therapy in RAS-mutated cancer (Fig 1).
The importance of feedback mechanisms across pathways in RAS-mutated cancer is highlighted by the failure of RAF inhibition in this subgroup and the paradoxical activation of the RAF/MEK/ERK pathway in cells expressing wild-type BRAF.13,64 Currently available RAF inhibitors tend to inhibit selected RAF dimers. However, next-generation RAF inhibitors, considered pan-RAF inhibitors, appear not to cause paradoxical activation of the RAF/MEK/ERK pathway and, thus, may be more effective in RAS-mutated disease.64 Similarly, MEK inhibition was considered a potential therapeutic target.73 However, to date, monotherapy with MEK inhibitors has not demonstrated clinical activity because of increased upstream signaling mediated via ERBB3 and FGFR1 leading to ERK signaling activation.13,74,75 Consequently, MEK inhibitors were evaluated in combination regimens, with early results indicating MEK inhibition may be synergistic with docetaxel in KRAS-mutated NSCLC.76 However, this was not borne out with a randomized comparative study.61 Finally, ERK inhibition has been investigated as a strategy.13 Although not specifically in NSCLC, results of an in vitro analysis suggested the ERK node may be the rate-limiting step in the RAF/MEK/ERK pathway. Direct ERK inhibition may prevent relief of negative feedback leading to ERK hyperactivation, which is observed with RAF/MEK inhibitors, and may prove a useful therapeutic approach.77 However, it is suggested that resistance will likely occur because of single amino acid mutations in the ERK DFG motif after exposure to ERK inhibition.78
Similarly, it appears that pan-PI3K inhibitors and dual PI3K/mTOR inhibitors inhibit growth of NSCLC cell lines in vitro. However, limited efficacy has been observed in clinical trials due to feedback mechanisms.79,80
Cyclin-dependent kinase 4/6 inhibitors, such as abemaciclib, have been reported to inhibit cell growth in vitro. Early results were encouraging in patients with KRAS-mutated NSCLC81,82; however, a phase III study comparing abemaciclib with erlotinib in previously treated patients failed to demonstrate efficacy.83
The focal adhesion kinase (FAK) pathway is another signaling axis for KRAS-mutated NSCLC. FAK is a master regulator of cell adhesion during cell motility and invasion, and several FAK inhibitors are under development.84 Preclinical models have demonstrated that KRAS-mutant cells with alterations in TP53 or CDKN2A are sensitive to FAK inhibition.85 In patients with KRAS-mutated NSCLC, the FAK inhibitor defactinib was reported to have modest clinical activity; however, efficacy was not associated with comutation (TP53 or CDKN2A) status.65
Small-molecule inhibitors of PARP impair the stabilization and restart of stalled DNA replication forks, trapping PARP on chromatin to achieve its cytotoxic effect. The combination with WEE1 inhibitors is associated with killing of 25%-40% of KRAS-mutant NSCLC cells,86 although clinical efficacy in KRAS-mutated disease is yet to be determined.
To overcome the increased upstream signaling observed when a single downstream effector pathway is targeted in KRAS-mutated cancer, combination therapy targeting multiple downstream pathways may be required.13,79 Combinations of MEK inhibitors and EGFR inhibitors have demonstrated growth inhibition of KRAS-mutated NSCLC cell lines in vitro and tumor regression in vivo in KRAS-mutated lung cancers, but in clinical trials the combination was associated with unacceptable toxicity without any clinical benefit.87 Combinations including PI3K inhibitors have proven effective against KRAS-mutated cell lines88,89 and in KRAS-mutated murine lung cancers90 and were initially considered as a potential approach for the treatment of KRAS-mutated NSCLC. However, combinations of MEK and PI3K inhibitors appear to be associated with unacceptable toxicity, and further clinical evaluation appears unlikely.79 In contrast, the combination of P13K inhibition with KRASG12C inhibition may offer a more acceptable therapeutic window in patients with KRAS G12C–mutated NSCLC.79
Finally, the Src homology 2 domain-containing PTP2 (SHP2) protein is required for RAS/ERK pathway activation, with evidence implicating SHP2 as an oncogenic driver.91 Inhibition of SHP2 is ineffective against KRAS-mutated cancer cell lines in vitro, but in vivo evidence suggests a role for SHP2 inhibition under growth factor–limiting conditions.92 Combined with MEK inhibition, SHP2 inhibition may have therapeutic potential in KRAS-mutated NSCLC.92
Synthetic lethal interaction partners of RAS are potentially important because they represent genes that act as modifiers of known oncogenes but have no effect on their wild-type counterparts.93 Metabolic synthetic lethality appears a possible approach,94-96 with SLC7A11 suppression being highly selective for KRAS mutant versus wild-type cell lines in vitro and leading to tumor regression in animal models.95 Another potential partner includes CDKN1A.97 The heterogeneity of mutant KRAS proteins likely extends to heterogeneity in synthetic lethality partners.97 Collateral genetic dependencies specific to mutant KRAS proteins may also be relevant to understand, and therapies targeting these dependencies may enhance KRASG12C inhibition.81
Direct Inhibitors of Mutant KRAS
The lack of success with direct targeting of KRAS proteins, downstream inhibition of KRAS effector pathways, and other strategies contributed to a focus on developing mutation-specific KRAS inhibitors.11,15 Given the crucial role of KRAS proteins in normal physiology, KRAS inhibition has potential for substantial toxicity.19 Inhibition of mutant KRAS proteins, however, should limit toxicity in healthy tissue and thus provide a therapeutic advantage over inhibition of wild-type proteins or the effector pathways.13
