

Abstract
Multitarget-directed ligands are a promising class of drugs for discovering innovative new therapies for difficult to treat diseases. In this study, we designed dual inhibitors targeting the human fatty acid amide hydrolase (FAAH) enzyme and human soluble epoxide hydrolase (sEH) enzyme. Targeting both of these enzymes concurrently with single target inhibitors synergistically reduces inflammatory and neuropathic pain; thus, dual FAAH/sEH inhibitors are likely to be powerful analgesics. Here, we identified the piperidinyl-sulfonamide moiety as a common pharmacophore and optimized several inhibitors to have excellent inhibition profiles on both targeted enzymes simultaneously. In addition, several inhibitors show good predicted pharmacokinetic properties. These results suggest that this series of inhibitors has the potential to be further developed as new lead candidates and therapeutics in pain management.
Introduction
Pain results from a wide variety of conditions and illnesses. Managing pain represents a unique challenge to health professionals, which requires multidisciplinary strategies.1, 2 The most effective analgesic drugs currently used to treat moderate-to-severe pain are opioid agonists (e.g., oxycodone).3 The endogenous opioid system is a part of the body’s natural defense network in the brain, and opioid-based drugs (opioids) interact with opioid receptors, which leads to pain relief.4 Prolonged use of opioids will eventually lead to tolerance, physical dependence, and addiction.5 Currently, the most commonly used non-opioid drugs in pain management are nonsteroidal anti-inflammatory drugs (NSAIDs).6 The anti-inflammatory action of NSAIDs is a result of their inhibitory effect on the cyclooxygenase (COX) enzymes, that are involved in the metabolism of arachidonic acid into pro-inflammatory prostaglandins.7 During inflammation, arachidonic acid (AA) is released from the membrane phospholipids by enzyme phospholipase A2 (PLA2) and converted to different inflammatory mediators (Figure 1).8, 9 NSAIDs can treat only mild and moderate pain and, in addition, there are a number of adverse effects associated with their use, but most common are dyspepsia, an increased risk of gastric ulcer, and an increased risk of myocardial infarction.10, 11 Currently there is a great demand for more effective therapeutics to treat acute and chronic pain, and only a few new drugs have been introduced to the market in the past years. One approach toward designing novel analgesics with improved efficacy and reduced adverse effects is the poly-pharmacological approach, i.e., to design a new, single drug that is able to modulate multiple molecular pathways that are involved in pain regulation.12
Fig. 1.

Metabolic pathways of Arachidonic acid and the role of enzymes fatty acid amide hydrolase and soluble epoxide hydrolase.
Soluble epoxide hydrolase (sEH) is a ubiquitous enzyme widely distributed throughout the body with the most concentrated expression in the liver, kidneys, lungs, and vascular tissues.13 This enzyme is selective for aliphatic epoxides of fatty acids, such as epoxyeicosatrienoic acids (EETs)14 (Figure 1). The EETs are one of the metabolic derivatives of AA.15 EETs exhibit vasodilatory effects in various arteries and have also been shown to possess analgesic and anti-inflammatory properties.16 AA is metabolized through three major enzymatic pathways: cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) pathways.14 The enzyme sEH mediates the addition of water to EETs, leading to the corresponding diols, dihydroxyeicosatrienoic acids (DHETs), which show diminished biological activity.17 Therefore, inhibition of the enzyme sEH causes an increase in EET concentration, which has beneficial therapeutic effects on pain and inflammation.8 Thus far, a large body of work has focused on a class of urea-based inhibitors for sEH (e.g. AUDA, TPPU) shown in Figure 2.18 TPPU demonstrated to be active against both acute inflammation and chronic pain conditions in several rodent and primate preclinical models.19-22
Fig. 2.
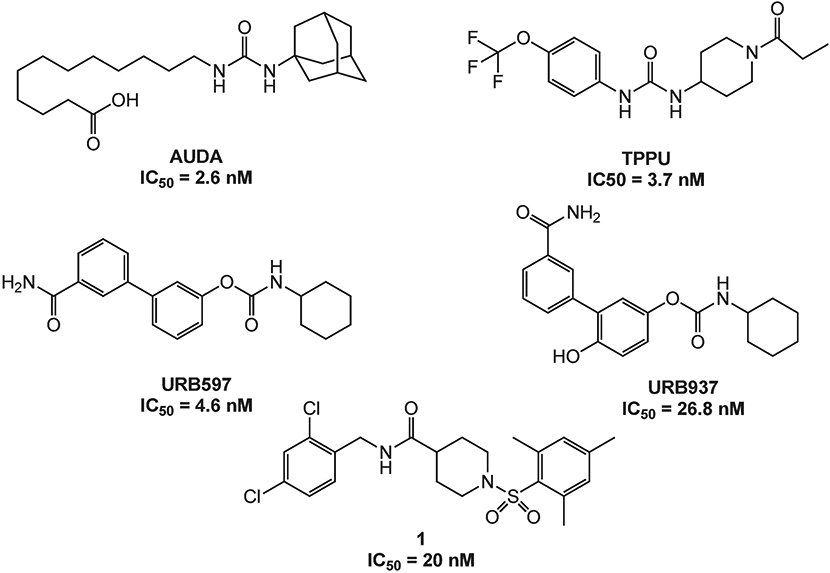
Known FAAH and sEH inhibitors.
FAAH is an integral membrane protein that hydrolyzes endocannabinoids.23 Endocannabinoids are endogenous lipid ligands that activate the cannabinoid receptors, CB1 and CB2.24 Two endocannabinoids have been identified in mammals, anandamide (AEA) and 2-arachidonoylglycerol (2-AG). FAAH terminates AEA signaling by hydrolyzing AEA to arachidonic acid and ethanolamine, two metabolites that do not activate cannabinoid receptors (Figure 1). The pharmacological inactivation of FAAH produces analgesic, anti-inflammatory, anxiolytic and mild antidepressant effects, without showing the undesirable side effects of direct cannabinoid receptor agonists, indicating that FAAH is a promising therapeutic target for pain management.25 Several classes of FAAH inhibitors have been reported, including carbamates (e.g., URB597, URB937 – Figure 2), ureas, and substrate-derived inhibitors.26, 27
A recent study by Sasso et al. (2015) showed that the combination of sEH inhibitor TPPU and FAAH inhibitor URB937 causes a significant synergistic reduction in pain behavior in two rodent models of pain.28 We believe that this synergy may be exploited therapeutically to achieve better pain control at lower drug dosages or to develop dual agents that simultaneously inhibit both FAAH and sEH activities. Previously we have described dual sEH/FAAH inhibitors that are potent on the human form of both enzymes but have poor metabolic stability and water solubility.29, 30
Here, we sought to take alternative approaches to design a new series of dual sEH/FAAH inhibitors that may have a more favorable physio-chemical profile to be used as a possible therapeutic. These inhibitors were designed by integrating a piperidine-sulfonamide-phenyl pharmacophore common to the medicinal chemistry of both selective sEH and FAAH inhibitors.
Results and Discussion
Design and Synthesis
Previously, we identified potent non-urea sEH inhibitors using high-throughput screening (HTS) in combination with structure-activity relationship (SAR) studies. These molecules were derivatives of isonipecotic acid (e.g., inhibitor 1, Figure 2). Our SAR studies showed that the pharmacophore for the sEH inhibitors should include a central sulfonamide moiety next to the piperidine ring. We also observed that the bulky, hydrophobic groups on the left-hand side of the molecules are positively correlated with inhibitory potency.31, 32 In addition, we were able to successfully co-crystallize one of the non-urea inhibitors with human sEH.33 Our docking experiments revealed that an amide functional group binds in the proximity of key amino acids needed for catalytic activity, two tyrosines (Y383 and Y466), and one aspartic acid (D335).34 The orientation of this amide moiety is similar to the orientation of the urea groups on urea sEH inhibitors (e.g., AUDA, Figure 2); thus, the amide group likely satisfies the same hydrogen bonding interactions with tyrosine and aspartic acid residues that contribute to highly potent urea inhibitors. Using information obtained from SAR studies in combination with molecular modeling and crystallography data, we were able to determine a particular pharmacophore for this series of sEH inhibitors (e.g., analog 2, shown in red, Figure 3) required to inhibit the sEH enzyme.
Fig. 3.

Design strategy for synthesis of dual sEH/FAAH inhibitors.
Wang et al. (2010) performed an HTS and identified the benzothiazole analog 3 (Figure 3) as a potent rat FAAH inhibitor having an IC50 of 18 nM.26 Their SAR studies indicated that the sulfonamide group, the piperidine ring, and benzothiazole on the left-hand side of the molecule were key components to their activity. The sulfonamide group likely forms hydrogen bonding interactions with the catalytic serine group, similar to the hydrogen-bonded network between the FAAH enzyme and the pyridyl nitrogen and oxazoyl oxygen of the α-keto-oxazole FAAH inhibitors.35 In addition, the modeling study performed by Wang et al. (2010) also indicated that the benzothiazole ring satisfies hydrophobic interactions within the hydrophobic binding pocket of the rat FAAH enzyme that confers extraordinary potency.26 We also identified several potent 4-phenylthiazole FAAH inhibitors that possess the piperidine moiety connected to the phenyl ring via a sulfonamide bond.36 These studies have shown that all three components, sulfonamide bond, piperidine ring and benzothiazole/4-phenylthiazole moieties, are important for the FAAH inhibition, but more work has to be done in order to access the particular relationship of each of these moieties with the inhibition potencies.
Overall, these extensive SAR studies indicate that modifications to the aromatic ring on the left- and right-hand sides of the pharmacophore (shown in red in Figure 3) should allow for improved sEH and FAAH inhibition. This data guided our design for the preparation of the dual inhibitors wherein modifications to the right side of the aromatic ring were carried out.
Since inhibition of each enzyme showed analgesic effect individually37, 38, and co-administration of sEH and FAAH inhibitors resulted in a significant synergistic reduction in pain behavior in animal models of pain28, we hypothesized that dual inhibitors will treat pain at a lower dose, and consequently with fewer side-effects. We decided to employ a Designed Multiple Ligand (DML) strategy12, 39 wherein inhibitors used the core pharmacophore (a phenyl ring connected to a piperidine moiety, which is connected to the sulfonamide bond) common to the sEH inhibitor 2 and the FAAH inhibitor 3 with modifications on the aromatic rings on either side. Figure 3 shows representative structures with the key pharmacophoric regions boxed. Given that the benzothiazole ring contributes to high FAAH inhibitory potency and bulky hydrophobic groups are well-tolerated in that position for sEH inhibitory potency32, 33, we kept this structure constant (general structure 4) while modifying aromatic groups bound to the sulfonamide group.
Following the established synthetic procedure34 shown in Scheme 1, we started from the readily available 2-(4-aminophenyl)benzothiazole and Boc-isonipecotic acid. EDC coupling yielded the amide 5, which was subjected to Boc-deprotection with trifluoroacetic acid (TFA) which provided the key amine intermediate 6. Coupling with different R-sulfonylchlorides furnished final compounds 4-1 to 4-30 in moderate yields (16-84%).
Scheme 1.
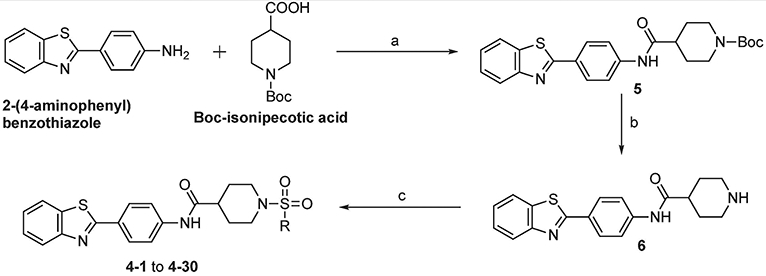
Reagents and conditions: (a) EDC, DCM, rt, 24 h, 48%; (b) TFA, DCM, rt, 24 h, 87%; (c) R-sulfonyl chloride, Et3N, DCM, rt, 24 h, 16–84%.
