

Abstract
The cAMP pathway is known to stabilize endothelial barrier function and maintain vascular physiology. The family of cAMP-response element binding (CREB)-regulated transcription coactivators (CRTC)1–3 activate transcription by targeting the basic leucine zipper domain of CREB. CRTC2 is a master regulator of glucose metabolism in liver and adipose tissue. However, the role of CRTC2 in endothelium remains unknown. The aim of this study was to evaluate the effect of CRTC2 on endothelial function. We focused the effect of CRTC2 in endothelial cells and its relationship with p190RhoGAP-A. We examined the effect of CRTC2 on endothelial function using a mouse aorta ring assay ex vivo and with photothrombotic stroke in endothelial cell-specific CRTC2-knock-out male mice in vivo. CRTC2 was highly expressed in endothelial cells and related to angiogenesis. Among CRTC1–3, only CRTC2 was activated under ischemic conditions at endothelial cells, and CRTC2 maintained endothelial barrier function through p190RhoGAP-A expression. Ser171 was a pivotal regulatory site for CRTC2 intracellular localization, and Ser307 functioned as a crucial phosphorylation site. Endothelial cell-specific CRTC2-knock-out mice showed reduced angiogenesis ex vivo, exacerbated stroke via endothelial dysfunction, and impaired neurologic recovery via reduced vascular beds in vivo. These findings suggest that CRTC2 plays a crucial protective role in vascular integrity of the endothelium via p190RhoGAP-A under ischemic conditions.
SIGNIFICANCE STATEMENT Previously, the role of CRTC2 in endothelial cells was unknown. In this study, we firstly clarified that CRTC2 was expressed in endothelial cells and among CRTC1–3, only CRTC2 was related to endothelial function. Most importantly, only CRTC2 was activated under ischemic conditions at endothelial cells and maintained endothelial barrier function through p190RhoGAP-A expression. Ser307 in CRTC2 functioned as a crucial phosphorylation site. Endothelial cell-specific CRTC2-knock-out mice showed reduced angiogenesis ex vivo, exacerbated stroke via endothelial dysfunction, and impaired neurologic recovery via reduced vascular beds in vivo. These results suggested that CRTC2 maybe a potential therapeutic target for reducing blood–brain barrier (BBB) damage and improving recovery.
Keywords: CREB, CRTC, endothelial cells, p190RhoGAP-A, stroke
Introduction
Enhancement of angiogenesis has been found to improve neurologic outcomes after ischemic stroke (Bang et al., 2008), and trials targeting angiogenesis have been performed or are currently in progress (Sobrino et al., 2007; Osanai et al., 2018). Recently, genome analysis covering fields of ischemic stroke is developing (Dykstra-Aiello et al., 2015; Tiedt et al., 2017; Bao et al., 2018; Cai et al., 2019; Wang et al., 2019). Using RNA sequencing, the involvement of blood–brain barrier (BBB) dysfunctions in neurologic diseases; seizure, multiple sclerosis, stroke, and traumatic brain injury have suggested possible treatments of neurologic diseases by the proper regulation of BBB (Munji et al., 2019). These reports also suggest that endothelial cells, major components of BBB and related to angiogenesis, play an important role in neurologic diseases and are promising therapeutic targets.
The cAMP pathway stabilizes endothelial barrier function, and cAMP-response element binding (CREB) protein activity is positively associated with the normal vasculature maintenance (Schauer et al., 2010). Under ischemic conditions, CREB is found to be activated vascular endothelial growth factor receptor 2 (VEGFR2) in neurons and bEnd.3 cells (Lee et al., 2010), suggesting enhancing angiogenesis in endothelial cells (Xu et al., 2019).
The family of CREB-regulated transcription coactivators (CRTCs) isoforms 1–3 have provided new insights into CREB activation (Conkright et al., 2003; Iourgenko et al., 2003). CREB activates the transcription of target genes when its Ser133 is phosphorylated (Gonzalez and Montminy, 1989). Protein kinase A is activated in the cAMP-signaling pathway and phosphorylates CREB at Ser133, thus enhancing its binding to coactivators CREB-binding protein (CBP) and p300 (Vo and Goodman, 2001), which recruit basal transcription machinery to CREB-containing promoters (Goodman and Smolik, 2000). Under basal conditions, CRTCs are phosphorylated, and present in the cytoplasm. Once dephosphorylated, in response to Ca2+ and cAMP signals, CRTCs move to the nucleus (Bittinger et al., 2004; Screaton et al., 2004). CRTCs activate transcription by targeting the basic leucine zipper domain of CREB in a phospho-Ser133-independent manner.
CRTC1 is abundantly expressed in brain, and plays an important role in neuronal survival after ischemia, neurodegenerative disease, or hippocampal long-term potentiation, as shown by us and other groups (España et al., 2010; Jeong et al., 2011; Sasaki et al., 2011). CRTC2 is the most abundant isoform in the liver and pancreatic β-cells and plays an important role in the regulation of gluconeogenesis and cell survival (Koo et al., 2005). Recent studies on CRTC have focused on their functions in cancer progression (Tasoulas et al., 2019); however, no studies thus far have looked at their role in angiogenesis. Recently, CRTC2 was found to promote cell migration and invasion in lung cancer cell line A594 (Shi et al., 2018), suggesting an involvement of CRTC2 in angiogenesis.
Previously, we and other groups reported that the activation of Rho-kinase, an effector of the small GTPase Rho family protein (Amano et al., 1996; van Nieuw Amerongen et al., 1998; Mehta and Malik, 2006; Spindler et al., 2010), in endothelial cells was observed in early phase of ischemia and led to microcirculatory disturbance and aggravated cerebral ischemia (Yagita et al., 2007). The Rho-kinase inhibitor Fasudil has been shown to partly improve patient's clinical outcome in a prospective double-blind trail (Shibuya et al., 2005), suggesting that the suppression of Rho signaling is a promising therapeutic target for cerebral ischemia.
p190RhoGAP is a negative regulator RhoA (Harrington et al., 2004; Holinstat et al., 2006), and its expression is regulated by CREB in endothelial cells (Chava et al., 2012); however, no direct evidence has not been reported for the involvement of p190RhoGAP and CRTC2 in alleviation of brain damages after ischemia.
In the present study, we examined the function of CRTC family in endothelial cells in vitro and analyzed the relationship of CRTC2 with p190RhoGAP-A. To examine more directly the role of CRTC in endothelial cells, we performed ex vivo aorta ring assay and in vivo experimental ischemic stroke using endothelial cell-specific CRTC2-knock-out mice.
Materials and Methods
Cell culture and transfection
Bovine brain microvasculature endothelial cells (BBMCs), bEnd.3 cells, and human umbilical vein endothelial cells (HUVECs) were used in this study. Cells in passages 4–8 were used for the experiments, and cells were cultured for 24 h at 37°C in a humidified atmosphere of 95% air and 5% CO2. BBMCs were plated onto a dish coated with collagen in medium (RPMI 1640, Wako) containing 10% fetal bovine serum (FBS; Invitrogen), 1% antibiotic-antimycotic (Invitrogen), and 10 ng/ml basic FGF (Roche Applied Science). The bEnd.3 cells were plated in a dish in medium (DMEM; WAKO) containing 10% FBS, 1% antibiotic-antimycotic, and 1% MEM non-essential amino acid Solution (NEAA; Thermo Fisher Scientific). HUVECs were plated onto a dish coated with collagen in medium (Humedia-EG; Kurabo). We also prepared mouse cortical neuron culture, as previously described (Sasaki et al., 2011).
An adenovirus-mediated reporter system was prepared. BBMCs, HUVECs, or bEnd.3 cells were transfected with purified adenovirus, including CRTC1–3 or control (EGFP), or miRNA targeting CRTC1–3 or control, and evaluated after 4–7 d.
Mouse primary endothelial cell culture
Mouse primary endothelial cell culture were prepared from the brain of three-week-old male endothelial cell-specific CRTC2-knock-out mice CRTC2flox/flox/Cre (CRTC2flox/flox/Cre+) mice or CRTC2 CRTC2flox/flox/Cre– mice. Removed the membranes and the blood vessels on the surface of the brain of the mouse, and shred tissue with scissors. The cells were dissociated with collagenase type 2 (1 mg/ml)-DMEM (papain dissociation system; Worthington) with DNase, and the cells were incubated at 37°C for 90 min with shaking. Added 10 ml of DMEM and centrifuged at 1000 × g for 10 min. Aspirated the supernatant, added 20 ml of 20% BSA-DMEM, and centrifuged at 1000 × g for 10 min. Pipetted off upper layer and aspirated middle layer, and then transferred the pellet to a 50-ml tube containing DMEM. Add 1 mg/ml collagenase/dispase (Sigma) with DNase to the mixture, pipetted, and incubated at 37°C for 45 min with shaking. Added 10 ml of DMEM and centrifuged at 1000 × g for 10 min. Aspirated the supernatant and added 1 ml of DMEM to suspend. Gently injected this suspension onto Percoll and centrifuged at 1000 × g for 10 min. Collected only the endothelial cell layer in the middle layer and poured into a tube containing 10 ml of DMEM. Then, centrifuged at 1000 × g for 10 min, washed once, and suspended in 2.5 ml of DMEM containing 10% FBS. Centrifuged for 10 min again, and cell were cultured in high-glucose DMEM/F-12 (Sigma) containing 10% fetal calf serum (FCS; Invitrogen), L-glutamine, insulin-transferring-selenium supplement (100×; Sigma), basic FGF (1.5 ng/ml; R&D), heparin (1 mg/ml; Sigma), 100-IU/ml penicillin, and 100 μg/ml streptomycin sulfate plated onto 60-mm dishes (Falcon, Becton Dickinson) coated with a mixture of Collagen Type IV (Sigma), fibronectin (Sigma), and sterile water at a ratio of 8:1:1, filtered through a 0.45-μm filter. The cells were cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2 and used after 7–10 d in vitro.
Tube-like capillary formation assay
The tube-like capillary formation assay was performed on Matrigel (Corning). Wells of four-well culture plates were coated with 100 µl of Matrigel and then seeded with cells (1 × 105 cells/ml) in reduced serum (5% FBS). After 18 h, tube-like capillary formation was observed. The tube length was determined by imaging software (ImageJ v1.44; National Institutes of Health).
Migration assay
After reaching confluency, a wound was generated by scratching with 200-µl tips. At 0 and 18 h after the scratch, the width of the wound was determined using ImageJ.
Proliferation assay
A proliferation assay was performed with a cell proliferation kit (Biological Industries Israel Beit Haemek Ltd.).
Permeability assay
A permeability assay was performed using cell culture inserts, as previously reported (Kanki et al., 2019). Briefly, HUVECs (1 × 104 cells/well) were seeded on the Sarsted TC-Inserts for 24 wells (Sarsted). Next day, we transfected each adenovirus and after 7 d we measured permeability. First, the medium of the insert was removed and phenol red-free medium containing lucifer yellow (1:1000, WAKO) was added, and absorbance was measured after 1 h (excitation 432 nm, emission 536 nm).
Western blotting
Cell samples were collected in lysis buffer [20 mm Tri-HCl, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, protease inhibitor cocktail (Complete Mini; Roche Applied Science) and phosphatase inhibitor cocktail (PhosSTOP; Roche Applied Science)], and centrifuged at 15,000 rpm for 20 min at 4°C. The supernatant was evaluated by Western blotting. Samples were also isolated from the mouse cortex. The protein (10 µg) was subjected to electrophoresis in a 7.5% Tris-HCl polyacrylamide gel, and then transferred to a polyvinylidene difluoride membrane (Immobilon P; Millipore). Blots were probed with the appropriate antibody and detected using a sheep anti-mouse or donkey anti-rabbit horseradish peroxidase-conjugated secondary antibody (GE HealthcarePharmacia Biotech), followed by enhanced chemiluminescence (GE HealthcarePharmacia Biotech). Digital images were obtained by densitometric scans of autoradiographs.
Antibodies
Antibodies against CRTC1 and CRTC3 (CST), CRTC2 (Proteintech Group), p190RhoGAP-A (CST), p190RhoGAP-B (CST), Rho-associated protein kinase (ROCK)1 (Abcam) were used at a 1:1000 dilution for the Western blot analysis and 1:500 dilution for immunohistochemistry. β-Actin (Sigma-Aldrich) was used at a 1:1000 dilution for Western blot analysis. Antibody against Drebrin (Wako) was used at a 1:500 for immunohistochemistry. FITC conjugate Lycopersicon esculentum lectin (Tomato Lectin; Vector Laboratories) was used at 1:500 dilution for the immunohistochemistry. We made antibodies targeting for each phosphate site Ser171, Ser275, and Ser307 in our laboratory, as previously described (Uebi et al., 2010). We used these phospho-CRTC2 antibodies at a 1:250 for Western blot analysis.
Quantitative real-time PCR analysis
Total RNA was extracted using the RNAqueous-4 PCR kit (Thermo Fisher Scientific). The cDNA was prepared by reverse transcription from 1 µg of total RNA using Superscript III and random primers (Invitrogen). The resultant cDNA was used for real-time PCR analyses with Platinum SYBR Green qPCR SuperMix (Invitrogen). One-hundredth of the reverse transcription products and standard plasmids were subjected to real-time PCR analysis (ABI Prism 7900 Sequence Detector System; Thermo Fisher Scientific) using human 36B4 as an internal control. The PCR program was as follows: 10 min of denaturation at 95°C, and then 40 cycles at 95°C for 15 s, 58°C for 30 s, and 72°C for 30 s. The specific primers used are listed in Table 1.
Table 1.
The primer sequences for qPCR
| Human 36B4 forward CCAACTGTTGCATCAG |
| Human 36B4 reverse GCCACAAAGGCAGATG |
| Human CRTC1 forward GCGCTGCACAATCAGAAGCA |
| Human CRTC1 reverse ATGGTGCCACTCCCGATCTG |
| Human CRTC2 forward TCCCCACACCTTCAACCACC |
| Human CRTC2 reverse GCTGCTGACACCTCAAAGCC |
| Human CRTC3 forward TCCAGCACCAGCCTGTTCAA |
| Human CRTC3 reverse TGCAGCTCCTCTTCCAGTGG |
| Mouse 36B4 forward TGTGTGTCTGCAGATCGGGT |
| Mouse 36B4 reverse TGGATCAGCCAGGAAGGCCT |
| Mouse CRTC1 forward CACCTGGCTCCTCTCC |
| Mouse CRTC1 reverse AGCTGCTGCTCCAGAG |
| Mouse CRTC2 forward TGACCTCACCAACCTG |
| Mouse CRTC2 reverse GTGAGTCATGGTGTGG |
| Mouse CRTC3 forward CAACATCCCAGCTGCT |
| Mouse CRTC3 reverse GATGCGTTGGGAACAG |
Oxygen glucose deprivation (OGD)
OGD was performed by placing cultures in a 37°C incubator housed in an anaerobic chamber as previously described (Sasaki et al., 2011).
