

Abstract
Oxaliplatin, a platinum-based chemotherapeutic drug, which is used as first-line treatment for some types of colorectal carcinoma, causes peripheral neuropathic pain in patients. In addition, an acute peripheral pain syndrome develop in almost 90% of patients immediately after oxaliplatin treatment, which is poorly understood mechanistically but correlates with incidence and severity of the later-occurring neuropathy. Here we investigated the effects of acute oxaliplatin treatment in a murine model, showing that male and female mice develop mechanical hypersensitivity 24 h after oxaliplatin treatment. Interestingly, we found that the levels of several lipids were significantly altered in nervous tissue during oxaliplatin-induced acute pain. Specifically, the linoleic acid metabolite 9,10-EpOME (epoxide of linoleic acid) as well as the lysophospholipids lysophosphatidylcholine (LPC) 18:1 and LPC 16:0 were significantly increased 24 h after oxaliplatin treatment in sciatic nerve, DRGs, or spinal cord tissue as revealed by untargeted and targeted lipidomics. In contrast, inflammatory markers including cytokines and chemokines, ROS markers, and growth factors are unchanged in the respective nervous system tissues. Importantly, LPC 18:1 and LPC 16:0 can induce Ca2+ transients in primary sensory neurons, and we identify LPC 18:1 as a previously unknown endogenous activator of the ligand-gated calcium channels transient receptor potential V1 and M8 (transient receptor potential vanilloid 1 and transient receptor potential melastatin 8) in primary sensory neurons using both pharmacological inhibition and genetic knockout. Additionally, a peripheral LPC 18:1 injection was sufficient to induce mechanical hypersensitivity in naive mice. Hence, targeting signaling lipid pathways may ameliorate oxaliplatin-induced acute peripheral pain and the subsequent long-lasting neuropathy.
SIGNIFICANCE STATEMENT The first-line cytostatic drug oxaliplatin can cause acute peripheral pain and chronic neuropathic pain. The former is causally connected with the chronic neuropathic pain, but its mechanisms are poorly understood. Here, we performed a broad unbiased analysis of cytokines, chemokines, growth factors, and ∼200 lipids in nervous system tissues 24 h after oxaliplatin treatment, which revealed a crucial role of lysophospholipids lysophosphatidylcholine (LPC) 18:1, LPC 16:0, and 9,10-EpOME in oxaliplatin-induced acute pain. We demonstrate for the first time that LPC 18:1 contributes to the activation of the ion channels transient receptor potential vanilloid 1 and transient receptor potential melastatin 8 in sensory neurons and causes mechanical hypersensitivity after peripheral injection in vivo. These findings suggest that the LPC-mediated lipid signaling is involved in oxaliplatin-induced acute peripheral pain.
Keywords: linoleic acid metabolites, lysophospholipid, oxaliplatin-induced pain, oxidized lipids, pain, TRPV1
Introduction
Chemotherapy-induced peripheral pain (CIPN) is a severe dose- and therapy-limiting side effect of cytostatic drugs (Windebank and Grisold, 2008; Park et al., 2013; Marmiroli et al., 2017). Oxaliplatin is widely used as a first-line treatment for colorectal carcinoma and can cause two different types of pain syndromes, an acute peripheral pain syndrome and a chronic neuropathic pain syndrome (Brewer et al., 2016). A large clinical trial revealed that an acute peripheral pain develops in up to 89% of patients after oxaliplatin treatment. The study also showed that 80% of the patients who already have acute peripheral pain have a significantly higher risk of development of a severe chronic neuropathy after oxaliplatin treatment (Argyriou et al., 2013; Pachman et al., 2015), showing a causal connection between acute peripheral pain and chronic peripheral neuropathic pain caused by oxaliplatin (Pachman et al., 2015).
The underlying molecular mechanisms leading to CIPN are still poorly understood. Nevertheless, it is known that ligand-gated ion channels, such as the transient receptor potential (TRP) channels, may contribute to oxaliplatin-induced neuropathic pain (Anand et al., 2010; Descoeur et al., 2011; Nassini et al., 2011; Zhao et al., 2012; Basso and Altier, 2017). TRP channels can be activated through chemical stimuli such as capsaicin (TRP vanilloid 1), allyl isothiocyanate (TRP ankyrin 1), and menthol (TRP melastatin 8). Several studies could also demonstrate that under pathophysiological conditions, TRP channel sensitization and activation caused by inflammatory mediators, such as cytokines, chemokines, and proinflammatory neuropeptides as well as endogenous lipid mediators, may lead to neurogenic inflammation and increased activity of peripheral sensory neurons and may be responsible for mechanical hypersensitivity in CIPN (Julius, 2013; Ji et al., 2014; Sisignano et al., 2016; Hohmann et al., 2017). Interestingly, several groups of signaling lipids, such as eicosanoids, linoleic acid metabolites, and sphingolipids have previously been demonstrated to contribute to pain hypersensitivity (Patwardhan et al., 2010; Sisignano et al., 2016; Hohmann et al., 2017; Ramsden et al., 2017; Stockstill et al., 2018; Zimmer et al., 2018).
We hypothesize that changes in neuronal plasticity and activity of sensory neurons occur early after oxaliplatin treatment. These alterations in neuronal activity may lead to mechanical hypersensitivity in oxaliplatin-induced acute peripheral pain and may prime the peripheral nervous system to cause increased occurrence and severity of the later occurring chronic neuropathy.
Materials and Methods
Animals
All animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (Kilkenny et al., 2010), and the recommendations of the Preclinical Pain Research Consortium for Investigating Safety and Efficacy Working Group (PPRECISE; Andrews et al., 2016), and were approved by the local Ethics Committees for Animal Research (Darmstadt, Germany).
The experimental C57BL/6NJ male and female mice were purchased from the commercial breeding company Janvier. TRPV1 (transient receptor potential vanilloid receptor 1) male knock-out mice were bred in-house and originally purchased from The Jackson Laboratory (B6.129 × 1.Trpv1tm1Jul/J). TRPM8 (transient receptor potential melastatin 8) male knock-out 8-week-old mice (B6.129P2-Trpm8tm1Jul/J) were purchased from The Jackson Laboratory. Food and water were available ad libitum. The animals housed on a 12 h light/dark cycle.
For behavioral experiments and targeted lipidomics analysis, 8- to 12-week-old male and female mice were used. For untargeted lipidomics, quantitative PCR (qPCR), and multiplex assay, 8- to 12-week-old adult mice were killed. For spinal cord electrophysiology [spontaneous EPSC (sEPSC) measurements], 5- to 6-week-old mice were used.
Oxaliplatin-induced neuropathic pain model
Oxaliplatin was dissolved in autoclaved deionized water as a stock solution of 3 mg/ml and diluted 1:4 in saline [sodium chloride 0.9% (v/v); Fresenius Kabi]. A dose of 3 mg/kg oxaliplatin or saline (vehicle) was injected intraperitoneally to C57BL/6NJ wild-type mice, as described previously (Nassini et al., 2011). Application of the model and the following tests is in accordance with the suggestions from the PPRECISE Working Group (Andrews et al., 2016).
Behavioral tests
To determine mechanical pain thresholds, the dynamic plantar test was performed. Mice were put on a wire mesh floor 1–2 h before the measurement of the mechanical pain threshold to allow accommodation. The mid-plantar hindpaw area was stimulated with ascending force reaching 5 × g after 10 s by using a Dynamic Plantar Aesthesiometer (Ugo Basile). The time between touching the mid-plantar hindpaw and a fast withdrawal response was recorded. The cutoff time was set to 20 s, as described previously (Sisignano et al., 2012).
The determination of cold hypersensitivity was assessed using the dynamic cold plate test and the acetone test. The dynamic cold plate test was performed as reported previously (Sisignano et al., 2016). Briefly, mice were put on a cooled steel plate. The plate temperature gradually decreased from 25 to 0°C at a rate of 12°C/min. The time was measured until nocifensive responses, such as fast paw withdrawal, flinching, and jumping, occurred. The cutoff time was set to 300 s.
The acetone evaporation test was modified according to Flatters and Bennett, 2004; (Flatters and Bennett, 2004). Briefly, animals were put on a wire mesh floor for accommodation 1–2 h before cold hypersensitivity measurement. On the center of the mid-plantar hindpaws, a small drop of acetone (Carl Roth) was applied. The nocifensive response of mice was scored using the following system: if the mice did not withdraw, flinch, or shake their hindpaw within the first 20 s after acetone application, the behavioral response was scored as 0. Once the mice responded to the acetone, they were observed again for 20 s and assessed into the following four different classes: (1) quick withdrawal, flick, or stamp of the paw; (1.5) prolonged withdrawal; (2) prolonged withdrawal and repeated flicking (>2) of the paw; and (3) repeated flicking of the paw with licking directly at the ventral side of the paw.