Biological Evaluation and Structure-activity relationship Studies
All synthesized analogs 4-1 to 4-30 were tested in vitro in both human sEH and human FAAH inhibition assays. The inhibition potencies of analyzed analogs against both enzymes are summarized in Table 1. Our initial SAR investigation started with the synthesis of the thiophene-2-yl analog, 4-1. This analog showed inhibition potency in the low nanomolar range for human FAAH enzyme (IC50 = 16 nM), but only moderate inhibition potency at the human sEH enzyme (IC50 = 420 nM). The introduction of bromine and chlorine atoms on the thiophene ring (analog 4-2) led to significantly diminished inhibition potencies at both enzymes. We decided to replace the thiophene ring with a phenyl ring, which allowed us to access many sterically and electronically diverse chemical groups, which could in turn improve inhibition profiles at both enzymes. The phenyl analog 4-3, showed excellent inhibition potency with the human FAAH enzyme, having an IC50 of 8.6 nM, but only low micromolar inhibition for human sEH enzyme (IC50 = 1100 nM). Fluoro-, chloro-, bromo- and methyl- groups placed at the ortho position (4-4, 4-5, 4-6, and 4-7, respectively) were all well tolerated in the human FAAH binding pocket and led to low nanomolar inhibition potency on human FAAH enzyme. The bulkier, electron-donating methoxy- group (4-8) was less potent for human FAAH (IC50 = 80 nM). Placement of chloro-, bromo-, or methyl-groups in the ortho position improved potency at the sEH enzyme relative to the unsubstituted inhibitor (4-3). This led to the best dual sEH/FAAH inhibitor of the series (4-5) with equally high potency for sEH (IC50 = 9.6 nM) and FAAH (IC50 = 7 nM). Next, we introduced the same replacements into the meta-position on the phenyl ring. We noticed that adding fluoro-, chloro-, bromo-, methyl- or methoxy- groups at the meta position (4-9, 4-10, 4-11, 4-12 and 4-13, respectively) had comparable potencies relative to the unsubstituted inhibitor (4-3). These inhibitors retain low nanomolar inhibition potencies for human FAAH enzyme with low potency at the human sEH enzyme (IC50s ranged from 620-2400 nM). Modification of the para position with fluoro-, chloro, bromo-, methyl- and methoxy- substitutions (4-14, 4-15, 4-16, 4-17 and 4-18, respectively) led to a loss of potency towards FAAH relative to the unsubstituted inhibitor (4-3). Surprisingly, the introduction of the 2,4-disubstitutions for fluoro-, chloro-, bromo- and methoxy- groups (4-19, 4-20, 4-21, 4-22 and 4-23, respectively) led to inhibitors with potency at both enzymes comparable to the same single ortho- substitutions and improved potency relative to para- substitutions. Thus, the benefit from ortho- substitutions is greater than the loss from para- substitutions. The fluoro- and methyl- 3,5-disubstitutions (4-24 and 4-25) had low potency at both enzymes (IC50s > 10,000 nM on FAAH and 1,000 nM on sEH). Additionally, the fluoro-, chloro-, methyl- and isopropyl- tri-substitutions (4-26, 4-27, 4-28 and 4-29, respectively) and the pentafluoro- substitution (4-30) had lower potency than the 2,4-disubstituted molecules. This suggests compounds that have high bulk are unfavored in the active sites of FAAH and sEH.
TABLE 1.
Fatty acid amide hydrolase (FAAH) and soluble epoxide hydrolase (sEH) inhibitory activities and docking scores of analogs
 |
|||||
|---|---|---|---|---|---|
| Compound | R | FAAH IC50 (nM) |
sEH IC50 (nM) |
Docking Score FAAH |
Docking Score sEH |
| URB 597 | - | 38 | - | −31.45 | −22.16 |
| AUDA | - | - | 2.6 | −22.18 | −25.83 |
| 4-1 | 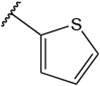 |
16 | 420 | −21.04 | −22.35 |
| 4-2 | 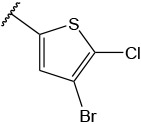 |
2700 | 5900 | −12.3 | −21.02 |
| 4-3 | 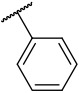 |
8.6 | 1100 | −23.77 | −30.32 |
| 4-4 | 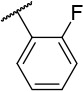 |
1.3 | 150 | −28.05 | −32.42 |
| 4-5 | 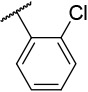 |
7 | 9.6 | −28.93 | −33.03 |
| 4-6 | 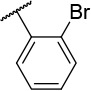 |
6.1 | 24 | −26.88 | −30.93 |
| 4-7 | 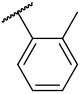 |
9.6 | 35 | −34.41 | −32.44 |
| 4-8 | 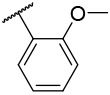 |
80 | 26 | −29.93 | −32.98 |
| 4-9 | 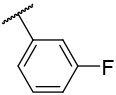 |
13.4 | 1700 | −35.20 | −34.19 |
| 4-10 | 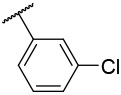 |
7.2 | 2400 | −32.98 | −29.41 |
| 4-11 | 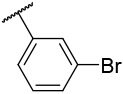 |
51.6 | 1100 | −32.17 | −29.25 |
| 4-12 | 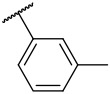 |
7 | 940 | −36.5 | −30.08 |
| 4-13 | 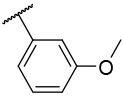 |
14.6 | 620 | −26.88 | −32.16 |
| 4-14 | 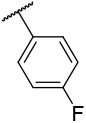 |
77 | 1300 | −19.99 | −28.91 |
| 4-15 | 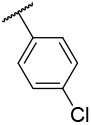 |
260 | 710 | −22.49 | −27.61 |
| 4-16 | 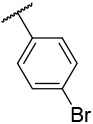 |
185 | 580 | −23.64 | −31.23 |
| 4-17 | 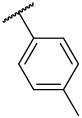 |
>10000 | 370 | −29.02 | −32.60 |
| 4-x18 | 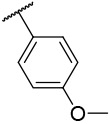 |
>10000 | 84 | −18.66 | −31.59 |
| 4-19 | 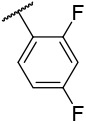 |
11.4 | 195 | −27.22 | −33.84 |
| 4-20 | 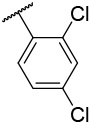 |
31.3 | 13.1 | −23.64 | −34.11 |
| 4-21 | 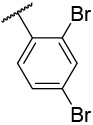 |
10 | 36 | −37.38 | −34.20 |
| 4-22 | 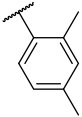 |
5.5 | 29.2 | −33.14 | −32.08 |
| 4-23 | 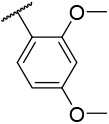 |
36.2 | 180 | −33.22 | −34.47 |
| 4-24 | 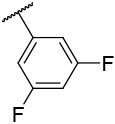 |
>10000 | 1100 | −36.69 | −32.39 |
| 4-25 | 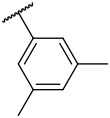 |
>10000 | 3600 | −41.62 | −33.18 |
| 4-26 | 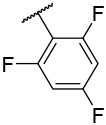 |
250 | 6600 | −26.88 | −30.58 |
| 4-27 | 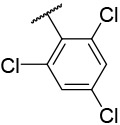 |
270 | 1300 | −33.37 | −29.05 |
| 4-28 | 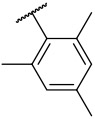 |
84 | 790 | −28.97 | −31.33 |
| 4-29 | 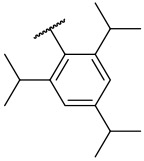 |
2233 | 840 | −18.61 | −24.48 |
| 4-30 | 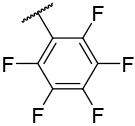 |
>10000 | 2670 | −37.58 | −27.90 |
Molecular Modeling Studies
Our design and evaluation of synthesized analogs were complemented with in silico experiments. Since the crystal structure of the human FAAH enzyme has not been reported, we built and evaluated a homology model for the human FAAH enzyme.36 We docked all synthesized analogs, 4-1 to 4-30, in both the human FAAH enzyme homology model and the human sEH enzyme crystal structure derived from PDB: 4HAI. Docking scores obtained in these experiments are shown in Table 1 and all non-covalent interactions are shown in Table 2A and Table 2B for FAAH and sEH enzymes, respectively. The ICM docking score represents unitless approximations of the binding free energy between the inhibitor and the enzyme where a lower docking score suggests a higher chance the inhibitor is bound to the enzyme.40 For the human FAAH enzyme, the low values for the potential energy (docking scores) were obtained for all the analogs that also show in vitro low nanomolar inhibition potencies (e.g., 4-7, 4-11, 4-12, 4-21, 4-23, etc.). Analysis of these values show that most of the obtained docking energies are correlated with inhibitory potency (R2 = 0.3255, p = 0.0023) with the exception of compounds with very poor potency on FAAH (IC50 > 10,000) (Figure 4A). This suggests that the potency of these inhibitors is primarily based on Van der Waals interactions between the enzyme active site and inhibitors. After visual inspection of the top binding modes of the most active inhibitors, we noticed that all docked compounds are located in the proximity of S241 and S217, both residues of the catalytic triad, S241-S217-K142, which is responsible for the hydrolytic cleavage of the amide bond of the substrate anandamide by the FAAH enzyme.25 We were able to define the most important residues within the binding pocket of the FAAH enzyme and tried to explain the increased in vitro inhibition potencies by observing and analyzing the type of contacts of the most active FAAH inhibitor identified in this study, 4-4, with the inhibitory potency of 1.3 nM, and other analogs with low nanomolar inhibition potencies, e.g., 4-5, 4-6, 4-10 and 4-22. The aromatic part of the inhibitor 4-4 is found to be embedded between several hydrophobic amino acid residues (F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531) and forms other important non-covalent interactions (M191, G216, S217, L380, L401, G485, M495), which we believe all contribute to high inhibitory potency of this analog (Table 2A). The best dual inhibitor identified herein, 4-5 has a very similar defined binding pocket, showing the importance of selected hydrophobic and other non-covalent interactions for the low nanomolar inhibition potency (Figure 5A, Figure 5B, Table 2A). The poor correlation of docking energies with in vitro experiments were observed with only three analogs, 4-24, 4-25 and 4-30, but visual inspection of these compounds within the binding pocket of the human FAAH homology model reveals the absence of several important contacts with residues, S241 and S217, that are present with the biologically active analogs, which could explain the lower inhibition potencies of these three analogs (Table 2A).
TABLE 2A.
The list of hydrogen bonds, hydrophobic interactions and other non-covalent interactions of analogs docked in human FAAH enzyme.