Immunohistochemistry
Endothelial cells were fixed immediately with 4% paraformaldehyde for 30 min. Mouse brains were removed and were immediately freezing. Then, 14 μm thick brain slide were prepared and the slide was fixed with 1% paraformaldehyde for 10 min. For double immunofluorescence, the slides with endothelial cells or mouse brain were incubated in PBS with 0.1% Triton X-100 at room temperature for 30 min. The slides were incubated in TBS containing 10% normal donkey serum at room temperature for 30 min. Sections were incubated with the appropriate primary antibody overnight at 4°C. For mouse primary endothelial cells, we used Can Get Signal Immunostain Solution A (TOYOBO). Subsequently, secondary donkey antibodies conjugated to FITC or rhodamine (1:200; Invitrogen) were used for 90 min at room temperature. Sections were then mounted with Vectashield (Vector Laboratories) and visualized or photographed with a confocal microscope (LSM-710; Zeiss) or a fluorescence microscope (BZ-X 800; Keyence).
Dual-Glo luciferase assay
Endothelial cells were infected with the purified adenovirus [adeno-GAL4-fusion CREB, CREB b-ZIP (−), CRTC1, CRTC2, CRTC3, or p300, and the Adeno-5×GAL4-luciferase reporter system (firefly) and adeno-TK-Luc (Renilla)]. The adenovirus containing a CMV promoter driving Renilla luciferase was used as an internal control (Promega) as previously described (Katoh et al., 2004, 2006).
Promoter-luciferase constructs
VEGF-A(−1853), VEGF-R1(−1160), VEGF-R2(−220), VEGF-R2(−780), VEGF-R3(−870), and p190RhoGAP promoter constructs were constructed in our lab. The PCR-amplified fragment spanned the region from −2950 to −1 relative to the translational start site of the human p190RhoGAP protein (Chava et al., 2012). We used mouse or human genomic DNA as the template for the PCR. Luciferase reporter genes were generated by inserting each promoter fragment upstream of the luciferase gene in pGL4 Basic (Promega), using the SacI and HindIII sites of the vector. Because plasmid-based reporters could not provide an adequately strong signal in endothelial cells, we prepared an adenovirus-mediated reporter system. Endothelial cells were transfected with each adeno luciferase-construct, and an adeno TK-pRL construct was used as an internal control. At 48 or 96 h after transfection, the cells were subjected to a luciferase assay.
Mice
The CRTC2-knock-out mice were a generous gift from Marc Montminy (The Salk Institute for Biological Studies, CA; Wang et al., 2010). CRTC2-Tek-Cre mice with endothelial CRTC2 deficiency were generated by crossing Tie2-Cre mice (C57BL/6J background) that express Cre recombinase specifically in the endothelium (C57BL/6-Tg(Tie2-Cxcl12)1Tng; RIKEN) with CRTC2flox/flox mice (from Kaestner KH, Perelman School of Medicine, University of Pennsylvania; Le Lay et al., 2009).
Mouse aorta ring assay and immunofluorescence
This assay was performed using Matrigel. We used eight-week-old male endothelial cell-specific CRTC2-knock-out mice CRTC2flox/flox/Tie2-Cre (CRTC2flox/flox/Cre+ mice) and the descending aorta. One millimeter of the descending aorta was cut and seeded in Matrigel, and then assessed for angiogenesis at 7 d later. Next, we performed immunofluorescence targeting for spouting out sites with aorta ring assay as previously described (Baker et al., 2011).
Photochemically induced thrombosis (PIT) model
Adult male mice (older than eight weeks) were anesthetized by isoflurane in an induction chamber in 100% oxygen. The skin along the midline of the scalp, from the eye level down to the neck, was incised and the skull was exposed. Photothrombotic vascular occlusion was induced by intraperitoneal injection with 0.2 ml of rose bengal (10 mg/ml) for 5 min. Next, 30 min of illumination was applied to the exposed skull using a cold light source (4.5-mm diameter fiber optic end; Hamamatsu Photonics K.K.) placed 2.2 mm lateral left of the bregma. The illuminated area included the motor (M1) and somatosensory (S1) cortices. After 30 min of illumination, the skin of the scalp was closed using surgical cyanoacrylate glue.
Determination of infarct volume
2,3,5-Triphenyltetrazolium chloride (TTC) staining was used to evaluate infarct volume, as previously described (Sasaki et al., 2011).
Behavior experiments
The rotarod test, cylinder test and grid walking test were used to evaluate neurologic deficit. (1) A rotarod test was performed as described previously (Lee et al., 2019). The time at which the mouse fell off the drum (latency to fall) was recorded. Mice were tested for three trials, and the mean time was calculated. (2) A cylinder test was performed, as previously reported (Monai et al., 2019). For each test, a mouse was placed inside the cylinder for 5 min and evaluated for lateralization of limb usage preference: R/L preference = [(R + both) – (L + both)]/[(R + both) + (L + both)]. (3) Grid walking test was performed as previously reported (Stokowska et al., 2017). For each test, a mouse was placed on the grid for 5 min, and the foot fault rate of the left forelimb was evaluated (foot fault/total foot × 100). We excluded the mice showing <30 steps for 5 min.
Blood glucose level
We measured blood glucose level using FORA (Research & Innovation Japan).
Enzyme activities and electrolytes
Mice were euthanized and blood sampling was performed by right ventricular injection. All enzymatic activities and electrolytes were measured using Comprehensive Diagnostic Profile (Abaxis).
In vivo model for analysis of BBB permeability
Mice were injected with 100 µl of 4% Evans Blue (Sigma-Aldrich) intravenously through the tail vein 24 h after PIT. After 1 h, the brain was removed. Images were obtained by microscopy (SZ-12; Olympus). Then, brain was separated into hemispheres ipsilateral and contralateral to the PIT lesion. Next, each hemisphere supplemented with 500 µl of foramide and transferred to a 55°C heat block and incubated for 24 h to extract Evans Blue from tissues. The foramide/Evans Blue mixture was centrifuged to pellet any remaining tissue fragments and absorbance was measured at 610 nm; 500 µl of formamide was used as a blank and Evans Blue extravasated per gram of tissue was determined.
Statistical analysis
The majority of data are presented as mean ± SEM or mean ± SD. Statistical analyses were performed using statistical software (JMP Pro V14.0.0). Statistical comparison of the means between the two groups was performed using a two-tailed unpaired Student's t test. Multiple group comparisons were performed using one-way ANOVA, followed by Tukey−Kramer post hoc analysis. Statistical significance was defined as p < 0.05.
Study approval
The animal experimental protocol was approved by the Institutional Animal Care and Use Committee of Osaka University Graduate School of Medicine.
Results
CRTC2 is expressed in endothelial cells and activated under ischemic conditions in endothelial cells
CRTC1 is enriched in the brain, while CRTC2 is expressed in the liver, spleen, and lymph nodes (Dormond et al., 2008; Liu et al., 2008; Hernandez et al., 2015). First, we examined the expression of CRTC1 and CRTC2 in the mouse brain. The adult-type drebrin is reported to be localized in the dendritic spines of rat forebrain neurons (Hayashi et al., 1996), then we performed immunohistochemical double staining for drebrin (a neuronal marker) or L. esculentum lectin (a blood vessel marker) and CRTC1 or CRTC2 in the mouse brain. CRTC1 was predominantly expressed on drebrin positive cells (Fig. 1A) and CRTC2 was less expressed on drebrin-positive cells (Fig. 1B). However, CRTC1 was not expressed in L. esculentum lectin-positive areas (Fig. 1C) and CRTC2 was mainly expressed in L. esculentum lectin-positive areas (Fig. 1D).
Figure 1.
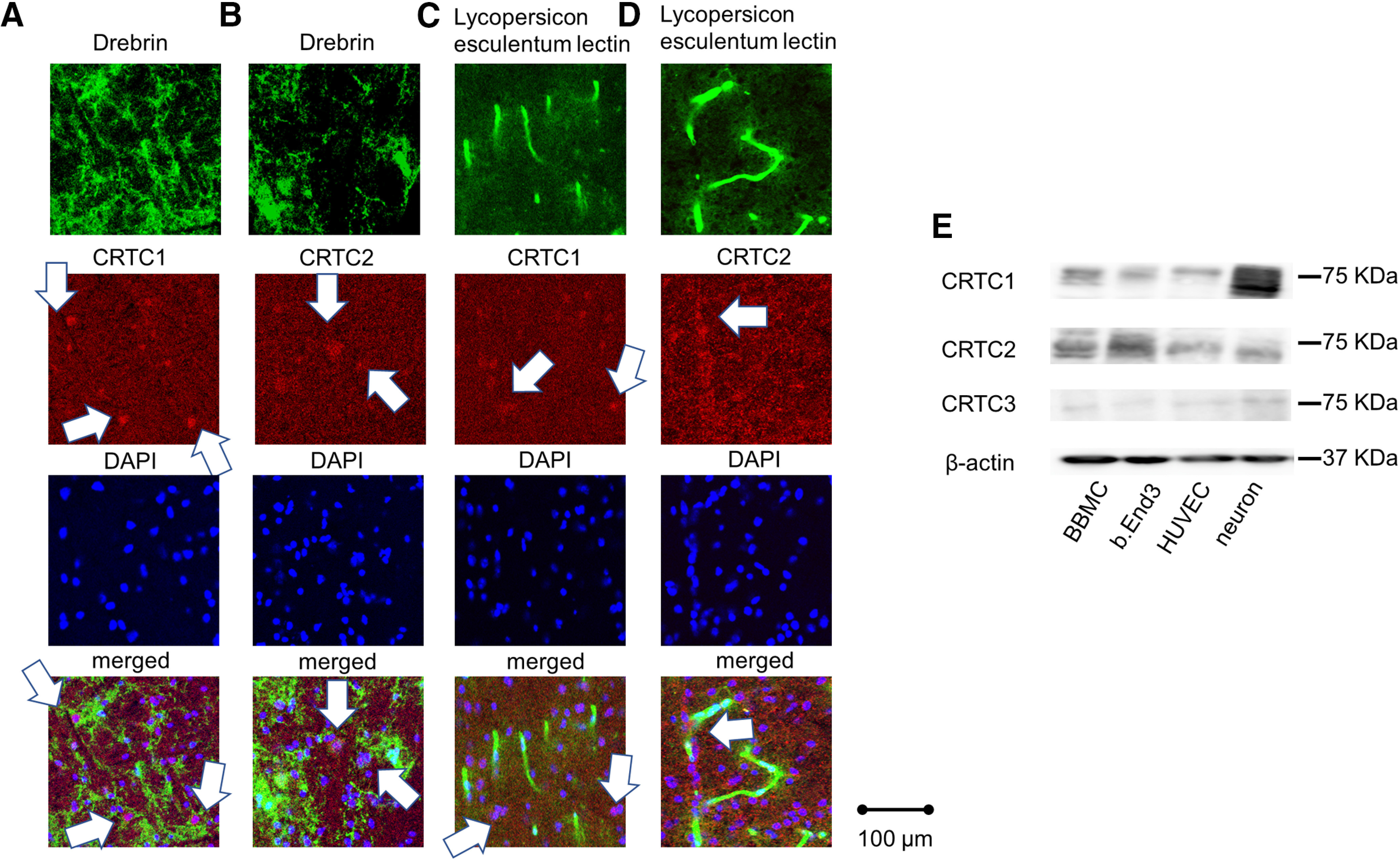
CRTC2 is expressed in endothelial cells. A, Drebrin staining following co-staining of endogenous CRTC1 in the C57B6 mouse cortex via immunofluorescence. B, Drebrin and CRTC2 visualized by immunofluorescence in the C57B6 mouse cortex. C, L. esculentum lectin and CRTC1 visualized by immunofluorescence in the C57B6 mouse cortex. D, L. esculentum lectin and CRTC2 visualized by immunofluorescence in the C57B6 mouse cortex. Scale bar: 100 µm. E, The expression of CRTC1–3 at BBMCs, bEnd.3 cells, HUVECs, or mouse primary culture neurons using Western blot analysis were shown.
Next, we examined the expression levels of CRTC1−3 in endothelial cells, including BBMC, bEnd.3 cells, and HUVECs compared with mouse primary cortical neurons by Western blot analysis. Western blotting showed prominent expression of CRTC2 in BBMCs, bEnd.3 cells, and HUVECs (Fig. 1E). The expression of CRTC1 was most abundant in mouse primary cortical neurons, as previously reported (Sasaki et al., 2011) and also detected in each endothelial cell, while only faint expression of CRTC3 was observed (Fig. 1E). These results also showed that CRTC2 was expressed in endothelial cells.
Then, we examined the intracellular distribution of CRTC1, CRTC2, or CRTC3 using a GFP fusion protein. We found that under basal conditions, endogenous CRTC1 and CRTC2 predominantly localized in the cytoplasm of HUVECs, whereas CRTC2, but not CRTC1, was translocated into the nucleus after OGD-reoxygenation (Fig. 2A,B; Takemori et al., 2009). We also found that CRTC3 predominantly localized in the nucleus under both basal conditions and ischemic conditions (Fig. 2A). Additionally, we examined the expression levels of CRTC family after OGD treatment by quantitative PCR. We found that only CRTC2 was significantly increased compared with controls (no treatment) after OGD treatment in HUVECs (Fig. 2C–E). We also performed quantitative PCR using bEnd.3 cells for CNS endothelial cells targeting for CRTC family. CRTC3 was almost not detected by quantitative PCR in bEnd.3 cells. Therefore, we determined that the expression of CRTC3 was very low in bEnd.3 cells. The results in bEnd.3 cells showed that only CRTC2 was more increased with controls after OGD treatment for CNS endothelial cells (Fig. 2F,G). These results showed that CRTC2 is expressed in endothelial cells and that only CRTC2 is activated under ischemic conditions in endothelial cells. Based on these findings, we examined the importance of CRTC2 in endothelial cell function.
Figure 2.