To investigate the effect of 10 and 50 μm lysophosphatidylcholine (LPC) 18:1 (1-oleoyl-2-hydroxy-sn-glycero-3-phosphocholine; Avanti Polar Lipids) we injected 10 µl of the lipid subcutaneously into the plantar surface of the left hindpaw of wild-type C57BL/6NJ mice using a 25 µl syringe (Hamilton). LPC 18:1 was first dissolved in ethanol (absolute 99.8%; Thermo Fisher Scientific) and then diluted in saline to reach the desired concentrations. As vehicle control, the corresponding volume of ethanol was added to saline and 10 µl of vehicle solution was injected subcutaneously into the plantar surface of the left hindpaw of control animals. The mechanical pain threshold was measured 0.5–6 h after LPC 18:1 or vehicle injection using the Dynamic Plantar Aesthesiometer (Ugo Basile; Brenneis et al., 2011).
Primary dorsal root ganglion cultures
Dorsal root ganglia (DRGs) were dissected and transferred into ice-cooled HBSS (Thermo Fisher Scientific) containing CaCl2 and MgCl2 as described previously (Sleigh et al., 2016). Dissected DRGs were treated with collagenase (500 U/ml; Sigma-Aldrich) and dispase (2.5 U/ml; Roche) for 75 min at 37°C. After removing the collagenase/dispase mixture, the DRGs were washed twice with 10 ml of Neurobasal medium (Thermo Fisher Scientific) containing 1% (v/v) Pen-Strep (Thermo Fisher Scientific) and 10% (v/v) of inactivated FCS (Thermo Fisher Scientific). Before washing the DRGs again twice with 10 ml of Neurobasal medium, they were incubated for 10 min at 37°C with 0.05% trypsin-EDTA (Thermo Fisher Scientific). Following this, the DRGs were mechanically dissociated and plated on poly-l-lysine (Sigma-Aldrich) precoated glass coverslips. After incubating the DRGs at 37°C for 2 h, 2 ml of Neurobasal medium containing 1% (v/v) Pen-Strep, 1% l-glutamine (Thermo Fisher Scientific), and B27 supplement (Thermo Fisher Scientific) were added and the DRGs were incubated over night at 37°C and 5% CO2 (Zimmer et al., 2018).
Cultivation of HEK293 cells
HEK293 cells were cultivated in 10 mm dishes with DMEM (1×)+GlutaMAX-I (Thermo Fisher Scientific) medium containing 10% FCS (Thermo Fisher Scientific) and 1% Pen-Strep (Thermo Fisher Scientific) at 37°C and 5% CO2.
HEK293 transfection for Ca2+ imaging
For Ca2+ imaging in vitro studies, a heterologous expression system with transient receptor potential ankyrin 1 (TRPA1)-, TRPV1-, and TRPM8-transfected HEK293 cells was used. Transfection of HEK293 cells was performed as previously described (Hohmann et al., 2017). Before HEK293 transfection, cells were plated on poly-l-lysine glass coverslips (two coverslips per one six-well dish) with a confluency of 40% and then incubated for 2 h at 37°C. Next, HEK293 cells were transfected by using 1 µg of pcDNA3.1(+)-(h)TRPA1, pCMV6-AC-GFP-(h)TRPV1 (OriGene Technologies), and pEGFP-C1-(r)TRPM8 as well as pCDNA3.1(+)GFP empty vector (Addgene) in 100 µl serum-free DMEM (1×)+GlutaMAX and 5 µl TurboFect (Thermo Fisher Scientific) per six-well coverslip dish. For DNA complex formation, TurboFect and plasmid–DNA mixtures were incubated for 20 min at RT. Then, 100 µl of transfection solution was added dropwise to each coverslip six-well dish after adding 2 ml of HEK293 medium containing 10% FCS (Thermo Fisher Scientific) and 1% Pen-Strep (Thermo Fisher Scientific) to the plated HEK293 cells. After 24 h, transfection was controlled by GFP detection by using an Axio Observer.Z1 Microscope (Carl Zeiss). For image acquisition, Zen 2.3 software (Carl Zeiss) was used. For Ca2+ imaging experiments, the cells were stained 48 h after transfection with Fura-2-AM (Biotium) for 40 min.
Calcium imaging
After staining the neurons with fura-2 AM (Biotium) for 45–60 min, the plated DRGs were washed twice with Ringer's solution, pH 7.3, containing 145 mm NaCl, 1.25 mm CaCl2·2 H2O, 1 mm MgCl2·6 H2O, 5 mm KCl, 10 mm d-glucose (all from Sigma-Aldrich) and 10 mm HEPES (AppliChem). Baseline measurements were performed using Ringer's solution. The DRGs were stimulated for 45–60 s with 1, 5, and 10 μm of LPC 18:1 and LPC 16:0 (1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine; both from Avanti Polar Lipids) or with their corresponding volume of vehicle (EtOH). For the identification of TRP channels reacting on 5 μm LPC 18:1, DRGs were costimulated with allyl isothiocyanate (AITC; 100 μm; TRPA1 agonist) for 30 s and with capsaicin (100 nm; TRPV1 agonist; both from Sigma-Aldrich) for 20 s. Between each stimulation, neurons were washed with Ringer's solution for 5–6 min. For further characterization of LPC 18:1-dependent responses of TRP channels, we performed in vitro Ca2+ imaging experiments using a heterologous expression system. We therefore transfected human TRPA1 [(h)TRPA1], (h)TRPV1, and rat TRPM8 [(r)TRPM8] as well as empty vector in HEK293 cells that were stimulated with 30 μm LPC 18:1 or vehicle for 60 s. Afterward, transfected HEK293 cells were stimulated with the TRP channel-specific agonists (TRPA1, 100 μm AITC; 30 s; TRPV1, 300 nm capsaicin, 20 s; TRPM8, 300 μm menthol, 30 s; all from Sigma-Aldrich). For TRP channel inhibition, neurons were preincubated for 2 min with 1 μm AMG 9810 (TRPV1 blocker; Tocris Bioscience), 20 μm HC 030031 (TRPA1 blocker; Tocris Bioscience), or 1 μm AMTB [N-(3-aminopropyl)-2-((3-methylbenzyl)oxy)-N-(thiophen-2-ylmethyl)benzamide hydrochloride] hydrochloride (TRPM8 blocker; Tocris Bioscience). Afterward, neurons were directly stimulated with 5 μm LPC 18:1. For proper TRP channel identification after LPC 18:1 and LPC 16:0 stimulation (45–60 s), only TRPV1 and TRPM8 channel and a combination of both inhibitors were used. For these experiments, neurons were stimulated with 5 μm LPC 18:1 or 5 μm LPC 16:0 before and after preincubation with the TRPV1 or TRPM8 channel inhibitors or both inhibitors together. For a functionality test of the TRP channel inhibitors, neurons were stimulated with the corresponding agonists of the TRP channels TRPV1 (100 nm capsaicin, 20 s; Sigma-Aldrich), TRPA1 (100 μm AITC, 30 s; Sigma-Aldrich), and TRPM8 (100 μm menthol, 30 s; Sigma-Aldrich) before and after preincubating the neurons with the corresponding TRP channel inhibitors. All stimulating compounds were dissolved in Ringer's solution to their final concentration. Ca2+ imaging measurement was performed using a DMI4000 B Microscope and the compact light source CTR550 HS (Leica). For perfusion, the ValveBank II system (AutoMate Scientific) was used.