| Compound | H-bonds | Hydrophobic interactions | Other non-covalent interactions |
|---|---|---|---|
| URB-597 | G485 | F192, S193, Y194, G239, F244, L401, L404, I407, M436, T488, V491, W531 | M191, G240, S241, F381, D403, R486, I530 |
| AUDA | G272 | M191, F192, S193, Y194, I238, G239, F244, V270, Q273, L278, V491 | S190, S217, S241, C269, Y271, E274 |
| 4-1 | N/A | F192, S193, Y194, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 |
M191, G216, S217, L380, L401, G485 |
| 4-2 | G485 | F192, S193, Y194, I238, G239, G240, S241, F244, L404, I407, V422, L429, L433, T488, V491, I530, W531 | M191, G216, S217, F381, L401, P484 |
| 4-3 | N/A | F192, S193, Y194, G239, S241, F244, L380, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | G216, S217, M191, I238, G240 |
| 4-4 | N/A | F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491 I530, W531 | M191, G216, S217, L380, L401, G485, M495 |
| 4-5 | N/A | F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491 I530, W531 | M191, G216, S217, L380, G485, M495 |
| 4-6 | N/A | F192, S193, Y194, I238, G239, L380, L404, I407, V422, L429, L433, M436, T488, V491, M495, I530, W531 | M191, S241, F244, F381, G485 |
| 4-7 | N/A | F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | M191, G216, S217, L380, L401, G485, M495 |
| 4-8 | N/A | F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | M191, G216, S217, L380, G485, M495 |
| 4-9 | N/A | F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | M191, G216, S217, L380, L401, G485 |
| 4-10 | N/A | F192, S193, Y194, I238, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | M191, G216, S217, L380, L401, G485 |
| 4-11 | G485 | F192, S193, Y194, I238, G239, G240, S241, F244, L404, I407, V422, L429, L433, T488, V491, I530, W531 | M191, G216, F381, L401, P484 |
| 4-12 | G485 | F192, S193, Y194, L380, F381, L404, I407, V422, L429, F432, L433, T488, V491, M495, I530, W531 | M191, T377, L401, D403, P484 |
| 4-13 | G485 | F192, S193, Y194, I238, G239, S241, F244, L404, I407, V422, L429, L433, T488, V491, I530, W531 | M191, S217, F381, L401, D403, P484 |
| 4-14 | N/A | F192, S193, Y194, G239, G240, S241, F244, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | M191 G216, S217, L380 |
| 4-15 | N/A | M191, F192, S193, Y194, I238, G239, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | S241, F244, M495 |
| 4-16 | N/A | M191, F192, S193, G239, L404, I407, V422, L429, F432, L433, M436, T488, I530, W531 | Y194, S217, S241, F244, G485, V491 |
| 4-17 | N/A | M191, F192, S193, I238, G239, L404, I407, V422, L429, F432, L433, M436, T488, I530, W531 | Y194, S217, S241, F244, G485, V491 |
| 4-18 | N/A | M191, F192, S193, I238, G239, L380, F381, L404, I407, V422, L429, F432, L433, M436, T488, I530, W531 | Y194, S241, F244, V491 |
| 4-19 | N/A | M191, F192, S193, I238, G239, L380, F381, L404, I407, V422, L429, F432, L433, M436, T488, V491, I530, W531 | Y194, S241, F244, M495 |
| 4-20 | N/A | M191, F192, S193, Y194, I238, G239, L404, I407, V422, L429, L433, M436, T488, I530, W531 | G216, S217, S241, F244, L401, G485, V491 |
| 4-21 | N/A | M191, F192, S193, Y194, I238, G239, L380, L404, I407, L429, L433, M436, T488, V491, I530, W531 | G216, S241, F244, G485 |
| 4-22 | G485 | F192, S193, Y194, G239, G240, S241, F244, L404, I407, V422, L429, L433, T488, V491, M495, I530, W531 | M191, G216, F381, P484 |
| 4-23 | N/A | M191, F192, S193, Y194, I238, G239, L380, F381, L404, I407, L429, L433, T488, V491, I530, W531 | S217, S241, F244, L401, P484, G485 |
| 4-24 | G485 | Y194, G239, F381, F432, L404, I407, V422, L429, F432, L433, T488, V491, M495, I530, W531 | F192, T377, L401, D403, P484 |
| 4-25 | G485 | F192, S193, Y194, L380, F381, L404, I407, V422, L429, F432, L433, T488, V491, M495, I530, W531 | M191, T377, L401, D403, P484 |
| 4-26 | N/A | M191, F192, S193, Y194, I238, G239, L380, L404, I407, V422, L429, F432, L433, M436, T488, I530, W531 | G216, S217, S241, F244, G485, V491, M495 |
| 4-27 | N/A | M191, F192, S193, Y194, I238, G239, L266, G268, Y271, T377, F381, L404, F432, M436, T488, V491, M495 | S190, S217, T236, G240, S241, K267, V270 |
| 4-28 | N/A | M191, F192, S193, Y194, I238, G239, L380, L404, I407, V422, L429, L433, M436 | G216, S217, S241, F244, P484, G485 |
| 4-29 | I238 | M191, F192, S193, Y194, G239, F244, L266, G268, Y271, L278, T377, L380, F381, L404, F432, M436, T488, V491, M495 | S190, S217, T236, D237, G240, S241, K267, C269, V270, L401, P484 |
| 4-30 | G485 | S193, Y194, G239, S376, T377, L380, F381, D403, L404, I407, V422, F432, L433, T488, V491, M495, I530, W531 | F192, L401, P484 |
TABLE 2B.
The list of hydrogen bonds, hydrophobic interactions and other non-covalent interactions of analogs docked in human sEH enzyme.
| Compound | H-bonds | Hydrophobic interactions | Other non-covalent interactions |
|---|---|---|---|
| URB-597 | L417, Y466 | F267, D335, W336, T360, Y383, M419, V498, L499, H524, W525 | Q384, L408, S415 |
| AUDA | Y466, H524, W525 | D335, W336, M339, T360, F381, Y383, S412, M419, F497, V498, L499 | F267, Q384, L408, R410, A411, K495, G523 |
| 4-1 | D335 | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, M469, N472, V498, L499, H524 | Q384, Y466, W525 |
| 4-2 | Y466 | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, M469, N472, V498, L499, H524, W525 | P268, D335, Q384, F387, S418 |
| 4-3 | D335 | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, M469, N472, V498, L499, H524 | Q384, Y466, W525 |
| 4-4 | Y466 | D335, W336, M339, T360, P361, I363, F381, Y383, L407, R410, S415, L417, M419, V498, L499, M503, H524, W525 | Q384, S407, A411, M469, D496 |
| 4-5 | D335 | F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524 | P268, Q384, Y466, W525 |
| 4-6 | D335 | F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524 | P268, Q384, Y466, W525 |
| 4-7 | N/A | D335, W336, M339, T360, P361, I363, F381, Y383, L408, R410, S415, L417, M419, Y466, V498, L499, M503, H524, W525 | F267, Q384, S407, A411, M469 |
| 4-8 | Y466 | W336, M339, T360, P361, I363, I375, F381, Y383, L408, R410, S415, L417, M419, V498, L499, M503, H524, W525 | D335, Q384, S407, A411, M469, D496 |
| 4-9 | Y466 | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, M469, V498, L499, H524 | D335, Q384, F387 |
| 4-10 | D335 | W336, M339, P361, I363, F381, Y383, L408, R410, S415, L417, M419, Y466, V498, L499, M503 | P268, Q384, Y466, W525 |
| 4-11 | Y466 | W336, M339, T360, P361, I363, F381, Y383, L408, R410, S415, L417, M419, V498, L499, M503, H524, W525 | D335, Q384, S407, A411, M469, D496 |
| 4-12 | Y466 | F267, D335, W336, M339, P371, I375, F381, Y383, Q384, L408, L417, M419, L428, Y466, M469, V498, L499, H524 | D335, Q384, F387 |
| 4-13 | N/A | D335, W336, M339, T360, P361, I363, F381, Y383, L408, R410, S415, L417, M419, Y466, V498, L499, M503, H524, W525 | F267, Q384, S407, A411, M469 |
| 4-14 | N/A | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, M469, N472, V498, L499, H524 | P268, D335, Q384, S418, Y466, W525 |
| 4-15 | N/A | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, Y466, M469, N472, V498, L499, H524 | P268, D335, Q384, S418, W525 |
| 4-16 | Y466 | F267, D335, W336, M339, P371, I375, F381, Y383, L408, L417, M419, M469, V498, L499, H524, W525 | D335, Q384, F387, S415, L428 |
| 4-17 | N/A | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, Y466, M469, N472, V498, L499, H524 | P268, D335, Q384, W473, W525 |
| 4-18 | Y466 | W336, M339, I363, I375, F381, Y383, L408, R410, S415, L417, M419, V498, L499, M503, H524, W525 | F267, D335, T360, Q384, S407, A411, D496 |
| 4-19 | D335 | F267, W336, M339, P371, I375, F381, F387, Y383, L408, L417, M419, L428, M469, N472, L499, V498, H524 | P268, Q384, S418, Y466, W525 |
| 4-20 | N/A | F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524 | P268, D335, Q384, S418, Y466, W525 |
| 4-21 | Y466 | F267, W336, M339, P371, I375, F381, Y383, L408, L417, M419, L428, M469, N472, V498, L499, H524, W525 | D335, Q384, F387, S415 |
| 4-22 | N/A | F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524 | P268, D335, Q384, Y466, W473, W525 |
| 4-23 | Y466 | W336, M339, P361, I363, I375, F381, Y383, L408, R410, S415, L417, M419, V498, L499, M503, H524, W525 | F267, D335, T360, Q384, S407, A411, D496 |
| 4-24 | Y466 | W336, M339, I375, F381, Y383, L408, R410, S415, L417, M419, M469, L499, V498, H524, W525 | D335, T360, Q384, S407, A411, N472, D496 |
| 4-25 | N/A | W336, M339, I375, F381, Y383, L408, R410, S415, L417, M419, Y466, M469, V498, L499, H524, W525 | F267, D335, T360, Q384, S407, A411, D496 |
| 4-26 | Y466 | W336, M339, T360, P361, I363, F381, Y383, L408, R410, S415, L417, M419, M469, V498, L499, M503, H524, W525 | F267, D335, Q384, S407, A411, D496 |
| 4-27 | N/A | F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524, W525 | P268, D335, Q384, S418, Y466 |
| 4-28 | D335 | F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524 | P268, Q384, S418, Y466 |
| 4-29 | Y466 | W336, M339, T360, P361, I363, P371, I375, F381, Y383, L408, R410, L417, M419, Y466, M469, V498, L499, M503, H524, W525 | D335, S374, Q384, S407, D496 |
| 4-30 | Y466 | W336, M339, T360, P361, I363, I375, F381, Y383, L408, R410, S415, L417, M419, M469, V498, L499, M503, H524, W525 | F267, D335, Q384, S407, A411, D496 |
Fig. 4A.

Correlation of inhibitory potency (−log(IC50)) vs. Docking Scores for FAAH enzyme.
Fig. 5A.

A 3D docking pose of the inhibitor 4–5 in the catalytic site of the human fatty acid amide hydrolase (FAAH) enzyme.
Fig. 5B.

2D representation for the lowest energy conformation of inhibitor 4–5 in the binding pocket of human FAAH. Green shading represents hydrophobic region; gray parabolas represent accessible surface for large areas; broken thick line around ligand shape indicates accessible surface; size of residue ellipse represents the strength of the contact.
The docking experiments of all synthesized inhibitors in the human sEH enzyme model revealed that most of the analogs are located in the proximity of key amino acids within the catalytic pocket that are involved in the hydrolysis of EETs. Compared with docking scores for the FAAH enzyme, the docking scores of sEH poorly correlated with sEH potency (Figure 4B). All potent sEH inhibitors (e.g. 4-5, 4-6, 4-7, 4-8, 4-20, 4-21 and 4-22) have good docking scores (Table 1), however, the analogs that were inactive in the in vitro inhibition assay, also have good docking scores in our docking experiments. This possibly indicates that sEH potency is primarily determined based on hydrogen bonding interactions with the catalytic residues rather than hydrophobic interactions with the binding pocket which probably is responsible for the good docking scores in this series of analogs. Our previous molecular modeling studies and X-ray crystallographic structure showed that two tyrosine residues (Y383 and Y466) and one aspartic acid residue (D335), located in the hydrolase catalytic pocket of sEH, are involved in hydrogen bonding with the inhibitors. One of the most potent sEH inhibitors identified in this study, and the best dual inhibitor, 4-5, is in the close proximity of these three amino acid residues (Figure 6A, Figure 6B, Table 2B). This inhibitor forms a hydrogen bond with D335 via amide bond, and has several important hydrophobic interactions (F267, W336, M339, P371, I375, F381, Y383, F387, L408, L417, M419, L428, M469, N472, V498, L499, H524), that, combined with other non-covalent interactions (P268, Q384, Y466, W525), contribute to the high inhibition potency of this compound. We also noticed that all low nanomolar sEH inhibitors identified in this study are docked in the proximity of the key amino acid residues, Y383, Y466, and D335, located in the catalytic site of the sEH enzyme (Table 2B), while inactive compounds lack this interaction.
Fig. 4B.

Correlation of inhibitory potency (−log(IC50)) vs. Docking Scores for sEH enzyme.
Fig. 6A.

A 3D docking pose of the inhibitor 4–5 in the catalytic site of the human soluble epoxide hydrolase (sEH) enzyme.
Fig. 6B.

2D representation for the lowest energy conformation of inhibitor 4–5 in the binding pocket of human sEH. Green shading represents hydrophobic region; gray parabolas represent accessible surface for large areas; broken thick line around ligand shape indicates accessible surface; size of residue ellipse represents the strength of the contact.
Finally, we looked at the binding poses of the potent dual inhibitors, 4-5, 4-6, 4-20, 4-21, and 4-22, within both, human FAAH and sEH enzyme binding pockets. We observed that the benzothiazole moiety of all aforementioned ligands is located in the proximity of the two hydrophobic residues, V422 and L433 in the human FAAH binding pockets, while this group probably interacts with one of the methionine residues (M419 or M469) within human sEH binding pocket.
