

Abstract
Immune checkpoint inhibitor (ICI) therapy has achieved remarkable clinical benefit in melanoma and non-small cell lung cancer (NSCLC). Tumor mutational signatures are the fingerprints of endogenous and exogenous factors that have acted throughout tumorigenesis and heterogeneity; however, their association with immune response in ICI-treated samples remains unclear. Here, we leveraged whole-exome sequencing (WES)-based mutational profiles combined with clinicopathologic characteristics from melanoma and NSCLC datasets to examine whether tumor genomic features contribute to clinical benefit of ICI treatment. Mutational data acquired from targeted next-generation sequencing (NGS) assays (MSK-IMPACT panels) were also employed for further corroboration. A mutational signature (known as age-related clock-like processing) characterized by enrichment of C>T mutations at NpCpG trinucleotides were identified to be associated with a worse prognosis and lower tumor mutation load (TML) in both WES and targeted NGS immunotherapy cohorts. We also analyzed gene transcriptomic profiles and identified immune regulation-related gene pathways that were significantly altered in samples with different clock-like signature grouping. Leucocyte subset analysis further revealed that clock-like signature was associated with the reduction of cytotoxic cell infiltration and elevation of regulatory T cells. Overall, our work re-annotated that the age-related clock-like signature was associated with worse prognosis and lower immune activity, offering opportunities to stratify patients into optimal immunotherapy plans based on genomic subtyping.
Keywords: clock-like signature, prognosis, melanoma, NSCLC, immune infiltration, tumor mutation load
Graphical Abstract
Tumor mutational signatures are the fingerprints of endogenous and exogenous factors that have acted throughout tumorigenesis and heterogeneity. Chong et al. revealed that the clock-like mutational signature was associated with mutation load and resistance to immune checkpoint inhibitor (ICI) treatment, offering opportunities to stratify patients into optimal immunotherapy plans based on genomic subtyping.
Introduction
Immune checkpoint inhibitor (ICI) therapy, such as anti-CTLA-4 and/or anti-PD-1 agents, have revolutionized the treatment of patients with advanced-stage skin cutaneous melanoma (SKCM) and non-small cell lung cancer (NSCLC).1,2 However, the durable benefit is limited to a minority of patients due to tumor heterogeneity and diverse immunogenicity. Recent studies elucidated that cancer genomic and transcriptome features can contribute to their prediction of response to ICI; for example, higher tumor mutation burden, PD-L1 expression, neoantigen quality, and T cell-inflamed gene levels were associated with clinical benefits from immunotherapy.3, 4, 5, 6 Further studies also found that tumors surrounded with a greater level of immune cell infiltration, particularly the cytotoxic cells (CD8+ T cells, natural killer [NK] cells, etc.) had a better prognosis and treatment response.7
Genomic profiling studies of whole-exome sequencing (WES) assays and hybridization capture-based next-generation sequencing (NGS) assays (targeted-NGS panels) have been frequently utilized to identify various genetic aberrations that provide insights into the biological heterogeneity and clinical implication in melanoma and NSCLC.8 The mutational signatures are the fingerprints of endogenous and exogenous factors that have acted throughout tumorigenesis and heterogeneity.9 Substitution of C>T at TpCpW (where W = A or T) correlated with over-activity of the APOBEC(apolipoprotein B mRNA editing catalytic polypeptide-like) RNA-editing enzyme and mediated the tumor immune activity in lung cancer.10,11 Prevalent C>T mutations at CpG dinucleotide via spontaneous deamination of 5-methylcytosine was associated with aging, a risk factor for cancer development.12 Recent genomic mutation studies also elucidated potential driver genetic mutation underlying tumor immune response.4,13,14 Acquired resistance to PD-1 blockade in patients with advanced melanoma can be associated with loss-of-function mutations in the genes encoding interferon-gamma receptor-associated JAK1 or JAK2.15 There is also evidence suggesting that KRAS mutation is a genetic marker of benefit from ICIs and emerging direct inhibitors of K-Ras, which will soon be combined with immunotherapy into clinical utilization.16
Considering melanoma and NSCLC possess higher mutational frequency and various mutational footprints, exploring their association with immune infiltration and prognostic prediction will provide potential novel targets for immunotherapy. The relationship between the specific mutational signature process (e.g., CC>AA and CC>TT dinucleotide substitutions) and the origin of pathogenesis (e.g., UV exposure and tobacco smoking) have been observed in melanoma and lung cancer;11,17,18 however, the association with immune response and prognostic prediction are still ambiguous in such tumors. Recent advances suggested a negative correlation between immune infiltration and clock-like mutational signature across specific cancer types (melanoma, breast and stomach cancers) from The Cancer Genome Atlas (TCGA) datasets,19,20 indicating that the common mutational process may have an impact on tumor immunogenicity.
Since somatic mutations can be applied to clinical practice, evaluation of larger patient series is essential to identify potential tumor genomic aberrations that predict immune response and to investigate whether patients have distinct clinical outcomes. Hence, our present study aimed to characterize mutational signatures driving the molecular subtypes with genetic prognosticators and immune activity in melanoma and NSCLC patients from published multi-omic studies. We consider these findings may be applicable for prognostic prediction and therapeutic guidance for melanoma and NSCLC immunotherapy.