Patch-clamp recordings in spinal cord slices
Mice (5–7 weeks old) were anesthetized with urethane (1.5–2.0 g/kg, i.p.). The lumbosacral spinal cord was quickly removed and placed in ice-cold ACSF dissection solution (sucrose 240 mm, NaHCO3 25 mm, KCl 2.5 mm, NaH2PO4 1.25 mm, CaCl2 0.5 mm, and MgCl2 3.5 mm). After spinal extraction under anesthesia, animals were killed by decapitation. Transverse spinal slices (400 μm) were prepared using a vibratome (catalog #VT1200S, Leica). The slices were incubated at 34°C for at least 60 min in ACSF (NaCl 126 mm, KCl 3 mm, MgCl2 1.3 mm, CaCl2 2.5 mm, NaHCO3 26 mm, NaH2PO4 1.25 mm, and glucose 11 mm) before recordings. The slices were placed in a recording chamber and completely submerged and perfused at a flow rate of 2–4 ml/min with ACSF. All extracellular solutions were constantly carbogenated (95% O2, 5% CO2). Lamina II neurons in lumbar segments were identified as a translucent band under a microscope (model BX51WIF, Olympus). As previously recorded (Chen et al., 2016; Sisignano et al., 2016), whole-cell voltage-clamp recordings were made from outer lamina II neurons by using patch pipettes fabricated from thin-walled, fiber-filled capillaries. The patch pipette solution used to record sEPSCs contained the following: K-gluconate 135 mm, KCl 5 mm, CaCl2 0.5 mm, MgCl2 2 mm, EGTA 5 mm, HEPES 5 mm, and Mg-ATP 5 mm, at pH 7.3 adjusted with KOH, 300 mOsm. The patch pipettes had a resistance of 4–8 m. The sEPSCs were recorded at a holding potential of −70 mV in the presence of 10 μm picrotoxin. Signals were acquired using a HEKA EPC-10 amplifier and Patchmaster software. sEPSC events were detected and analyzed using Mini Analysis (Synaptosoft).
Calcitonin gene-related peptide-ELISA
Calcitonin gene-related peptide (CGRP) release was measured using a CGRP enzyme immune assay kit (Bertin Bioreagent) and performed according to the manufacturer recommendations. Briefly, DRGs of C57BL/6NJ mice were dissected and cultivated as described previously. The cultivated DRGs were then resuspended in Neurobasal medium and the resuspended DRGs were plated on a poly-l-lysine-coated (Sigma-Aldrich) 48-well plate at 250 µl/well. After incubating the DRGs for 2 h at 37°C, 300 µl of Neurobasal medium (Thermo Fisher Scientific) were added to the DRGs. After incubation for 24 h at 37°C, DRGs were stimulated for 15 min at 37°C with 250 µl of capsaicin (400 nm), LPC 18:1 (5 μm; Avanti Polar Lipids), vehicle (DMSO + ethanol), and capsaicin (400 nm) + LPC 18:1 (5 μm) to determine the CGRP release.
Quantitative real-time PCR
RNA of dissected DRGs from vehicle- and oxaliplatin-treated animals was isolated using the Invitrogen mirVana miRNA Isolation Kit (Thermo Fisher Scientific) and following the manufacturer instructions. For cDNA syntheses, the First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) was used. Finally, the qPCR analysis of calcium-independent phospholipase A2 (iPLA2), cytosolic phospholipase A2 (cPLA2), 5-lipoxygenase (LOX), 15-LOX, 12-LOX, cyclooxygenase-2 (COX-2), CYPs (cytochrome P450s), soluble epoxide hydrolase (sEH), tumor necrosis factor-α (TNF-α), nerve growth factor (NGF), interferon-γ (IFN-γ), NADPH oxidase 2 (NOX2), NOX4, xanthine dehydrogenase (XDH), inducible nitric oxide synthase (iNOS), activating transcription factor 3 (ATF3), matrix metalloprotease 3 (MMP3), and MMP9 transcripts was performed using assay primers for the TaqMan system (Thermo Fisher Scientific) on a QuantStudio 5 Real-Time PCR system (Thermo Fisher Scientific) and the software QuantStudio Design and Analysis Software version 1.4.3 (Thermo Fisher Scientific). Expression analysis was performed using the ΔΔC(T)-method (Livak and Schmittgen, 2001).
Multiplex assay
For Invitrogen ProcartaPlex Multiplex Immunoassay (Thermo Fisher Scientific) isolated proteins of spinal cords and DRGs of six vehicle and six oxaliplatin-treated animals were used after 24 h. For protein isolation, dissected DRGs were lysed using 150 µl of lysis buffer consisting of PhosphoSafe (Merck) and 1× protease inhibitor (Roche). For dissected spinal cords, 200 µl of lysis buffer was used. Then, tissue was homogenized with a Sonopuls Sonicator (Bandelin) by using energy of 6 × 10% for 10 s two times. During sonication, tissue was held in an ice-cooled water beaker. Afterward, samples were centrifuged for 10 min at 10,000 rpm and supernatant was collected. For protein concentration measurement, a Bradford assay was performed. The ProcartaPlex multiplex immunoassay was performed according to manufacturer instructions. Briefly, standards of all analytes were prepared in a 1:4 serial dilution in 200 µl of Universal Assay Buffer/lysis buffer (1:1, v/v). Afterward, 100 µl of the mixed magnet bead vials Mix B and Mix D were added into each well. Then, the 96-well plate was placed for 2 min on a magnet holder for magnetic bead accumulation. After removing the liquid, the wells were washed with 150 µl of wash buffer. Fifty microliters of samples, standards, and blanks were added into the respective wells. The plate was incubated for 2 h at room temperature (RT) on an orbital shaker (NeoLab) at 180 rpm and washed for three times as described above. Then, 25 µl of detection antibody mixture was added into each well. Again, the plate was washed three times after incubating the plate for 30 min at room temperature on an orbital plate shaker at 180 rpm. This step was repeated after adding 50 µl of Streptavidin Phycoerythrin (SAPE) into each well. Afterward, the plate was prepared for the Luminex instrument (Bio-Rad) by adding 120 µl of reading buffer into each well and incubating the 96-well plate a last time for 5 min on an orbital shaker at 180 rpm and room temperature.
Immunohistochemistry
Immunohistochemistry was modified as previously reported (Liu et al., 2019). Mice were killed and the spinal canal was opened, beginning at the neck. Afterward, the spinal cord of the whole lumbar L1–L2 segment was carefully dissected. Spinal cord of vehicle- and oxaliplatin-treated animals were used. The L1–L2 spinal cord segments were transferred into a 20% sucrose (Sigma-Aldrich) solution for 24 h at 4°C. Then the spinal cord was embedded vertically, into Tissue-Tek Cryomold molds (Sakura Finetek Europe) in Tissue-Tek O.C.T. Compound (Sakura Finetek Europe). Afterward, the spinal cord tissue was sliced into 12 µm sections using the Cryostat (catalog #CM30505, Leica). The sliced spinal cord tissue was fixed with a 2% PFA (Carl Roth) in PBS (Thermo Fisher Scientific) solution for 15 min. After washing the slices with PBS, the spinal cord tissue was permeabilized with a 0.1% Triton X-100 (Sigma-Aldrich) in PBS solution for 10 min. For nonspecific binding of antibodies, the spinal cord tissue was blocked with a 3% BSA in PBS solution for 2 h at RT. Next, spinal cord slices were incubated with the primary antibody glial fibrillary acidic protein (GFAP; 1:1000; rabbit; catalog #NB300-141, Novus Biologicals) in a 1% BSA in PBS solution at 4°C overnight. The incubation with the secondary antibody anti-rabbit Cy3 (1:1000; sheep; catalog #C2306-1ML, Sigma-Aldrich) in a 1% BSA in PBS solution occurred at RT for 1 h in the dark. Nuclear staining was performed by incubating spinal cord tissue for 8 min with the fluorescent dye DAPI (1:1000; Carl Roth).
Fluorescence images were captured with an Axio Observer.Z1 microscope (catalog #3834002187, Carl Zeiss) and the compact light source HXP 120 (catalog #091580751055, Lighting and Electronics Jena). For image acquisition, Zen 2.3 software (Carl Zeiss) was used.