In Silico ADMET Studies
Based on the encouraging biological results described above, we performed some selected absorption, distribution, metabolism, excretion, and toxicology (ADMET) predictions to assess their drug-like properties. Unfavorable ADMET properties have been identified as a major cause of drug candidate failure in the pharmaceutical industry.41, 42 In silico ADMET prediction represents the use of computer modeling software to understand structure-property relationships and predicts the in vivo behavior of potential drug candidates in the human body.43
Using the ICM-Chemist-Pro tool, we started our investigations (see Table 3) by calculating simple physicochemical descriptors, such as cLogP and aqueous solubility (cLogS). It has been shown previously that these parameters are good predictors for drug candidate permeability.44, 45 High logP values usually mean poor absorption/barrier penetration and is also an integral part of the well-known Lipinski Rule of 5 prediction.46 All synthesized analogs in this study, 4-1 to 4-30, are in the agreement with Lipinski Rule of 5 in terms of cLogP, the number of hydrogen bond acceptors and the number of hydrogen bond donors. Several inhibitors have molecular weights that exceed 500 g/mol, but these numbers are very close to 500 Da and as the prediction rule states, potential orally active drugs should not violate more than one of the four criteria. Therefore, we believe that these inhibitors represent excellent candidates for future follow-up SAR studies and drug development. In addition, another important rule that predicts oral bioavailability, Veber’s Rule, states that drug candidates will have good oral bioavailability if the number of rotatable bonds present in a molecule is less than 10.47 All compounds synthesized in this study has 6 to 9 rotatable bonds. The logS for aqueous solubility of a molecule, expressed in mol/L, helps to predict oral absorption. The acceptable range for logS is between −6.5 and −0.5 moles/liter. In this study, all synthesized analogs showed values close to, but below, −6.5 mol/L, suggesting a moderate aqueous solubility. Improving solubility of these potential drug candidates, if needed, can be addressed in the future guided design of follow-up inhibitors or in the drug formulation process. The next important parameter to consider for the drug design and development is permeability, and we assessed it using the Caco-2 prediction tool.48 The ICM-Chemist-Pro Caco-2 prediction scores higher than −5 suggest a highly permeable drug candidate, while scores of below −6 represent a poorly permeable compound. As shown in Table 3, all compounds synthesized in this study have predicted scores for Caco-2 between −5 and −6, suggesting moderately permeable drug candidates. We also wanted to predict the plasma half-life of synthesized dual inhibitors. Our analysis showed that inhibitors 4-26, 4-27 and 4-28 have the longest predicted half-life in hours (10.10 hours) whereas the inactive compound 4-2 had the shortest predicted half-life of 1.2 h. The most potent inhibitors identified in this study, 4-5, 4-6, 4-20, 4-21, and 4-22 showed a moderate predicted half-life of 3.2 to 5.7 hours. As part of our Tox studies, we analyzed several important toxicology descriptors, LD50, hERG inhibition, and the Tox score. Prediction of the hERG inhibition is performed because pharmacological blockade of the hERG channel results in a severe life-threatening cardiac side effect possibly causing sudden death, leading to the withdrawal of many drugs from the development process.49, 50 The ICM-Chemist-Pro tool predicts that scores above or equal to 0.5 will probably exhibit some hERG inhibition at 100 μM or less, while compounds with predicted values below 0.5 will likely not be hERG inhibitors. No compounds synthesized in this study exceeded a value of 0.5. As part of the toxicity study predictions, we calculated the Tox score for each compound synthesized. This value represents the identification of potentially toxic parts/bi-products (during metabolism) of the molecule. The only compound that showed a toxicity score was analog 4-30. Finally, we calculated a “drug-like” properties for each synthesized compound. This is a purely empirical value and is based on several factors calculated above. The scores are on a scale of −1 to 1 and should not exceed 1. Only two compounds, 4-15 and 4-29, slightly exceed a value of 1. In addition, these two analogs show moderate and low inhibition profiles against both enzymes, respectively, and will not be considered for future follow up studies.
TABLE 3.
Predicted ADMET properties of synthesized analogs
| Compound | Mol. Wt. | cLogP | cLogS | Drug Likeness |
Bad Groups |
# of Rot Bonds |
# of H-B Acceptors |
# of H-B Donors |
Caco2 | Half-Life (hours) |
hERG Inhibition |
Tox Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| URB-597 | 338.407 | 4.022 | −6.168 | 1.008 | 0 | 6 | 5 | 3 | −4.985 | 0.85 | 0.714 | 2 |
| AUDA | 392.584 | 6.107 | −6.679 | −0.016 | 0 | 15 | 5 | 3 | −4.760 | 2.838 | 0.601 | 0.878 |
| 4-1 | 483.619 | 5.317 | −6.918 | 0.584 | 0 | 6 | 8 | 1 | −5.12 | 2.05 | 0.106 | 0 |
| 4-2 | 596.957 | 6.757 | −8.513 | 0.740 | 0 | 6 | 8 | 1 | −5.348 | 1.213 | 0.106 | 0 |
| 4-3 | 477.597 | 5.386 | −7.327 | 0.553 | 0 | 6 | 8 | 1 | −5.055 | 2.873 | 0.149 | 0 |
| 4-4 | 495.587 | 5.535 | −7.804 | 0.541 | 0 | 6 | 8 | 1 | −5.252 | 3.20 | 0.255 | 0 |
| 4-5 | 512.039 | 5.980 | −7.900 | 0.644 | 0 | 6 | 8 | 1 | −5.180 | 3.205 | 0.182 | 0 |
| 4-6 | 556.493 | 6.117 | −8.070 | 0.396 | 0 | 6 | 8 | 1 | −5.276 | 3.21 | 0.106 | 0 |
| 4-7 | 491.624 | 5.667 | −7.664 | 0.428 | 0 | 6 | 8 | 1 | −5.216 | 3.71 | 0.182 | 0 |
| 4-8 | 507.623 | 5.356 | −7.277 | 0.382 | 0 | 7 | 9 | 1 | −5.217 | 4.47 | 0.182 | 0 |
| 4-9 | 495.587 | 5.655 | −7.506 | 0.595 | 0 | 6 | 8 | 1 | −5.289 | 7.20 | 0.255 | 0 |
| 4-10 | 512.039 | 6.100 | −8.086 | 0.658 | 0 | 6 | 8 | 1 | −5.231 | 7.44 | 0.182 | 0 |
| 4-11 | 556.493 | 6.237 | −8.273 | 0.409 | 0 | 6 | 8 | 1 | −5.319 | 7.44 | 0.106 | 0 |
| 4-12 | 491.624 | 5.787 | −7.519 | 0.379 | 0 | 6 | 8 | 1 | −5.289 | 7.26 | 0.182 | 0 |
| 4-13 | 507.623 | 5.476 | −7.339 | 0.625 | 0 | 7 | 9 | 1 | −5.225 | 5.78 | 0.182 | 0 |
| 4-14 | 495.587 | 5.655 | −7.844 | 0.911 | 0 | 6 | 8 | 1 | −5.298 | 3.23 | 0.255 | 0 |
| 4-15 | 5112.039 | 6.100 | −8.285 | 1.013 | 0 | 6 | 8 | 1 | −5.141 | 3.23 | 0.182 | 0 |
| 4-16 | 556.493 | 6.237 | −8.476 | 0.722 | 0 | 6 | 8 | 1 | −5.270 | 3.24 | 0.106 | 0 |
| 4-17 | 491.624 | 5.787 | −7.783 | 0.498 | 0 | 6 | 8 | 1 | −5.237 | 3.46 | 0.182 | 0 |
| 4-18 | 507.623 | 5.476 | −7.587 | 0.817 | 0 | 7 | 9 | 1 | −5.195 | 4.20 | 0.182 | 0 |
| 4-19 | 513.577 | 5.805 | −8.010 | 0.511 | 0 | 6 | 8 | 1 | −5.343 | 5.28 | 0.357 | 0 |
| 4-20 | 546.481 | 6.695 | −8.826 | 0.6212 | 0 | 6 | 8 | 1 | −5.346 | 5.28 | 0.182 | 0 |
| 4-21 | 635.389 | 6.967 | −9.177 | 0.362 | 0 | 6 | 8 | 1 | −5.397 | 5.29 | 0.106 | 0 |
| 4-22 | 505.651 | 6.068 | −7.953 | −0.018 | 0 | 6 | 8 | 1 | −5.347 | 5.87 | 0.274 | 0 |
| 4-23 | 537.649 | 5.447 | −7.40 | 0.385 | 0 | 8 | 10 | 1 | −5.262 | 6.69 | 0.371 | 0 |
| 4-24 | 513.577 | 5.925 | −7.285 | 0.662 | 0 | 6 | 8 | 1 | −5.370 | 5.56 | 0.357 | 0 |
| 4-25 | 505.651 | 6.188 | −7.451 | 0.719 | 0 | 6 | 8 | 1 | −5.382 | 5.00 | 0.274 | 0 |
| 4-26 | 531.568 | 5.954 | −8.057 | 0.943 | 0 | 6 | 8 | 1 | −5.391 | 10.10 | 0.310 | 0 |
| 4-27 | 580.923 | 7.289 | −9.402 | 0.9648 | 0 | 6 | 8 | 1 | −5.426 | 10.10 | 0.310 | 0 |
| 4-28 | 519.678 | 6.349 | −7.788 | 0.772 | 0 | 6 | 8 | 1 | −5.361 | 10.10 | 0.255 | 0 |
| 4-29 | 603.840 | 8.500 | −10.319 | 1.060 | 0 | 9 | 8 | 1 | −5.396 | 3.90 | 0.342 | 0 |
| 4-30 | 567.549 | 6.013 | −9.372 | 0.738 | 0 | 6 | 8 | 1 | −5.263 | 4.45 | 0.342 | 2.408 |
Conclusion
We successfully designed, synthesized, and identified several potent dual FAAH/sEH inhibitors. Inhibitors 4-5, 4-6, 4-20, 4-21, and 4-22 were identified as the most potent compounds in the benzothiazole series against both enzymes. Molecular modeling studies of these compounds revealed important residues within the catalytic sites of both enzymes that are responsible for the low nanomolar potencies of these inhibitors. This knowledge will be used in future design and follow-up SAR studies. In addition, these selected dual inhibitors showed acceptable predicted ADMET properties, and further study is currently underway to test their stability in liver microsome assays. In addition, the inhibitors of both enzymes have been individually investigated as potential therapeutics in many diseases, i.e., cardiovascular51, pulmonary52, 53, and Parkinson’s disease54, 55. In this study, we observed several novel compounds that have inhibitor activity for one of these two enzymes. Compounds that were found to inhibit only one particular enzyme, sEH (e.g., 4-18) or FAAH (e.g., 4-4), will be very useful to further develop as novel therapeutics for the aforementioned diseases and other disorders beyond pain.
Material and Methods
All solvents and reagents were obtained from Sigma–Aldrich, Matrix Scientific, TCI, and Acrōs Organic and used without further purification. Analytical thin-layer chromatography (TLC) was performed on aluminum plates precoated with silica gel, also obtained from Sigma–Aldrich. Column chromatography was carried out on Merck 938S silica gel. Proton and carbon NMR spectra were recorded with a Bruker 400 MHz NMR spectrometer. Spectra were referenced to the residual solvent peak: proton chemical shifts are reported relative to the residual solvent peak (chloroform = 7.26 ppm or dimethyl sulfoxide = 2.50 ppm) as follows: chemical shift (δ), proton ID, multiplicity (s = singlet, bs = broad singlet, d = doublet, bd = broad doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, integration, coupling constant(s) in Hz). Carbon chemical shifts are reported relative to the residual deuterated solvent signals (chloroform = 77.2 ppm, or dimethyl sulfoxide = 39.5 ppm). All compounds described were of > 95% purity. Purity was confirmed by high-resolution liquid chromatography mass spectrometer (ThermoFisher Scientific system). Elution was isocratic with water (30%, +0.1% formic acid) and acetonitrile (70%, +0.1% formic acid) at a flow rate of 0.4 mL/min. Melting points were measured with a MEL-TEMP II melting point apparatus and are reported uncorrected. Human recombinant FAAH enzyme (Item No. 100101183, Batch No. 0523867) and human recombinant sEH enzyme (Item No. 10011669) were obtained from Cayman Chemical. Molecular modeling studies and docking experiments were performed using ICM Pro Molsoft software.