Results
Mutational signatures operative in aggregated melanoma immunotherapy cohort
The workflow is displayed in Figure 1. Somatic mutational profiles were acquired and uniformly analyzed from previous WES datasets of 216 melanoma patients. A median of 223 mutations per sample (range, 1–6,945) in a total of 138,182 coding somatic mutations were identified in this aggregated melanoma cohort (Figures S1A–S1C). To gain further insights into the mutational processes operative in melanoma patients, we delineated the mutation signatures from the somatic mutation data. The overall mutational pattern in melanoma was mainly dominated by C>T mutations (Figure S1D). Subsequently, we extracted three mutational signatures (i.e., signatures 1, 7, and 11) with varying mutational activities (Figure 2A; Figure S2A) and re-annotated against the Catalogue of Somatic Mutations in Cancer (COSMIC) signature nomenclature by using cosine similarity analysis (Figure S2B). The proportion and activities of the three extracted signatures in each melanoma sample are illustrated in Figure 2B and Table S1. We observed that signature 1 (clock-like mutational process, 38.7%) was dominated by C>T transitions at NpCpG trinucleotides that were associated with age-related accumulation of spontaneous deamination of 5-methylcytosine in most cancer types. The signature 7 (UV signature, 48.2%) was associated with large numbers of CC>TT dinucleotide mutations at dipyrimidines, possibly due to exposure to UV light. Signature 11 (alkylating agents signature, 13.1%), which was characterized by a strong transcriptional strand-bias for C>T substitutions, indicating that mutations occurred on guanine, exhibited a mutational pattern resembling that of alkylating agent treatment.21
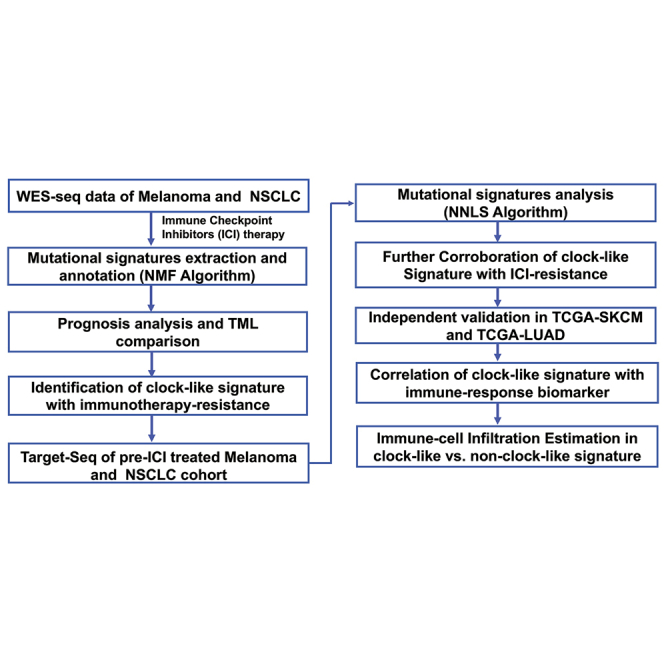
Figure 1.

The workflow of this study
The clock-like mutational signature attenuates tumor response to immune checkpoint inhibitors by contributing to exclusion of immune activation and immune infiltration in melanoma and NSCLC immunotherapy cohorts (both in WES-Seq and Target-Seq tumors).
Figure 2.

The clock-like signature is associated with the immune non-response in the ICI-treated melanoma cohort
(A) The mutational activities of corresponding extracted mutational signatures (clock-like signature, UV signature, and alkylating-agents signature, named as COSMIC signature). The trinucleotide base mutation types are on the x axes, whereas y axes show the percentage of mutations in the signature attributed to each mutation type. (B) Mutational exposures (number of mutations) were attributed to each mutation signature. (C) Kaplan-Meier survival analysis classified by mutational clock-like signature status. p value was calculated by log-rank test. (D) Multivariate Cox regression analysis of clock-like signature with age, grade, drug type, and clinical stage taken into account. (E and F) Square data markers indicate estimated hazard ratios. Error bars represent 95% CIs. Schematics of mutational proportion (E) and counts (F) of the six base substitution types (C>A, C>G, C>T, T>C, T>A, and T>G), which were classified by the clock-like signature grouping in each of melanoma samples. (G) TML was calculated and compared by clock-like mutation signature status. p value was calculated by Wilcoxon rank-sum test. Error bars represented the 95% confidence interval.
To investigate whether the extracted mutational signature was associated with immune response/resistance, we followed the previous procedure22 and stratified the extracted mutational signature into binary variables (i.e., clock-like versus non-clock-like signature: 1 versus 0) model for further analysis. Among the three signatures, we observed that patients with the clock-like signature were markedly associated with worse survival outcomes in the melanoma cohort (log-rank test, p = 0.007; hazard ratio [HR], 1.61 [95% confidence interval (CI): 1.14–2.28]; Figure 2C). This association remained statistically significant after controlling for age, sex, monoclonal antibody type, and clinical stage (HR, 1.93 [95% CI: 1.24–2.98], p = 0.003; Figure 2D). We further investigated the relationship between clock-like signature and immune response and found samples with clock-like signature were more likely clustered in a subgroup of immune resistance outcome (Fisher’s exact test, odds ratio [OR] = 3.18, p = 0.004). The distribution of six single-nucleotide variant (SNV) mutational patterns between clock-like and non-clock-like signature grouping also exhibits a significant difference; on average, the clock-like signature subset presented fewer C-to-T transitions at dipyrimidines (Figures 2E and 2F). In addition, we compared the clock-like signature with tumor mutation load (TML) and found a significantly lower TML in patients with the clock-like signature (Wilcoxon rank-sum test, log2 median TML, 5.6 versus 8.8, p < 0.001; Figure 2G).