Liquid chromatography-tandem mass spectrometry of prostanoids and linoleic acid metabolites
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of prostanoids in sciatic nerve, DRG, and dorsal spinal cord tissue samples was in general performed as described previously (Tarighi et al., 2019). Briefly, tissue was dissected, weighted, and immediately transferred into liquid nitrogen. Upon dissection of spinal cord tissue, the ventral and dorsal parts were carefully separated. Tissue samples were stored at −80°C until lipids were extracted. For lipid extraction, tissue samples were completely homogenized with 200 µl of water/ethanol (3:1, v/v). The homogenized tissue samples were extracted using liquid–liquid extraction with 2 × 600 µl of ethyl acetate after adding 100 µl of EDTA and 20 µl of internal standard solution (PGE2-d4, PGD2-d4, TXB2-d4, PGF2α-d4, 6-keto PGF1α-d4, all purchased from Cayman Chemicals) and 100 µl 0.15 m EDTA solution. The extraction step was repeated, organic layers were collected, evaporated at 45°C under a gentle stream of nitrogen, and reconstituted with 50 µl of acetonitrile/water/formic acid (20:80:0.0025, v/v) and then injected into the LC-MS/MS system. For measurement of the linoleic acid metabolites stock solutions with 25 µg/ml analytes 9,10-EpOME, 9,10-DiHOME, 12,13-EpOME, 12,13-DiHOME, 13-HODE (hydroxyoctadecadienoic acid), and 9-HODE as well as the deuterated internal standards 9,10-EpOME-d4 9,10-DiHOME-d4, 12,13-EpOME-d4, 12,13-DiHOME-d4, 13-HODE-d4, and 9-HODE-d4 were prepared in methanol/butylated hydroxytoluene (BHT; 100:0.1 v/v). Sample pretreatment was performed using liquid–liquid extraction with ethyl acetate. The combined organic phases were removed at a temperature of 45°C under a gentle stream of nitrogen. The residues were reconstituted with 50 µl of methanol/water/BHT (50:50:10^4, v/v/v), and then injected into the LC-MS/MS system. For LC-MS/MS analysis, an Agilent 1200 LC system coupled to a hybrid triple quadrupole linear ion trap mass spectrometer QTrap 5500 (Sciex) equipped with a Turbo-V-source operating in negative electrospray ionization mode was used. Chromatographic separation of prostanoids was done using a Synergi Hydro-RP column (2.0 × 150 mm, 4 µm particle size; Phenomenex), coupled with a precolumn of the same material; 0.0025% formic acid and acetonitrile with 0.0025% formic acid served as mobile phases. Chromatographic separation was achieved under gradient conditions. The gradient program started with 90% A for 1 min, then mobile phase A was decreased to 60% within 1 min, held for 1 min, further decreased to 50% within 1 min, and held for 2 min. Within 2 min, mobile phase A was further decreased to 10% and held for 1 min. Within 1 min, the initial conditions were restored, and the column was re-equilibrated for 6 min. Mass spectrometric parameters were as follows: ionspray voltage, −4500 V; source temperature, 500°C; curtain gas, 35 psi; nebulizer gas, 40 psi; and Turboheater gas, 60 psi. Both quadrupoles were running at unit resolution. For the chromatographic separation of the linoleic acid metabolites, a Gemini NX C18 column and precolumn (150 × 2 mm inner diameter, 5 µm particle size, and 110 Å pore size; Phenomenex) were used. A linear gradient was used at a flow rate of 0.5 ml/min with a total run time of 17.5 min. Mobile phase A consists of water/ammonia (100:0.05, v/v), and mobile phase B of acetonitrile/ammonia (100:0.05, v/v). The gradient changed from 85% A to 10% within 12 min. These conditions were held for 1 min. Then, the mobile phase shifted back to 85% A within 0.5 min, and it was maintained for 4 min to re-equilibrate the column. For the analysis of the lipids, Analyst Software 1.6.3 and Multiquant Software 3.0.2 (both from Sciex) were used, using the internal standard method (isotope-dilution mass spectrometry).
LC- quadruple time of flight MS
The LC-quadruple time of flight MS (QTOFMS) measurement was performed as described previously (Hahnefeld et al., 2020). Briefly, tissue samples of spinal cord, sciatic nerve, and DRGs were weighed and adjusted to a tissue concentration of 0.025 mg/µl with water/ethanol (3:1, v/v). Shredding was performed on a Mixer Mill MM 400 (Retsch) with a frequency of 25/s for 2.5 min and four zirconium dioxide grinding seeds per tube. Forty microliters of the resulting homogenate was extracted using 150 µl of methanol (LC-MS grade; Carl Roth) containing one internal standard per lipid class (Avanti Polar Lipids), 500 µl methyl-tert-butyl-ether (MTBE; HPLC grade; Carl Roth) and 125 µl of 50 mm ammonium formate (for mass spectrometry; Sigma-Aldrich). After re-extraction of the lower phase with 200 µl of MTBE/methanol/water (10:3:2.5, v/v/v), the combined upper organic phases were split into two aliquots for measurement in positive and negative ionization modes, respectively. The samples were dried under a nitrogen stream at 45°C and stored at −20°C. To verify the system stability, six replicates of the pooled homogenate per tissue type were extracted and analyzed as quality control samples. Before injection, the samples were dissolved in 120 µl of methanol. The chromatographic separation was performed on a Nexera X2 system (Shimadzu) using a Zorbax RRHD Eclipse Plus C8 1.8 µm 50 × 2.1 mm inner diameter column (Agilent), which was maintained at 40°C. A 17 min linear gradient was applied with a flow rate of 0.3 ml/min and 10 mm ammonium formate containing 0.1% formic acid (98–100%; AppliChem) in water and 0.1% formic acid in acetonitrile/isopropanol (2:3, v/v; acetonitrile, ultra LC-MS grade, Biosolve; isopropanol, Carl Roth) as mobile phases. For the negative ion mode, the mobile phase A was changed to 1 mm ammonium formate and 0.1% formic acid in water. The samples were analyzed on a TripleTOF 6600 (Sciex) using electrospray ionization operated in positive and negative modes. The acquisition method consisted of a scan ranging from 100 to 1000 mass-to-charge ratio (m/z) and six data-dependent spectra per cycle with a mass range from 50 to 1000 m/z and a collision energy of ±40 V with a 20 V spread.
Experimental design and statistical analysis
Throughout the whole study, all efforts were made to minimize bias. For the initial screen, a broad library of cytokines, chemokines, and growth factors was analyzed, and the use of untargeted and unbiased LC-QTOFMS allowed simultaneous analysis of >200 individual lipids in the nervous system tissue samples. Moreover, all animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institute of Health and the ARRIVE guidelines (Kilkenny et al., 2010) and the recommendations of the PPRECISE (Andrews et al., 2016), involving statistical power analysis and internal validity, as well as experimental blinding to reduce the risk of bias.
For behavior experiments a minimum number of six male or female C57BL/6NJ mice at the age of 8–12 weeks per group were measured.
For LC-QTOFMS data acquisition and processing Analyst TF 1.71 and MultiQuant 3.0.2 software were used with an extraction width of 0.01 Da and a retention time window of 20 s. The peak areas were normalized with a scale factor calculated from the median of the peak area ratio for each sample and a set reference sample for all peaks of the same ionization mode with a peak area >1% of the largest peak of the reference sample. A quality control of the respective tissue type was used as a reference sample. Normalization with one internal standard per lipid class showed comparable results, but the correction with the median peak ratio allowed for better visualization. Compound identification was performed with MasterView 1.1 software using a 5 ppm mass tolerance, isotopic distribution and by comparison of the MS/MS spectra to a custom build library crosschecked with LIPID MAPS (http://www.lipidmaps.org), METLIN (http://metlin.scripps.edu) and the Human Metabolome Database (HMDB, version 4.0). All software was obtained from Sciex.
All data are represented as the mean SEM. Group sizes were carefully selected according to the recommendations by the guidelines mentioned above. To determine statistically significant differences in all behavioral experiments two-way ANOVA for repeated measures was used followed by post hoc Bonferroni correction. For in vitro experiments comparing more than two groups one-way ANOVA was performed and for comparing only two groups two-tailed t test with Welch's correction was used. Data with a p value <0.05 was considered as statistically significant. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software).
Results
Mechanical threshold, cytokines, chemokines, and growth factors in oxaliplatin-induced acute pain
To investigate acute effects of oxaliplatin, we first assessed the mechanical threshold of vehicle- and oxaliplatin-treated male and female mice (n = 6–9 animals/group) in a well described and reproducible model (3 mg/kg, i.p.; Nassini et al., 2011) using the dynamic plantar test. We determined the paw withdrawal latency directly after intraperitoneal injection of oxaliplatin or vehicle at 2, 4, 6, 24, 26, 48, and 72 h. The oxaliplatin-treated male as well as female animals showed mechanical hypersensitivity in comparison to the vehicle mice starting 24 h after treatment (male, p = 0.0048; female, p < 0.0001; two-way ANOVA and Bonferroni post hoc test), which maintained until the end of the experiment at 72 h (male: 26 h, p = 0.0003; 48 h, p = 0.0003; 72 h, p = 0.0005; female: 26 h, p = 0.0004; 48 h, p = 0.0004; 72 h, p < 0.0001; two-way ANOVA and Bonferroni post hoc test; Fig. 1A). We could not observe any difference in the mechanical thresholds comparing male and female animals (p > 0.9999). Likewise, no difference between 24 and 72 h after oxaliplatin treatment could be assessed (Fig. 1A). Using the cold plate test and the acetone test, we also investigated the effect of acute oxaliplatin treatment on cold pain perception (n = 6 male animals/group). However, we could not detect any cold hypersensitivity after treatment (Extended Data Fig. 1-1A,B).