Experimental
Chemistry
General procedure for the preparation of benzothiazole-phenyl analogs
The mixture of N-Boc-4-piperidinecarboxylic acid (250 mg, 1.09 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, EDC (252 mg, 1.31 mmol) and a catalytic amount of 4-dimethylaminopyridin, DMAP were dissolved in anhydrous tetrahydrofuran, THF (40 mL). The reaction mixture was stirred at room temperature for 1.5 hours under an argon atmosphere. Then, 2-(4-aminophenyl) benzothiazole (197 mg, 0.87 mmol) was added to the stirring solution. The reaction mixture was stirred at room temperature for 48 hours under an argon atmosphere. After removal of the solvent under reduced pressure, the residue was dissolved in ethyl acetate (40 mL). The mixture was transferred to a separatory funnel and the organic layer was washed with an aqueous solution of 1 M hydrochloric acid, HCl (3x25 mL), followed by an aqueous solution of saturated sodium bicarbonate, NaHCO3 (25 mL), dried over anhydrous sodium sulfate, Na2SO4, filtered and concentrated. The crude product 5 was purified by flash chromatography (1:4 ethyl acetate/hexane solvent system) and recrystallized from diethyl ether: tert-butyl 4-((4-(benzo[d]thiazol-2-yl)phenyl)carbamoyl)piperidine-1-carboxylate, 5 white solid, yield 70% (0.87 mmol, 267 mg), mp: 238-240 °C. 1H NMR (400 MHz, CDCl3) δ ppm 8.05 – 8.02 (m, 3H), 7.88 (d, J= 7.2 Hz, 1H), 7.67 (d, J= 8.4 Hz, 2H), 7.62 (s, 1H), 7.47 (t, J= 7.2 Hz, 1H), 7.36 (t, J= 6.8 Hz, 1H), 4.17 (bs, 1H), 2.77 (t, J= 11.2 Hz, 2H), 2.43 – 2.37 (m, 1H), 1.89 (d, J= 10.8 Hz, 2H), 1.79 – 1.70 (m, 3H), 1.47 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 172.9, 167.5, 154.8, 154.2, 140.4, 135.0, 129.6, 128.5, 126.4, 125.2, 123.1, 121.7, 119.9, 79.9, 44.5, 28.7, 28.5 ppm.
The amide 5 (250 mg, 0.57 mmol) was dissolved in anhydrous dichloromethane, DCM (10 mL), and stirred in an ice bath at 0°C. Trifluoroacetic acid, TFA (1 mL, 11.4 mmol) was added dropwise into the solution and the reaction mixture was stirred at room temperature for 24 hours under an argon atmosphere. Following concentration in vacuo, the crude product was triturated with diethyl ether and filtered. The product 6 was obtained as a TFA salt and used for next step without further purification. A small amount was free-based and used for NMR analysis: N-(4-(benzo[d]thiazol-2-yl)phenyl)piperidine-4-carboxamide, 6 pale green solid, yield 91% (0.57 mmol, 234 mg), mp: 242-244 °C. 1H NMR (400 MHz, DMSO-d6) δ ppm 10.31 (s, 1H), 8.11 (d, J= 8 Hz, 1H), 8.02 (t, J= 9.2 Hz, 3H), 7.81 (d, J= 8.8 Hz, 2H), 7.52 (t, J= 8.4 Hz, 1H), 7.43 (t, J= 8 Hz, 1H), 4.23 (bs, 1H), 3.14 (d, J= 12.4 Hz, 2H), 2.68 (t, J= 12.4 Hz, 2H), 2.59 – 2.55 (m, 1H), 1.82 (d, J= 11.6 Hz, 2H), 1.65 (q, J= 12, 11.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.5, 166.9, 153.6, 142.1, 134.2, 127.9, 127.4, 126.5, 125.2, 122.5, 122.2, 119.3, 44.1, 42.1, 27.5 ppm.
The amine 6, as a TFA salt (100 mg, 0.22 mmol), was dissolved in anhydrous DCM (20 mL), and stirred in an ice bath at 0°C. Triethylamine (0.19 mL, 1.33 mmol) was added and the reaction mixture was warmed to room temperature. Corresponding benzenesulfonyl chloride (0.33 mmol) was added to the reaction mixture and the reaction mixture was stirred for 48 hours at room temperature under an argon atmosphere. Next, the mixture was transferred to a separatory funnel where the organic layer was washed with an aqueous solution of saturated NaHCO3 (30 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated. The crude product was purified by flash chromatography (1:4 ethyl acetate/hexane solvent system) and recrystallized from diethyl ether.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-(thiophen-2-ylsulfonyl)piperidine-4-carboxamide, 4-1 was obtained as an off-white solid in the amount of 18 mg (17% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.12 (d, J= 7.6 Hz, 1H), 8.07-8.00 (m, 4H), 7.78 (d, J= 8.8 Hz, 2H), 7.67 (d, J= 2.4 Hz, 1H), 7.52 (t, J= 8 Hz, 1H), 7.44 (t, J= 12 Hz, 1H), 7.31 (t, J= 5.2 Hz, 1H),3.68 (d, J=12 Hz, 2H), 2.47-2.38 (m, 3H), 1.95 (d, J= 14 Hz, 2H), 1.70 (q, J=16, 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 167.0, 153.7, 142.0, 135.5, 134.3, 133.8, 133.0, 128.4, 128.0, 127.6, 126.6, 125.5, 122.6, 122.3, 119.4, 45.4, 41.4, 27.5 ppm. HRMS-ESI+: calculated for C23H21N3O3S3 + H: 484.0823; Found: 484.0818.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((4-bromo-5-chlorothiophen-2-yl)sulfonyl)piperidine-4-carboxamide, 4-2 was obtained as an off-white solid in the amount of 55 mg (42% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.12 (d, J= 7.2 Hz, 1H), 8.03 (t, J=8.8Hz, 3H), 7.80 (t, J= 22.4 Hz, 2H), 7.53 (t, J= 7.2 Hz, 1H), 7.44 (t, J= 6.8 Hz, 1H), 3.69 (d, J= 12 Hz, 2H), 2.62 (t, J= 12 Hz, 2H), 1.97 (d, J= 10.4 Hz, 2H), 1.74 (q, J= 11.6, 9.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 167.0, 153.6, 141.6, 134.9, 133.9, 133.5, 132.2, 127.9, 127.1, 126.6, 125.3, 122.6, 122.2, 119.4, 112.1, 45.3, 41.4, 27.5 ppm. HRMS-ESI+: calculated for C23H19BrClN3O3S3 + H: 595.9539; Found: 595.9529.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-(phenylsulfonyl)piperidine-4-carboxamide, 4-3 was obtained as an off-white solid in the amount of 40 mg (38% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.01 (t, J= 9.2 Hz, 3H), 7.79-7.65 (m, 5H), 7.52 (t, J= 6.8 Hz, 1H), 7.43 (t, J= 8 Hz, 1H), 3.69 (d, J= 12 Hz, 2H), 2.36 (t, J= 9.6 Hz, 3H), 1.87 (d, J= 10.4 Hz, 2H), 1.69 (q, J= 11.6, 9.2 Hz, 2H).13C NMR (100 MHz, DMSO-d6): δ 173.0, 167.0, 153.7, 142.0, 135.7, 134.3, 133.3, 129.5, 128.1, 127.5, 126.7, 125.3, 122.7, 122.3, 119.4, 45.4, 41.6, 27.4 ppm. HRMS-ESI+: calculated for C25H23N3O3S2 + H: 478.1259; Found: 478.1252.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2-fluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-4 was obtained as an off-white solid in the amount of 51 mg (46% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.84-7.77 (m, 4H), 7.53, (t, J= 8.4 Hz, 2H), 7.44 (q, J= 9.2, 7.2 Hz, 2H), 3.75 (d, J= 12.8 Hz, 2H), 2.66 (t, J= 11.6 Hz, 2H), 2.48-2.45 (m, 1H), 1.92 (d, J= 10.4 Hz, 2H), 1.65 (q, J= 11.6, 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 159.3, 156.9, 153.6, 141.9, 135.9, 135.8, 134.2, 130.8, 127.9, 127.5, 126.5, 125.2, 124.8, 122.5, 122.2, 119.3, 117.6, 117.4, 54.8, 44.8, 41.4, 27.7 ppm. HRMS-ESI+: calculated for C25H22FN3O3S2 + H: 496.1165; Found: 496.1154.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2-chlorophenyl)sulfonyl)piperidine-4-carboxamide, 4-5 was obtained as an off-white solid in the amount of 93 mg (83% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.02 (t, J= 8.8 Hz, 4H), 7.79 (d, J= 8.8 Hz, 2H), 7.73 (q, J= 8, 10 Hz, 2H), 7.59-7.50 (m, 2H), 7.43 (t, J= 8 Hz, 1H), 3.78 (d, J= 12.8 Hz, 2H), 2.84 (t, J= 12.4 Hz, 2H), 2.57-2.52 (m, 1H), 1.89 (d, J= 13.2 Hz, 2H), 1.65 (q, J=15.6, 12 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 178.8, 173.0, 166.9, 153.6, 141.9, 135.9, 134.5, 134.2, 132.3, 131.5, 130.9, 127.9, 127.8, 127.5, 126.6, 125.2, 122.5, 122.2, 119.4, 99.5, 44.6, 41.7, 27.9 ppm. HRMS-ESI+: calculated for C25H22ClN3O3S2 + H: 512.0869; Found: 512.0858.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2-bromophenyl)sulfonyl)piperidine-4-carboxamide, 4-6 was obtained as an off-white solid in the amount of 96 mg (78% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.11 (t, J= 6.8 Hz, 1H), 8.02 (t, J= 10.4 Hz, 4H), 7.91-7.89 (m, 1H), 7.79 (d, J= 7.6 Hz, 2H), 7.64-7.49 (m, 3H), 7.45-7.40 (m, 1H), 3.76 (d, J= 12.8 Hz, 2H), 2.86 (t, J= 12.0 Hz, 2H), 2.55 (d, J= 11.6 Hz, 1H), 1.89 (d, J= 12.0 Hz, 2H), 1.63 (q, J= 11.2, 11.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 153.6, 142.0, 137.5, 135.8, 134.4, 134.2, 131.6, 128.2, 127.9, 127.4, 126.5, 125.2, 122.5, 122.2, 119.5, 119.3, 44.6, 41.7, 27.9 ppm. HRMS-ESI+: calculated for C25H22BrN3O3S2 + H: 556.0364; Found: 556.0356.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-(o-tolylsulfonyl)piperidine-4-carboxamide, 4-7 was obtained as an off-white solid in the amount of 91 mg (84% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H), 8.11 (d, J= 7.6 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.83 (d, J= 8 Hz, 1H), 7.79, (d, J= 8.8 Hz, 2H), 7.60-7.50 (m, 2H), 7.48-7.41 (m, 3H), 7.78 (d, J= 8.8 Hz, 2H), 7.67 (d, J= 2.4 Hz, 1H), 7.52 (t, J= 8 Hz, 1H), 3.66 (d, J= 12.4 Hz, 2H), 2.70 (t, J= 12 Hz, 2H), 2.58 (s, 3H), 1.90 (d, J= 10.4 Hz, 2H), 1.68-1.59 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 177.0, 173.1, 167.0, 153.7, 142.1, 137.3, 135.9, 134.3, 133.1, 133.0, 132.9, 129.6, 128.0, 128.0, 127.6, 126.6, 126.5, 126.4, 125.3, 125.2, 122.6, 122.3, 119.4, 114.7, 110.1, 44.4, 41.9, 27.9, 20.2 ppm. HRMS-ESI+: calculated for C26H25N3O3S2 + H: 492.1416; Found: 492.1407.