The identification of a hidden pocket adjacent to switch II in GDP-bound KRAS9 has renewed interest in direct targeting of KRAS mutant proteins (Fig 3).63,98 Covalent binding of small molecules to KRAS mutant proteins appears to lead to changes in the switch I‒ or switch II‒binding regions, interfering with the switch functions and affecting the binding of GEFs to the GDP-bound protein, thereby preventing conversion of the mutant protein to the GTP-bound active state.98 These small-molecule inhibitors are KRAS mutant protein–specific, with initial focus on KRASG12C proteins. Cysteine 12 in KRASG12C protein offers the potential for disulfide covalent binding with small-molecule inhibitors, maintaining the mutant KRAS in its inactive GDP-bound state.63,98 However, early KRASG12C inhibitors were not sufficiently potent or were too unstable for further development.63 AMG 510 is the most clinically advanced of the covalent small-molecule inhibitors of KRASG12C mutant proteins.99,100 AMG 510 selectively and irreversibly binds to cysteine 12 in a small pocket, the H95 groove, on the mutated KRASG12C protein.101 AMG 510 has been shown to inhibit SOS-catalyzed nucleotide exchange in mutant KRASG12C, resulting in in vitro inhibition of KRAS signaling and impaired viability of KRAS G12C-mutated cell lines, but not wild-type cells.102,103 Early phase I results support the tolerability and antitumor efficacy of a small-molecule inhibitor of KRASG12C, but caution in result interpretation is urged until survival advantages for direct inhibitors of mutant proteins are confirmed in large-scale randomized trials.104 The benefit of direct KRASG12C inhibition is further supported by preliminary evidence that a second covalent inhibitor of KRASG12C, MRTX849, is associated with regression and objective responses in patients with KRAS G12C-mutated NSCLC.105 Clinical trials are also underway evaluating other oral KRASG12C inhibitors, LY3499446 (ClinicalTrials.gov identifier: NCT04165031) and JNJ-74699157 (ClinicalTrials.gov identifier: NCT04006301), in patients with KRAS G12C-mutated advanced solid tumors, including NSCLC. Similarly, strategies to inhibit KRASG12D are also being developed and offer promise for future treatment options.106
FIG 3.

A three-dimensional representation of KRAS in the guanosine diphosphate–bound state showing the hidden pocket.
Resistance to direct targeting of RAS proteins does occur and may be due to genetic alterations in the nucleotide exchange function or adaptive mechanisms in either downstream pathways or in newly expressed KRASG12C.79,107-111 For example, newly expressed KRASG12C may stay in an active state (ie, resistant to inhibition) as a result of EGFR or AURK signaling pathways and mediated by PTPN11/SHP2 recruiting SOS1 to promote conversion to the active state.111 Thus, combination therapies are likely to provide greater benefit than direct targeting of KRASG12C alone. In addition, increased understanding of the biologic diversity underpinning KRAS-mutated NSCLC will allow for improved identification of patient subgroups most likely to benefit from a specific targeted treatment.99 The presence of STK11/LKBI comutations, for example, appears to have an important role in the resistance of NSCLC to checkpoint inhibitors. Thus, combining checkpoint inhibitors with the evolving new direct inhibitors of mutant KRAS proteins may shift the balance away from an immune-suppressive tumor microenvironment to allow effective antitumor immunity110 and provide a new improved approach to treating the disease.99 The combination of KRAS and SHP2 inhibitors offers potential synergistic activity, as described above, and is worthy of being assessed clinically.101,111 Furthermore, MEK inhibitors and SHP2 inhibitors were found to be synergistic in KRAS-mutant cancer cell lines, including NSCLC lines, making this combination also worthy of additional exploration.92,112,113 The SHP2 inhibitors, TNO155 (Novartis) and RMC-4630 (Revolution Medicine), are currently in active phase I/II clinical trials as monotherapy (ClinicalTrials.gov identifiers: NCT03114319 and NCT03634982, respectively) and in combination with a KRASG12C inhibitor (ClinicalTrials.gov identifiers: NCT04330664 and NCT04185883) and a MEK inhibitor (ClinicalTrials.gov identifier: NCT03989115), respectively. Similarly, combinations of KRAS inhibitors with inhibitors of the ErbB family including EGFR, PI3K, AKT, and MEK1/2 have been evaluated in vitro and are also of interest.101 Triple-drug regimens are also explored, aimed to disrupt compensatory feedback loops to avoid the onset of resistance, while minimizing additive toxicities.114
In conclusion, the new class of KRASG12C inhibitors is currently undergoing clinical assessment with promising early results. This represents a major breakthrough against a molecular subtype of NSCLC present in a significant proportion of patients with no currently available targeted treatment options. Extensive clinical and translational evaluation of AMG 510 and other KRAS inhibitors is being conducted in patients with KRAS G12C mutation in ongoing studies. Efforts to develop novel combination approaches building on AMG 510 are already being undertaken. These developments prompt the inclusion of KRAS testing in the molecular testing strategies for advanced lung adenocarcinoma.
ACKNOWLEDGMENT
We thank Paul Cifuentes, Beloo Mirakhur, and I-Fen Chang, of Amgen, Thousand Oaks, CA, for critical review of the manuscript; Noella Moua Vang and Jonathan Aspe of Amgen, Thousand Oaks, CA, for operational assistance; Lee Hohaia, PharmD, CMPP, and James Balwit, MS, CMPP (ICON, North Wales, PA, whose work was funded by Amgen, and Yang Li, PhD of Amgen, for medical writing assistance in the preparation of this manuscript.