Identify driver genes associated with clock-like signature
We combined OncodriveCLUST and MutsigCV algorithms to identify driver genes that significantly mutated in melanoma and associated with the clock-like process. First, four potential driver genes (e.g., BRAF, NRAS, HRAS, and PPP6C) in pooled melanoma datasets were identified by the two algorithms (Figure 3A). The mutational landscape of these potential tumor driver genes with different variant classification in melanoma samples is illustrated in Figures 3B and S3A. We further analyzed the association between driver gene mutation rate and clock-like signature and found that alterations in BRAF and PPP6C were more likely clustered in the non-clock-like signature subgroup (Fisher’s exact test, BRAF: p = 0.004; PPP6C: p = 0.003; Figure 3C).
Figure 3.

Melanoma driver gene mutation associated with clock-like signature status
(A) Driver genes with mutation clustering detected by OncodriveCLUST. (B) Mutational landscape of the five driver genes in the melanoma cohort with different colors indicating different variant classifications. (C) Forest plot of the gene mutation frequency associated with clock-like signature status. p value was calculated by Fisher’s exact test. Driver genes also identified by the MutsigCV algorithm are highlighted with the upper right asterisk.
Clock-like signature in pooled NSCLC immunotherapy cohort
Next, we sought to further investigate the association of the clock-like signature with immune response modulation in the NSCLC cohort. A median of 171 mutations per sample (range, 11–1,311) in a total of 70,950 coding somatic mutations were identified in this aggregated NSCLC cohort. The overall mutational pattern was mainly dominated by C>T and C>A mutations (Figure S4). We next extracted mutational signatures from lung cancer with varying mutational activities (Figures 4A and 4B; Table S2) and annotated against the COSMIC signature nomenclature (Figure S5). The extracted mutational signatures in NSCLC samples included signature 1 (clock-like signature activities, 36.5%), signature 4 (smoking signature activities, 40.9%), and signature 7 (UV signature activities, 22.6%).
Figure 4.

The clock-like signature identified the immune non-responders from the aggregated NSCLC immunotherapy cohort
(A) Mutational exposures (number of mutations) were attributed to each mutation signature. (B) The mutational activities of corresponding extracted mutational signatures (smoking signature, UV signature, and clock-like signature, named as COSMIC signature). The trinucleotide base mutation types are on the x axes, whereas y axes show the percentage of mutations in the signature attributed to each mutation type. (C) Kaplan-Meier survival analysis classified by clock-like signature status. p value was calculated by log-rank test. (D) Multivariate Cox regression analysis of clock-like signature with age, grade, drug type, and PD-L1 expression taken into account. p value was inferred by multivariate Cox regression model. (E) Tumor mutation load was calculated and compared in different clock-like mutation signature groupings. p value was calculated by Wilcoxon rank-sum test. (F) Cancer driver genes with mutation clustering detected by OncodriveCLUST. Genes also identified by MutsigCV algorithm are highlighted with the upper right asterisk. (G) Mutation landscape of the four driver genes in the NSCLC cohort. Error bars represented the 95% confidence interval.
We also observed that patients with the clock-like signature were significantly associated with worse survival prognosis (log-rank test, p = 0.003; HR, 2.01 [95% CI: 1.27–3.19]; Figure 4C) even after adjusting for age, sex, monoclonal antibody type, and PD-L1 expression (HR, 1.98 [95% CI: 1.21–3.24], p = 0.006; Figure 4D). The six SNV mutational patterns between different clock-like signature subgroups exhibited a lower proportion of C>A but a higher proportion of C>T substitutions in the clock-like signature (Figure S6). Samples with clock-like signature occurred predominantly in the immune non-response group (Fisher’s exact test, OR = 2.92, p = 0.033). In addition, we compared the clock-like signature with TML and found significantly lower TML in patients with clock-like signature (Wilcoxon rank-sum test, log2 median TML, 6.86 versus 7.71, p = 0.004; Figure 4E). KRAS was identified as a potential lung cancer driver gene in immunotherapy datasets with combined oncogene detection algorithms (Figure 4F; Figure S3B). We further analyzed and confirmed that samples with KRAS mutation were clustered in the non-clock-like signature subgroup (Fisher’s exact test, p = 0.006; Figure 4G).
Further corroboration of clock-like signature characteristics in the targeted-sequencing cohort
Currently, the majority of precision oncology platforms use NGS of targeted gene panels (e.g., MSK-IMPACT) to test the genomic mutation with immune response. To gain further insights into the mutational processes operative in targeted NGS panel datasets, we utilized the SigMA analysis tool based on the nonnegative-least-squares (NNLS) algorithm to decompose the mutation signatures from melanoma and NSCLC samples treated with ICI and whose tumors underwent MSK-IMPACT panel sequencing. The 96-type trinucleotide mutational context of 253 targeted-sequencing melanoma samples (t-melanoma) and 339 targeted-sequencing NSCLC (t-NSCLC) samples are delineated and illustrated in Figures S7A and S7B. Clock-like mutational signature coupled with homologous recombination deficiency (HRD) signature and microsatellite instability (MSI) signature were both detected in t-melanoma and t-NSCLC cohorts (Figures S7C and S7D; Tables S3 and S4). The percentage of mutational activities attributed to the clock-like signature was 17.8% in t-melanoma and 40.2% in t-NSCLC samples. We also divided the patients into clock-like versus non-clock-like signature subgroups based on the clock-like mutational contribution and analyzed the relationship with patient survival. Samples with clock-like signature were significantly associated with a worse prognosis in both t-melanoma and t-NSCLC cohorts (log-rank test, t-melanoma: p = 0.009; Figure 5A; t-NSCLC: p = 0.023; Figure 5B). This association remained significant after taking into account age, gender, drug type, and metastasis status (Cox proportional hazards model, t-melanoma: HR, 1.67 [95% CI: 1.10–2.52], p = 0.016; Figure 5C; t-NSCLC: HR, 1.39 [95% CI: 1.06–1.83], p = 0.017; Figure 5D). As TML played a vital role in predicting immune response to ICI therapy, we compared the mutational burden between clock-like and non-clock-like signature subgroups. Likewise, tumor samples with the clock-like mutational signature exhibited significantly lower TML than those without this signature (Wilcoxon rank-sum test, t-melanoma: 5.58 versus 21.64, p < 0.001; Figure 5E; t-NSCLC: 5.10 versus 10.82, p < 0.001; Figure 5F).