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2-methoxyphenyl)sulfonyl)piperidine-4-carboxamide, 4-8 was obtained as a white solid in the amount of 61 mg (54% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.11 (d, J= 7.6 Hz, 1H), 8.02 (t, J= 8.4 Hz, 3H), 7.78 (t, J= 8.8 Hz, 3H), 7.65 (t, J= 7.6 Hz, 1H), 7.53 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 7.2 Hz, 1H), 7.27 (d, J= 7.6 Hz, 1H), 7.11 (t, J= 7.6 Hz, 1H), 3.91 (s, 3H), 3.74 (d, J= 10.4 Hz, 2H), 2.68 (t, J= 12 Hz, 2H), 1.87 (d, J= 13.6 Hz, 2H), 1.61 (t, J= 12.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.1, 166.9, 156.6, 153.6, 142.0, 134.8, 134.2, 130.6, 127.9, 126.5, 126.0, 124.8, 123.4, 122.5, 122.2, 120.2, 119.3, 113.1, 55.9, 44.9, 41.9, 28.1 ppm. HRMS-ESI+: calculated for C26H25N3O4S2 + H: 508.1365; Found: 508.1354.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((3-fluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-9 was obtained as an off-white solid in the amount of 79 mg (72% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.12 (d, J= 7.6 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.79-7.71 (m, 3H), 7.65-7.62 (m, 3H), 7.53 (t, J= 6.8 Hz, 1H), 7.44 (t, J= 8.0 Hz, 1H), 3.72 (d, J= 12 Hz, 2H), 2.42 (q, J= 12.0, 9.6 Hz, 3H), 1.91 (d, J= 10.8 Hz, 2H), 1.66 (q, J= 11.6, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 167.0, 160.6, 153.6, 142.0, 137.5, 134.3, 131.9, 128.0, 127.5, 126.6, 125.3, 123.7, 122.6, 122.3, 120.5, 119.4, 114.3, 45.3, 41.4, 27.6 ppm. HRMS-ESI+: calculated for C25H22FN3O3S2 + H: 496.1165; Found: 496.1154.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((3-chlorophenyl)sulfonyl)piperidine-4-carboxamide, 4-10 was obtained as an off-white solid in the amount of 81 mg (72% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.04 (q, J= 2.4, 8.8 Hz, 2H), 7.84-7.69 (m, 4H), 7.52 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 8 Hz, 1H), 3.71 (d, J= 12 Hz, 2H), 2.46-2.38 (m, 3H), 1.91 (d, J= 10.8 Hz, 2H), 1.70-1.61 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 141.9, 137.8, 134.2, 134.1, 133.1, 131.5, 127.9, 127.5, 126.8, 126.5, 126.1, 125.2, 122.5, 122.2, 119.4, 99.5, 45.3, 41.4, 27.5 ppm. HRMS-ESI+: calculated for C25H22ClN3O3S2 + H: 512.0869; Found: 512.0858.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((3-bromophenyl)sulfonyl)piperidine-4-carboxamide, 4-11 was obtained as an off-white solid in the amount of 75 mg (61% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.04-7.95 (m, 5H), 7.91 (s, 1H), 7.78 (t, J= 10.0 Hz, 3H), 7.64 (t, J= 8 Hz, 1H), 7.53 (t, J= 8 Hz, 1H), 7.44 (t, J= 8 Hz, 1H), 3.71 (d, J= 11.6 Hz, 2H), 2.44 (t, J= 11.6 Hz, 3H), 1.91 (d, J= 11.6 Hz, 2H), 1.67 (t, J= 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 136.0, 134.2, 131.6, 129.5, 127.9, 126.5, 122.5, 122.4, 122.2, 119.3, 45.2, 41.3, 27.5 ppm. HRMS-ESI+: calculated for C25H22BrN3OS2 + H: 556.0364; Found: 556.0356.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-(m-tolylsulfonyl)piperidine-4-carboxamide, 4-12 was obtained as an off-white solid in the amount of 53 mg (49% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.01 (t, J= 8.8 Hz, 3H), 7.76 (d, J= 8.8 Hz, 2H), 7.58 (s, 1H), 7.57-7.50 (m, 4H), 7.43 (t, J= 8 Hz, 1H), 3.67 (d, J= 12 Hz, 2H), 2.43 (s, 3H), 2.38 – 2.31 (m, 3H) 1.90 (d, J= 10.4 Hz, 2H) 1.70 – 1.62 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 174.5, 172.9, 166.9, 159.0, 153.6, 152.5, 150.8, 150.6, 145.0, 143.3, 141.9, 139.1, 135.4, 134.2, 133.7, 129.1, 127.9, 127.5, 127.4, 126.5, 126.0, 125.2, 124.5,122.5, 122.2, 119.3, 116.7, 115.2, 110.4 ppm. HRMS-ESI+: calculated for C26H25N3O3S2 + H: 492.1416; Found: 492.1407.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((3-methoxyphenyl)sulfonyl)piperidine-4-carboxamide, 4-13 was obtained as a white solid in the amount of 59 mg (47% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 8.11 (d, J= 8 Hz, 1H), 8.01 (t, J= 8.8 Hz, 3H), 7.76 (d, J= 8.8 Hz, 2H), 7.58 (t, J= 8.4 Hz, 1H), 7.52 (t, J= 7.2 Hz, 1H), 7.429 (t, J= 7.2 Hz, 1H), 7.31 (q, J= 8 Hz, 6.8 Hz, 2H), 7.22 (s, 1H), 3.85 (s, 3H), 3.69 (d, J= 12 Hz, 2H), 2.38 (t, J= 11.6 Hz, 3H), 1.90 (d, J= 11.6 Hz, 2H), 1.66 (q, J= 11.6 Hz, 9.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.6, 166.7, 159.9, 153.1, 142.5, 136.9, 134.1, 130.6, 127.9, 127.6, 126.1, 125.7, 122.5, 122.2, 119.3, 188.9, 112.2 55.6, 45.3, 41.4, 27.5 ppm. HRMS-ESI+: calculated for C26H25N3O4S2 + H: 508.1365; Found: 508.1354.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((4-fluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-14 was obtained as an off-white solid in the amount of 67 mg (61% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.12 (d, J= 7.2 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.86 (q, J= 5.2, 4.0 Hz, 2H), 7.77 (d, J= 8.8 Hz, 2H), 7.51-7.49 (m, 3H), 7.44 (t, J= 8.0 Hz, 1H), 3.69 (d, J= 11.6 Hz, 2H), 2.39 (t, J= 9.6 Hz, 3H), 1.91 (d, J= 10.8 Hz, 2H), 1.67 (q, J= 11.6, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 163.2, 153.6, 141.9, 134.2, 132.0, 130.5, 130.4, 127.9, 127.5, 126.5, 125.2, 122.5, 122.2, 119.3, 116.7, 116.4, 45.2, 41.4, 27.5 ppm. HRMS-ESI+: calculated for C25H22FN3O3S2 + H: 496.1165; Found: 496.1154.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((4-chlorophenyl)sulfonyl)piperidine-4-carboxamide, 4-15 was obtained as an off-white solid in the amount of 84 mg (75% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.12 (d, J= 7.6 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.79 (q, J= 9.2, 12, Hz, 6H), 7.53 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 8 Hz, 2H), 3.68 (d, J= 12 Hz, 2H), 2.42 (q, J= 12, 9.2 Hz, 3H), 1.91 (d, J= 11.2 Hz, 2H), 1.69 (q, J= 11.6, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.3, 167.5, 138.5, 133.9, 129.6, 129.3, 127.9, 122.6, 122.3, 119.4, 45.2, 41.8, 27.5 ppm. HRMS-ESI+: calculated for C25H22ClN3O3S2 + H: 512.0869; Found: 512.0858.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((4-bromophenyl)sulfonyl)piperidine-4-carboxamide, 4-16 was obtained as an off-white solid in the amount of 38 mg (31% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.11 (d, J= 76 Hz, 1H), 8.01 (t, J= 8.8 Hz, 3H), 7.88 (d, J= 8.8 Hz,2H), 7.77 (d, J= 8.8 Hz, 2H), 7.71 (d, J= 8.8 Hz, 2H), 7.52 (t, J= 8.4 Hz, 1H), 7.43 (t, J= 8.0 Hz, 1H), 3.67 (d, J= 12.0 Hz, 2H), 2.43-2.36 (m, 3H), 1.90 (d, J= 13.6 Hz, 2H), 1.66 (q, J= 15.2, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 141.9, 135.0, 134.2, 132.4, 129.3, 127.9, 127.5, 127.1, 126.5, 122.5, 122.2, 119.3, 45.1, 41.3, 27.5 ppm. HRMS-ESI+: calculated for C25H22BrN3OS2 + H: 556.0364; Found: 556.0355.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-tosylpiperidine-4-carboxamide, 4-17 was obtained as an off-white solid in the amount of 73 mg (68% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 8.11 (d, J= 8 Hz, 1H), 8.01 (t, J= 8.8 Hz, 3H), 7.76 (d, J= 9.2 Hz, 2H), 7.65 (d, J= 8 Hz, 2H), 7.52 (t, J= 6.8 Hz, 1H), 7.44 (q, J= 8.2 Hz, 3H), 3.65 (d, J= 12 Hz, 2H), 2.42 (s, 3H), 2.31 (q, J= 11.6, 9.6 Hz, 3H), 1.89 (d, J= 10.4 Hz, 2H), 1.66 (q, J= 11.2, 9.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6: δ 173.0, 166.9, 153.6, 143.4, 141.9, 134.2, 132.6, 129.8, 127.9, 127.4, 127.4, 126.5, 125.2, 122.2, 119.3 ppm. HRMS-ESI+: calculated for C26H25N3O3S2 + H: 492.1416; Found: 492.1409.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((4-methoxyphenyl)sulfonyl)piperidine-4-carboxamide, 4-18 was obtained as a white solid in the amount of 54 mg (48% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 8.11 (d, J= 8 Hz, 1H), 8.01 (t, J= 8.8 Hz, 3H), 7.7 (d, J= 8.8 Hz, 2H), 7.70 (d, J= 8.8 Hz, 2H), 7.52 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 7.2 Hz, 1H), 7.17 (d, J= 8.8 Hz, 2H), 3.86 (s, 3H), 3.64 (d, J= 12 Hz, 2H), 2.38-2.28 (m, 3H), 1.89 (d, J= 10.8 Hz, 2H), 1.66 (q, J= 11.2 Hz, 9.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 162.6, 153.6, 141.9, 134.2, 129.6, 127.9, 122.5, 122.2, 119.3, 114.5, 55.7, 45.2, 41.4, 27.5 ppm. HRMS-ESI+: calculated for C26H25N3O4S2 + H: 508.1365; Found: 508.1356.