Figure 1.

Oxaliplatin-induced acute pain and lipid synthesis in nervous system tissue. A, Time course of the paw withdrawal latency (PWL) in seconds after dynamic plantar test of wild-type male and female mice after intraperitoneal injection of vehicle or oxaliplatin (3 mg/kg). The arrow at 24 h indicates the point in time of the subsequent lipidomics analysis. Data represent the mean SEM of 6–9 mice/group; male: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; female: ###p < 0.001; ####p < 0.0001; two-way ANOVA and Bonferroni's post hoc test (Extended Data Figure 1-1). Determination of cold allodynia in oxaliplatin-induced acute peripheral pain in male mice. B, Heat map showing the results of an unbiased LC-QTOFMS measurement from sciatic nerve, DRGs, and spinal cord 24 h after oxaliplatin or vehicle treatment from five mice per nervous system tissue. The numbers in brackets depict the amount of measured lipid species of each lipid subgroup. C, Distribution of free fatty acids of different chain lengths in sciatic nerve (blue), DRGs (red), and spinal cord (green) after oxaliplatin or vehicle treatment. D, Schematic overview of lipid oxygenase enzymes catalyzing arachidonic acid and linoleic acid metabolites 24 h after oxaliplatin treatment. E–I, Concentrations of prostanoids and OLAMs in sciatic nerve, DRGs, and spinal cord 24 h after intraperitoneal injection of vehicle and oxaliplatin (3 mg/kg) in male and female animals. Data represent the mean SEM of four to six mice per group; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA and Tukey's post hoc test (Extended Data Figure 1-4). Further prostanoid and linoleic acid metabolite concentrations 24 h after oxaliplatin treatment in male and female mice (Extended Data Figure 1-5). Concentrations of prostanoids in male and female mice 72 h after oxaliplatin treatment (Extended Data Figure 1-2). Analysis of inflammatory cytokines, chemokines, and growth factors (Extended Data Figure 1-3). Examination of dorsal spinal cord astrocytes with GFAP 24 h after oxaliplatin-induced acute peripheral pain. J, Expression levels of 5-LOX, 12-LOX, COX2, CYP2j6, and sEH in DRGs 24 h after intraperitoneal injection of oxaliplatin (3 mg/kg) or vehicle. The transcripts of the lipid oxygenases 15-LOX, CYP3a11, CYP2C37, CYP2C38, and CYP2C39 were measured but could not be detected. Data represent the mean SEM from DRGs of 5–13 mice/group and transcript; one-way ANOVA and Dunnett's multiple comparison test. ns, Not significant, n.d., not detectable.
Cold allodynia in oxaliplatin-induced acute peripheral pain. A, Cold-score time course after intraperitoneal injection of oxaliplatin (3 mg/kg) and vehicle. B, Paw withdrawal latency in seconds up to 48 h after intraperitoneal injection of oxaliplatin (3 mg/kg) and vehicle. Data represent the mean SEM of 6 mice/group; two-way ANOVA and Bonferroni's post hoc test. Download Figure 1-1, TIF file (148.7KB, tif) .
Involvement of chemokines, growth factors, and cytokines during oxaliplatin-induced acute peripheral pain. A–O, Measurement of different cytokines and chemokines in DRGs and spinal cords 24 h after oxaliplatin (3 mg/kg, i.p.) treatment. Granulocyte macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-2, TNF-α, and macrophage inflammatory protein 2 (MIP-2) were measured but not detectable. Data represent the mean SEM from DRGs and spinal cord of three to six mice per group; one-way ANOVA and Sidak's post hoc test. P, Relative expression levels of NGF, NOX2, NOX4, XDH, iNOS, ATF3, and MMP9 transcripts in DRGs of oxaliplatin-treated animals after 24 h. The transcripts TNF-α, IFN-γ, and MMP3 were measured but could not be detected. Data represent the mean ± SEM from DRGs of four to nine mice per group and transcript; one-way ANOVA and Dunnett's multiple comparison test. IL, Interleukin. Download Figure 1-2, TIF file (707KB, tif) .
Immunohistochemistry of dorsal spinal cord astrocytes with GFAP 24 h after oxaliplatin or vehicle treatment. A, Representative GFAP immunohistochemistry image of the dorsal spinal cord captured with 50× magnification. B, Representative GFAP immunohistochemistry image of the marked dotted square area in A with 100× magnification. C, Representative GFAP immunohistochemistry image of the marked dotted square area in B with 200× magnification. Data represent a representative image of spinal cord slices. Download Figure 1-3, TIF file (4.1MB, tif) .
Lipid syntheses of prostanoids and linoleic acid metabolites in oxaliplatin-induced acute peripheral pain after 24 h. A, B, Concentration in picograms per milligram of the prostanoids PGF2α and 6-keto PGF1α in sciatic nerve, DRGs, and spinal cord 24 h after intraperitoneal injection of vehicle and oxaliplatin (3 mg/kg) in male and female mice. C–F, Concentrations of linoleic acid-derived metabolites (12,13-EpOME, 12,13-DiHOME, 13-HODE, and 9-HODE) in sciatic nerve, DRGs, and spinal cord 24 h after intraperitoneal injection of vehicle and oxaliplatin in male and female mice. Data represent the mean SEM from DRGs, sciatic nerve, and spinal cord of 4–6 mice/group; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA and Tukey's post hoc test. Download Figure 1-4, TIF file (511.5KB, tif) .
Lipid syntheses of prostanoids in oxaliplatin-induced acute peripheral pain after 72 h. A–F, Concentration in picograms per milligram of the prostanoids PGE2, PGD2, TXB2, PGF2α, and 6-keto PGF1α in sciatic nerve, DRGs, and spinal cord 72 h after intraperitoneal injection of vehicle and oxaliplatin (3 mg/kg) in male and female mice. Data represent the mean SEM from DRGs, sciatic nerve, and spinal cord of three to five mice per group; *p < 0.05; ***p < 0.001; one-way ANOVA and Tukey's post hoc test. Download Figure 1-5, TIF file (289.7KB, tif) .
We next investigated the role of typical inflammatory mediators by performing a broad chemokine, growth factor, and cytokine analysis with dissected DRGs and spinal cords of oxaliplatin-treated male animals (24 h). The simultaneous analysis of 20 different cytokines, growth factors, and chemokines revealed no differences between vehicle and oxaliplatin-treated animals in DRGs and spinal cord (Extended Data Fig. 1-2A–O). Proinflammatory cytokines like TNF-α and IFN-γ were even under the limit of quantification and could not be detected. The expression analysis of the NGF transcript, which can be triggered by TNF-α and IL-1β (Gadient et al., 1990), revealed no difference compared with the vehicle-treated mice (Extended Data Fig. 1-1P). Moreover, we analyzed the mRNA expression level of enzymes producing reactive oxygen species (ROS) such as NOX2 and NOX4, iNOS, XDH as well as the neuronal stress marker ATF3 (Hoetzenecker et al., 2011) and MMP3 and MMP9, all of which are known to contribute to both neuronal stress and neuropathic pain (Ji et al., 2009; Salvemini et al., 2011; Yowtak et al., 2011). While the MMP3 transcript could not be detected, the transcripts of other ROS-responsible enzymes could be determined, but no differences between oxaliplatin- and vehicle-treated animals could be observed (Extended Data Fig. 1-2P). As previously reported, astrocyte activation occurred after paclitaxel acute pain syndrome and may contribute in proinflammatory cytokine release (Liu et al., 2019). However, it is controversial, whether or not astrocyte activation in the spinal cord occurs also after oxaliplatin treatment (Robinson et al., 2014; Makker et al., 2017). Using immunohistochemical staining of astrocytes with GFAP in dorsal spinal cord of oxaliplatin-treated mice, in our model we could not detect any differences in astrocytes activation after 24 h compared with vehicle-treated animals (Extended Data Fig. 1-3A–C).