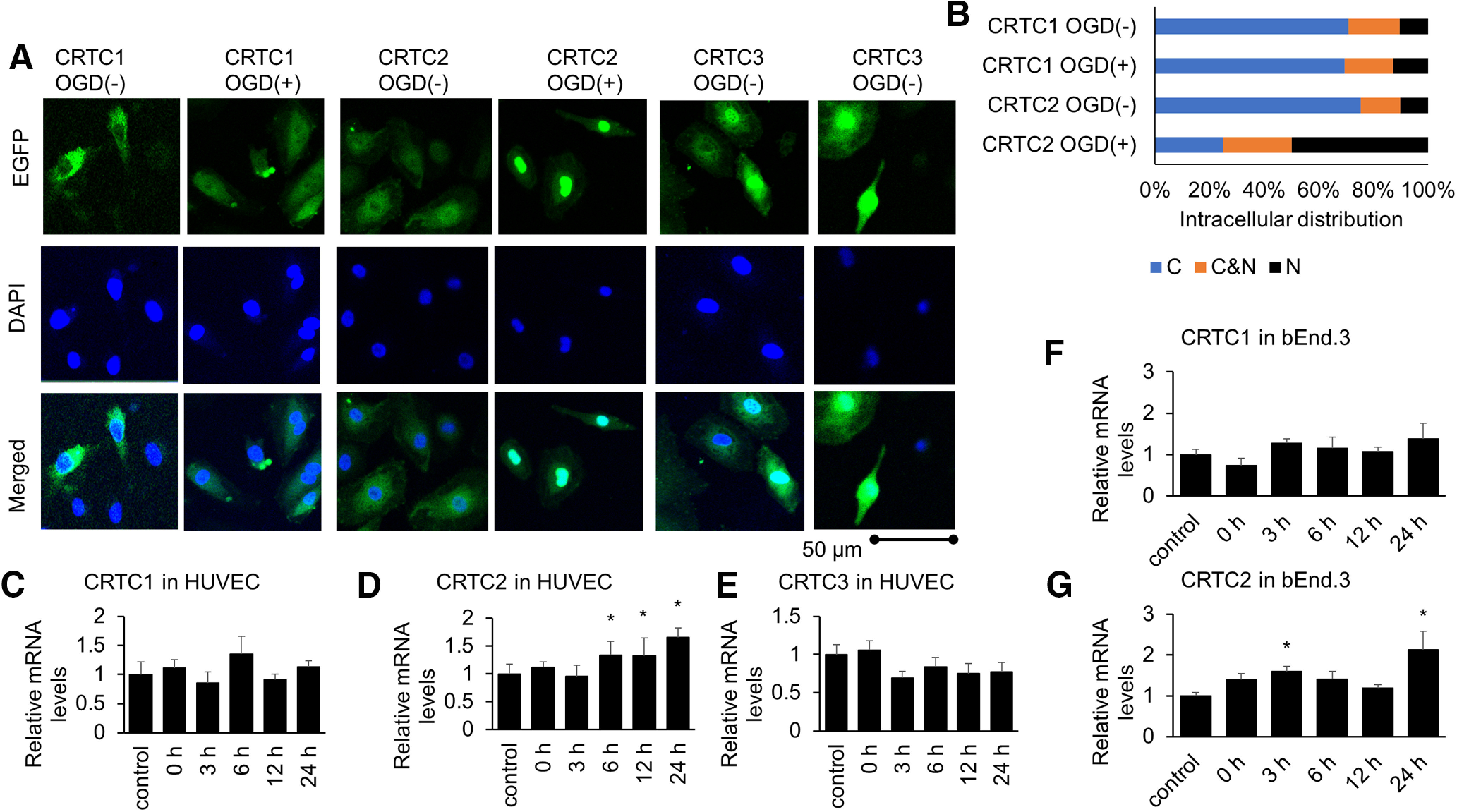
Among CRTC1–3, only CRTC2 is activated under ischemic conditions in endothelial cells. A, Endogenous CRTC1, CRTC2, or CRTC3 protein visualized by immunofluorescence in HUVECs under basal conditions and after OGD treatment. DAPI was used to stain the nucleus. Each CRTC (CRTC1, CRTC2, or CRTC3)/DAPI in HUVECs was shown. Scale bar: 50 µm. B, HUVEC cells were classified into three categories located in cytoplasma (C), in both cytoplasma and the nucleus (C&N), and in the nucleus (N) under normal or ischemic conditions. C–E, The mRNA levels of CRTC1−3 in HUVECs using quantitative PCR (qPCR) at the indicated periods after OGD treatment (n = 5–8). F, G, The mRNA levels of CRTC1 and CRTC2 in bEnd.3 cells using qPCR at the indicated periods after OGD treatment (n = 5 or 6). Error bars represent mean ± SEM. For C–G, *p < 0.05 compared with control (no treatment) by two-tailed unpaired Student's t test. C, Control = 1 ± 0.22, n = 6; 0 h = 1.11 ± 0.15, n = 7, p = 0.6456; 3 h = 0.85 ± 0.19, n = 8, p = 0.5107; 6 h = 1.45 ± 0.15, n = 5, p = 0.0905; 12 h = 0.91 ± 0.1, n = 5, p = 0.7347; 24 h = 1.13 ± 0.11, n = 5, p = 0.6032. D, Control = 1 ± 0.08, n = 5, 0 h = 1.45 ± 0.13, n = 7, p = 0.1957; 3 h = 1.24 ± 0.26, n = 8, 0.4652; 6 h = 1.85 ± 0.2, n = 5, p = 0.0409; 12 h = 1.87 ± 0.4, n = 6, p = 0.0240; 24 h = 2.14 ± 0.22, n = 5, p = 0.0038. E, Control = 1 ± 0.13, n = 6; 3 h = 1.06 ± 0.12, n = 7, p = 0.6783; 3 h = 0.69 ± 0.09, n = 8, p = 0.0527, 6 h = 0.89 ± 0.09, n = 5, p = 0.5549; 12 h = 0.75 ± 0.13, n = 5, p = 0.1966. F, Control = 1 ± 0.13, n = 6; 0 h = 0.74 ± 0.17, n = 6, p = 0.2885; 3 h = 1.28 ± 0.11, n = 5, p = 0.4320; 6 h = 1.15 ± 0.28, n = 5, p = 0.7334; 12 h = 1.07 ± 0.11, n = 6, p = 0.9533; 24 h = 1.38 ± 0.38, n = 5, p = 0.2715. G, Control = 1 ± 0.08, n = 6, 0 h = 1.39 ± 0.15, n = 6, p = 0.1974; 3 h = 1.60 ± 0.13, n = 6, 0.0477; 6 h = 1.41 ± 0.19, n = 6, p = 0.1697; 12 h = 1.19 ± 0.08, n = 6, p = 0.5570; 24 h = 2.13 ± 0.45, n = 5, p = 0.0007.
CRTC2 enhanced endothelial function in vitro
To examine the effect of CRTC2 on endothelial function, we performed overexpression of CRTC1, CRTC2, CRTC3, or EGFP (control) and knock-down of CRTC1, CRTC2, or CRTC3 compared with control (miR-Cont) using adenoviral-mediated gene transduction. Importantly, only overexpression of CRTC2 significantly reduced paracellular permeability (CRTC1: p = 0.2120; CRTC2: p = 0.0002; CRTC3: p = 0.8728; Fig. 3A). Also, only knock-down of CRTC2 was increased paracellular permeability (CRTC1: p = 0.9844; CRTC2: p = 0.0464, CRTC3: p = 0.9877; Fig. 3B). These results directly showed that CRTC2 enhanced endothelial function in vitro. Next, we examined the effect of CRTC2 on angiogenesis performed overexpression of CRTC2 (Fig. 3C) or knock-down of CRTC2 (Fig. 3D) using adenovirus system. Overexpression of CRTC2 enhanced tube-like capillary formation compared with EGFP (p < 0.001; Fig. 3E,F) and knock-down of CRTC2 (miR-CRTC2) led to reduced tube length compared with controls (miR-Cont; p = 0.0484; Fig. 3G. Additionally, overexpression of CRTC2 also significantly potentiated migration (p < 0.001; Fig. 3H,I) and knock-down of CRTC2 also led to reduced migration (p = 0.0023; Fig. 3J). However, overexpression of CRTC2 did not alter endothelial cell proliferation (p = 0.1963; Fig. 3K). Additionally, to investigate the role of CRTC2 under ischemic conditions, we performed permeability assay under ischemic conditions. Under ischemic conditions, the treatment of OGD increased paracellular permeability, but overexpression of CRTC2 reduced paracellular permeability under ischemic conditions (Fig. 3L) and knock-down of CRTC2 aggravated paracellular permeability under ischemic conditions (Fig. 3M). These findings suggest that CRTC2 plays an important role in angiogenesis in vitro.
Figure 3.
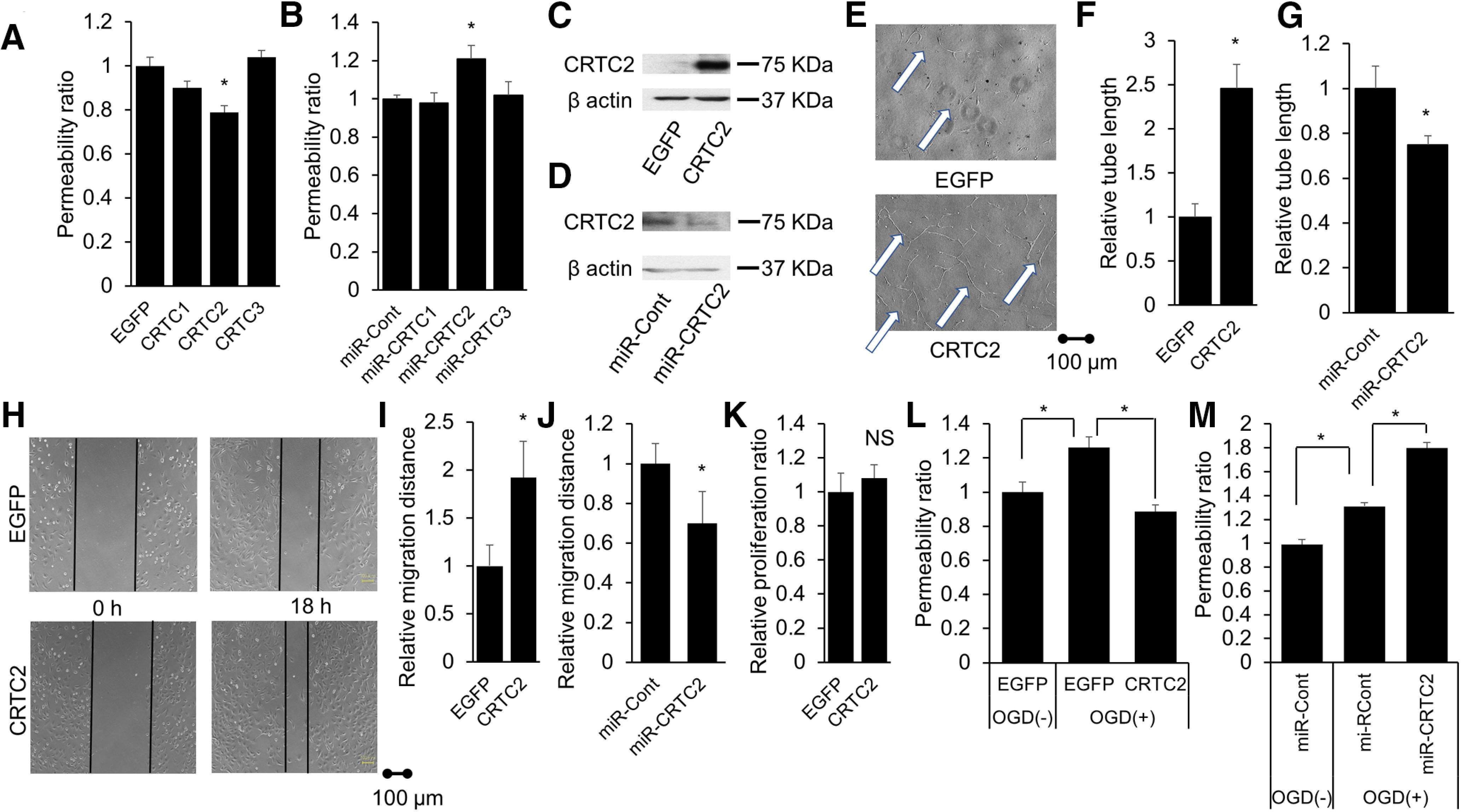
CRTC2 plays an important protective role in endothelial function. A, Quantitative results of the relative permeability ratio (n = 6–10). B, Quantitative results of the relative permeability ratio (n = 9). C, The expression level of CRTC2 in BBMCs overexpression of CRTC2 compared with those transfected with EGFP (control) is shown. D, The expression level of CRTC2 in BBMCs transfected with LacZ-specific (miR-Cont) or CRTC2-specific (miR-CRTC2) miRNAi-expressing adenovirus is shown. E, Representative image of tube-like capillary formation in Matrigel by BBMCs transfected with EGFP or CRTC2. Scale bar: 100 µm. F, Quantitative results of the relative tube length (n = 6). G, Quantitative results of relative tube length (n = 6). H, Representative migration assay image of BBMCs. Scale bar: 100 µm. I, Quantitative results of the relative migration distance (n = 5). J, Quantitative results of relative migration distance (n = 6). K, Quantitative results of the relative proliferation ratio (n = 6). L, Quantitative results of the relative permeability ratio under ischemic conditions (n = 5 or 6). M, Quantitative results of the relative permeability ratio under ischemic conditions (n = 5 or 6). Error bars represent mean ± SEM. For A, B, L, M, *p < 0.05 by one-way ANOVA, followed by Tukey−Kramer post hoc analysis. For F, G, I, J, *p < 0.05 compared with EGFP by two-tailed unpaired Student's t test. For K, NS indicates no significant change by two-tailed unpaired Student's t test.
CREB-CRTC signaling facilitated VEGF pathway
In Figure 4, we examined the involvement of the CREB-CRTC pathway in VEGF-A and kinase insert domain receptor (KDR) in vascular endothelial cells. CRTC1 and CRTC2 is differently activated by KCl or Forskolin. To clarify which KCl or Forskolin is the activator of CRTC1/2 in endothelial cells, we performed transactivation assay using Forskolin and KCL. First, CRTC1 is most activated by co-stimulation with KCL and Forskolin, as previously reported (Ch'ng et al., 2012). Furthermore, CRTC1 translocates into the nucleus by KCl stimulation (Ch'ng et al., 2012). We also demonstrated that Ca2+ influx via NMDA receptors, especially NR2A, causes activation of CTRC1. That is, CRTC1 is activated by KCl or Ca2+ stimulation (Sasaki et al., 2011). On the other hand, it has also been reported that CRTC2 is more strongly activated in Forskolin than in KCl (Blanchet et al., 2015) and CRTC2 is less activated by KCl alone (Blanchet et al., 2015). These results suggested that CRTC1 is KCl or Ca2+ dependent, while CRTC2 is cAMP dependent. Our results showed that activation of CRTC2 by Forskolin was stronger than activation of CRTC1 by co-treatment of KCl and Forskolin in endothelial cells (Fig. 4A). VEGF-A is a representative cytokine derived from vascular endothelium and involved in angiogenesis. VEGF-A was also activated by Forskolin but not KCl (Fig. 4B). These results also showed that cAMP pathway is important for CRTC2 pathway.