Figure 5.

Clock-like mutational signature extracted from the targeted NGS-panel sequencing datasets also associated with immune non-responding
SigMA analysis tool was utilized to decompose the mutational signatures. (A and B) Kaplan-Meier survival analysis was utilized to analyze clock-like mutational signature grouping in t-melanoma and t-NSCLC cohorts. p value was calculated by log-rank test. (C and D) Forest plot representation of the multivariate Cox regression model with adjustment for confounding clinical factors. p value was inferred by multivariate Cox regression model. (E and F) Tumor mutation load was assessed by clock-like mutation signature status in t-melanoma and t-NSCLC. p value was calculated by Wilcoxon rank-sum test. Error bars represented the 95% confidence interval.
Immune-related pathways and immunocyte infiltration associated with clock-like signature
We further investigated the potential mechanism behind the mutagenesis of the clock-like signature. Gene set enrichment analysis (GSEA) on melanoma gene expression (RNA) profile (GEP) against REACTOME datasets revealed that enrichment of genes involved in interferon-gamma, PD-1 signaling, immune cytokine, and nuclear factor κB (NF-κB ) pathway was significantly altered in the different clock-like signature subgroups (Figure 6A; Figure S8). To ascertain the association of the clock-like mutational signature with immune cell infiltration, we composed a heatmap with the single sample GSEA (ssGSEA) method to visualize the relative abundance of 28 infiltrating immune cell populations (Figure 6B). Anti-tumor leucocyte subsets, including effector memory CD4+, CD8+ T cells, and natural killer T cells, were markedly decreased in samples with the clock-like mutational footprint. To further validate and characterize the clock-like signature, we also performed the mutational signature analysis with Maftools package on TCGA-SKCM genomic data. Signature 7 (UV exposure), signature 11 (alkylating agents), signature 1 (Clock-like), and signature 17 (etiology unknown but previously identified in melanoma) were extracted and annotated against the COSMIC signature nomenclature (Figures S9A and S9B; Table S5). Clock-like signature accounts for 22.7% of mutational activities and stratified the melanoma samples into clock-like signature and non-clock-like subgroups (Figure S9C). Patients with clock-like signature were characterized by a significantly lower TML (Figure S9D). We also further analyzed the immune response-related biomarker in the TCGA-SKCM cohort. The T cell-inflamed GEP algorithm was utilized and found a lower inflamed score in the clock-like signature subgroup (Figure S9E). TIDE (Tumor Immune Dysfunction and Exclusion) was significantly higher in the clock-like subtype compared with the non-clock-like subtype (Figure S9F).
Figure 6.

Significantly enriched pathways and immune infiltration alteration with clock-like signature
(A) Top enriched pathways in distinct clock-like signature activity groups (0 versus 1) from the melanoma cohort were assessed by using the GSEA algorithm. (B) Single-sample gene set enrichment analysis identifying the relative infiltration of immune cell subpopulations for melanoma samples with available RNA-sequencing data. (C) The relative abundance of tumor-infiltrating leukocytes (TILs) with clock-like signature grouping in lung adenocarcinoma from TCGA datasets was estimated by the CIBERSORT algorithm. Leucocyte subsets with statistically significant differences are highlighted in red. (D) Distribution of TIDE immune response prediction scores in lung adenocarcinoma (TCGA cohort) stratified by clock-like signature grouping. (E) Distribution and association of T cell-inflamed GEP in clock-like versus non-clock-like signature subgroups. p value was calculated by Wilcoxon rank-sum test. Error bars represented the 95% confidence interval.
As gene expression data of the NSCLC immunotherapy cohort were unavailable, we curated and analyzed the genomic and transcriptomic data from the TCGA lung adenocarcinoma (TCGA-LUAD) dataset. Clock-like (36.1%), APOBEC (21.4%), and smoke signatures (42.5%) were extracted and annotated from the LUAD cohort (Figures S10A and S10B; Table S6). We further stratified the LUAD tumors into clock-like signature and non-clock-like subtype based on mutational signature activities. Intriguingly, the clock-like signature was not significantly associated with LUAD patients’ prognosis (log-rank test, p = 0.93; Figure S10C), implying its specific prediction efficacy of response to immunotherapy but not simple treatment.
Moreover, we evaluated (using the Cell Type Identification by Estimating Relative Subsets of RNA Transcripts[CIBERSORT] algorithm) the abundance of tumor-infiltrating lymphocyte in the LUAD tumor microenvironment with the GEP data. We found that CD8+ T cells, CD4+ memory-activated T cells, and macrophages M1 were less enriched in the mutational clock-like signature subgroup, whereas CD4+ memory resting T cells, monocytes, dendritic cells resting, and mast cells resting were more enriched in this subgroup (p < 0.05; Figure 6C). In addition, the tumor immune escape prediction score of TIDE was significantly higher in the clock-like subtype compared with the non-clock-like subtype (TIDE, 0.033 versus −0.006, p = 0.004; Figure 6D). The T cell-inflamed GEP algorithm also revealed a lower inflamed score in the clock-like signature subgroup (GEP scores, 2.9 versus 3.1, p = 0.036; Figure 6E). These results indicated that the clock-like mutational signature markedly associated with the exclusion of immune infiltration in melanoma and NSCLC.