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4-difluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-19 was obtained as an off-white solid in the amount of 28 mg (25% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.12 (d, J= 7.6 Hz, 1H), 8.03 (t, J= 8.8 Hz, 3H), 7.90 (q, J= 8.4, 6.4 Hz, 1H), 7.79 (d, J= 8.8 Hz, 2H), 7.64 (t, J= 10.8 Hz, 1H), 7.53 (t, J= 7.2 Hz, 1H), 7.44 (t, J= 8.0 Hz, 1H), 7.36 (t, J= 8.0 Hz, 1H), 3.74 (d, J= 12.0 Hz, 2H), 2.68 (t, J= 12.0 Hz, 2H), 1.92 (d, J= 10.8 Hz, 2H), 1.65 (q, J= 11.6, 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 141.9, 134.2, 126.5, 125.2, 122.5, 122.2, 119.3, 54.8, 44.7, 41.5, 27.7 ppm. HRMS-ESI+: calculated for C25H21F2N3O3S2 + H: 514.1071; Found: 514.1065.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4-difluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-20 was obtained as an off-white solid in the amount of 58 mg (48% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.05-7.94 (m, 4H), 7.81 (s, 1H), 7.79 (d, J= 9.2 Hz, 2H), 7.67 (d, J= 10.8 Hz, 1H), 7.52 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 8 Hz, 1H), 3.77 (d, J= 12.8 Hz, 2H), 2.86 (t, J= 12.6 Hz, 2H), 2.54 (t, J= 11.6 Hz, 1H), 1.89 (d, J= 10.8 Hz, 2H), 1.65 (q, J= 12, 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 153.6, 141.9, 138.4, 134.2, 132.9, 132.2, 131.7, 127.9, 127.5, 126.6, 125.2, 122.6, 122.2, 119.4, 44.7, 41.6, 27.9 ppm. HRMS-ESI+: calculated for C25H21Cl2N3O3S2 + H: 546.0480; Found: 546.0476.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4-dibromophenyl)sulfonyl)piperidine-4-carboxamide, 4-21 was obtained as an off-white solid in the amount of 39 mg (28% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.19 (s,1H), 8.12 (d, J= 7.6 Hz, 1H), 8.04 (d, J= 7.6 Hz, 3H), 7.95 (d, J= 8.4 Hz, 1H), 7.85-7.79 (m, 3H), 7.53 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 7.6 Hz, 1H), 3.75 (d, J= 11.2 Hz, 2H), 2.88 (t, J= 12.0 Hz, 2H), 2.58-2.55 (m, 1H), 1.89 (d, J= 13.2 Hz, 2H), 1.64 (q, J= 10.8, 12.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 153.6, 141.8, 137.6, 137.0, 134.2, 133.0, 131.3, 127.9, 127.1, 126.5, 125.0, 122.5, 122.2, 120.7, 119.3, 44.6, 41.6, 27.9 ppm. HRMS-ESI+: calculated for C25H21Br2N3O3S2 + H: 633.9469; Found: 633.9461.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4-dimethylphenyl)sulfonyl)piperidine-4-carboxamide, 4-22 was obtained as an off-white solid in the amount of 83 mg (75% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.78 (d, J= 8.8 Hz, 2H), 7.71 (d, J= 8 Hz, 1H), 7.52 (t, J= 6.8 Hz, 1H) 7.43 (t, J= 8 Hz, 1H), 7.24 (t, J= 11.2 Hz, 1H), 3.63 (d, J= 12.4 Hz, 2H), 2.66 (t, J= 12 Hz, 2H), 2.53 (s, 3H), 2.35 (s, 3H), 1.89 (d, J= 10.4 Hz, 2H), 1.62 (q, J= 12, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 153.6, 143.3, 141.9, 137.0, 134.2, 133.4, 132.7, 129.8, 127.9, 127.4, 126.8, 126.5, 125.2, 122.5, 122.2, 119.3, 44.2, 41.8, 27.8, 20.7, 20.0 ppm. HRMS-ESI+: calculated for C27H27N3O3S2 + H: 506.1572; Found: 506.1566.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4-dimethoxyphenyl)sulfonyl)piperidine-4-carboxamide, 4-23 was obtained as an off-white solid in the amount of 73 mg (62% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.11 (d, J= 8 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.79 (d, J= 8.8 Hz, 2H), 7.67, (d, J= 8.4 Hz, 1H), 7.52 (t, J= 8 Hz, 1H), 7.43 (t, J= 8 Hz, 1H), 6.75 (s, 1H), 6.66 (d, J= 8.8 Hz, 1H), 3.87 (d, J= 11.6 Hz, 3H), 3.69 (d, J= 10 Hz, 2H), 2.62 (t, J= 10.4, 2H), 2.44 (d, J= 11.2, 1H), 1.86 (d, J= 10.8, 2H), 1.60 (q, J= 11.6 Hz, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.4, 167.1, 164.5, 158.4, 153.8, 142.2, 134.4, 132.7, 128.1, 127.6, 126.7, 122.7, 122.4, 119.5, 118.2, 105.2, 99.7, 56.2, 55.9, 45.1, 42.2, 28.3 ppm. HRMS-ESI+: calculated for C27H27N3O5S2 + H: 538.1470; Found: 538.1462.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((3,5-difluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-24 was obtained as an off-white solid in the amount of 60 mg (53% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.12 (d, J= 7.2 Hz, 1H), 8.03 (t, J= 8.8 Hz, 3H), 7.80-7.71 (m, 3H), 7.57-7.52 (m, 3H), 7.44 (t, J= 8.0 Hz, 1H), 3.75 (d, J= 12.4 Hz, 2H), 2.55-2.41 (m, 4H), 1.92 (d, J= 10.4 Hz, 2H), 1.67 (q, J= 11.6, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 141.9, 134.2, 127.9, 127.5, 126.5, 125.2, 122.5, 122.2, 119.3, 111.3, 111.0, 45.2, 41.3, 27.5 ppm. HRMS-ESI+: calculated for C25H21F2N3O3S2 + H: 514.1071; Found: 514.1064.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((3,5-dimethylphenyl)sulfonyl)piperidine-4-carboxamide, 4-25 was obtained as an off-white solid in the amount of 70 mg (63% yield): 1H NMR (400 MHz, DMSO-d6 δ 10.13 (s, 1H), 8.12 (d, J= 7.2 Hz, 1H), 8.01 (t, J= 8.8 Hz, 3H), 7.76 (d, J= 9.2 Hz, 2H), 7.52 (t, J= 7.2 Hz, 1H), 7.43 (t, J= 8 Hz, 1H), 7.36 (d, J= 6 Hz, 3H), 3.66 (d, J= 11.6 Hz, 2H), 2.38 (s, 6H), 2.33 (t, J= 10 Hz, 3H), 1.90 (d, J= 10.8 Hz, 2H), 1.66 (q, J= 15.6, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 153.6, 141.9, 138.9, 135.2, 134.4, 134.2, 127.9, 127.4, 126.5, 125.2, 124.8, 122.5, 122.2, 119.3, 45.3, 41.5, 27.5, 20.7 ppm. HRMS-ESI+: calculated for C27H27N3O5S2 + H: 538.1470; Found: 538.1462.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4,6-trifluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-26 was obtained as an off-white solid in the amount of 19 mg (16% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.24 (s, 1H), 8.12 (d, J= 7.2 Hz, 1H), 8.03 (t, J= 8.8 Hz, 3H), 7.79 (d, J= 8.8 Hz, 2H), 7.54-7.50 (m, 3H), 7.43 (t, J= 8.0 Hz, 1H), 3.77 (d, J= 12.4 Hz, 2H), 2.77 (t, J= 11.2 Hz, 2H), 1.95 (d, J= 10.4 Hz, 2H), 1.67 (q, J= 11.6, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 141.9, 134.2, 127.9, 127.5, 126.5, 125.2, 122.5, 122.2, 119.4, 44.5, 41.3, 27.6 ppm. HRMS-ESI+: calculated for C25H22F3N3O3S2 + H: 532.0976; Found: 532.0968.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4,6-trichlorophenyl)sulfonyl)piperidine-4-carboxamide, 4-27 was obtained as an off-white solid in the amount of 65 mg (51% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.11 (d, J= 8 Hz, 1H), 8.03 (t, J= 8.8 Hz, 3H), 7.93 (s, 2H), 7.79 (d, J= 8.8 Hz, 2H), 7.52 (t, J= 8.8 Hz, 1H), 7.45 (t, J= 8 Hz, 1H), 3.84 (d, J= 13.2 Hz, 2H), 2.98 (t, J= 12 Hz, 2H), 2.58 (t, J=11.2 Hz, 1H), 1.91 (d, J= 11.2 Hz, 2H), 1.61 (t, J= 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.9, 166.9, 153.6, 141.9, 135.4, 134.1, 131.5, 129.6, 126.6, 122.5, 122.2, 119.4, 45.8, 44.6, 41.7, 27.9 ppm. HRMS-ESI+: calculated for C25H20Cl3N3O3S2 + H: 580.0090; Found: 580.0081.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-(mesitylsulfonyl)piperidine-4-carboxamide, 4-28 was obtained as an off-white solid in the amount of 72 mg (63% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.11 (d, J= 7.2 Hz, 1H), 8.02 (t, J= 8.8 Hz, 3H), 7.79 (d, J= 8.8 Hz, 2H), 7.52 (t, J= 6.8 Hz, 1H), 7.43 (t, J= 8.4 Hz, 1H), 7.09 (s, 2H), 3.52 (d, J= 12.8 Hz, 2H), 2.77 (t, J= 12.4 Hz, 2H), 2.56 (s, 6H), 2.28 (s, 3H), 1.88 (d, J= 10.8 Hz, 2H), 1.58 (q, J= 16, 9.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 173.3, 167.1, 153.8, 142.6, 142.2, 139.8, 136.0, 134.4, 132.0, 131.8, 130.0, 128.1, 127.7, 126.7, 125.4, 122.7, 122.4, 119.5, 43.4, 42.3, 27.8, 22.9, 22.5, 20.6 ppm. HRMS-ESI+: calculated for C28H29N3O3S2 + H: 520.1729; Found: 520.1721.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((2,4,6-triisopropylphenyl)sulfonyl)piperidine-4-carboxamide, 4-29 was obtained as an off-white solid in the amount of 92 mg (69% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 8.11 (d, J= 7.6 Hz, 1H), 8.02 (t, J= 8.8 Hz, 2H), 7.79 (d, J= 8.8 Hz, 2H), 7.52 (t, J= 6.8 Hz, 1H) 7.43 (t, J= 7.2 Hz, 1H), 7.29 (s, 2H), 4.14-4.07 (m, 2H), 3.53 (d, J =12 Hz, 2H), 2.97-2.90 (m, 1H), 2.79 (t, J= 10.4 Hz, 2H) 2.56 (t, J= 10.8 Hz, 1H), 1.93 (d, J=10.8 Hz, 2H), 1.60 (q, J= 11.6, 9.6 Hz, 2H), 1.21 (d, J= 6.8 Hz, 18H). 13C NMR (100 MHz, DMSO-d6): 13C NMR (100 MHz, DMSO-d6): δ 173.0, 166.9, 154.1, 153.0. 151.0, 142.0, 134.2, 129.8, 127.9, 127.4, 126.5, 125.0, 123.8, 122.5, 122.2, 119.3, 43.1, 41.9, 33.3, 28.7, 27.6, 24.5, 23.3 ppm. HRMS-ESI+: calculated for C34H41N3O3S2 + H: 604.2668; Found: 604.2658.
N-(4-(benzo[d]thiazol-2-yl)phenyl)-1-((perfluorophenyl)sulfonyl)piperidine-4-carboxamide, 4-30 was obtained as an off-white solid in the amount of 95 mg (76% yield): 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.12 (d, J= 7.6 Hz, 1H), 8.03 (t, J= 8.8 Hz, 3H), 7.80 (d, J= 8.8 Hz, 2H), 7.53 (t, J= 6.8 Hz, 1H), 7.44 (t, J= 6.8 Hz, 1H), 3.78 (d, J= 11.6 Hz, 2H), 2.85 (t, J= 11.2 Hz, 2H), 1.99 (d, J= 10.8 Hz, 2H), 1.71 (q, J= 12.0, 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 172.8, 166.9, 153.6, 141.9, 134.2, 127.9, 127.5, 126.5, 122.5, 122.2, 119.4, 44.5, 41.2, 27.6 ppm. HRMS-ESI+: calculated for C25H18F5N3O3S2 + H: 568.0788; Found: 568.0780.