Figure 4.
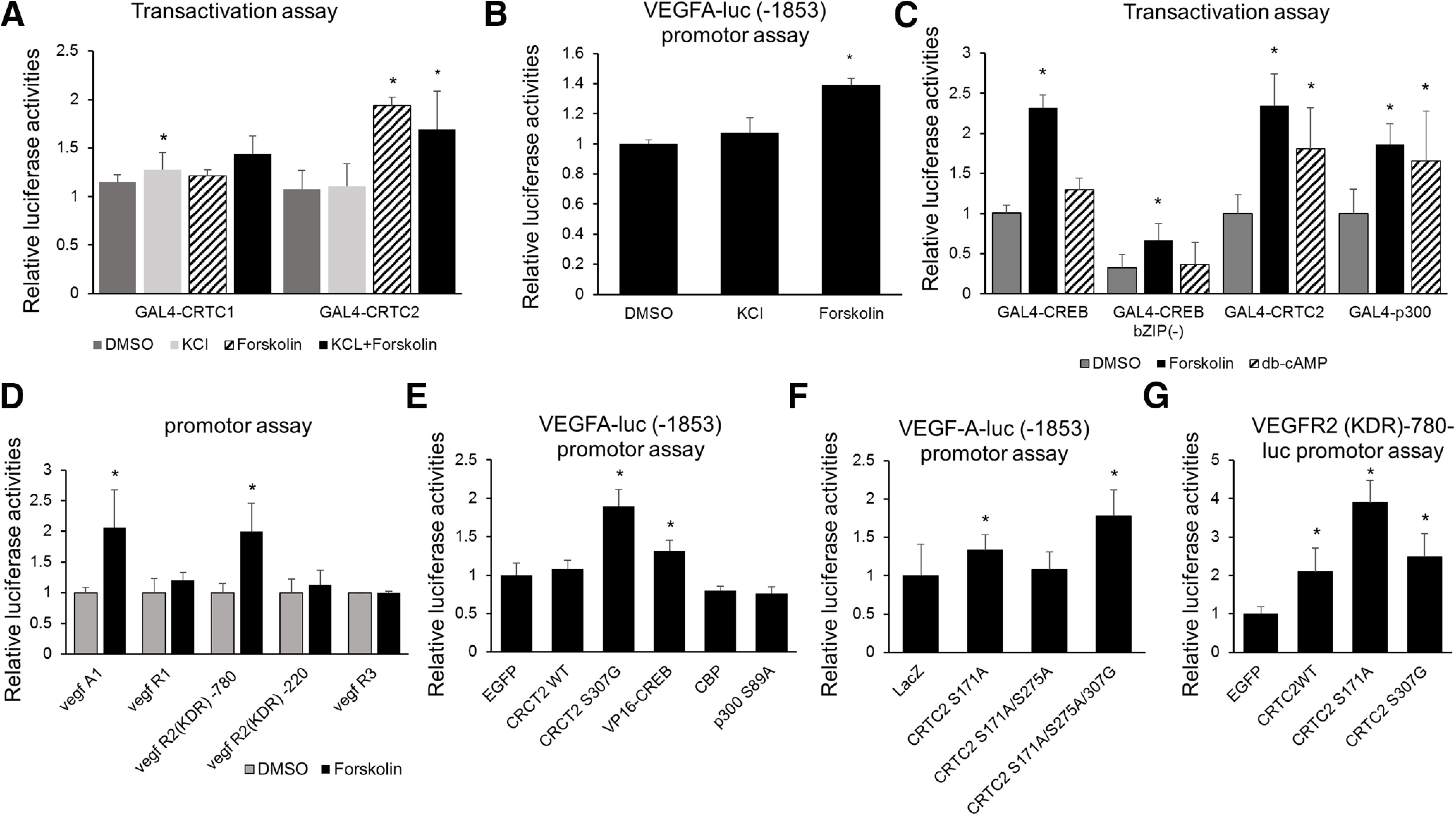
Forskolin enhances CRTC2 activity and VEGF-A/VEGF-R2 promoter activity. A, Transient assay of bEnd.3 cells cotransfected with adeno-GAL4-fusion CRTC1 or CRTC2 and the adeno-5×GAL4-luciferase reporter with the internal reporter Adeno-TK-rluc. The specific transfection activities of either CRTC1 or CRTC2 were expressed as the fold activation of the empty adeno-GAL4 vector. Effect of KCl, Forskolin and Forskolin +KCl on GAL4 luciferase reporter activity are shown. B, Transient assay of bEnd.3 cells expressing the VEGF-A promoter (1853 bp) luciferase-adenovirus. Effects of KCl or Forskolin on luciferase reporter activity are shown. C, The bEnd.3 cells were cotransfected with adeno-GAL4-fusion CREB, CREB b-ZIP (–), CRTC2, or p300 and the adeno-5×GAL4-luciferase reporter with the internal reporter Adeno-TK-rluc. The bEnd.3 cells were treated with forskolin, dibutyryl-cAMP, or DMSO for 6 h, and a luciferase assay performed. D, The bEnd.3 cells were transiently transfected with the VEGF-A(−1853), VEGF-R1(−1160), VEGF-R2(−220), VEGF-R2(−780), VEGF-R3(−870)-luciferase adenovirus, and ad-TK-rluc. At 48 h postinfection, cells were exposed to forskolin, and after 12 h, a luciferase assay was performed. E, The bEnd.3 cells were transfected with the each EGFP, CRTC2WT, CRTC2-S307G, VP16-CREB, CBP, p300 S89A adenovirus vector and VEGF-A promoter (1853 bp) luciferase adenovirus. After 72 h, a luciferase assay was done. F, The bEnd.3 cells were cotransfected with adenovirus expressing the LacZ-, CRTC2-S171A, CRTC2-S171A/S275A double mutant, or CRTC2-S171A/S275A/S307A triple mutant, with the VEGF-A promoter (1853 bp) luciferase adenovirus. At 72 h postinfection, a luciferase assay was performed. Error bars represent mean ± SD. G, The bEnd.3 cells were transfected with the each EGFP, CRTC2WT, CRTC2-S171A, and CRTC2-S307G adenovirus vector and VEGF-R2 (KDR; −780)-luciferase adenovirus. After 72 h, a luciferase assay was done. Luciferase assay data are presented as mean ± SD. For A–G, *p < 0.05 by one-way ANOVA, followed by Tukey−Kramer post hoc analysis.
Next, to examine the reactivity of vascular endothelial cells by cAMP stimulation, the transactivation activity of transcription factors or coactivator was examined using a reporter assay using GAL4-fusion CREB, CRTC2, or p300 (Fig. 4C). The activity of the full-length CREB was enhanced after forskolin treatment, but bZIP-less CREB at a lower level. CRTC2 activity also significantly increased following forskolin treatment, suggesting that CRTCs may contribute to the Ser133-independent activation of CREB in response to forskolin treatment (Fig. 4C). The activities of both CREB and the CREB co-activator CRTCs were enhanced by forskolin, suggesting that both the CREB-CBP pathway (Ser133-dependent) and CREB-CRTC pathway (Ser133-independent) may contribute to angiogenesis after forskolin treatment. Next, among VEGF and VEGF receptor, cAMP-dependent signals were examined by their promoter activities of VEGF-A, VEGF-R1, VEGF-R2, and VEGF-R3 (Fig. 4D). As a result, VEGFA-VEGFR2 (KDR) was activated by cAMP pathway (Fig. 4D). Furthermore, to accurately investigate whether VEGF-A was CRTC2 dependent, the VEGF-A promoter activity was examined using various CRTC2 mutants including CRTC2 S307G (CRTC2 constitutive active; Uebi et al., 2010; Fig. 4E). We found that VEGF-A promoter activity increased in response to the overexpression of either CRTC2 S307G or VP16-CREB (Fig. 4E). We previously clarified the importance of Ser171, Ser275, and Ser307 for CRTC2 function in COS-7 cells and the mouse liver. Consistent with COS-7 cells, phosphorylation at Ser171 or Ser307, but not at Ser275, regulated VEGF-A promoter activity in endothelial cells (Fig. 4F). To investigate the relation between CRTC2 and VEGFR2, we performed VEGFR2 (KDR) luciferase promotor assay using overexpression of CRTC2 in vascular endothelial cells (Fig. 4G). The overexpression of CRTC2 wild-type (WT), CRTC2 S171A and CRTC2 S307G lead to the activation of VEGFR2 (KDR) promotor activity (Fig. 4G). These results showed that CRTC2 regulates the activation of VEGF-A/VEGFR-2 in endothelial cells.
We then examined the intracellular distribution of CRTC2 using a GFP fusion protein and found that Ser171 and Ser307 in CRTC2 were related to its nuclear localization in bEnd.3 cells and HUVECs (Fig. 5A–D). CRTC2 was retained in the nucleus after truncation of the nuclear export signal (NES) containing Ser275 (275–280; Fig. 5A–D). A triple mutation at Ser171, Ser275, and Ser307 prominently increased the nuclear distribution of CRTC2 (Fig. 5E,F).
Figure 5.
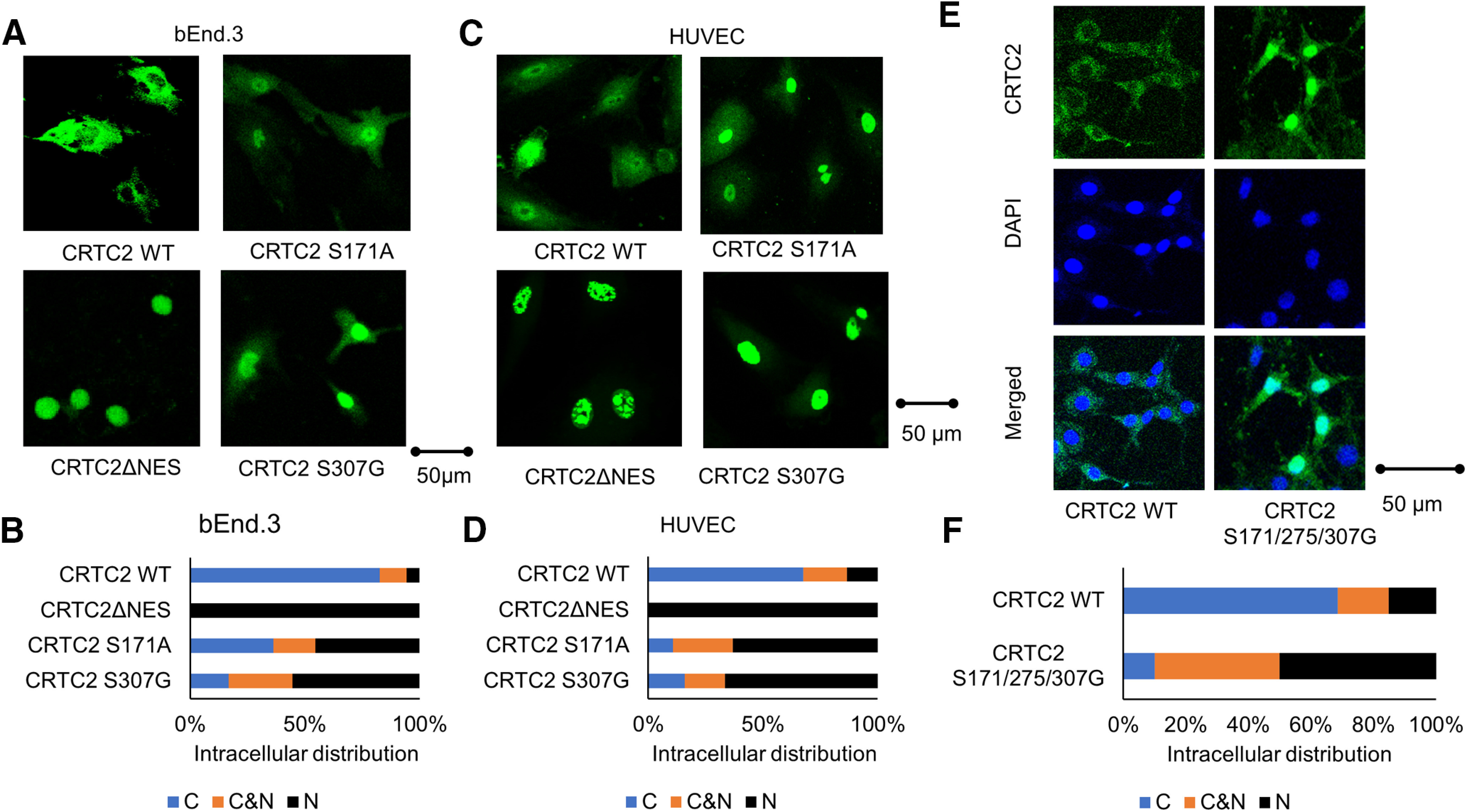
Ser307 in CRTC2 is related to its nuclear localization. A, The bEnd.3 cells were cotransfected with the each CRTC2WT, CRTC2 S307G, CRTC2ΔNES, CRTC2 S171A adenovirus vector. CRTC2 protein was visualized by immunofluorescence. Scale bar: 50 µm. B, Four types of bEnd.3 cells were classified into three categories located in cytoplasma (C), in both cytoplasma and the nucleus (C&N), and in the nucleus (N). C, Diagrams of CRTC2 mutants used for assays of intracellular distribution. The HUVECs transformed with pEGFP-CRTC2 WT, CRTC2 S307G, CRTC2 S171A, or CRTC2 ΔNES adenovirus vector. Scale bar: 50 µm. D, Four types of HUVEC cells were classified into three categories located in cytoplasma (C), in both cytoplasma and the nucleus (C&N), and in the nucleus (N). E, The bEnd.3 cells were cotransfected with adenovirus expressing CRTC2 WT- or CRTC2 containing a triple mutation at Ser171, Ser275, and Ser307. The localization of CRTC2 was examined by immunofluorescence. Scale bar: 50 µm. F, Two types of bEnd.3 cells were classified into three categories located in cytoplasma (C), in both cytoplasma and the nucleus (C&N), and in the nucleus (N).
CRTC2 regulates p190RhoGAP expression
We examined the involvement of CRTC2 in the regulation of p190RhoGAP-A expression. The dominant-active CRTC2 mutants, VP16-CREB and CRTC2 S307G, significantly increased the expression of p190RhoGAP-A (Fig. 6A). On the other hand, CRTC2 knocked down by adenovirus-mediated CRTC2-specific miRNA resulted in a decreased expression of p190RhoGAP-A levels (Fig. 6B), and the overexpression of a dominant-negative CRTC2 (DN-CRTC2, N-terminal 56 amino acids) strongly inhibited the expression of p190RhoGAP-A (Fig. 6C). Additionally, we performed Western blot analysis using endothelial cells. Overexpression of CRTC2 lead to the increased expression level of p190RhoGAP-A and VEGFR2 (Fig. 6D,E) and knock-down of CRTC2 also lead to the reduced expression level of p190RhoGAP-A and VEGFR2 (Fig. 6D,F).