Lipids are associated with oxaliplatin-induced acute pain
The chemokine, growth factor and cytokine screen revealed that the oxaliplatin-induced acute peripheral pain differs mechanistically from classical inflammatory pain. Since endogenous lipid mediators are known to contribute to pain hypersensitivity (Piomelli and Sasso, 2014), we next performed a broad unbiased LC-QTOFMS screen with DRGs, sciatic nerves, and spinal cords of oxaliplatin- and vehicle-treated animals after 24 h, allowing us to detect several hundred different lipids simultaneously. The analysis showed that free fatty acids were especially increased in DRGs of oxaliplatin-treated animals (Fig. 1B,C). We next determined the concentrations of the prostanoids, which can be synthesized from the polyunsaturated free fatty acid (20:4; Fig. 1D) using LC-MS/MS. Furthermore, these lipids are known to be increased during inflammation and can contribute to pain hypersensitivity (Chen et al., 2013; Piomelli and Sasso, 2014). In addition, similar to the cytokines and chemokines, prostanoids were not increased after oxaliplatin treatment. Instead, we could surprisingly observe that prostaglandin E2 (PGE2), PGD2, and thromboxane B2 (TXB2) were even significantly decreased in DRGs of oxaliplatin-treated male animals (PGE2, p = 0.0128; PGD2, p = 0.0073; TXB2, p = 0.0479; one-way ANOVA and Tukey's post hoc test; Fig. 1E–G). For female animals, a similar result could be observed 24 h after oxaliplatin treatment. Compared with the vehicle, PGE2 and PGD2 were significantly decreased in female mice after oxaliplatin treatment (PGE2, p = 0.0047; PGD2, p = 0.002; one-way ANOVA and Tukey's post hoc test). While TXB2 was significantly decreased in male mice, the TXB2 concentration was not altered when compared with vehicle in female mice. Consequently, the TXB2 concentration in female mice differs significantly from that in male mice 24 h after oxaliplatin treatment (p = 0.0077; one -way ANOVA and Tukey's post hoc test). The concentrations of PGF2α and 6-keto PGF1α were not altered in oxaliplatin-treated male as well as female animals 24 h after oxaliplatin treatment (Extended Data Fig. 1-4A,B).
In addition, we measured the concentrations of prostanoids of male and female mice 72 h after oxaliplatin treatment where a stable mechanical hypersensitivity could be observed. As already observed for PGD2 and PGE2, 24 h after oxaliplatin treatment, these two prostaglandins were significantly decreased in DRGs of male and female animals 72 h after oxaliplatin treatment (male: PGD2, p < 0.0001; PGE2, p < 0.0001; female: PGD2, p < 0.0001; PGE2, p = 0.0009; one-way ANOVA and Tukey's post hoc test; Extended Data Fig. 1-5A,B). In male mice, PGD2 was also significantly decreased in spinal cord when compared with vehicle (p = 0.0216; one-way ANOVA and Tukey's post hoc test), and TXB2 was significantly decreased only in DRGs of female animals (p = 0.0137; one-way ANOVA and Tukey's post hoc test; Extended Data Fig. 1-5C). For PGF2α and 6-keto PGF1α, no alteration in their concentrations could be observed in male or female mice 24 or 72 h after oxaliplatin treatment (Extended Data Fig. 1-5D,E). In accordance with the chemokine and cytokine screen, these findings support our conclusion that the acute peripheral pain after oxaliplatin treatment differs mechanistically from a classical inflammatory pain. Moreover, we could observe that the levels of the free fatty acid linoleic acid (18:2) was strongly increased in DRGs 24 h after oxaliplatin treatment (p = 0.0204; multiple t test; Fig. 1C). We therefore also investigated the concentrations of oxidized linoleic acid metabolites (OLAMs) in nervous system tissue by LC-MS/MS 24 h after oxaliplatin treatment from male and female animals (Fig. 1D). We found that 9,10-EpOME was significantly increased (male, p = 0.0007; female, p < 0.0001; one-way ANOVA and Tukey's post hoc test), whereas its metabolite 9,10-DiHOME was significantly decreased (male, p < 0.0001; female, p < 0.0001; one way-ANOVA and Tukey's post hoc test) in DRGs of oxaliplatin-treated male and female mice (Fig. 1H,I). Interestingly, we could not observe any differences in the concentrations of other linoleic acid metabolites such as HODEs between the DRGs of vehicle- and oxaliplatin-treated male animals (Extended Data Fig. 1-4C–F). However, for female mice significantly increased 12,13-EpOME and significantly decreased 12,13-DiHOME concentrations could be observed in DRGs after oxaliplatin treatment when compared with vehicle- and male-treated mice (12,13-EpOME: vehicle vs oxaliplatin female, p < 0.0001; oxaliplatin male vs oxaliplatin female, p < 0.0001; 12-13-DiHOME: vehicle vs oxaliplatin female, p = 0.0361; oxaliplatin male vs oxaliplatin female, p = 0.0189; one-way ANOVA and Tukey's post hoc test; Extended Data Fig. 1-4C,D), and 13-HODE was also significantly decreased in female mice after oxaliplatin treatment (vehicle vs oxaliplatin female, p = 0.0034; oxaliplatin male vs oxaliplatin female, p = 0.0004; one-way ANOVA and Tukey's post hoc test; Extended Data Fig. 1-4E). However, as already shown for male mice, 9-HODE was also not altered in DRGs of female mice after oxaliplatin treatment (Extended Data Fig. 1-4F).
Because of the different lipid concentrations 24 h after oxaliplatin treatment, we next investigated the mRNA expression of the related lipid oxygenases (COX-2, LOX, CYPs) in DRGs of oxaliplatin-treated animals (Fig. 1D,J). However, we could not observe any differences in the expression of any of the lipid oxygenases after oxaliplatin treatment (Fig. 1J). Likewise, we analyzed the mRNA expression level of the sEH2, which is responsible for the degradation of epoxides to diols (Imig and Hammock, 2009). The mRNA expression analysis revealed no differences in the expression of the sEH transcript in DRGs of oxaliplatin-treated animals (Fig. 1J). These results indicate, in agreement with the LC-QTOFMS measurement, that the elevated 9,10-EpOME concentrations in DRGs after oxaliplatin treatment are most likely because of increased abundance and availability of the substrate linoleic acid rather than increased expression and activity of lipid oxygenases.
Lysophosphatidylcholines and free fatty acid metabolites are associated with oxaliplatin-induced acute pain
Apart from the free fatty acids, LPCs were also strongly increased in sciatic nerve, DRGs, and spinal cord after oxaliplatin treatment (Fig. 1B). Specifically, the subgroup of LPCs was the most abundant lipid group, which could be detected among the lysophospholipid family (Fig. 2A). The LPC subgroup consists of several LPC species that differ in their chain length and double-bound position. Each LPC species has distinct properties, and species may have different functions (Law et al., 2019; Liu et al., 2020). Hence, we next analyzed the 19 different lipid species of the LPC subgroup. The specific analysis of the lipid species of the most abundant lipid family, the LPC subgroup, revealed that in sciatic nerve, as well as in DRGs, LPC 18:1 was significantly increased after oxaliplatin treatment (sciatic nerve, p = 0.0258; DRGs, p < 0.0001; one-way ANOVA and Sidak's post hoc test; Fig. 2B). In spinal cord, LPC 16:0 was significantly increased (p = 0.0252; one-way ANOVA and Sidak's post hoc test; Fig. 2C).
Figure 2.

Increased lysophosphatidylcholine levels. A, Distribution of lysophospholipid species in sciatic nerve, DRGs, and spinal cord after oxaliplatin treatment (the number in the pie chart corresponds to the amount of detected lipid species per lipid subgroup). B, C, LPC 18:1 and 16:0 in nervous system tissue after oxaliplatin or vehicle treatment. Data represent the mean SEM from DRGs, sciatic nerve, and spinal cord of five mice per group; *p < 0.05; ***p < 0.001; one-way ANOVA and Sidak's post hoc test. D, Schematic image of iPLA2- and cPLA2-derived phosphatidylcholine lipids. E, Relative expression level of iPLA2 and cPLA2 transcript in DRGs. Data represent the mean SEM from DRGs of four to five mice per group and transcript; **p < 0.01; ***p < 0.001; one-way ANOVA and Dunnett's multiple-comparison test. For all experiments, tissue from intraperitoneally injected vehicle or oxaliplatin (3 mg/kg) mice was used after 24 h. LP, Lysophosphatidyl; C, choline; E, ethanolamine; I, inositol; G, glycerol; S, serine.