Appendix 1
Clinicopathological Features of Non–Small-Cell Lung Cancer With KRAS Mutations
KRAS-mutated non–small-cell lung cancer (NSCLC) occurs with an equal sex distribution across men and women (Dacic S, et al: Mod Pathol 23:159-168, 2010). KRAS mutations are more frequent in patients with a current or previous smoking history, with 93% of patients with KRAS mutations being smokers,22 although in one series up to 30% of patients with a KRAS mutation had never smoked.36 In contrast, up to 73%22,36 of patients with EGFR-mutant disease (Midha A, et al: Am J Cancer Res 5:2892-2911, 2015) had never smoked, contributing to EGFR-mutant disease occurring more frequently in women than men (Dacic S, et al: Mod Pathol 23:159-168, 2010; Midha A, et al: Am J Cancer Res 5:2892-2911, 2015; Hsiao SH, et al: Mol Clin Oncol 2:252-258, 2014). KRAS mutations occurred more frequently in older patients (Lee B, et al: Oncotarget 7:23874-23884, 2016). KRAS-mutant tumors are more likely to be large and poorly differentiated, with a solid pattern and of a mucinous type (Lee B, et al: Oncotarget 7:23874-23884, 2016; Kakegawa S, et al: Cancer 117:4257-4266, 2011).
KRAS-mutated NSCLC appears to be heterogeneous; of the three prevalent KRAS mutations, KRAS G12C mutations are more likely than KRAS G12D and KRAS G12V mutations to be nonmucinous (Lee B, et al: Oncotarget 7:23874-23884, 2016). KRAS G12C mutations are significantly more likely to be present in women with KRAS-mutant lung cancer (43.4%) than in men (32.7%, P < .01); no significant difference was observed between women and men for other subtypes of KRAS mutations.23 Some studies have reported that tobacco carcinogens appear to be associated with transverse mutations such as KRAS G12C mutations,23 although this association has not always been reported to be significant (Kakegawa S, et al: Cancer 117:4257-4266, 2011). It appears that women are more susceptible to tobacco carcinogens, which may explain the higher incidence of KRAS G12C mutations in women.23 Similarly, transversion mutations are more likely to be associated with disease with bronchioloalveolar features (Kakegawa S, et al: Cancer 117:4257-4266, 2011).
Finally, KRAS G12C mutation may be associated with poorer survival than other KRAS mutations, although results are not consistent across analyses (Table 2).22,27,34,55,115,116
APPENDIX 2
Effect of Comutations on Prognosis
Recent evidence suggests that mutations in genes (ie, KEAP1/NFE2L2) encoding proteins involved in the stress response pathway may represent a highly aggressive, rapidly progressing subgroup of non–small-cell lung cancer (NSCLC; Nadal E, et al: J Thorac Oncol 14:1881-1883, 2019), regardless of smoking history, EGFR status, ALK status, and KRAS (Goeman F, et al: J Thorac Oncol 14:1924-1934, 2019). Deregulation of the KEAP1/NFE2L2 pathway appears to lead to resistance to various treatments (eg, tyrosine kinase inhibitors, immune checkpoint inhibitors) and rapid proliferation of cancer cells (Goeman F, et al: J Thorac Oncol 14:1924-1934, 2019). KEAP1/NFE2L2 mutations co-occur with KRAS mutations but appear to be mutually exclusive with TP53 mutations, a mutation leading to alteration in the DNA damage response machinery (Goeman F, et al: J Thorac Oncol 14:1924-1934, 2019). Survival results were worse in patients with KRAS and STK11 mutation versus those with double wild-type disease,22 or KRAS26 or STK11 mutations alone (Bange E et al: JCO Precis Oncol 10.1200/PO.18.00326). It is suggested that the STK11 mutation alongside the KRAS mutation results in augmented downstream KRAS signaling and increased tumorigenesis (Bange E et al: JCO Precis Oncol 10.1200/PO.18.00326).
Effect of Comutations on Clinical Outcomes
Concurrent mutations may also have a negative overall impact on response to treatment. For example, the presence of concurrent KEPA1/NEF2L2 mutations was associated with a shorter overall survival in patients with KRAS mutations treated with immune checkpoint inhibitor monotherapy (ie, nivolumab or pembrolizumab; Arbour KC, et al: Clin Cancer Res 24:334-340, 2018).
Similarly, in preliminary results STK11/LKB1 mutations co-occurred with KRAS mutations in approximately one-third of 49 patients from a single study site.56 Across 17 study sites and nearly 500 patients, STK11/LKB1 mutations were associated with significantly shorter progression-free survival (PFS) and overall survival in patients treated with chemo-immunotherapy (ie, pemetrexed/carboplatin/pembrolizumab)56 than those with STK11/LKB1–wild-type tumors (Skoulidis F, et al: J Clin Oncol 37, 2019 [suppl 15; abstr 102]). Similarly, PFS was shorter in those with STK11/LKB1 and KRAS comutations from a group of 174 patients from a multicenter trial treated with immune checkpoint inhibitor monotherapy (ie, nivolumab).57 In contrast, patients with TP53 and KRAS comutations were more sensitive to nivolumab, with a PFS similar to those with only KRAS mutations.57 Furthermore, patients from the Lung Adjuvant Cisplatin Evaluation (LACE) database receiving adjuvant chemotherapy with cisplatin and with KRAS and TP53 comutations had approximately 2.5 times worse overall survival than patients with double wild-type disease (Shepherd FA, et al: J Clin Oncol 35:2018-2027, 2017). In contrast, KRAS mutation status was not predictive of survival after adjuvant therapy in the absence of a TP53 comutation.52
NF1 loss in KRAS-mutant disease is associated with focal adhesion kinase-1 hyperactivation and phosphoserine aminotransferase upregulation, enhancing tumor cell viability and having the potential to be associated with resistance to KRAS inhibitors (Wang X, et al: EMBO Mol Med 11:e9856 2019).