Figure 6.
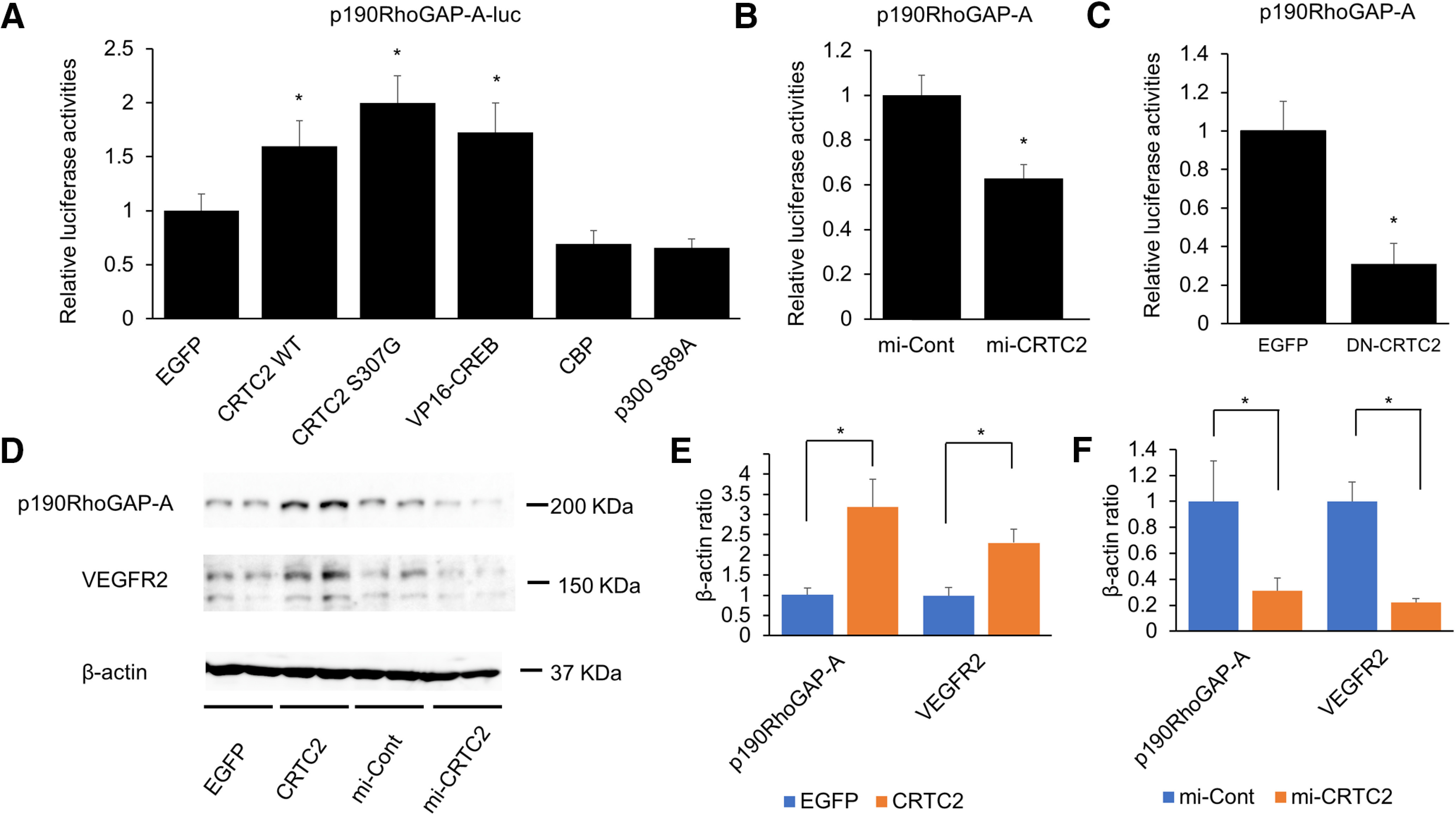
CRTC2 positively regulates p190RhoGAP-A expression. A, The bEnd.3 cells were cotransfected with either the EGFP-, CRTC2-WT-, CRTC2-S307G mutant-, VP16-CREB-, CBP-, or p300 S89A-expressing adenovirus, and p190RhoGAP promoter luciferase-adenovirus. At 72 h postinfection, a luciferase assay was performed. B, The bEnd.3 cells were cotransfected with LacZ-specific (mi-Cont) or CRTC2-specific (mi-CRTC2) miRNAi-expressing adenovirus, and the p190RhoGAP-A promoter luciferase-adenovirus. C, The bEnd.3 cells were transfected with the p190RhoGAP-A promoter luciferase adenovirus. At 4 d postinfection, a luciferase assay was performed in combination with the overexpression of adeno-EGFP or adeno-DN-CRTC2. D, Western blot analysis of the expression level of p190RhoGAP-A and VEGFR2 in bEnd.3 cells transfected with EGFP, CRTC2-WT, mi-Cont or mi-CRTC2 expressing adenovirus. E, Quantitative results of p190RhoGAP-A/β-actin ratio and VEGFR2/β-actin ratio (n = 3 or 4). F, Quantitative results of p190RhoGAP-A/β-actin ratio and VEGFR2/β-actin ratio (n = 3 or 4). Luciferase assay data are presented as mean ± SD. Quantitative results of Western blot analysis are presented as mean ± SEM. For A, *p < 0.05 by one-way ANOVA, followed by Tukey−Kramer post hoc analysis. For B, C, *p < 0.05 compared with mi-Cont or EGFP by two-tailed unpaired Student's t test. For E, F, *p < 0.05 compared with control (EGFP or mi-Cont) by two-tailed unpaired Student's t test.
p190RhoGAP is initially reported to RhoA negatively regulator and recently the dysregulation of RhoGAPs is thought to be related with CNS disease (Huang et al., 2017) and ROCK1 is downstream target of RhoA (Kümper et al., 2016; Lu et al., 2020).
Then, we examined the levels of CRTC2 phosphorylation, p190RhoGAP-A expression and ROCK1 expression under ischemic conditions (Fig. 7A,B). CRTC2 was hyperphosphorylated just after the lethal ischemia (OGD 9 h; Fig. 7C) and gradually dephosphorylated, and then CRTC2 remained dephosphorylated monitored by band shift of CRTC2 protein. We further found that all three phosphorylation sites involved in nuclear translocation, Ser171, Ser275, and Ser307, remained dephosphorylated using site specific anti-phospho-Ser antibodies of CRTC2. Dephosphorylated CRTC2 moved into the nucleus and was subsequently degraded by lethal ischemia. After lethal ischemia (∼9 h OGD), the level of p190RhoGAP-A protein was reduced compared with control (no treatment; Fig. 7C,D) and induced activation of ROCK1 (Fig. 7C,D). These results suggested that p190RhoGAP-A was decreased by lethal ischemia, resulting in activation of ROCK1.
Figure 7.
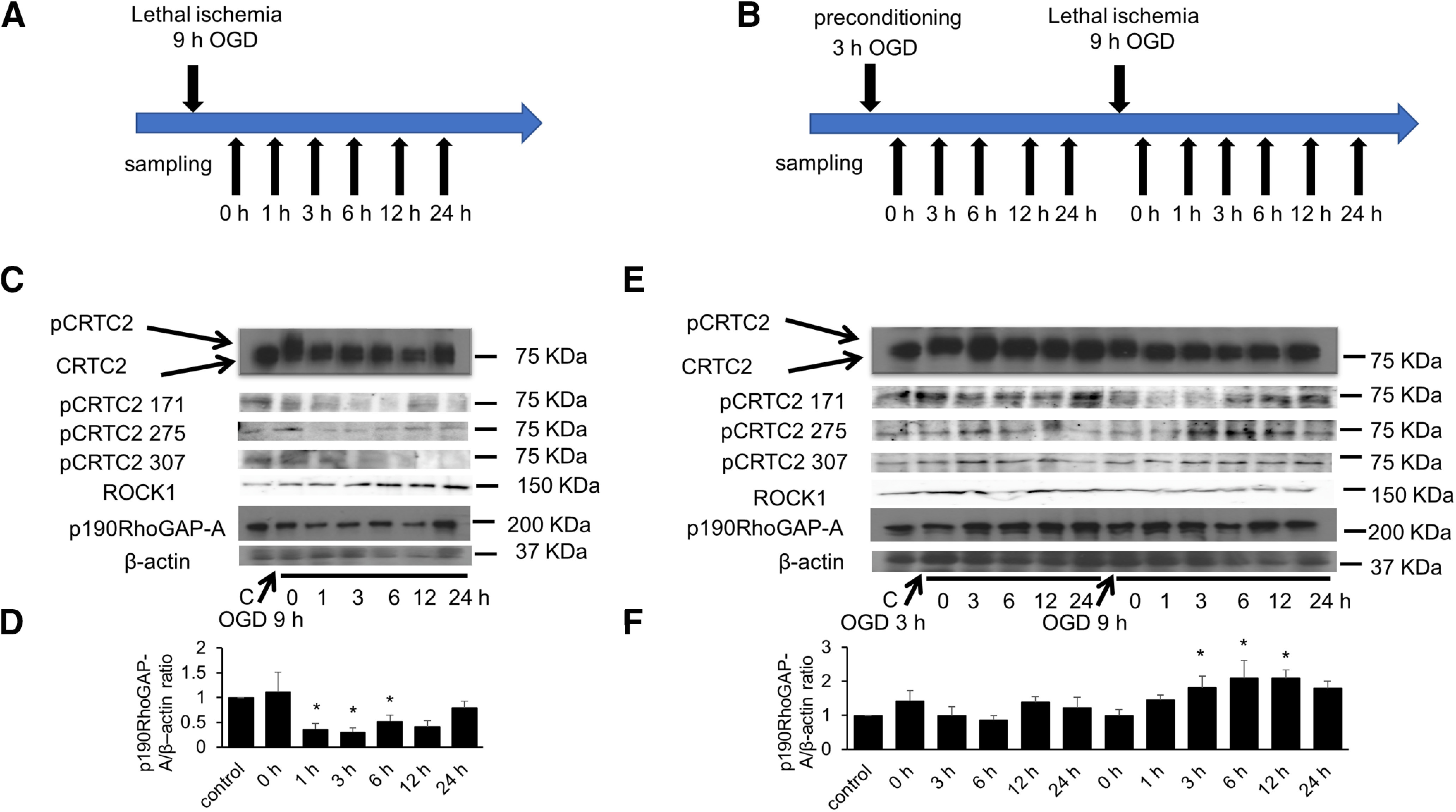
CRTC2 is related with the expression of p190RhoGAP-A under ischemic conditions. A, Experimental procedures. The cells were subjected to lethal ischemia (OGD 9 h). B, Experimental procedures. The cells were subjected to 3 h OGD before lethal ischemia (OGD 9 h). C, Western blot analysis of the phosphorylation of CRTC2 and the expression level of p190RhoGAP-A and ROCK1 in bEnd.3 cells at the indicated periods following OGD. D, Quantitative results of p190RhoGAP-A/β-actin ration are shown (n = 5). E, Western blot analysis of the phosphorylation of CRTC2 and the expression level of p190RhoGAP-A in bEnd.3 cells at the indicated periods following OGD or preconditioning OGD. F, Quantitative results of p190RhoGAP-A/β-actin ration are shown (n = 4). Quantitative results of Western blot analysis are presented as mean ± SEM. For D, F, *p < 0.05 compared with control by two-tailed unpaired Student's t test. D, 0 h, p = 0.6739; 1 h, p = 0.0216; 3 h, p = 0.0125; 6 h, p = 0.0783; 12 h, p = 0.0331; 24 h, p = 0.7357. F, 0 h (after OGD 3 h), p = 0.2968; 3 h (after OGD 3 h), p = 0.9970; 6 h (after OGD 3 h), p = 0.7349; 12 h (after OGD 3 h), p = 0.3374; 24 h (after OGD 3 h), p = 0.5648; 0 h (after 3 h + 9 h), p = 0.9925 1 h (after OGD 3 h + 9 h), p = 0.2552; 3 h (after OGD 3 h + 9 h), p = 0.0448; 6 h (after OGD 3 h + 9 h), p = 0.0085; 12 h (after OGD 3 h + 9 h), p = 0.0092; 24 h (after OGD 3 h + 9 h), p = 0.0523.
Ischemia preconditioning, along with hypothermia, provides a definitive neuroprotective effect. Ischemic preconditioning has neuroprotection through multiple pathways. We then examined the effect of ischemic preconditioning on endothelial cells. Like lethal ischemia, sublethal ischemia (3 h OGD) weakly increased the level of phospho-CRTC2, which was maintained even after reoxygeneration (at an early phase, ∼6 h; Fig. 7E). Interestingly, sublethal ischemia resulted in a resistance to subsequent lethal ischemia (the second 9 h OGD). When sublethal ischemia was loaded, dephosphorylation of CRTC2, including in Ser171, Ser275, and Ser307, occurred in the early phase of ischemia after subsequent lethal ischemia, and then CRTC2 phosphorylation was restored in the late phase of ischemia because CRTC2 was not degraded unlike lethal ischemia. Restoration of CRTC2 phosphorylation in this non-fatal ischemia might reflect CRTC2 shuttling of the nucleus and cytoplasm, but not CRTC2 degradation. In ischemic preconditioning, enhanced transactivational activity of CRTC2 maintained p190RhoGAP-A levels, which resulted in not activating of ROCK1 (Fig. 7E,F). These results suggested that activation of p190RhoGAP-A by CRTC2-CREB in the vascular endothelium provides a protective effect on vascular endothelial cells under ischemic conditions.