Discussion
In this study, WES-based genomic meta-analysis of 216 melanoma samples and 113 NSCLC samples from previous immunotherapy cohorts was performed and delineated extracted mutational signatures and driver mutations that were associated with survival prediction and immune response. We revealed that the signature of the clock-like mutational process was associated with tumor progression and immune suppression in melanoma and NSCLC and confirmed these findings in another type of sample, whose tumors underwent NGS of targeted gene panels (MSK-IMPACT). We further identified that the clock-like signature was associated with lower lymphocyte infiltration and suppressed immune modulation process, suggesting that cancers with the clock-like signature may remodel the tumor microenvironment (TME) and escape from immune surveillance.
Immune phenotypes in determining the prognosis of various types of cancers were increasingly recognized23, 24, 25, 26 and highlighted baseline levels of tumor-infiltrating lymphocytes (e.g., CD8+, CD3+ T cells, etc.) markedly correlated with the likelihood of immune response and survival outcome.27 Our analyses also revealed that TME characterization of the clock-like mutational footprint was characterized by less immune cell infiltration, indicating tumors with this mutational signature may represent the immune “cold” status. A recent study pointed out elderly patients with lung adenocarcinoma were characterized by impaired cytolytic molecule expression and loss of clonal neoantigens and associated with the accumulation of immunosuppressive elements.28 In this research, we identified patients with aging-related mutational signatures were associated with decreased T lymphocyte infiltration and immunogenicity, further demonstrated by restrained expression of pathways related to interferon-gamma and immune cytokine signaling. Moreover, clock-like signature activities inversely correlated with the anti-CTLA-4/PD-1 therapy immune response predictor TIDE and T cell-inflamed GEP,7 suggesting the clock-like signature may predict the failure of ICI treatment.
Comprehensive knowledge of the mutated driver genes underlying human cancers is a critical foundation for cancer diagnostics, therapeutics, and selection of rational combination therapies.29 Here, we used OncodriveCLUST and MutSigCV algorithms followed by further filter criteria to re-annotate mutations and identified that tumor driver genes BRAF and PPP6C in melanoma and KRAS in NSCLC were negatively associated with the clock-like signature. Mutations in these genes were more clustered in non-clock-like signature subgroups, demonstrating its specific immune response to ICI treatment. BRAF is the most prevalent mutation gene in melanoma; an association between cancer patients’ age and mortality with distinct mutation status in BRAF V600E was observed and independent of other clinicopathologic risk factors.30,31 PPP6C, a catalytic subunit of protein phosphatase 6 (PP6), is also frequently mutated in melanoma.32 PP6 participates in the innate immune defense against viruses mediated by RIG-I signaling,33 whose mutation may cause chromosome instability and DNA damage owing to dysregulated cell cycle-regulated kinase Aurora-A.34 KRAS mutation in NSCLC is correlated with an inflammatory tumor microenvironment and tumor immunogenicity, resulting in superior response to PD-1/PD-L1 inhibitors.35 Moreover, age is reported to be associated with different subtypes of EGFR and KRAS mutations in lung cancer patients,36 and perturbations induced by Ras-mediated signaling are sufficient to deregulate the mammalian circadian clock.37
Recent advances reported that genomic mutational signatures were associated with clinical prognostic and treatment response. Trucco et al.38 classified the melanoma samples depending on the predominance of UV radiation signature and identified UV signature surrogated as prognostic for survival outcome. There was evidence suggested that the APOBEC mutational signature has potential to predict immune response for anti-PD-1 therapy in NSCLC.11 In our genomic meta-analysis, we identified that the clock-like signature was associated with shortening survival outcomes among melanoma and lung cancer patients treated with ICI, regardless of profiling the genome with whole-exome or targeted gene-panel sequencing platforms. Moreover, the clock-like signature was significantly negatively associated with TML and tumor immune infiltration. Recent studies indicated that short-term local chemotherapy may induce a more favorable tumor microenvironment and enhance the anti-PD-1 antitumor immunity.39,40 Therefore, we speculated that patients with the clock-like signature after induction treatments of controlled chemotherapy may improve the clinical benefit of ICI therapy.
The main limitation of this research was using the public dataset from different cohorts, which have somewhat heterogeneous data processing and may introduce a batch effect in the final mutation lists. In addition, RNA-sequencing (RNA-seq) results of NSCLC samples were obtained from TCGA dataset, instead of the original ICI treatment cohort, which may not fully explain the effects of immunotherapy. As a result, the association between mutational patterns and gene expression, including analysis of T-lymphocyte infiltration and oncogenic pathways, need further validation.
In this study, we assembled and characterized genomic data and clinical information from melanoma and NSCLC patients treated by ICI to determine whether the tumor genetic landscape affects the clinical benefit. This study indicated that the clock-like mutational signature was associated with prognosis of ICI treatment by integrating WES cohorts and targeted-gene panel sequencing cohorts, demonstrating the complex interplay of host and tumor in the immune response. In addition, we identified that the clock-like signature correlated with decreased TML and lessened leucocyte infiltration, further corroborating its prediction efficacy of immune resistance to ICI agents. Therefore, the clock-like signature may act as a biomarker for prognostic estimation that guides the application of immunotherapies. However, the mechanisms underlying the association between clock-like signature and the prognostic outcome are still unclear, and further studies in other cancers are warranted.