APPENDIX 3
Molecularly Driven Subgroups of Non–Small-Cell Lung Cancer: Testing and Treatment
Molecular testing, an essential first step in the diagnostic work-up of advanced-stage non–small-cell lung cancer (NSCLC), supports the selection of optimal targeted therapies for patients harboring certain driver mutations/gene rearrangements (Osmani L, et al: Semin Cancer Biol 52:103-109, 2018; Kalemkerian GP, et al: J Clin Oncol 36:911-919, 2018; Planchard D, et al: Ann Oncol 29:iv192-iv237, 2018; National Comprehensive Cancer Network: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf). Of patients with lung cancer who received personalized therapy matched to their mutational profile, nearly 80% had a clinical benefit,4 with longer survival experienced by those receiving targeted therapy (Aisner DL, et al: Clin Cancer Res 24:1038-1047, 2018), underlining the importance of molecular testing and targeted treatment (Pennell NA, et al: Am Soc Clin Oncol Educ Book 39:531-542, 2019).
Currently, EGFR, ALK, and ROS1 molecular testing is recommended for patients with advanced adenocarcinoma in most European countries and in the United States (Planchard D, et al: Ann Oncol 29:iv192-iv237, 2018; Lindeman NI, et al: J Thorac Oncol 13:323-358, 2018; Liebs S, et al: Cancer Med 8:3761-3769, 2019; Hanna N, et al: J Clin Oncol 35:3484-3515, 2017). As newer targeted therapies become available, testing for BRAF V600E mutations and NTRK-gene fusions is also recommended (Kalemkerian GP, et al: J Clin Oncol 36:911-919, 2018; Planchard D, et al: Ann Oncol 29:iv192-iv237, 2018; Lindeman NI, et al: J Thorac Oncol 13:323-358, 2018; Liebs S, et al: Cancer Med 8:3761-3769, 2019). In addition, agents targeting MET, RET, and ERBB2 (HER2) have demonstrated promising efficacy and are anticipated to gain regulatory approval in the upcoming months. As the list of targetable alterations has grown, many institutions have adopted more comprehensive next-generation sequencing (NGS) panels to detect less common targetable alterations as well as avoid the need for repeat biopsies due to exhaustion of tissue from sequential testing.
In patients with NSCLC, EGFR, ALK, ROS1, and BRAF mutations are often mutually exclusive and tend not to overlap with KRAS and other driver mutations (Sholl LM, et al: J Thorac Oncol 10:768-777, 2015). Molecular testing at the time of acquired resistance to targeted therapies is also recommended to select appropriate salvage therapy for certain driver mutations (Planchard D, et al: Ann Oncol 29:iv192-iv237, 2018; National Comprehensive Cancer Network: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf; Hanna N, et al: J Clin Oncol 35:3484-3515, 2017).
In addition to identifying the presence of oncogenic driver mutations, testing is also conducted to ascertain programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) receptor expression levels associated with NSCLC (Planchard D, et al: Ann Oncol 29:iv192-iv237, 2018; National Comprehensive Cancer Network: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf). Despite the significant clinical impact of conducting molecular testing, the uptake continues to be suboptimal in the routine clinical practice setting (Mason C, et al: J Clin Pathw 4:49-54, 2018).
Finally, KRAS testing should also be included as part of any expanded testing panel able to detect a wide range of mutations, such as NGS, and offered to all patients with advanced lung adenocarcinoma (Kalemkerian GP, et al: J Clin Oncol 36:911-919, 2018; National Comprehensive Cancer Network: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf; Lindeman NI, et al: J Thorac Oncol 13:323-358, 2018; Kim ES, et al: J Thorac Oncol 14:338-342, 2019). Outside of clinical trial scenarios, specific targeted therapies for patients with an identified KRAS mutation are not effective; hence, single-gene testing is not current practice (Kalemkerian GP, et al: J Clin Oncol 36:911-919, 2018).
SUPPORT
Supported by Amgen.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Administrative support: Tony S. Mok
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Provision of study material or patients: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Targeting KRAS-Mutant Non–Small-Cell Lung Cancer: One Mutation at a Time, With a Focus on KRAS G12C Mutations
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Timothy F. Burns
Consulting or Advisory Role: AbbVie/Stemcentrx, Blueprint Medicines, Novartis, Thermo Fisher Scientific
Hossein Borghaei
Honoraria: Bristol Myers Squibb, Celgene, Axiom Biotechnologies, Pfizer
Consulting or Advisory Role: Bristol Myers Squibb, Lilly, Celgene, Genentech, Pfizer, Boehringer Ingelheim, EMD Serono, Trovagene, Novartis, Merck, AstraZeneca, Genmab, Regeneron, Cantargia, BioNTech, AbbVie, PharmaMar, Takeda, Amgen, HUYA Bioscience International, Sonnetbio, Rgenix
Research Funding: Millennium (Inst), Merck (Inst), Celgene (Inst), Bristol Myers Squibb (Inst), Lilly (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Lilly, Clovis Oncology, Celgene, Genentech, Novartis, Merck, Amgen
Other Relationship: University of Pennsylvania, Takeda, Incyte
Suresh S. Ramalingam
Consulting or Advisory Role: Amgen, Boehringer Ingelheim, Celgene, Genentech/Roche, Lilly/ImClone, Bristol Myers Squibb, AstraZeneca, AbbVie, Merck, Takeda, Tesaro, Nektar, Loxo
Research Funding: AbbVie (Inst), Bristol Myers Squibb (Inst), Pfizer (Inst), Merck (Inst), AstraZeneca/MedImmune (Inst), Vertex (Inst), Takeda (Inst), EMD Serono (Inst), Genmab (Inst), Advaxis (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Tony S. Mok
Employment: The Chinese University of Hong Kong
Leadership: Sanomics, Hutchison MediPharma, AstraZeneca
Stock and Other Ownership Interests: Sanomics, Hutchison MediPharma
Honoraria: AstraZeneca, Roche/Genentech, Lilly, Bristol Myers Squibb, Boehringer Ingelheim, Novartis, Merck Sharp & Dohme, Pfizer, Merck Serono, SFJ Pharmaceuticals Group, ACEA Biosciences, Vertex, Celgene, Ignyta, Fishawack Facilitate, Takeda, Janssen, Hutchison MediPharma