Endothelial cell-specific CRTC2-knock-out mice show reduced angiogenesis ex vivo
CRTC2 is expressed in the liver and pancreatic cells, and plays an important functional role (Koo et al., 2005; Liu et al., 2008). In the present study, we focused on the function of CRTC2 in endothelial cells, and we used endothelial cell-specific CRTC2-knock-out mice. First, we cultured primary endothelial cells from CRTC2flox/flox/Tie2-Cre mice (CRTC2flox/flox/Cre+ mice) and performed immunofluorescence using CRTC2 and CD31 (endothelial cell marker) antibody. The expression of CRTC2 was not detected at primary endothelial cells from CRTC2flox/flox/Cre+ mice (Fig. 8A). Then, we examined the effect of CRTC2 on angiogenesis ex vivo using a mouse aorta ring assay for endothelial cell-specific CRTC2-knock-out mice (Burbridge and West, 2001; Shimada et al., 2004). On day 7 of culture, CRTC2flox/flox/Cre+ mice showed reduced sprouting vessels from the aortas compared with CRTC2flox/flox/Cre– control mice (p = 0.0051; Fig. 8B,C). These results show that angiogenesis was impaired in endothelial cell-specific CRTC2-knock-out mice. We also performed immunofluorescence using p190RhoGAP-A antibody targeting for sprouting out sites with aorta ring assay. The expression of p190RhoGAP-A at sprouting out sites from CRTC2flox/flox/Cre+ mice showed less expression compared with from CRTC2flox/flox/Cre− control mice (Fig. 8D,E). Additionally, we examined the expression level of p190RhoGAP-A and VEGFR2 in primary endothelial cells under ischemic conditions. After OGD treatment, p190RhoGAP-A and VEGFR2 showed less expression at primary endothelial cells from CRTC2flox/flox/Cre+ mice compared with from CRTC2flox/flox/Cre– control mice (Fig. 8F,G). We also performed Western blot analysis using primary endothelial cells. The expression level of p190RhoGAP-A and VEGFR2 were lower at primary endothelial cells from CRTC2flox/flox/Cre+ mice compared with from CRTC2flox/flox/Cre– control mice under ischemic conditions (Fig. 8H,I). These data suggested that CRTC2 is related to angiogenesis, and that CRTC2 regulates p190RhoGAP-A and VEGFR2 expression in endothelial cells.
Figure 8.
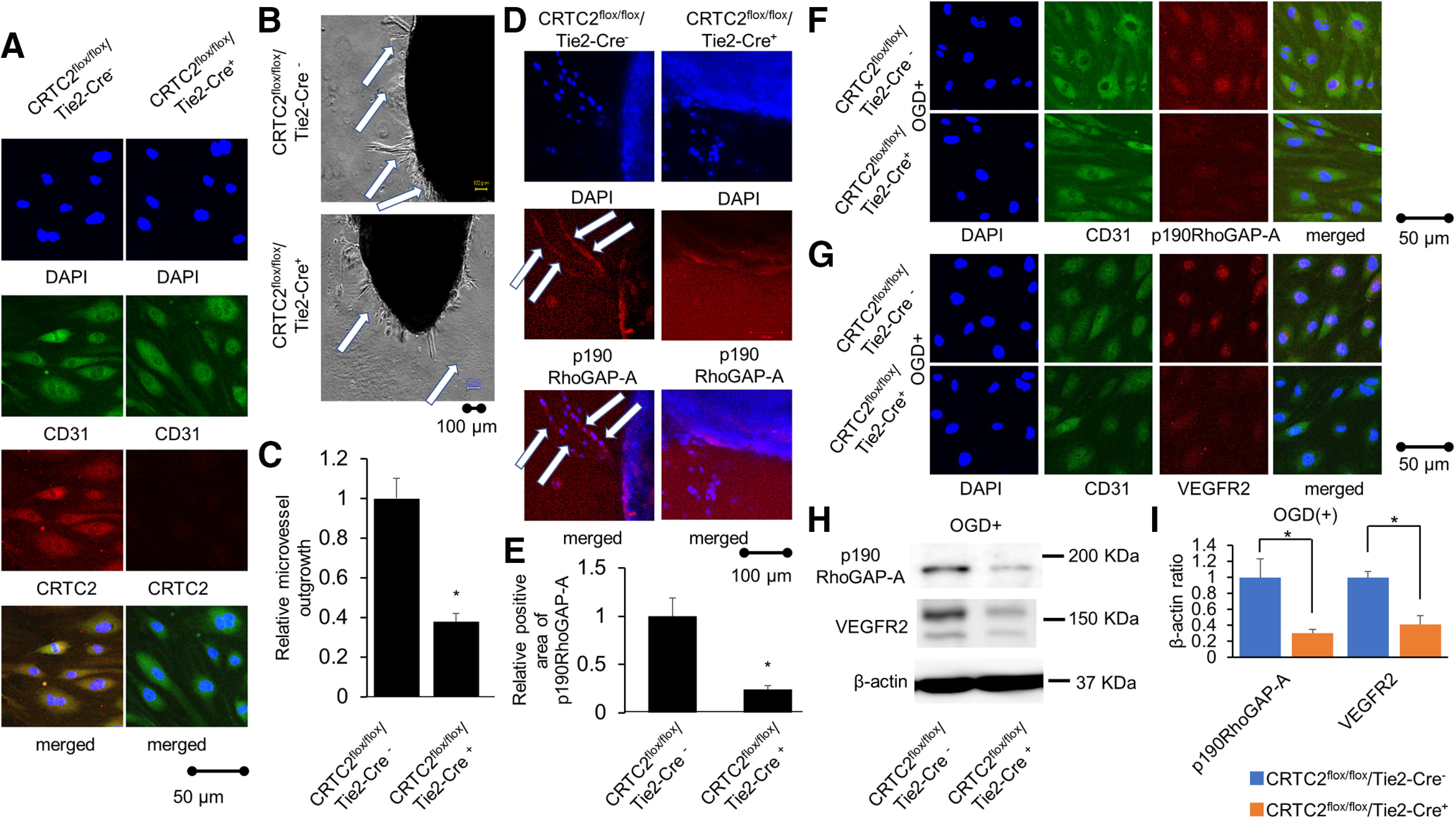
Endothelial cell-specific CRTC2-knock-out mice showed reduced angiogenesis. A, The panels showed CRTC2 staining following co-staining of CD31 via immunofluorescence at primary endothelial cells from CRTC2flox/flox/Cre+ mice (right panels) compared with CRTC2flox/flox/Cre– control mice (left panels). Scale bar: 50 µm. B, Representative images of the mouse aorta ring assay using endothelial cell-specific CRTC2-knock-out mice. Scale bar: 100 µm. C, Quantitative results of relative microvessel outgrowth. Results are presented as mean ± SEM (n = 5); *p < 0.05 compared with CRTCflox/flox/Cre– mice by two-tailed unpaired Student's t test. D, The panels showed p190RhoGAP-A staining via immunofluorescence at sprouting sites with aorta ring assay from CRTC2flox/flox/Cre+ mice (right panels) compared with CRTC2flox/flox/Cre– control mice (left panels). Scale bar: 100 µm. E, Quantitative results of relative positive area of p190RhoGAP-A ratio are shown. Results are presented as mean ± SEM (n = 3); *p < 0.05 compared with CRTC2flox/flox/Cre− mice by two-tailed unpaired Student's t test. F, The panels showed p190RhoGAP-A staining following co-staining of CD31 via immunofluorescence at primary endothelial cells after OGD treatment from CRTC2flox/flox/Cre+ mice (lower panels) compared with CRTC2flox/flox/Cre– control mice (upper panels). Scale bar: 50 µm. G, The panels showed VEGFR2 staining following co-staining of CD31 via immunofluorescence at primary endothelial cells after OGD treatment from CRTC2flox/flox/Cre+ mice (lower panels) compared with CRTC2flox/flox/Cre– control mice (upper panels). Scale bar: 50 µm. H, Western blot analysis of the expression level of p190RhoGAP-A, VEGFR2, and β-actin in mouse primary endothelial cells under ischemic conditions. I, Quantitative results of p190RhoGAP-A/β-actin ratio (n = 4) and VEGFR2/β-actin ratio (n = 4). Quantitative results of Western blot analysis are presented as mean ± SEM; *p < 0.05 compared with CRTC2flox/flox/Cre– by two-tailed unpaired Student's t test.
Endothelial cell-specific CRTC2-knock-out mice show more severe neurologic deficit and impaired recovery after photothrombotic stroke
CRTC2 is related to glucose metabolism in the liver and under fasting, and CRTC2 is translocated into the nucleus and induces glucogenesis by glucagon in the liver (Koo et al., 2005). Thus, we examined the blood glucose level and other enzymatic activities and electrolytes at 1 d after fasting. Endothelial cell-specific CRTC2-knock-out mice showed no significant change in blood glucose levels (n = 5; Table 2) or other enzymatic activities and electrolytes (n = 5; Table 3). Next, to elucidate the effect of CRTC2 on endothelial function, the endothelial cell-specific mice were subjected to PIT. At 1 d after PIT, the endothelial cell-specific CRTC2-knock-out mice showed a larger infarct volume compared with CRTC2flox/flox/Cre− control mice (p = 0.0297; Fig. 9A,B). We performed neurologic examination at 1−28 d after PIT, and endothelial cell-specific CRTC2-knock-out mice showed a more severe neurological deficit and impaired neurologic recovery from the acute phase to the chronic phase (Fig. 9C–E).
Table 2.
Blood glucose level of CRTC2flox/flox/Cre− or CRTC2flox/flox/Cre+ knock-out mice after fasting
| Time after fasting | CRTC2flox/flox/Tie2-Cre– | CRTC2flox/flox/Tie2-Cre+ | p value |
|---|---|---|---|
| 0 h | 205.6 ± 16.4 | 192.2 ± 13.1 | 0.4366 |
| 6 h | 179 ± 11.9 | 211.4 ± 13 | 0.0678 |
| 24 h | 88.4 ± 4.9 | 120.2 ± 9.2 | 0.0727 |
Data are presented as mean ± SEM (n = 5); the p value was calculated by a t test.
Table 3.
The results of other enzymes at 1 d after fasting in CRTC2flox/flox/Cre− or CRTC2flox/flox/Cre+ knock-out mice
| CRTC2flox/flox/Tie2-Cre– | CRTC2flox/flox/Tie2-Cre+ | p value | |
|---|---|---|---|
| ALB | 4.62 ± 0.13 | 4.44 ± 0.12 | 0.476 |
| ALP | 75.6 ± 2.58 | 92.4 ± 13.9 | 0.267 |
| ALT | 70.6 ± 9.54 | 51.6 ± 4.87 | 0.114 |
| AMY | 1257.2 ± 167.33 | 1190 ± 76.5 | 0.728 |
| T-Bil | 0.24 ± 0.02 | 0.24 ± 0.02 | 1.000 |
| BUN | 27.2 ± 1.85 | 24.4 ± 1.32 | 0.254 |
| Ca | 11.26 ± 0.12 | 11.32 ± 0.15 | 0.763 |
| P | 13.84 ± 1.27 | 13.48 ± 0.47 | 0.797 |
| Na | 161 ± 1 | 160.4 ± 0.81 | 0.654 |
| TP | 6.26 ± 0.12 | 6.14 ± 0.07 | 0.423 |
| Glob | 1.62 ± 0.07 | 1.7 ± 0.18 | 0.686 |
Data are presented as mean ± SEM (n = 5); the p value was calculated by a t test.
Figure 9.
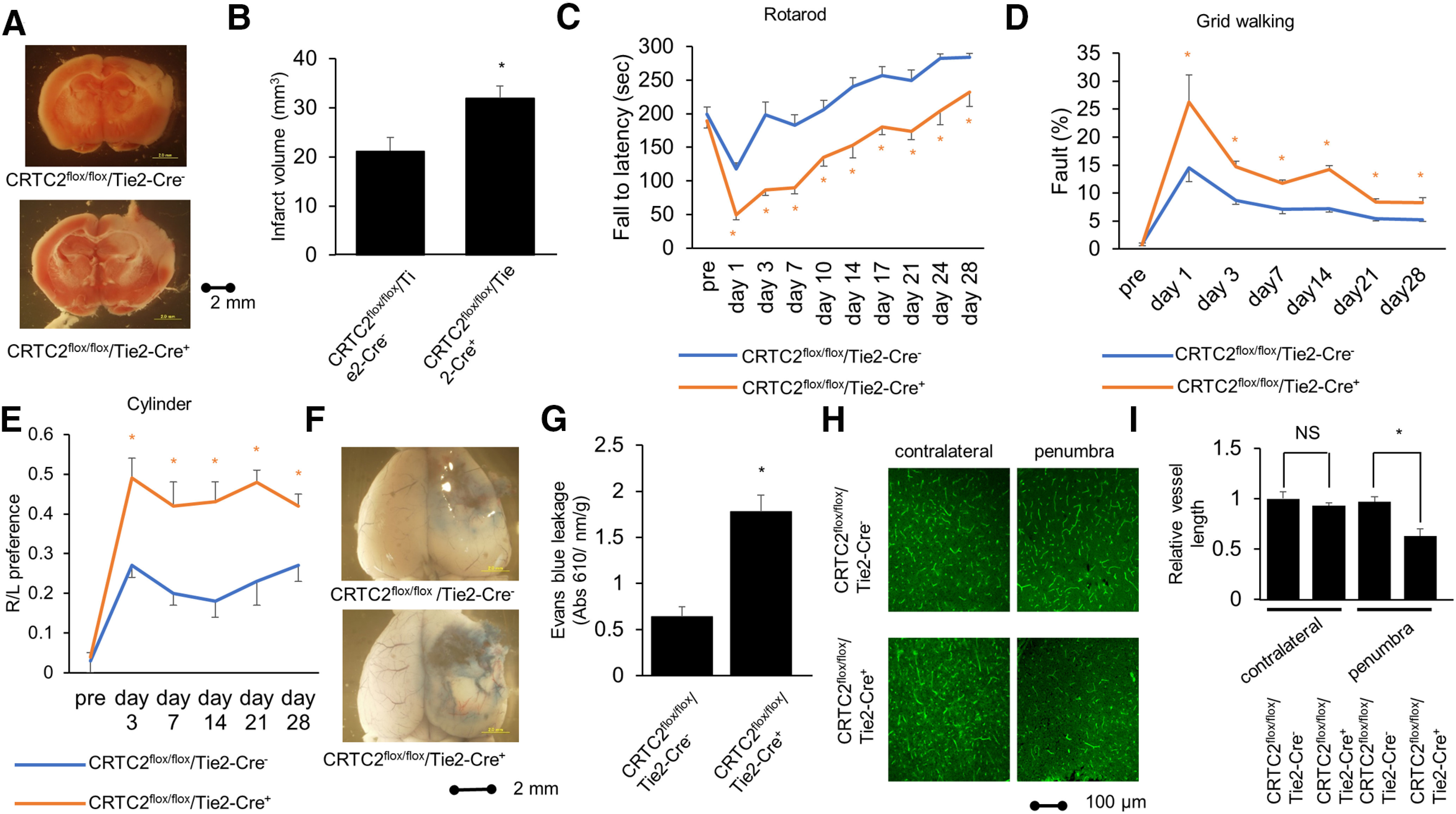
Endothelial cell-specific knock-out of CRTC2 mice show more severe ischemic stroke and impaired neurologic recovery. A, Representative images of TTC staining after PIT of CRTC2flox/flox/Cre− or CRTC2flox/flox/Cre+ knock-out mice. Scale bar: 2 mm. B, Quantitative results of infarct volume of CRTC2flox/flox/Cre– or CRTC2flox/flox/Cre+ knock-out mice (n = 5 or 7). C, Rotarod test data (n = 8–11). D, Grid walking test data (n = 7 or 8). E, Cylinder test data (n = 6–8). F, Photomicrograph of Evans Blue leakage at 1 d after PIT. Scale bar: 2 mm. G, Quantitative results of Evans Blue leakage of CRTC2flox/flox/Cre– or CRTC2flox/flox/Cre+ knock-out mice (n = 4). H, Representative image of immunostaining in the contralateral lesion and penumbra lesion of the brain at 7 d after PIT. The vessel appears green with L. esculentum lectin antibody staining. Scale bar: 100 µm. I, Quantitative results of the length of blood vessels in contralateral lesion or penumbra lesion at CRTC2flox/flox/Cre– or CRTC2flox/flox/Cre+ knock-out mice (n = 3 or 4). Error bars represent mean ± SEM. For C, D, E, G, I, *p < 0.05 compared with CRTC2flox/flox/Cre− mice by two-tailed unpaired Student's t test. For, I, NS indicates no significant change by two-tailed unpaired Student's t test.