Materials and Methods
Genomic data and clinical information
Somatic mutational profiles of the WES platform were curated from previous immunotherapy studies, including a total of 216 melanoma samples41, 42, 43 and 113 NSCLC samples.4,13,14 Whole-exome capture libraries were constructed using the Agilent SureSelect All Exon V2/V4 kit and sequenced on a HiSeq 2000/2500 platform (Illumina) to generate a goal of 174× mean target coverage (range, 32–380). We re-annotated all of the previously called somatic mutations by Oncotator44 against the hg19 reference genomics database and generated a total of 138,182 and 70,950 somatic mutations belonging to melanoma and NSCLC cohorts. Gene expression data were obtained from supplementary materials of the aforementioned melanoma study (n = 117) and TCGA datasets. Genomic profile of targeted NGS assay (MSK-IMPACT, 341/410 panels) and clinical details of ICI-treated melanoma (n = 253) and NSCLC (n = 339) samples were obtained from Samstein et al.45 Detailed clinical information including age, gender, stage, drug type, lymph node status, survival time, and immune response status were also collected from these studies and are listed in Tables S1–S6. All of the extracted DNA and RNA for sequencing were obtained from the pre-treatment and primary tumor tissues. This study was approved by the Shandong Provincial Hospital Affiliated to Shandong First Medical University Institutional Review Board, which waived additional informed consent because all data used in this study were obtained from public databases. Participants in the original genomic studies provided informed consent.
Identification of cancer driver genes
We first explored the cancer driver genes by using the OncodriveCLUST46 algorithm and further validated and filtered the gene sets by MutSigCV29 algorithm. OncodriveCLUST is designed to identify cancer genes with a significant bias toward mutation clustering within the protein sequence. MutSigCV measures the significant enrichment of non-silent somatic mutations in a gene by addressing mutational context-specific background mutation rates. Candidate driver genes were required to meet these criteria: statistically significant in two algorithms (both q < 0.1 in MutSigCV and OncodriveCLUST), and expressed in the human Cancer Cell Lines Encyclopedia (CCLE, https://portals.broadinstitute.org/ccle).47
Deciphering mutational signature operative in the genome
We used R packages Maftools proposed by Mayakonda et al.48 to extract mutational signatures from the aggregated WES genomic data of melanoma and NSCLC cohorts. The ExtractSignatures function from Maftools factorized the mutation portrait matrix into two nonnegative matrices, “signatures” and “contributions,” where “signatures” represented mutational processes and “contributions” represented the corresponding mutational activities. This framework is based on Bayesian variant nonnegative matrix factorization, and it can automatically determine the optimal number of extracted mutational signatures. SigMA49 is a signature analysis tool optimized to detect the specific mutational signatures from hybrid capture panels (e.g., MSK-IMPACT assay) according to their known tumor-specific signature composition. Multivariate analysis with gradient boosting machines yields a final score for the presence of signatures, combining likelihood with cosine similarity and exposure of signature with the NNLS algorithm. Mutational signatures were annotated by calculating cosine similarity against 30 validated mutational signatures in COSMIC, Version 2.50
GSEA and network analysis
The functional pathway-level changes in samples with different clock-like signature subgroups (0 versus 1) were evaluated by the R packages limma51 and fgsea. Specifically, the expression data were background corrected and quantile normalized, and probe sets were summarized using robust multi-array average (RMA) with the affy R package. Subsequently, the normalized expression data was then fed into lmFit and eBayes functions to calculate the differential statistics with the limma package. The logFC produced by limma was used as input to perform GSEA against the REACTOME reference gene set (download from MSigDB database v7.1). The enrichment plot obtained from the fast gene set enrichment analysis algorithm was implemented in the Bioconductor R package fgsea.
CIBERSORT estimation
CIBERSORT (https://cibersort.stanford.edu/index.php)52 was used to conduct an estimation of the abundances of immune cell subsets in each sample. The machine-learning-derived algorithm uses RNA-seq data against the LM22 signature (including 22 human hematopoietic cell phenotypes) with 1,000 permutations that produce an empirical p value for the deconvolution using Monte Carlo sampling. Cases with the output of p <0.05 indicated that the inferred fractions of immune cell populations are accurate and eligible for further analysis.
Quantifying the TIDE and T cell-inflamed GEP
The TIDE algorithm53 (http://tide.dfci.harvard.edu/) uses a set of gene expression markers to estimate two distinct mechanisms of tumor immune evasion, including dysfunction of tumor infiltration cytotoxic T lymphocytes (CTL) and exclusion of CTL by immunosuppressive factors. Patients with higher TIDE prediction scores represent the greater potential of tumor immune escape; therefore, these patients would obtain poorer immunotherapy response. T cell-inflamed GEP proposed by Ayers et al.54 was used to predict clinical response to PD-1 blockade. The GEP was composed of 18 inflammatory genes associated with antigen presentation, chemokine expression, cytotoxic activity, and adaptive immune resistance.
Immune cell infiltration estimation with ssGSEA
ssGSEA55 was employed to quantify the infiltration level of 28 immune cell subsets that are over-represented in the tumor microenvironment. Special feature gene panels for each immune cell type were curated from a recent publication.7 The relative abundance of each immune cell type was represented by an enrichment score in R package GSVA. The ssGSEA score was normalized to unity distribution from 0 to 1 for each immune cell type. The bio-similarity of the immune cell filtration was estimated by multidimensional scaling (MDS) and a Gaussian fitting model.