Consulting or Advisory Role: AstraZeneca, Roche/Genentech, Lilly, Merck/Serono, Bristol Myers Squibb, Pfizer, Boehringer Ingelheim, Novartis, Clovis Oncology, Vertex, SFJ Pharmaceuticals Group, ACEA Biosciences, Merck Sharp & Dohme, geneDecode, Oncogenex, Celgene, Ignyta, Cirina, Hutchison MediPharma
Research Funding: AstraZeneca (Inst), Boehringer Ingelheim (Inst), Pfizer (Inst), Novartis (Inst), SFJ Pharmaceuticals Group (Inst), Roche (Inst), Merck Sharp & Dohme (Inst), Clovis Oncology (Inst), Bristol Myers Squibb (Inst), Xcovery (Inst)
Solange Peters
Honoraria: Roche (Inst), Bristol Myers Squibb (Inst), Novartis (Inst), Pfizer (Inst), MSD (Inst), AstraZeneca (Inst), Boehringer Ingelheim, Takeda (Inst), Illumina (Inst)
Consulting or Advisory Role: Roche/Genentech (Inst), Novartis (Inst), Bristol Myers Squibb (Inst), Pfizer (Inst), MSD (Inst), Amgen (Inst), AstraZeneca (Inst), Janssen (Inst), Regeneron (Inst), Merck Serono (Inst), Boehringer Ingelheim (Inst), Takeda (Inst), Lilly (Inst), AbbVie (Inst), Bayer (Inst), Biocartis (Inst), Debiopharm Group (Inst), Illumina (Inst), PharmaMar, Sanofi (Inst), Seattle Genetics (Inst), Blueprint Medicines (Inst), Daiichi Sankyo (Inst), Incyte (Inst), Bioinvent (Inst), Clovis Oncology (Inst), Vaccibody (Inst)
Research Funding: Roche (Inst), Bristol Myers Squibb (Inst), MSD (Inst), Amgen (Inst), Lilly (Inst), AstraZeneca (Inst), Pfizer (Inst), Illumina (Inst), Merck Serono (Inst), Novartis (Inst), Biodesix (Inst), Boehringer Ingelheim (Inst), Iovance Biotherapeutics (Inst)
Travel, Accommodations, Expenses: Roche, Bristol Myers Squibb, MSD, Sanofi, Incyte
Uncompensated Relationships: JTO Deputy Editor, ESMO President 20/21, ETOP Scientific Coordinator, Annals of Oncology associate editor (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1. doi: 10.3322/caac.21492. Bray F, Ferlay J, Soerjomataram I, et al: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394-424, 2018 [Erratum: CA Cancer J Clin 70:313, 2020] [DOI] [PubMed] [Google Scholar]
- 2. American Cancer Society: Cancer facts & figures 2019. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf.
- 3.Ferrer I, Zugazagoitia J, Herbertz S, et al. KRAS-mutant non-small cell lung cancer: From biology to therapy. Lung Cancer. 2018;124:53–64. doi: 10.1016/j.lungcan.2018.07.013. [DOI] [PubMed] [Google Scholar]
- 4.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mok T, Camidge DR, Gadgeel SM, et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann Oncol. 2020;31:1056–1064. doi: 10.1016/j.annonc.2020.04.478. [DOI] [PubMed] [Google Scholar]
- 6.Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41–50. doi: 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- 7. doi: 10.1038/nature13385. Cancer Genome Atlas Research Network: Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543-550, 2014 [Erratum: Nature 559:E12, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Román M, Baraibar I, López I, et al. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol Cancer. 2018;17:33. doi: 10.1186/s12943-018-0789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15:771–785. doi: 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- 10.Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wittinghofer A, Waldmann H. Ras-A molecular switch involved in tumor formation. Angew Chem Int Ed Engl. 2000;39:4192–4214. doi: 10.1002/1521-3773(20001201)39:23<4192::AID-ANIE4192>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 13.Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol. 2018;15:709–720. doi: 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- 14.McCormick F. Progress in targeting RAS with small molecule drugs. Biochem J. 2019;476:365–374. doi: 10.1042/BCJ20170441. [DOI] [PubMed] [Google Scholar]
- 15.Lindsay CR, Jamal-Hanjani M, Forster M, et al. KRAS: Reasons for optimism in lung cancer. Eur J Cancer. 2018;99:20–27. doi: 10.1016/j.ejca.2018.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Brunelli L, Caiola E, Marabese M, et al. Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget. 2014;5:4722–4731. doi: 10.18632/oncotarget.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci USA. 2019;116:15823–15829. doi: 10.1073/pnas.1904529116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. doi: 10.1038/nm.4333. Zehir A, Benayed R, Shah RH, et al: Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23:703-713, 2017 [Erratum: Nat Med 23:1004, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrido P, Olmedo ME, Gómez A, et al. Treating KRAS-mutant NSCLC: Latest evidence and clinical consequences. Ther Adv Med Oncol. 2017;9:589–597. doi: 10.1177/1758834017719829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clinical Lung Cancer Genome Project (CLCGP) Network Genomic Medicine (NGM) A genomics-based classification of human lung tumors. Sci Transl Med. 2013;5:209ra153. doi: 10.1126/scitranslmed.3006802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langer CJ. Roles of EGFR and KRAS mutations in the treatment of patients with non–small-cell lung cancer. P&T. 2011;36:263–279. [PMC free article] [PubMed] [Google Scholar]