To evaluate the BBB permeability at 1 d after PIT, we injected Evans blue intravenously. The endothelial cell-specific CRTC2-knock-out mice showed more BBB leakage compared with CRTC2flox/flox/Cre− control mice (p = 0.0014; Fig. 9F,G). Next, we evaluated the vascular beds at 7 d after PIT. In contralateral lesion, the endothelial cell-specific CRTC2-knock-out mice showed same vascular beds with CRTC2flox/flox/Cre− control mice, however, the endothelial cell-specific CRTC2-knock-out mice showed less vascular beds compared with CRTC2flox/flox/Cre− control mice in the penumbra lesion (p = 0.0361; Fig. 9H,I), which suggested that impairment of poststroke angiogenesis was occurred in the endothelial cell-specific CRTC2-knock-out mice. These results showed that CRTC2 plays a critical role in vascular network after cerebral infarction (Fig. 10).
Figure 10.
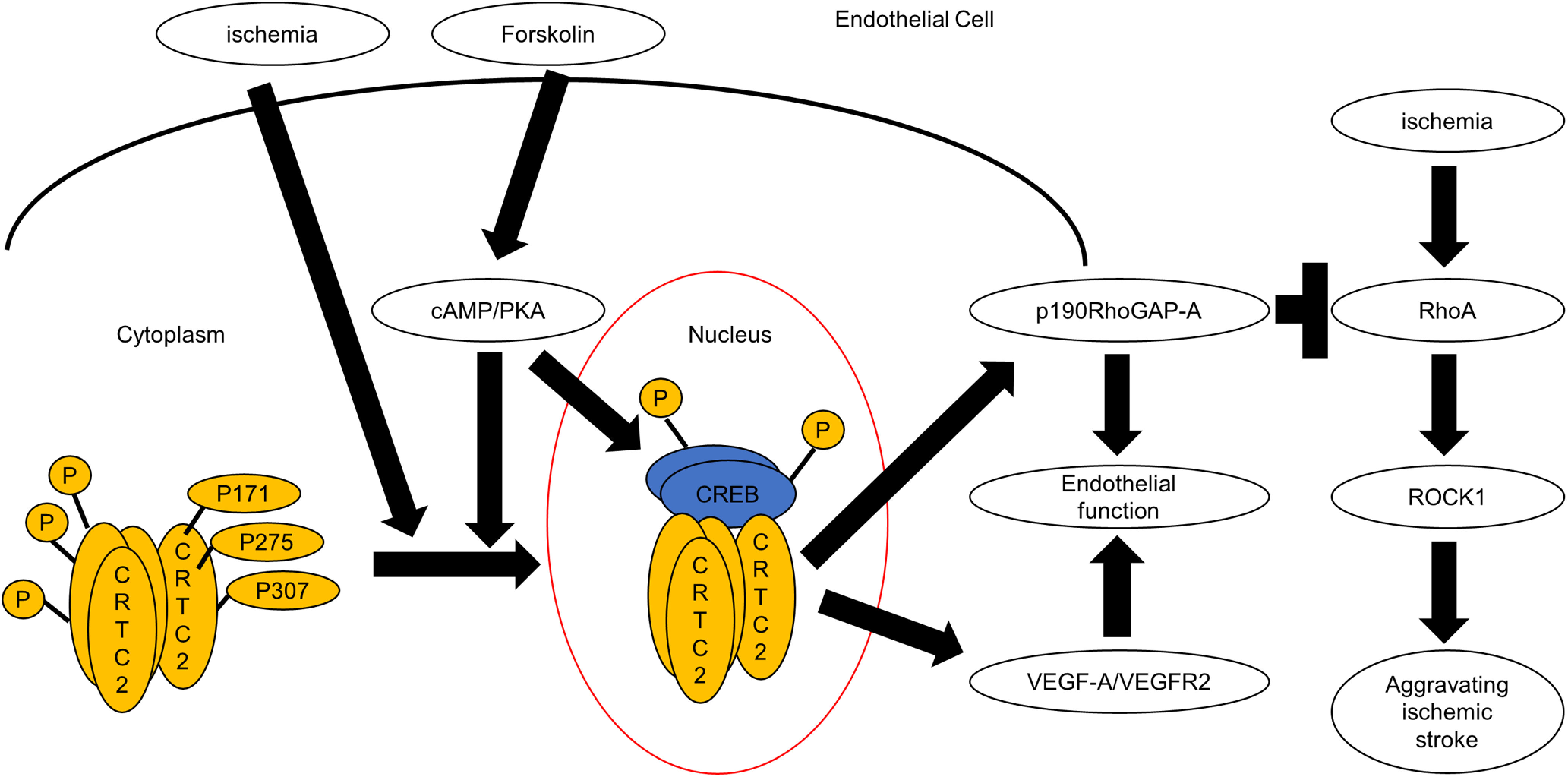
The model figure of the role of CRTC2 in endothelial cells. Under normal condition, CRTC2 is phosphorylated at three points, Ser171, Ser 275, Ser307 in cytoplasm. Forskolin induces activation of CRTC2 and CREB pathway via cAMP/PKA pathway. Ischemia induces dephosphorylation of CRTC2 at mainly Ser171 and Ser307 and leads to its translocation to the nucleus. Then, CRTC2 activates VEGF-A/VEGF-R2 pathway and sustains p190RhoGAP-A pathway and leads to the enhancement of endothelial functions. Ischemia also induces the activation of RhoA and ROCK1, and this activation leads to aggravate ischemia. This pathway is regulated negatively by p190RhoGAP-A. These results suggest that CRTC2 played a very important protective role via p190RhoGAP-A under ischemic conditions.
Discussion
The present study investigated the role of CRTC2 in endothelial cells. CRTC2 was expressed in endothelial cells and was related to angiogenesis and vascular integrity. Among CRTC1–3, only CRTC2 was activated under ischemic conditions. We found that CRTC2 maintained the barrier function of endothelial cells related to p190RhoGAP-A. We also showed that endothelial cell-specific CRTC2-knock-out mice aggravated ischemic stroke and impaired neurologic recovery accompanied by increased BBB permeability and reduced blood vessels. Overall, these findings suggest that CRTC2 plays a crucial role in angiogenesis and endothelial function, especially under ischemic conditions.
CRTC2 is a master regulator of glucose metabolism (Koo et al., 2005; Le Lay et al., 2009) and insulin sensitivity (Wang et al., 2010). CRTC2 has also been shown to be involved in obesity via regulation of endoplasmic reticulum stress (Han et al., 2015) and triglyceride homeostasis (Wang et al., 2010; Owen et al., 2015).
However, the physiological importance of CRTC2 in endothelial cells is unclear. This prompted us to investigate whether CRTC2 affects vascular integrity and barrier function in endothelial cells. As previously reported, cooperative regulation of the intracellular distribution of CRTC2 by Ser171 and Ser275 was also observed in COS-7 cells. However, mutations at Ser171 and Ser275 failed to inhibit the salt-induced kinase (SIK)1-mediated nuclear export of CRTC2 (Uebi et al., 2010). In a previous study, we investigated the molecular mechanism by which SIK1 induces the nuclear export of CRTC2, and we identified Ser307 in CRTC2 to be another important phosphorylation site (Uebi et al., 2010). Disruption of Ser307 completely inhibited the SIK1-induced nuclear export of CRTC2 in COS-7 cells (Uebi et al., 2010). Consistent with COS-7 cells and HepG2 cells, we found that Ser307 in CRTC2 is a crucial phosphorylation site for CRTC2 activation in endothelial cells. However, CRTC2 phosphorylation, such as Ser171, Ser275, and Ser307, nuclear translocation, and CRTC2 transactivational activity are not always correlated (Uebi et al., 2010). Therefore, further consideration will be needed.
Recent studies have demonstrated that the cAMP pathway enhances barrier functions, partly via Rap1-mediated activation of Rac1 and Cdc42, as well as by inhibition of RhoA. The small GTPase RhoA plays an important role in inducing endothelial cell contraction, thereby enhancing endothelial barrier function (van Nieuw Amerongen et al., 1998; Spindler et al., 2010). RhoA activity mediated by p190RhoGAP is a key step in endothelial barrier restoration signaling after increased endothelial permeability (Harrington et al., 2004; Holinstat et al., 2006). CREB dysfunction using CREB shRNAi or a dominant negative-CREB mutant (dn-CREB) markedly inhibits endothelial barrier function through downregulation of p190RhoGAP-A expression (Chava et al., 2012). Our results show that CRTC2 directly regulates the transcription of p190RhoGAP-A expression both in vitro and in vivo using endothelial cell-specific CRTC2-knock-out mice, resulting in decreased barrier function in endothelial cells. Further, increased vascular permeability by VEGF exacerbated ischemic brain injury and edema, while enhanced p190RhoGAP-A expression through CREB signaling attenuated this destructive effect.
In contrast to the decrease in p190RhoGAP-A protein in lethal ischemia, we found that OGD preconditioning upregulated p190RhoGAP-A and protected vascular endothelial permeability in endothelial cells. Importantly, we observed decreased angiogenesis and endothelial barrier function in endothelial cell-specific CRTC2-knock-out mice. Angiogenesis is important for brain edema and hemorrhagic changes after the early phase of ischemic stroke, and in neurologic recovery after the delayed phase of ischemic stroke (Bang et al., 2008). Comparably, we found that endothelial cell-specific CRTC2-knock-out mice showed more severe ischemic stroke in the early phase and impaired neurologic recovery.
We found the expression of ROCK1 was decreased after lethal ischemia correlated with the expression of p190RhoGAP-A and preconditioning lead to not decrease the expression of ROCK1 after lethal ischemia. The expression of RhoA and downstream molecule ROCK1 are upregulated after ischemia (Brabeck et al., 2003; Shimokawa et al., 2016; Liang et al., 2018; Zhong et al., 2019; Lu et al., 2020). The inhibition of ROCK1 leads to reduce neurologic deficit in experimental MCAO model mice (Li and Liu, 2019; Xu et al., 2020). Additionally, the overactivation of RhoA is related to increased BBB permeability in experimental MCA model (ElAli et al., 2011). Based on these findings, we think that the CRTC2-CREB-p190RhoGAP-A pathway is neuroprotective through inhibition of vascular permeability under ischemic conditions.
Because not only CRTC2 but also CRTC1 is also expressed in endothelial cells, it will be necessary to examine the function of CRTC1 in endothelial cells in further studies. Since the importance of CREB in vascular smooth muscle cells has been reported previously, the function of CRTC in vascular smooth muscle cells should be studied.
Importantly, p190RhoGAP-A enhances endothelial barrier function, thereby stabilizing the integrity of blood vessel walls. Herein, we demonstrated that the CREB coactivator CRTC2 enhanced endothelial barrier function through p190RhoGAP-A gene regulation. The CRTC-p190RhoGAP-A pathway plays a pivotal role in vascular integrity. Our findings suggest that the upstream regulators of the CRTC pathway, such as SIKs (Uebi et al., 2010; Sasaki et al., 2011), may be therapeutic targets for the treatment of abnormal vascular permeability after neurologic disorders including stroke.
Footnotes
Part of this work was supported by Ministry of Education, Culture, Sports, Science, and Technology, Japan, Grant-in-Aid for Challenging Exploratory Research 16K15317 (to T.S.) and Grant-in-Aid for Scientific Research (B) 16H04924 (to S.M.) and by the Smoking Research Foundation (T.S.). We thank the generous gift of CRTC2-knock-out mice and many helpful discussions with Prof. Marc Montminy (Salk Institute for Biologic Studies), the generous gift of CRTC2flox/flox mice and many helpful discussions with Prof. Klaus Kaestner (Perelman School of Medicine, University of Pennsylvania), and Y. Kurano for secretarial assistance and Edanz Group for editing a draft of this manuscript.
The authors declare no competing financial interests.