Statistical analyses
The statistical analyses in this study were generated by R-3.6.1. The extracted mutational signatures were stratified as binary variables (i.e., 0 and 1) according to activity in the multivariate model. Quantitative data are presented as medians. The continuous variables between groups were compared by the Wilcoxon rank-sum test. The association between mutational signatures and the immune response status was tested by Fisher’s exact test. Kaplan-Meier survival analysis and Cox proportional hazards model were used to analyze the association between mutational signatures and prognosis with the R survival package (Survminer 0.4.6). All comparisons were two-sided with an alpha level of 0.05, and the Benjamini-Hochberg method was applied to control the false discovery rate (FDR) for multiple hypothesis testing.56
Acknowledgments
This study was supported by grants from Key Research and Development Program of Shandong Province, China (No. 2019JZZY010104 and No. 2019GSF108146); Academic Promotion Programme of Shandong First Medical University, China (No. 2019QL021); Special Foundation for Taishan Scholars Program of Shandong Province, China (No. ts20190978); and the Research Incubation Funding of Shandong Provincial Hospital, China. The authors thank Prof. Xiangchun Li and Dr. Yichen Yang from Tianjin Medical University Cancer Institute and Hospital for analysis coding guidance and valuable suggestions. They also thank Ms. Claire Anderson for language editing.
Author Contributions
All authors reviewed the manuscript and agreed to the submission. H.C. and W.C. designed the project. H.C., Z.W., S.J., Z.F., F.Y.D., H.W., Y.L., and W.C. provided administrative, technical, or material support. H.C., W.C., L.S., J.L., and Y.C. performed statistical analysis. W.C. and H.C. wrote the manuscript. H.C. and W.C. revised the paper.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.10.033.
Supplemental Information
References
- 1.Brahmer J., Reckamp K.L., Baas P., Crinò L., Eberhardt W.E., Poddubskaya E., Antonia S., Pluzanski A., Vokes E.E., Holgado E. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolchok J.D., Chiarion-Sileni V., Gonzalez R., Rutkowski P., Grob J.J., Cowey C.L., Lao C.D., Wagstaff J., Schadendorf D., Ferrucci P.F. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizvi H., Sanchez-Vega F., La K., Chatila W., Jonsson P., Halpenny D., Plodkowski A., Long N., Sauter J.L., Rekhtman N. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018;36:633–641. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellmann M.D., Nathanson T., Rizvi H., Creelan B.C., Sanchez-Vega F., Ahuja A., Ni A., Novik J.B., Mangarin L.M.B., Abu-Akeel M. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell. 2018;33:843–852.e4. doi: 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cristescu R., Mogg R., Ayers M., Albright A., Murphy E., Yearley J., Sher X., Liu X.Q., Lu H., Nebozhyn M. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H., Yang M., Wang Q., Song F., Li X., Chen K. The new identified biomarkers determine sensitivity to immune check-point blockade therapies in melanoma. OncoImmunology. 2019;8:1608132. doi: 10.1080/2162402X.2019.1608132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 8.Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogozin I.B., Pavlov Y.I., Goncearenco A., De S., Lada A.G., Poliakov E., Panchenko A.R., Cooper D.N. Mutational signatures and mutable motifs in cancer genomes. Brief. Bioinform. 2018;19:1085–1101. doi: 10.1093/bib/bbx049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanton C., McGranahan N., Starrett G.J., Harris R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015;5:704–712. doi: 10.1158/2159-8290.CD-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H., Chong W., Teng C., Yao Y., Wang X., Li X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci. 2019;110:2348–2356. doi: 10.1111/cas.14113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov L.B., Jones P.H., Wedge D.C., Sale J.E., Campbell P.J., Nik-Zainal S., Stratton M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anagnostou V., Smith K.N., Forde P.M., Niknafs N., Bhattacharya R., White J., Zhang T., Adleff V., Phallen J., Wali N. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017;7:264–276. doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaretsky J.M., Garcia-Diaz A., Shin D.S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D.Y., Abril-Rodriguez G., Sandoval S., Barthly L. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adderley H., Blackhall F.H., Lindsay C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine. 2019;41:711–716. doi: 10.1016/j.ebiom.2019.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connor A.A., Denroche R.E., Jang G.H., Timms L., Kalimuthu S.N., Selander I., McPherson T., Wilson G.W., Chan-Seng-Yue M.A., Borozan I. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017;3:774–783. doi: 10.1001/jamaoncol.2016.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desrichard A., Kuo F., Chowell D., Lee K.W., Riaz N., Wong R.J., Chan T.A., Morris L.G.T. Tobacco Smoking-Associated Alterations in the Immune Microenvironment of Squamous Cell Carcinomas. J. Natl. Cancer Inst. 2018;110:1386–1392. doi: 10.1093/jnci/djy060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budczies J., Seidel A., Christopoulos P., Endris V., Kloor M., Győrffy B., Seliger B., Schirmacher P., Stenzinger A., Denkert C. Integrated analysis of the immunological and genetic status in and across cancer types: impact of mutational signatures beyond tumor mutational burden. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2018.1526613. e1526613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H., Chong W., Yang X., Zhang Y., Sang S., Li X., Lu M. Age-related mutational signature negatively associated with immune activity and survival outcome in triple-negative breast cancer. OncoImmunology. 