- 22.El Osta B, Behera M, Kim S, et al. Characteristics and outcomes of patients with metastatic KRAS-mutant lung adenocarcinomas: The Lung Cancer Mutation Consortium experience. J Thorac Oncol. 2019;14:876–889. doi: 10.1016/j.jtho.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dogan S, Shen R, Ang DC, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res. 2012;18:6169–6177. doi: 10.1158/1078-0432.CCR-11-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riely GJ, Kris MG, Rosenbaum D, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biernacka A, Tsongalis PD, Peterson JD, et al. The potential utility of re-mining results of somatic mutation testing: KRAS status in lung adenocarcinoma. Cancer Genet. 2016;209:195–198. doi: 10.1016/j.cancergen.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheffler M, Ihle MA, Hein R, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J Thorac Oncol. 2019;14:606–616. doi: 10.1016/j.jtho.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Aredo JV, Padda SK, Kunder CA, et al. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer. 2019;133:144–150. doi: 10.1016/j.lungcan.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das BR, Bhaumik S, Ahmad F, et al. Molecular spectrum of somatic EGFR and KRAS gene mutations in non small cell lung carcinoma: Determination of frequency, distribution pattern and identification of novel variations in Indian patients. Pathol Oncol Res. 2015;21:675–687. doi: 10.1007/s12253-014-9874-7. [DOI] [PubMed] [Google Scholar]
- 29.Veldore VH, Patil S, Satheesh CT, et al. Genomic profiling in a homogeneous molecular subtype of non-small cell lung cancer: An effort to explore new drug targets. Indian J Cancer. 2015;52:243–248. doi: 10.4103/0019-509X.175843. [DOI] [PubMed] [Google Scholar]
- 30.Jia XL, Chen G. EGFR and KRAS mutations in Chinese patients with adenosquamous carcinoma of the lung. Lung Cancer. 2011;74:396–400. doi: 10.1016/j.lungcan.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Jia Y, Jiang T, Li X, et al. Characterization of distinct types of KRAS mutation and its impact on first-line platinum-based chemotherapy in Chinese patients with advanced non-small cell lung cancer. Oncol Lett. 2017;14:6525–6532. doi: 10.3892/ol.2017.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li M, Liu L, Liu Z, et al. The status of KRAS mutations in patients with non-small cell lung cancers from mainland China. Oncol Rep. 2009;22:1013–1020. doi: 10.3892/or_00000529. [DOI] [PubMed] [Google Scholar]
- 33.Fathi Z, Mousavi SAJ, Roudi R, et al. Distribution of KRAS, DDR2, and TP53 gene mutations in lung cancer: An analysis of Iranian patients. PLoS One. 2018;13:e0200633. doi: 10.1371/journal.pone.0200633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nadal E, Chen G, Prensner JR, et al. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J Thorac Oncol. 2014;9:1513–1522. doi: 10.1097/JTO.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 35. Thein K, Banks K, Saam J, et al: The prevalence of KRASG12C mutations utilizing circulating tumor DNA (ctDNA) in 80,911 patients with cancer. J Clin Oncol 38, 2020 (suppl 15; abstr 3547) [Google Scholar]
- 36.Zheng D, Wang R, Zhang Y, et al. The prevalence and prognostic significance of KRAS mutation subtypes in lung adenocarcinomas from Chinese populations. OncoTargets Ther. 2016;9:833–843. doi: 10.2147/OTT.S96834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falk AT, Yazbeck N, Guibert N, et al. Effect of mutant variants of the KRAS gene on PD-L1 expression and on the immune microenvironment and association with clinical outcome in lung adenocarcinoma patients. Lung Cancer. 2018;121:70–75. doi: 10.1016/j.lungcan.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Liu C, Zheng S, Jin R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95–105. doi: 10.1016/j.canlet.2019.10.027. [DOI] [PubMed] [Google Scholar]
- 40.Dong ZY, Zhong WZ, Zhang XC, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23:3012–3024. doi: 10.1158/1078-0432.CCR-16-2554. [DOI] [PubMed] [Google Scholar]
- 41.Calles A, Liao X, Sholl LM, et al. Expression of PD-1 and its ligands, PD-L1 and PD-L2, in smokers and never smokers with KRAS-mutant lung cancer. J Thorac Oncol. 2015;10:1726–1735. doi: 10.1097/JTO.0000000000000687. [DOI] [PubMed] [Google Scholar]
- 42.Chen N, Fang W, Lin Z, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66:1175–1187. doi: 10.1007/s00262-017-2005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeanson A, Tomasini P, Souquet-Bressand M, et al. Efficacy of immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer (NSCLC) J Thorac Oncol. 2019;14:1095–1101. doi: 10.1016/j.jtho.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 44. Herbst RS, Lopes G, Kowalski DM, et al: Association of KRAS mutation status with response to pembrolizumab monotherapy given as first-line therapy for PD-L1 positive advanced nonsquamous NSCLC in KEYNOTE-042. Presented at the ESMO Immuno-Oncology Congress, Geneva, Switzerland, December 11-14, 2019 (abstr 359) [Google Scholar]
- 45.Martinez-Marti A, Felip E, Matito J, et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib-resistant-advanced non-small-cell lung cancer (NSCLC) Ann Oncol. 2017;28:2451–2457. doi: 10.1093/annonc/mdx396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulivi P, Delmonte A, Chiadini E, et al. Gene mutation analysis in EGFR wild type NSCLC responsive to erlotinib: Are there features to guide patient selection? Int J Mol Sci. 2014;16:747–757. doi: 10.3390/ijms16010747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ku BM, Bae YH, Lee KY, et al. Entrectinib resistance mechanisms in ROS1-rearranged non-small cell lung cancer. Invest New Drugs. 2020;38:360–368. doi: 10.1007/s10637-019-00795-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caiola E, Salles D, Frapolli R, et al. Base excision repair-mediated resistance to cisplatin in KRAS(G12C) mutant NSCLC cells. Oncotarget. 2015;6:30072–30087. doi: 10.18632/oncotarget.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bordi P, Tiseo M, Rofi E, et al. Detection of ALK and KRAS mutations in circulating tumor DNA of patients with advanced ALK-positive NSCLC with disease progression during crizotinib treatment. Clin Lung Cancer. 2017;18:692–697. doi: 10.1016/j.cllc.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 50.Garassino MC, Marabese M, Rusconi P, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235–237. doi: 10.1093/annonc/mdq680. [DOI] [PubMed] [Google Scholar]
- 51. Seymour L, Le Teuff G, Brambilla E, et al: LACE-Bio: Validation of predictive and/or prognostic immunohistochemistry/histochemistry-based biomarkers in resected non-small-cell lung cancer. Clin Lung Cancer 20:66-73.e6, 2019. [DOI] [PubMed]