Moreover, we determined the expression level of the phospholipases that are responsible for the release of LPC (iPLA2) or free fatty acids (cPLA2) from complex lipids in DRGs 24 h after oxaliplatin treatment (Lehr and Griessbach, 2000; Fig. 2D). We found that both phospholipases showed increased expression in DRGs 24 h after oxaliplatin treatment (iPLA2, p = 0.0089; cPLA2, p = 0.0008; one-way ANOVA and Dunnett´s post hoc test; Fig. 2E). This result is in accordance with the performed LC-QTOFMS as well as the LC-MS/MS measurements, where we observed increased levels of free fatty acids and lysophosphatidylcholines in the lipid families as well as an increased 9,10-EpOME concentration 24 h after oxaliplatin treatment. Additionally, the increased mRNA expression level of the lipid-releasing enzymes strengthens the presumption of an increased release and abundance of linoleic acid which can be reflected by the increased 9,10-EpOME concentrations.
LPC 16:0 and LPC 18:1 cause calcium transients in primary sensory neurons
Because of the significant increase of LPC 18:1 in sciatic nerve and DRGs (sciatic nerve, p = 0.0258; DRGs, p < 0.0001; one-way ANOVA) as well as of LPC 16:0 in spinal cord after oxaliplatin treatment (p = 0.0252; one-way ANOVA and Sidak's post hoc test), and the reason that it has been shown previously that 9,10-EpOME has a sensitizing effect on TRPV1 (Sisignano et al., 2016), we started to characterize the effect of LPC 16:0 and LPC 18:1 in sensory neurons using Ca2+ imaging. Stimulating sensory neurons for a short time (45 s) with LPC 16:0 as well as LPC 18:1, we could observe a calcium transient at all tested concentrations (1, 5, and 10 μm) compared with their corresponding vehicle (LPC 16:0, n = 18–34 neurons; 1 μm, p = 0.0055; 5 μm, p = 0.0007; 10 μm, p = 0.0058; LPC 18:1: n = 17–128 neurons; 1 μm, p = 0.0022; 5 μm, p < 0.0001; 10 μm, p = 0.0004; one-way ANOVA and Sidak's post hoc test; Fig. 3A,B, Extended Data Fig. 3-1A,B). We also determined the number of responding neurons after stimulation with LPC 16:0 and LPC 18:1, respectively, and observed that ∼1.5% of the sensory neurons reacted after stimulation with 1 and 5 μm LPC 16:0 and that ∼2% of the sensory neurons reacted using 10 μm LPC 16:0 (Extended Data Fig. 3-1C). Moreover, only 10 μm LPC 16:0 was capable of significantly increasing the number of responding sensory neurons compared with the vehicle (n = 33 independent measurements; p = 0.0011; one-way ANOVA and Sidak's post hoc test). Repeating the experiment with LPC 18:1, we could detect that ∼6% of the sensory neurons reacted after stimulation with 5 μm LPC 18:1 (n = 12–54 independent measurements; p = 0.0044; one-way ANOVA and Sidak's post hoc test). Even after stimulation with 1 and 10 μm LPC 18:1, the sensory neurons showed a reaction. However, at these tested concentrations there was no difference when compared with the corresponding vehicle (n = 12–54 independent measurements; Fig. 3C). In summary, both LPC 16:0 and LPC 18:1 seem to cause a transient Ca2+ influx in sensory neurons.
Figure 3.

LPC 18:1 causes calcium transients in sensory neurons and TRP channel-transfected HEK293 cells. A, Representative Ca2+ influx in primary sensory neurons after stimulation with LPC 18:1 (1, 5, and 10 μm; 45 s). Neurons were identified by stimulating the primary DRG culture with 50 mm KCl for 60 s. B, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 18:1 (1, 5, and 10 μm; 45 s) as well as their vehicle (EtOH). Data represent the mean SEM from n = 17–128 neurons/condition; **p < 0.01; ***p < 0.001; one-way ANOVA and Sidak's post hoc test. C, Percentage of neurons responding to LPC 18:1 (1, 5, and 10 μm; 45 s) as well as their corresponding vehicle (EtOH). Data represent the mean SEM of n = 12–54 measurements/condition; **p < 0.01; one-way ANOVA and Sidak's post hoc test (Extended Data Figure 3-1). Calcium transients in sensory neurons are evoked by LPC 16:0. D, Representative Ca2+ response in primary sensory neurons after stimulation with 5 μm LPC 18:1 for 45 s and costimulation of sensory neurons with AITC (100 μm; 40 s) and capsaicin (cap; 100 nm; 20 s). Neurons were identified by stimulating the primary sensory neurons with 50 mm KCl for 60 s. E, Percentage of neurons responding to LPC 18:1, AITC, and cap determined by KCl-responsive neurons. Data represent the mean SEM of n = 16 measurements. F, H, J, Representative Ca2+ influx in (h)TRPA1, (h)TRPV1, and (r)TRPM8 as well as empty vector-transfected HEK293 cells after stimulation with 30 μm LPC 18:1 for 60 s or vehicle. (h)TRPA1-, (h)TRPV1-, and (r)TRPM8-positive transfected HEK293 cells were identified by stimulating the transfected HEK293 cells with the TRP channel-specific agonists (TRPA1, 100 μm AITC for 30 s; TRPV1, 300 nm capsaicin for 20 s; TRPM8, 300 μm menthol for 30 s). G, I, K, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 18:1, vehicle, and TRP channel agonists in (h)TRPA1-, (h)TRPV1-, and (r)TRPM8-transfected HEK293 cells. Data represent the mean SEM from n = 18–196 transfected HEK293 cells/condition and n = 4–25 independent measurements; **p < 0.01; ***p < 0.001; one-way ANOVA and Sidak's post hoc test (Extended Data Figure 3-2). Transfection control of (h)TRPA1, (h)TRPV1, and (r)TRPM8 as well as empty vector-transfected HEK293 cells.
LPC 16:0 causes calcium transients in sensory neurons. A, Representative Ca2+ influx in primary sensory neurons after stimulation with LPC 16:0 (1, 5, and 10 µM; 45 s). Neurons were identified by stimulating the primary DRG culture with 50 mM KCl for 60 s. B, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 16:0 (1, 5, and 10 µM; 45 s) as well as their vehicle (EtOH). Data represent the mean SEM from n = 18–34 neurons/condition; **p < 0.01; ***p < 0.001; one-way ANOVA and Sidak's post hoc test. C, Percentage of neurons responding to LPC 16:0 (1, 5, and 10 µM; 45 s) as well as their corresponding vehicle (EtOH). Data represent the mean SEM of n = 22–44 measurements/condition; **p < 0.01; one-way ANOVA and Sidak's post hoc test. Download Figure 3-1, TIF file (135.9KB, tif) .
Transfection control of (h)TRPA1-, (h)TRPV1-, and (r)TRPM8-transfected, as well as empty vector-transfected HEK293 cells. Representative images of empty vector-, (h)TRPA1-, (h)TRPV1-, and (r)TRPM8-transfected HEK293 cells. The left panels show the phase contrast of transfected-HEK293 cells. The middle panels show the GFP signal of transfected HEK293 cells. The right panels show the merged images of the GFP signal and the phase of transfected HEK293 cells. The images were captured with 50× magnification. Download Figure 3-2, TIF file (2.2MB, tif) .
Next, we started to identify the subpopulation of the sensory neurons that respond to LPC 18:1. Therefore, we costimulated the neurons with the TRPA1 agonist AITC (100 μm; 40 s) and the TRPV1 agonist capsaicin (100 nm; 20 s) after stimulating the neurons for a short time (45 s) with 5 μm LPC 18:1 (Fig. 3D). As previously shown ∼7% of all neurons responding to 50 mm KCl showed a transient Ca2+ influx after 5 μm LPC 18:1 stimulation (n = 16 independent measurements; Fig. 3E). Moreover, the costimulation measurement revealed that the neurons reacting on LPC 18:1 also responded predominantly to the selective TRPV1 agonist capsaicin (Fig. 3E).