References
- Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K (1996) Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271:648–650. 10.1126/science.271.5249.648 [DOI] [PubMed] [Google Scholar]
- Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D'Amico G, Jones DT, Vojnovic B, Hodivala-Dilke K (2011) Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc 7:89–104. 10.1038/nprot.2011.435 [DOI] [PubMed] [Google Scholar]
- Bang OY, Saver JL, Buck BH, Alger JR, Starkman S, Ovbiagele B, Kim D, Jahan R, Duckwiler GR, Yoon SR, Vinuela F, Liebeskind DS, UCLA Collateral Investigators (2008) Impact of collateral flow on tissue fate in acute ischaemic stroke. J Neurol Neurosurg Psychiatry 79:625–629. 10.1136/jnnp.2007.132100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao MH, Szeto V, Yang BB, Zhu SZ, Sun HS, Feng ZP (2018) Long non-coding RNAs in ischemic stroke. Cell Death Dis 9:281. 10.1038/s41419-018-0282-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M (2004) Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol 14:2156–2161. 10.1016/j.cub.2004.11.002 [DOI] [PubMed] [Google Scholar]
- Blanchet E, Van de Velde S, Matsumura S, Hao E, LeLay J, Kaestner K, Montminy M (2015) Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep 10:1149–1157. 10.1016/j.celrep.2015.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabeck C, Mittelbronn M, Bekure K, Meyermann R, Schluesener HJ, Schwab JM (2003) Effect of focal cerebral infarctions on lesional RhoA and RhoB expression. Arch Neurol 60:1245–1249. 10.1001/archneur.60.9.1245 [DOI] [PubMed] [Google Scholar]
- Burbridge MF, West DC (2001) Rat aortic ring: 3D model of angiogenesis in vitro. Methods Mol Med 46:185–204. 10.1385/1-59259-143-4:185 [DOI] [PubMed] [Google Scholar]
- Cai Y, Zhang Y, Ke X, Guo Y, Yao C, Tang N, Pang P, Xie G, Fang L, Zhang Z, Li J, Fan Y, He X, Wen R, Pei L, Lu Y (2019) Transcriptome sequencing unravels potential biomarkers at different stages of cerebral ischemic stroke. Front Genet 10:814. 10.3389/fgene.2019.00814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chava KR, Tauseef M, Sharma T, Mehta D (2012) Cyclic AMP response element-binding protein prevents endothelial permeability increase through transcriptional controlling p190RhoGAP expression. Blood 119:308–319. 10.1182/blood-2011-02-339473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Uzgil B, Lin P, Avliyakulov NK, O'Dell TJ, Martin KC (2012) Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150:207–221. 10.1016/j.cell.2012.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M (2003) TORCs: transducers of regulated CREB activity. Mol Cell 12:413–423. 10.1016/j.molcel.2003.08.013 [DOI] [PubMed] [Google Scholar]
- Dormond O, Contreras AG, Meijer E, Datta D, Flynn E, Pal S, Briscoe DM (2008) CD40-induced signaling in human endothelial cells results in mTORC2- and Akt-dependent expression of vascular endothelial growth factor in vitro and in vivo. J Immunol 181:8088–8095. 10.4049/jimmunol.181.11.8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra-Aiello C, Jickling GC, Ander BP, Zhan X, Liu D, Hull H, Orantia M, Ho C, Stamova B (2015) Intracerebral hemorrhage and ischemic stroke of different etiologies have distinct alternatively spliced mRNA profiles in the blood: a pilot RNA-seq study. Transl Stroke Res 6:284–289. 10.1007/s12975-015-0407-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElAli A, Doeppner TR, Zechariah A, Hermann DM (2011) Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke 42:3238–3244. 10.1161/STROKEAHA.111.615559 [DOI] [PubMed] [Google Scholar]
- España J, Valero J, Miñano-Molina AJ, Masgrau R, Martín E, Guardia-Laguarta C, Lleó A, Giménez-Llort L, Rodríguez-Alvarez J, Saura CA (2010) beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci 30:9402–9410. 10.1523/JNEUROSCI.2154-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59:675–680. 10.1016/0092-8674(89)90013-5 [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14:1553–1577. [PubMed] [Google Scholar]
- Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, Deng H, Wang Y (2015) The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 524:243–246. 10.1038/nature14557 [DOI] [PubMed] [Google Scholar]
- Harrington EO, Newton J, Morin N, Rounds S (2004) Barrier dysfunction and RhoA activation are blunted by homocysteine and adenosine in pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol 287:L1091–L1097. 10.1152/ajplung.00421.2003 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Ishikawa R, Ye LH, He XL, Takata K, Kohama K, Shirao T (1996) Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J Neurosci 16:7161–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez JB, Chang C, LeBlanc M, Grimm D, Le Lay J, Kaestner KH, Zheng Y, Montminy M (2015) The CREB/CRTC2 pathway modulates autoimmune disease by promoting Th17 differentiation. Nat Commun 6:7216. 10.1038/ncomms8216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D (2006) Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem 281:2296–2305. 10.1074/jbc.M511248200 [DOI] [PubMed] [Google Scholar]
- Huang GH, Sun ZL, Li HJ, Feng DF (2017) Rho GTPase-activating proteins: regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol Cell Neurosci 80:18–31. 10.1016/j.mcn.2017.01.007 [DOI] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, et al. (2003) Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA 100:12147–12152. 10.1073/pnas.1932773100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR 3rd, Bordone L, Guarente L, Krainc D (2011) Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med 18:159–165. 10.1038/nm.2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki H, Sasaki T, Matsumura S, Yokawa S, Yukami T, Shimamura M, Sakaguchi M, Furuno T, Suzuki T, Mochizuki H (2019) β-Arrestin-2 in PAR-1-biased signaling has a crucial role in endothelial function via PDGF-β in stroke. Cell Death Dis 10:100. 10.1038/s41419-019-1375-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Takemori H, Min L, Muraoka M, Doi J, Horike N, Okamoto M (2004) Salt-inducible kinase-1 represses cAMP response element-binding protein activity both in the nucleus and in the cytoplasm. Eur J Biochem 271:4307–4319. 10.1111/j.1432-1033.2004.04372.x [DOI] [PubMed] [Google Scholar]
- Katoh Y, Takemori H, Lin X-Z, Tamura M, Muraoka M, Satoh T, Tsuchiya Y, Min L, Doi J, Miyauchi A, Witters LA, Nakamura H, Okamoto M (2006) Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J 273:2730–2748. 10.1111/j.1742-4658.2006.05291.x [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437:1109–1111. 10.1038/nature03967 [DOI] [PubMed] [Google Scholar]
- Kümper S, Mardakheh FK, McCarthy A, Yeo M, Stamp GW, Paul A, Worboys J, Sadok A, Jørgensen C, Guichard S, Marshall CJ (2016) Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. Elife 5:e12994. 10.7554/eLife.12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HT, Chang YC, Tu YF, Huang CC (2010) CREB activation mediates VEGF-A's protection of neurons and cerebral vascular endothelial cells. J Neurochem 113:79–91. 10.1111/j.1471-4159.2010.06584.x [DOI] [PubMed] [Google Scholar]
- Lee SW, Jang MS, Jeong SH, Kim H (2019) Exploratory, cognitive, and depressive-like behaviors in adult and pediatric mice exposed to controlled cortical impact. Clin Exp Emerg Med 6:125–137. 10.15441/ceem.18.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay J, Tuteja G, White P, Dhir R, Ahima R, Kaestner KH (2009) CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis. Cell Metab 10:55–62. 10.1016/j.cmet.2009.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu B (2019) ROCK inhibitor Y-27632 protects rats against cerebral ischemia/reperfusioninduced behavioral deficits and hippocampal damage. Mol Med Rep 20:3395–3405. 10.3892/mmr.2019.10584 [DOI] [PubMed] [Google Scholar]
- Liang H, Liao M, Zhao W, Zheng X, Xu F, Wang H, Huang J (2018) CXCL16/ROCK1 signaling pathway exacerbates acute kidney injury induced by ischemia-reperfusion. Biomed Pharmacother 98:347–356. 10.1016/j.biopha.2017.12.063 [DOI] [PubMed] [Google Scholar]
- Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J 3rd, Olefsky J, Guarente L, Montminy M (2008) A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456:269–273. 10.1038/nature07349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Wen J, Chen Z (2020) Distinct roles of ROCK1 and ROCK2 on the cerebral ischemia injury and subsequently neurodegenerative changes. Pharmacology 105:3–8. 10.1159/000502914 [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB (2006) Signaling mechanisms regulating endothelial permeability. Physiol Rev 86:279–367. 10.1152/physrev.00012.2005 [DOI] [PubMed] [Google Scholar]
- Monai H, Wang X, Yahagi K, Lou N, Mestre H, Xu Q, Abe Y, Yasui M, Iwai Y, Nedergaard M, Hirase H (2019) Adrenergic receptor antagonism induces neuroprotection and facilitates recovery from acute ischemic stroke. Proc Natl Acad Sci USA 116:11010–11019. 10.1073/pnas.1817347116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munji RN, Soung AL, Weiner GA, Sohet F, Semple BD, Trivedi A, Gimlin K, Kotoda M, Korai M, Aydin S, Batugal A, Cabangcala AC, Schupp PG, Oldham MC, Hashimoto T, Noble-Haeusslein LJ, Daneman R (2019) Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat Neurosci 22:1892–1902. 10.1038/s41593-019-0497-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osanai T, Houkin K, Uchiyama S, Minematsu K, Taguchi A, Terasaka S (2018) Treatment evaluation of acute stroke for using in regenerative cell elements (TREASURE) trial: rationale and design. Int J Stroke 13:444–448. 10.1177/1747493017743057 [DOI] [PubMed] [Google Scholar]
- Owen BM, Mangelsdorf DJ, Kliewer SA (2015) Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab 26:22–29. 10.1016/j.tem.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Takemori H, Yagita Y, Terasaki Y, Uebi T, Horike N, Takagi H, Susumu T, Teraoka H, Kusano K, Hatano O, Oyama N, Sugiyama Y, Sakoda S, Kitagawa K (2011) SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB. Neuron 69:106–119. 10.1016/j.neuron.2010.12.004 [DOI] [PubMed] [Google Scholar]
- Schauer IE, Knaub LA, Lloyd M, Watson PA, Gliwa C, Lewis KE, Chait A, Klemm DJ, Gunter JM, Bouchard R, McDonald TO, O'Brien KD, Reusch JE (2010) CREB downregulation in vascular disease: a common response to cardiovascular risk. Arterioscler Thromb Vasc Biol 30:733–741. 10.1161/ATVBAHA.109.199133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR 3rd, Takemori H, Okamoto M, Montminy M (2004) The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119:61–74. 10.1016/j.cell.2004.09.015 [DOI] [PubMed] [Google Scholar]
- Shi S, Luo W, Zhang R, Wang C, Zheng Y, Song Y, Wang R, Zhang L, Zhang L, Li W, Luo Z (2018) CRTC2 promotes non-small cell lung cancer A549 migration and invasion in vitro. Thorac Cancer 9:136–141. 10.1111/1759-7714.12550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E, Fasudil Ischemic Stroke Study G, Fasudil Ischemic Stroke Study Group (2005) Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci 238:31–39. 10.1016/j.jns.2005.06.003 [DOI] [PubMed] [Google Scholar]
- Shimada T, Takeshita Y, Murohara T, Sasaki K, Egami K, Shintani S, Katsuda Y, Ikeda H, Nabeshima Y, Imaizumi T (2004) Angiogenesis and vasculogenesis are impaired in the precocious-aging klotho mouse. Circulation 110:1148–1155. 10.1161/01.CIR.0000139854.74847.99 [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Sunamura S, Satoh K (2016) RhoA/Rho-kinase in the cardiovascular system. Circ Res 118:352–366. 10.1161/CIRCRESAHA.115.306532 [DOI] [PubMed] [Google Scholar]
- Sobrino T, Hurtado O, Moro MÁ, Rodríguez-Yáñez M, Castellanos M, Brea D, Moldes O, Blanco M, Arenillas JF, Leira R, Dávalos A, Lizasoain I, Castillo J (2007) The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke 38:2759–2764. 10.1161/STROKEAHA.107.484386 [DOI] [PubMed] [Google Scholar]
- Spindler V, Schlegel N, Waschke J (2010) Role of GTPases in control of microvascular permeability. Cardiovasc Res 87:243–253. 10.1093/cvr/cvq086 [DOI] [PubMed] [Google Scholar]
- Stokowska A, Atkins AL, Morán J, Pekny T, Bulmer L, Pascoe MC, Barnum SR, Wetsel RA, Nilsson JA, Dragunow M, Pekna M (2017) Complement peptide C3a stimulates neural plasticity after experimental brain ischaemia. Brain 140:353–369. 10.1093/brain/aww314 [DOI] [PubMed] [Google Scholar]
- Takemori H, Katoh Hashimoto Y, Nakae J, Olson EN, Okamoto M (2009) Inactivation of HDAC5 by SIK1 in AICAR-treated C2C12 myoblasts. Endocr J 56:121–130. 10.1507/endocrj.k08e-173 [DOI] [PubMed] [Google Scholar]
- Tasoulas J, Rodon L, Kaye FJ, Montminy M, Amelio AL (2019) Adaptive transcriptional responses by CRTC coactivators in cancer. Trends Cancer 5:111–127. 10.1016/j.trecan.2018.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedt S, Prestel M, Malik R, Schieferdecker N, Duering M, Kautzky V, Stoycheva I, Böck J, Northoff BH, Klein M, Dorn F, Krohn K, Teupser D, Liesz A, Plesnila N, Holdt LM, Dichgans M (2017) RNA-Seq identifies circulating miR-125a-5p, miR-125b-5p, and miR-143-3p as potential biomarkers for acute ischemic stroke. Circ Res 121:970–980. 10.1161/CIRCRESAHA.117.311572 [DOI] [PubMed] [Google Scholar]
- Uebi T, Tamura M, Horike N, Hashimoto YK, Takemori H (2010) Phosphorylation of the CREB-specific coactivator TORC2 at Ser(307) regulates its intracellular localization in COS-7 cells and in the mouse liver. Am J Physiol Endocrinol Metab 299:E413–E425. 10.1152/ajpendo.00525.2009 [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Draijer R, Vermeer MA, van Hinsbergh VW (1998) Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res 83:1115–1123. 10.1161/01.RES.83.11.1115 [DOI] [PubMed] [Google Scholar]
- Vo N, Goodman RH (2001) CREB-binding protein and p300 in transcriptional regulation. J Biol Chem 276:13505–13508. 10.1074/jbc.R000025200 [DOI] [PubMed] [Google Scholar]
- Wang Y, Inoue H, Ravnskjaer K, Viste K, Miller N, Liu Y, Hedrick S, Vera L, Montminy M (2010) Targeted disruption of the CREB coactivator Crtc2 increases insulin sensitivity. Proc Natl Acad Sci USA 107:3087–3092. 10.1073/pnas.0914897107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang X, Liu J, Zhao X, Yu H, Cai Y (2019) A systems analysis of the relationships between anemia and ischemic stroke rehabilitation based on RNA-Seq data. Front Genet 10:456. 10.3389/fgene.2019.00456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Liang M, Zhang Y, Huang K, Wang C (2019) Endothelial FAM3A positively regulates post-ischaemic angiogenesis. EBioMedicine 43:32–42. 10.1016/j.ebiom.2019.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Xiao P, Fan S, Chen Y, Huang W, Chen X, Liu G, Dang C, Zeng J, Xing S (2020) Blockade of Nogo-A/Nogo-66 receptor 1 (NgR1) inhibits autophagic activation and prevents secondary neuronal damage in the thalamus after focal cerebral infarction in hypertensive rats. Neuroscience 431:103–114. 10.1016/j.neuroscience.2020.02.010 [DOI] [PubMed] [Google Scholar]
- Yagita Y, Kitagawa K, Sasaki T, Terasaki Y, Todo K, Omura-Matsuoka E, Kaibuchi K, Hori M (2007) Rho-kinase activation in endothelial cells contributes to expansion of infarction after focal cerebral ischemia. J Neurosci Res 85:2460–2469. 10.1002/jnr.21375 [DOI] [PubMed] [Google Scholar]
- Zhong Y, Yu C, Qin W (2019) LncRNA SNHG14 promotes inflammatory response induced by cerebral ischemia/reperfusion injury through regulating miR-136-5p/ROCK1. Cancer Gene Ther 26:234–247. 10.1038/s41417-018-0067-5 [DOI] [PMC free article] [PubMed] [Google Scholar]