- 52.Shepherd FA, Domerg C, Hainaut P, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31:2173–2181. doi: 10.1200/JCO.2012.48.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 54.Ludovini V, Bianconi F, Pistola L, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6:707–715. doi: 10.1097/JTO.0b013e31820a3a6b. [DOI] [PubMed] [Google Scholar]
- 55.Zer A, Ding K, Lee SM, et al. Pooled analysis of the prognostic and predictive value of KRAS mutation status and mutation subtype in patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. J Thorac Oncol. 2016;11:312–323. doi: 10.1016/j.jtho.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 56. Skoulidis F, Elamin Y, Lam V, et al: Impact of STK11/LKB1 genomic alterations on clinical outcomes with chemo-immunotherapy in non-squamous NSCLC. Presented at IASLC 19th World Conference on Lung Cancer, Toronto, Ontario, Canada, September 23-26, 2018. [Google Scholar]
- 57.Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gadgeel S, Rodriguez-Abreu D, Felip E, et al: KRAS mutation status and efficacy in KEYNOTE-189: Pembrolizumab plus chemotherapy vs placebo plus chemotherapy as first-line therapy for metastatic nonsquamous NSCLC. Presented at ESMO Immuno-Oncology Congress, Geneva, Switzerland, December 11-14, 2019. [Google Scholar]
- 59.Riely GJ, Johnson ML, Medina C, et al. A phase II trial of salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6:1435–1437. doi: 10.1097/JTO.0b013e318223c099. [DOI] [PubMed] [Google Scholar]
- 60.Blumenschein GR, Jr, Smit EF, Planchard D, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC) Ann Oncol. 2015;26:894–901. doi: 10.1093/annonc/mdv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jänne PA, van den Heuvel MM, Barlesi F, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: The SELECT-1 randomized clinical trial. JAMA. 2017;317:1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gandara DR, Leighl N, Delord JP, et al. A phase 1/1b study evaluating trametinib plus docetaxel or pemetrexed in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2017;12:556–566. doi: 10.1016/j.jtho.2016.11.2218. [DOI] [PubMed] [Google Scholar]
- 63. Janes MR, Zhang J, Li LS, et al: Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172:578-589.e17, 2018. [DOI] [PubMed]
- 64.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17:676–691. doi: 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerber DE, Camidge DR, Morgensztern D, et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer. 2020;139:60–67. doi: 10.1016/j.lungcan.2019.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell. 2016;165:643–655. doi: 10.1016/j.cell.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jackson JH, Cochrane CG, Bourne JR, et al. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci USA. 1990;87:3042–3046. doi: 10.1073/pnas.87.8.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kohl NE, Mosser SD, deSolms SJ, et al. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science. 1993;260:1934–1937. doi: 10.1126/science.8316833. [DOI] [PubMed] [Google Scholar]
- 69.Liu M, Sjogren AK, Karlsson C, et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc Natl Acad Sci USA. 2010;107:6471–6476. doi: 10.1073/pnas.0908396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Whyte DB, Kirschmeier P, Hockenberry TN, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 71.Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 72.Hillig RC, Sautier B, Schroeder J, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci USA. 2019;116:2551–2560. doi: 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 74.Kitai H, Ebi H, Tomida S, et al. Epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS-mutant lung cancer. Cancer Discov. 2016;6:754–769. doi: 10.1158/2159-8290.CD-15-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kitai H, Ebi H. Key roles of EMT for adaptive resistance to MEK inhibitor in KRAS mutant lung cancer. Small GTPases. 2017;8:172–176. doi: 10.1080/21541248.2016.1210369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jänne PA, Shaw AT, Pereira JR, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: A randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 77.Morris EJ, Jha S, Restaino CR, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 78.Jha S, Morris EJ, Hruza A, et al. Dissecting therapeutic resistance to ERK inhibition. Mol Cancer Ther. 2016;15:548–559. doi: 10.1158/1535-7163.MCT-15-0172. [DOI] [PubMed] [Google Scholar]
- 79.Misale S, Fatherree JP, Cortez E, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. 2019;25:796–807. doi: 10.1158/1078-0432.CCR-18-0368. [DOI] [PubMed] [Google Scholar]
- 80.Caiola E, Brunelli L, Marabese M, et al. Different metabolic responses to PI3K inhibition in NSCLC cells harboring wild-type and G12C mutant KRAS. Oncotarget. 2016;7:51462–51472. doi: 10.18632/oncotarget.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. doi: 10.1126/scisignal.aaw9450. Lou K, Steri V, Ge AY, et al: KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal 12: eaaw9450, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goldman JW, Shi P, Reck M, et al. Treatment rationale and study design for the JUNIPER study: A randomized phase III study of abemaciclib with best supportive care versus erlotinib with best supportive care in patients with stage IV non-small-cell lung cancer with a detectable KRAS mutation whose disease has progressed after platinum-based chemotherapy. Clin Lung Cancer. 2016;17:80–84. doi: 10.1016/j.cllc.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 83.Goldman JW, Mazieres J, Barlesi F, et al. A randomized phase 3 study of abemaciclib versus erlotinib in previously treated patients with stage IV NSCLC with KRAS mutation: JUNIPER. J Clin Oncol. 2018;36(suppl 15; abstr 9025) [Google Scholar]