sEH and FAAH IC50 assay conditions Human FAAH enzyme inhibition assay
Measurement of sEH potency was performed using cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyloxyran-2-yl) methyl carbonate (CMNPC) as the fluorescent substrate.56 Human sEH (1 nM) was incubated with the inhibitor for 5 min in pH 7.0 Bis–Tris/HCl buffer (25 mM) containing 0.1 mg/mL of bovine serum albumin (BSA) at 30 °C prior to substrate introduction ([S] = 5 μM). Activity was determined by monitoring the appearance of 6-methoxy-2-naphthaldehyde over 10 min by fluorescence detection with an excitation wavelength of 330 nm and an emission wavelength of 465 nm. Reported IC50 values are the average of the three replicates with at least two data points above and at least two below the IC50. Measurement of FAAH potency was performed using the substrate N-(6-methoxypyridin-3-yl) octanamide (OMP) ([S]final=50 μM) in sodium phosphate buffer (0.1 M, pH = 8, 0.1 mg/mL BSA). Progress of the reaction was measured by fluorescence detection of 6-methoxypyridin-3-amine at an excitation wavelength of 303 nm and an emission wavelength of 394 nm at 37 °C by the use of microplate reader (Molecular Devices., CA, USA). All experiments were run in triplicate, and values reported as average +/− SD. The substrate OMP was synthesized following a previously reported synthetic procedure and reaction conditions.36
Molecular modeling
For the docking studies of the dual sEH/FAAH inhibitors, a crystal structure of human soluble epoxide hydrolase complexed with N-cycloheptyl-1-(mesitylsulfonyl)piperidine-4-carboxamide (PDBfile: 4HAI)33 and a homology model of human FAAH enzyme36 were used. PDB file 4HAI was first converted to an ICM file and the inhibitor N-cycloheptyl-1-(mesitylsulfonyl) piperidine-4-carboxamide was removed. Docking experiments were performed following the program guidelines. ICM scores were obtained after this procedure. ADMET properties for all synthesized target analogs were calculated using the ICM Chemist program.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number SC2GM135020 and, in part, by a grant from the National Institute of Environmental Health Sciences (NIEHS) Grant R35ES030443, and NIEHS Superfund Research Program P42 ES004699. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Instrumentation support was provided by the National Science Foundation MRI (CHE1726903) for acquisition of an UPLC-MS.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riedel W; Neeck G, Nociception, pain, and antinociception: current concepts. Z Rheumatol 2001, 60 (6), 404–15. [DOI] [PubMed] [Google Scholar]
- 2.Lynch ME; Watson CP, The pharmacotherapy of chronic pain: a review. Pain Res Manag 2006, 11 (1), 11–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmidt-Hansen M; Bennett MI; Arnold S; Bromham N; Hilgart JS, Efficacy, tolerability and acceptability of oxycodone for cancer-related pain in adults: an updated Cochrane systematic review. BMJ Support Palliat Care 2018, 8 (2), 117–128. [DOI] [PubMed] [Google Scholar]
- 4.Valentino RJ; Volkow ND, Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43 (13), 2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cicero TJ; Ellis MS, The prescription opioid epidemic: a review of qualitative studies on the progression from initial use to abuse. Dialogues Clin Neurosci 2017, 19 (3), 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blobaum AL; Marnett LJ, Structural and functional basis of cyclooxygenase inhibition. J Med Chem 2007, 50 (7), 1425–41. [DOI] [PubMed] [Google Scholar]
- 7.Malkowski MG; Ginell SL; Smith WL; Garavito RM, The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 2000, 289 (5486), 1933–7. [DOI] [PubMed] [Google Scholar]
- 8.Wagner KM; McReynolds CB; Schmidt WK; Hammock BD, Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther 2017, 180, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inceoglu B; Jinks SL; Ulu A; Hegedus CM; Georgi K; Schmelzer KR; Wagner K; Jones PD; Morisseau C; Hammock BD, Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A 2008, 105 (48), 18901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naesdal J; Brown K, NSAID-associated adverse effects and acid control aids to prevent them: a review of current treatment options. Drug Saf 2006, 29 (2), 119–32. [DOI] [PubMed] [Google Scholar]
- 11.Bjorkman DJ; Kimmey MB, Nonsteroidal anti-inflammatory drugs and gastrointestinal disease: pathophysiology, treatment and prevention. Dig Dis 1995, 13 (2), 119–29. [DOI] [PubMed] [Google Scholar]
- 12.Morphy R; Rankovic Z, Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 2005, 48 (21), 6523–43. [DOI] [PubMed] [Google Scholar]
- 13.Enayetallah AE; French RA; Thibodeau MS; Grant DF, Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem 2004, 52 (4), 447–54. [DOI] [PubMed] [Google Scholar]
- 14.Imig JD; Hammock BD, Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 2009, 8 (10), 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spector AA; Fang X; Snyder GD; Weintraub NL, Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 2004, 43 (1), 55–90. [DOI] [PubMed] [Google Scholar]
- 16.Nithipatikom K; Gross GJ, Review article: epoxyeicosatrienoic acids: novel mediators of cardioprotection. J Cardiovasc Pharmacol Ther 2010, 15 (2), 112–9. [DOI] [PubMed] [Google Scholar]
- 17.Yang T; Peng R; Guo Y; Shen L; Zhao S; Xu D, The role of 14,15-dihydroxyeicosatrienoic acid levels in inflammation and its relationship to lipoproteins. Lipids Health Dis 2013, 12, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen HC; Hammock BD, Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem 2012, 55 (5), 1789–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostermann AI; Herbers J; Willenberg I; Chen R; Hwang SH; Greite R; Morisseau C; Gueler F; Hammock BD; Schebb NH, Oral treatment of rodents with soluble epoxide hydrolase inhibitor 1-(1-propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea (TPPU): Resulting drug levels and modulation of oxylipin pattern. Prostaglandins & Other Lipid Mediators 2015, 121, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulu A; Appt S; Morisseau C; Hwang S; Jones P; Rose T; Dong H; Lango J; Yang J; Tsai H; Miyabe C; Fortenbach C; Adams M; Hammock B, Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. British Journal of Pharmacology 2012, 165 (5), 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner KM; Atone J; Hammock BD, Soluble epoxide hydrolase inhibitor mediated analgesia lacks tolerance in rat models. Brain Research 2020, 1728, 146573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner K; Lee KSS; Yang J; Hammock BD, Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. European Journal of Pain 2017, 21 (3), 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palermo G; Rothlisberger U; Cavalli A; De Vivo M, Computational insights into function and inhibition of fatty acid amide hydrolase. Eur J Med Chem 2015, 91, 15–26. [DOI] [PubMed] [Google Scholar]
- 24.Palermo G; Bauer I; Campomanes P; Cavalli A; Armirotti A; Girotto S; Rothlisberger U; De Vivo M, Keys to Lipid Selection in Fatty Acid Amide Hydrolase Catalysis: Structural Flexibility, Gating Residues and Multiple Binding Pockets. PLoS Comput Biol 2015, 11 (6), e1004231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn K; Johnson DS; Cravatt BF, Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov 2009, 4 (7), 763–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X; Sarris K; Kage K; Zhang D; Brown SP; Kolasa T; Surowy C; El Kouhen OF; Muchmore SW; Brioni JD; Stewart AO, Synthesis and evaluation of benzothiazole-based analogues as novel, potent, and selective fatty acid amide hydrolase inhibitors. J Med Chem 2009, 52 (1), 170–80. [DOI] [PubMed] [Google Scholar]
- 27.Seierstad M; Breitenbucher JG, Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem 2008, 51 (23), 7327–43. [DOI] [PubMed] [Google Scholar]
- 28.Sasso O; Wagner K; Morisseau C; Inceoglu B; Hammock BD; Piomelli D, Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol Res 2015, 97, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kodani SD; Bhakta S; Hwang SH; Pakhomova S; Newcomer ME; Morisseau C; Hammock BD, Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase. Bioorg Med Chem Lett 2018, 28 (4), 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodani SD; Wan D; Wagner KM; Hwang SH; Morisseau C; Hammock BD, Design and Potency of Dual Soluble Epoxide Hydrolase/Fatty Acid Amide Hydrolase Inhibitors. ACS Omega 2018, 3 (10), 14076–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y; Liu Y; Gong G; Smith DH; Yan F; Rinderspacher A; Feng Y; Zhu Z; Li X; Deng SX; Branden L; Vidovic D; Chung C; Schurer S; Morisseau C; Hammock BD; Landry DW, Discovery of potent non-urea inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett 2009, 19 (8), 2354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pecic S; Deng SX; Morisseau C; Hammock BD; Landry DW, Design, synthesis and evaluation of non-urea inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett 2012, 22 (1), 601–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pecic S; Pakhomova S; Newcomer ME; Morisseau C; Hammock BD; Zhu Z; Rinderspacher A; Deng SX, Synthesis and structure-activity relationship of piperidine-derived non-urea soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett 2013, 23 (2), 417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pecic S; Zeki AA; Xu X; Jin GY; Zhang S; Kodani S; Halim M; Morisseau C; Hammock BD; Deng SX, Novel piperidine-derived amide sEH inhibitors as mediators of lipid metabolism with improved stability. Prostaglandins Other Lipid Mediat 2018, 136, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boger DL; Miyauchi H; Du W; Hardouin C; Fecik RA; Cheng H; Hwang I; Hedrick MP; Leung D; Acevedo O; Guimaraes CR; Jorgensen WL; Cravatt BF, Discovery of a potent, selective, and efficacious class of reversible alpha-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem 2005, 48 (6), 1849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilt SR; Rodriguez M; Le TNH; Baltodano EV; Salas A; Pecic S, Design, microwave-assisted synthesis, biological evaluation and molecular modeling studies of 4- phenylthiazoles as potent fatty acid amide hydrolase inhibitors. Chemical Biology & Drug Design 2020, 95 (5), 534–547. [DOI] [PubMed] [Google Scholar]
- 37.Wagner K; Inceoglu B; Gill SS; Hammock BD, Epoxygenated Fatty Acids and Soluble Epoxide Hydrolase Inhibition: Novel Mediators of Pain Reduction. Journal of Agricultural and Food Chemistry 2011, 59 (7), 2816–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piomelli D; Sasso O, Peripheral gating of pain signals by endogenous lipid mediators. Nature Neuroscience 2014, 17 (2), 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gattrell WT; Sambrook Smith CP; Smith AJ, An example of designed multiple ligands spanning protein classes: dual MCH-1R antagonists/DPPIV inhibitors. Bioorg Med Chem Lett 2012, 22 (7), 2464–9. [DOI] [PubMed] [Google Scholar]
- 40.Schapira M; Totrov M; Abagyan R, Prediction of the binding energy for small molecules, peptides and proteins. J Mol Recognit 1999, 12 (3), 177–90. [DOI] [PubMed] [Google Scholar]
- 41.Takebe T; Imai R; Ono S, The Current Status of Drug Discovery and Development as Originated in United States Academia: The Influence of Industrial and Academic Collaboration on Drug Discovery and Development. Clin Transl Sci 2018, 11 (6), 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dowden H; Munro J, Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov 2019, 18 (7), 495–496. [DOI] [PubMed] [Google Scholar]
- 43.Lombardo F; Desai PV; Arimoto R; Desino KE; Fischer H; Keefer CE; Petersson C; Winiwarter S; Broccatelli F, In Silico Absorption, Distribution, Metabolism, Excretion, and Pharmacokinetics (ADME-PK): Utility and Best Practices. An Industry Perspective from the International Consortium for Innovation through Quality in Pharmaceutical Development. J Med Chem 2017, 60 (22), 9097–9113. [DOI] [PubMed] [Google Scholar]
- 44.Clark DE, In silico prediction of blood-brain barrier permeation. Drug Discov Today 2003, 8 (20), 927–33. [DOI] [PubMed] [Google Scholar]
- 45.Kelder J; Grootenhuis PD; Bayada DM; Delbressine LP; Ploemen JP, Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res 1999, 16 (10), 1514–9. [DOI] [PubMed] [Google Scholar]
- 46.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 1997, 23 (1), 3–25. [DOI] [PubMed] [Google Scholar]
- 47.Veber DF; Johnson SR; Cheng HY; Smith BR; Ward KW; Kopple KD, Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002, 45 (12), 2615–23. [DOI] [PubMed] [Google Scholar]
- 48.van Breemen RB; Li Y, Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol 2005, 1 (2), 175–85. [DOI] [PubMed] [Google Scholar]
- 49.Jing Y; Easter A; Peters D; Kim N; Enyedy IJ, In silico prediction of hERG inhibition. Future Med Chem 2015, 7 (5), 571–86. [DOI] [PubMed] [Google Scholar]
- 50.Lee HM; Yu MS; Kazmi SR; Oh SY; Rhee KH; Bae MA; Lee BH; Shin DS; Oh KS; Ceong H; Lee D; Na D, Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinformatics 2019, 20 (Suppl 10), 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou C; Huang J; Li Q; Nie J; Xu X; Wang DW, Soluble Epoxide Hydrolase Inhibition Protected against Angiotensin II-induced Adventitial Remodeling. Scientific Reports 2017, 7 (1), 6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tao W; Li PS; Yang LQ; Ma YB, Effects of a Soluble Epoxide Hydrolase Inhibitor on Lipopolysaccharide-Induced Acute Lung Injury in Mice. PLoS One 2016, 11 (8), e0160359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wenzel D; Matthey M; Bindila L; Lerner R; Lutz B; Zimmer A; Fleischmann BK, Endocannabinoid anandamide mediates hypoxic pulmonary vasoconstriction. Proceedings of the National Academy of Sciences 2013, 110 (46), 18710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ren Q; Ma M; Yang J; Nonaka R; Yamaguchi A; Ishikawa K.-i.; Kobayashi K; Murayama S; Hwang SH; Saiki S; Akamatsu W; Hattori N; Hammock BD; Hashimoto K, Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proceedings of the National Academy of Sciences 2018, 115 (25), E5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pasquarelli N; Porazik C; Bayer H; Buck E; Schildknecht S; Weydt P; Witting A; Ferger B, Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson's disease. Neurochem Int 2017, 110, 14–24. [DOI] [PubMed] [Google Scholar]
- 56.Jones PD; Wolf NM; Morisseau C; Whetstone P; Hock B; Hammock BD, Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem 2005, 343 (1), 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.