2020;9:1788252. doi: 10.1080/2162402X.2020.1788252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H., Chong W., Wu Q., Yao Y., Mao M., Wang X. Association of LRP1B Mutation With Tumor Mutation Burden and Outcomes in Melanoma and Non-small Cell Lung Cancer Patients Treated With Immune Check-Point Blockades. Front. Immunol. 2019;10:1113. doi: 10.3389/fimmu.2019.01113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efstathiou J.A., Mouw K.W., Gibb E.A., Liu Y., Wu C.L., Drumm M.R., da Costa J.B., du Plessis M., Wang N.Q., Davicioni E. Impact of Immune and Stromal Infiltration on Outcomes Following Bladder-Sparing Trimodality Therapy for Muscle-Invasive Bladder Cancer. Eur. Urol. 2019;76:59–68. doi: 10.1016/j.eururo.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahajan U.M., Langhoff E., Goni E., Costello E., Greenhalf W., Halloran C., Ormanns S., Kruger S., Boeck S., Ribback S. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2018;155:1625–1639.e2. doi: 10.1053/j.gastro.2018.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Karn T., Jiang T., Hatzis C., Sänger N., El-Balat A., Rody A., Holtrich U., Becker S., Bianchini G., Pusztai L. Association Between Genomic Metrics and Immune Infiltration in Triple-Negative Breast Cancer. JAMA Oncol. 2017;3:1707–1711. doi: 10.1001/jamaoncol.2017.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y., Yang M., Ng D.M., Haleem M., Yi T., Hu S., Zhu H., Zhao G., Liao Q. Multi-omics Data Analyses Construct TME and Identify the Immune-Related Prognosis Signatures in Human LUAD. Mol. Ther. Nucleic Acids. 2020;21:860–873. doi: 10.1016/j.omtn.2020.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 28.Gong Z., Jia Q., Chen J., Diao X., Gao J., Wang X., Zhu B. Impaired Cytolytic Activity and Loss of Clonal Neoantigens in Elderly Patients With Lung Adenocarcinoma. J. Thorac. Oncol. 2019;14:857–866. doi: 10.1016/j.jtho.2019.01.024. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen X., Zhu G., Liu R., Viola D., Elisei R., Puxeddu E., Fugazzola L., Colombo C., Jarzab B., Czarniecka A. Patient Age-Associated Mortality Risk Is Differentiated by BRAF V600E Status in Papillary Thyroid Cancer. J. Clin. Oncol. 2018;36:438–445. doi: 10.1200/JCO.2017.74.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang M., Wei H., Wang Y., Deng J., Tang Y., Zhou L., Guo G., Tong A. Targeted Disruption of V600E-Mutant BRAF Gene by CRISPR-Cpf1. Mol. Ther. Nucleic Acids. 2017;8:450–458. doi: 10.1016/j.omtn.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodis E., Watson I.R., Kryukov G.V., Arold S.T., Imielinski M., Theurillat J.P., Nickerson E., Auclair D., Li L., Place C. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan P., He L., Cui J., Qian C., Cao X., Lin M., Zhu Q., Li Y., Xing C., Yu X. Assembly of the WHIP-TRIM14-PPP6C Mitochondrial Complex Promotes RIG-I-Mediated Antiviral Signaling. Mol. Cell. 2017;68:293–307.e5. doi: 10.1016/j.molcel.2017.09.035. [DOI] [PubMed] [Google Scholar]
- 34.Hammond D., Zeng K., Espert A., Bastos R.N., Baron R.D., Gruneberg U., Barr F.A. Melanoma-associated mutations in protein phosphatase 6 cause chromosome instability and DNA damage owing to dysregulated Aurora-A. J. Cell Sci. 2013;126:3429–3440. doi: 10.1242/jcs.128397. [DOI] [PubMed] [Google Scholar]
- 35.Liu C., Zheng S., Jin R., Wang X., Wang F., Zang R., Xu H., Lu Z., Huang J., Lei Y. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95–105. doi: 10.1016/j.canlet.2019.10.027. [DOI] [PubMed] [Google Scholar]
- 36.Lee B., Lee T., Lee S.H., Choi Y.L., Han J. Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6,595 lung cancers. Oncotarget. 2016;7:23874–23884. doi: 10.18632/oncotarget.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Relógio A., Thomas P., Medina-Pérez P., Reischl S., Bervoets S., Gloc E., Riemer P., Mang-Fatehi S., Maier B., Schäfer R. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004338. e1004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trucco L.D., Mundra P.A., Hogan K., Garcia-Martinez P., Viros A., Mandal A.K., Macagno N., Gaudy-Marqueste C., Allan D., Baenke F. Ultraviolet radiation-induced DNA damage is prognostic for outcome in melanoma. Nat. Med. 2019;25:221–224. doi: 10.1038/s41591-018-0265-6. [DOI] [PubMed] [Google Scholar]
- 39.Voorwerk L., Slagter M., Horlings H.M., Sikorska K., van de Vijver K.K., de Maaker M., Nederlof I., Kluin R.J.C., Warren S., Ong S. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat. Med. 2019;25:920–928. doi: 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 40.Mathios D., Kim J.E., Mangraviti A., Phallen J., Park C.K., Jackson C.M., Garzon-Muvdi T., Kim E., Theodros D., Polanczyk M. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016;8:370ra180. doi: 10.1126/scitranslmed.aag2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., Hodi F.S., Martin-Algarra S., Mandal R., Sharfman W.H. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramos A.H., Lichtenstein L., Gupta M., Lawrence M.S., Pugh T.J., Saksena G., Meyerson M., Getz G. Oncotator: cancer variant annotation tool. Hum. Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samstein R.M., Lee C.H., Shoushtari A.N., Hellmann M.D., Shen R., Janjigian Y.Y., Barron D.A., Zehir A., Jordan E.J., Omuro A. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51:202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamborero D., Gonzalez-Perez A., Lopez-Bigas N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013;29:2238–2244. doi: 10.1093/bioinformatics/btt395. [DOI] [PubMed] [Google Scholar]
- 47.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mayakonda A., Lin D.C., Assenov Y., Plass C., Koeffler H.P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–1756. doi: 10.1101/gr.239244.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gulhan D.C., Lee J.J., Melloni G.E.M., Cortés-Ciriano I., Park P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019;51:912–919. doi: 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 50.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.