- 84.Gilbert-Ross M, Konen J, Koo J, et al. Targeting adhesion signaling in KRAS, LKB1 mutant lung adenocarcinoma. JCI Insight. 2017;2:e90487. doi: 10.1172/jci.insight.90487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang KJ, Constanzo JD, Venkateswaran N, et al. Focal adhesion kinase regulates the DNA damage response and its inhibition radiosensitizes mutant KRAS lung cancer. Clin Cancer Res. 2016;22:5851–5863. doi: 10.1158/1078-0432.CCR-15-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parsels LA, Karnak D, Parsels JD, et al. PARP1 trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res. 2018;16:222–232. doi: 10.1158/1541-7786.MCR-17-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carter CA, Rajan A, Keen C, et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann Oncol. 2016;27:693–699. doi: 10.1093/annonc/mdw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang ZB, Huang J, Xie C, et al. Combined use of PI3K and MEK inhibitors synergistically inhibits lung cancer with EGFR and KRAS mutations. Oncol Rep. 2016;36:365–375. doi: 10.3892/or.2016.4770. [DOI] [PubMed] [Google Scholar]
- 89.Broutin S, Stewart A, Thavasu P, et al. Insights into significance of combined inhibition of MEK and m-TOR signalling output in KRAS mutant non-small-cell lung cancer. Br J Cancer. 2016;115:549–552. doi: 10.1038/bjc.2016.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu J, Zeng LF, Shen W, et al. Targeting SHP2 for EGFR inhibitor resistant non-small cell lung carcinoma. Biochem Biophys Res Commun. 2013;439:586–590. doi: 10.1016/j.bbrc.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mainardi S, Mulero-Sánchez A, Prahallad A, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 2018;24:961–967. doi: 10.1038/s41591-018-0023-9. [DOI] [PubMed] [Google Scholar]
- 93.Downward J. RAS synthetic lethal screens revisited: Still seeking the elusive prize? Clin Cancer Res. 2015;21:1802–1809. doi: 10.1158/1078-0432.CCR-14-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kerr EM, Martins CP. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018;285:28–41. doi: 10.1111/febs.14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu K, Li K, Lv J, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2020;130:1752–1766. doi: 10.1172/JCI124049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Feng J, Jiang W, Liu Y, et al. Blocking STAT3 by pyrvinium pamoate causes metabolic lethality in KRAS-mutant lung cancer. Biochem Pharmacol. 2020;177:113960. doi: 10.1016/j.bcp.2020.113960. [DOI] [PubMed] [Google Scholar]
- 97.Pang X, Liu M. Defeat mutant KRAS with synthetic lethality. Small GTPases. 2017;8:212–219. doi: 10.1080/21541248.2016.1213783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patricelli MP, Janes MR, Li LS, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 99.Adderley H, Blackhall FH, Lindsay CR. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine. 2019;41:711–716. doi: 10.1016/j.ebiom.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lanman BA, Allen JR, Allen JG, et al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63:52–65. doi: 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 101.Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 102. Rex K, Saiki AY, Sun J-R, et al: In vivo characterization of AMG 510 - a potent and selective KRASG12C covalent small molecule inhibitor in preclinical KRASG12C cancer models. Presented at AACR Annual Meeting, Atlanta, GA, March 29-April 3, 2019. [Google Scholar]
- 103. Saiki AY, Gaida K, Rex K, et al: Discovery and in vitro characterization of AMG 510 - a potent and selective covalent small molecule inhibitor of KRASG12C. Presented at: AACR Annual Meeting, Atlanta, GA, March 29-April 3, 2019. [Google Scholar]
- 104.Lindsay CR, Blackhall FH. Direct Ras G12C inhibitors: Crossing the Rubicon. Br J Cancer. 2019;121:197–198. doi: 10.1038/s41416-019-0499-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sakamoto K, Kamada Y, Sameshima T, et al. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem Biophys Res Commun. 2017;484:605–611. doi: 10.1016/j.bbrc.2017.01.147. [DOI] [PubMed] [Google Scholar]
- 107.Lito P, Solomon M, Li LS, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hata AN, Shaw AT. Resistance looms for KRASG12C inhibitors. Nat Med. 2020;26:169–170. doi: 10.1038/s41591-020-0765-z. [DOI] [PubMed] [Google Scholar]
- 109.Ryan MB, Fece de la Cruz F, Phat S, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin Cancer Res. 2020;26:1633–1643. doi: 10.1158/1078-0432.CCR-19-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Maldegem F, Downward J. Mutant KRAS at the heart of tumor immune evasion. Immunity. 2020;52:14–16. doi: 10.1016/j.immuni.2019.12.013. [DOI] [PubMed] [Google Scholar]
- 111.Xue JY, Zhao Y, Aronowitz J, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421–425. doi: 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lu H, Liu C, Velazquez R, et al. SHP2 inhibition overcomes RTK-mediated pathway reactivation in KRAS-mutant tumors treated with MEK inhibitors. Mol Cancer Ther. 2019;18:1323–1334. doi: 10.1158/1535-7163.MCT-18-0852. [DOI] [PubMed] [Google Scholar]
- 113.Fedele C, Ran H, Diskin B, et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov. 2018;8:1237–1249. doi: 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. doi: 10.1126/scitranslmed.aaw7999. Molina-Arcas M, Moore C, Rana S, et al: Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Transl Med 11:eaaw7999, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Svaton M, Fiala O, Pesek M, et al. The prognostic role of KRAS mutation in patients with advanced NSCLC treated with second- or third-line chemotherapy. Anticancer Res. 2016;36:1077–1082. [PubMed] [Google Scholar]
- 116.Izar B, Zhou H, Heist RS, et al. The prognostic impact of KRAS, its codon and amino acid specific mutations, on survival in resected stage I lung adenocarcinoma. J Thorac Oncol. 2014;9:1363–1369. doi: 10.1097/JTO.0000000000000266. [DOI] [PubMed] [Google Scholar]