To further investigate the effects of LPC 18:1 on TRP channels, in vitro Ca2+ imaging experiments were performed in a heterologous expression system with TRPA1-, TRPM8-, and TRPV1-transfected HEK293 cells (Extended Data Fig. 3-2). Stimulating the TRP channel-transfected HEK293 cells for 60 s with 30 μm LPC 18:1, we could observe a significant transient Ca2+ influx in TRPV1- and TRPM8-positive transfected HEK293 cells compared with vehicle (TRPV1: n = 10–19 independent measurements; vehicle vs LPC 18:1, p = 0.0003; TRPM8: n = 12–19 independent measurements; vehicle vs LPC 18:1 p < 0.0001; one-way ANOVA and Sidak's post hoc test; Fig. 3H–K). In TRPA1-positive transfected HEK293 cells, no change in Ca2+ influx could be detected when compared with vehicle and LPC 18:1. In contrast, the vehicle caused a significant Ca2+ influx compared with LPC 18:1 (n = 6 independent measurements; vehicle vs LPC 18:1, p = 0.0052; one-way ANOVA and Sidak's post hoc test; Fig. 3F,G). TRP channel-positive transfected HEK293 cells were determined by the specific TRP channel agonist response (Fig. 3H–K, light gray bar; TRPV1 capsaicin: 300 nm, 20 s; TRPA1 AITC: 100 μm, 30 s; TRPM8 menthol: 300 μm, 30 s). The in vitro Ca2+ imaging analysis with TRP channel-transfected HEK293 cells confirms our previous observations in primary sensory neurons where we could identify LPC 18:1 as a putative TRPV1 channel activator. In addition, the TRPM8 channel could be identified as a second TRP channel that could be activated by LPC 18:1.
LPC 18:1 causes TRPV1- and TRPM8-dependent calcium transients in sensory neurons
For further LPC 18:1 characterization, we incubated sensory neurons with selective inhibitors for TRPA1 (HC 030031) and TRPV1 (AMG 9810) as well as for TRPM8 (AMTB hydrochloride; Gavva et al., 2005; Eid et al., 2008; Lashinger et al., 2008; Fig. 4A, Extended Data Fig. 4-1A–F). In this experimental setting, we could observe that there was no effect on the Ca2+ influx with LPC 18:1 when incubating the sensory neurons with the TRPA1 inhibitor (n = 17–18 neurons, n = 7–8 independent measurements; Fig. 4A–C). However, inhibition of TRPV1 and TRPM8 showed a significant reduction of LPC-mediated Ca2+ influx (TRPV1: n = 16–25 neurons; Δ ratio, p < 0.0001; n = 8–12 independent measurements; percentage of responding neurons, p = 0.0003; TRPM8: n = 11–25 neurons; Δ ratio, p < 0.0001; n = 8–12 independent measurements; percentage of responding neurons, p < 0.0001; one-way ANOVA and Sidak's post hoc test; Fig. 4B,C). Analyzing the mRNA expression of TRPA1, TRPV1, and the TRPM8 transcripts in DRGs of oxaliplatin-treated animals revealed no changes in the mRNA expression level (Extended Data Fig. 4-2A). Investigating the CGRP release of DRGs neurons after incubation only with LPC 18:1 and LPC 18:1 together with capsaicin, we could see that LPC 18:1 had no sensitizing effect on the TRPV1 channel (Extended Data Fig. 4-2B).
Figure 4.
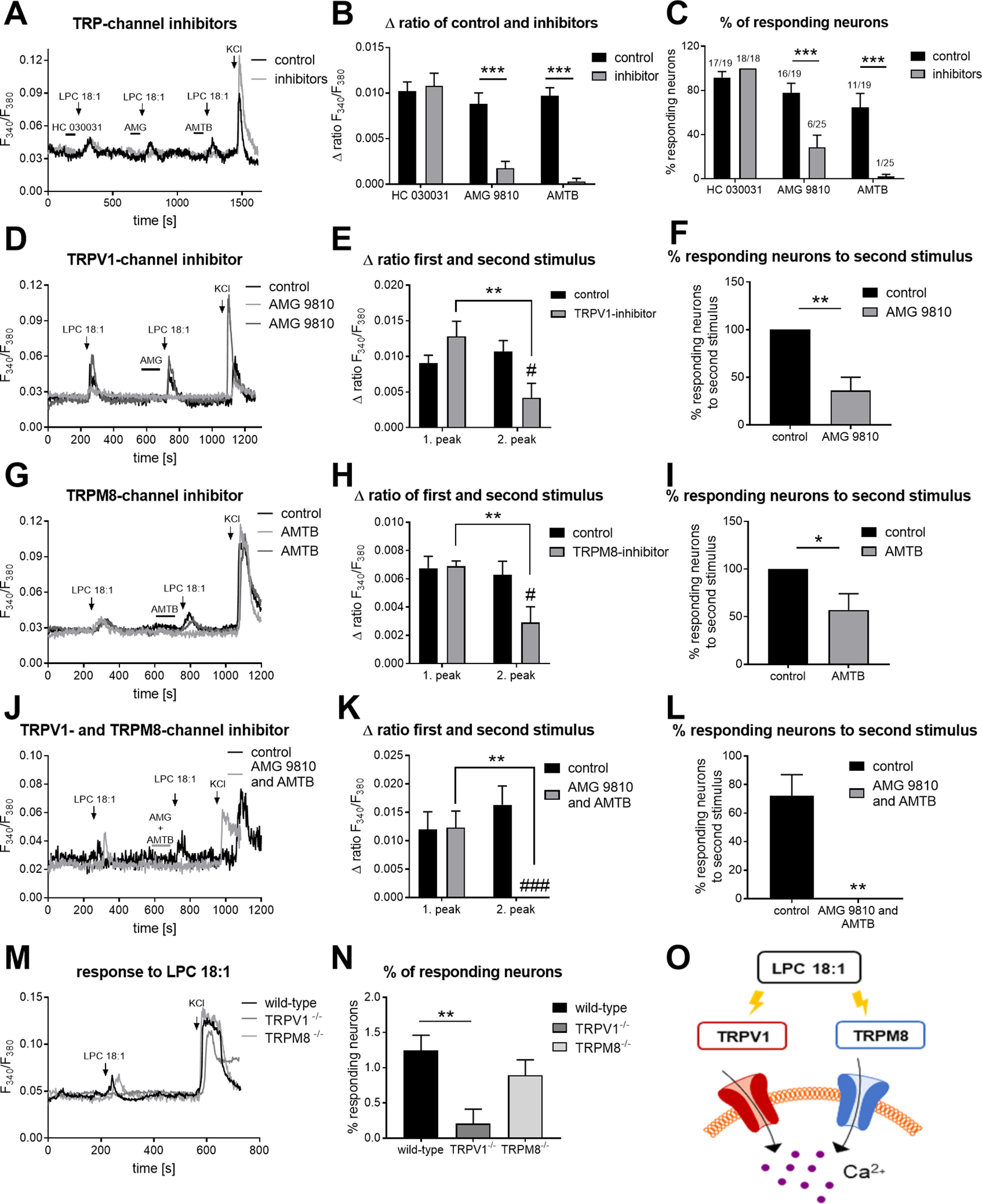
LPC 18:1 activates TRPV1 and TRPM8. A, Representative Ca2+ influx in primary sensory neurons after 2 min of preincubation with the TRPA1 inhibitor (20 μm; HC 030031), TRPV1 inhibitor (1 μm; AMG 9810), and TRPM8 inhibitor (1 μm; AMTB hydrochloride) before LPC 18:1 (5 μm; 45 s) stimulation. B, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 18:1 after TRPA1, TRPV1, and TRPM8 channel inhibition. Data represent the mean SEM from n = 11–25 neurons/condition; ***p < 0.001; one way-ANOVA and Sidak's post hoc test. C, Percentage of neurons responding to LPC 18:1 after TRPA1, TRPV1, and TRPM8 channel inhibition. Data represent the mean SEM of n = 7–12 measurements per condition; ***p < 0.001; one-way ANOVA and Sidak's post hoc test. D, G, J, Representative Ca2+ response in primary sensory neurons after stimulation with 5 μm LPC 18:1 for 45 s prior and after TRPV1, TRPM8, or TRPV1 and TRPM8 channel inhibition. E, H, K, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 18:1 after TRPV1, TRPM8, or TRPV1 and TRPM8 channel inhibition. Data represent the mean SEM from n = 9–21 neurons/condition; **p < 0.01; #p < 0.01; ###p < 0.001; one-way ANOVA and Sidak's post hoc test. F, I, L, Percentage of neurons responding to LPC 18:1, after TRPV1, TRPM8, or TRPV1 and TRPM8 channel inhibition. Data represent the mean SEM of n = 7–13 measurements; *p < 0.05; **p < 0.01; two-tailed unpaired t test with Welch's correction (Extended Data Figure 4-1). Functional testing of the effects of TRPA1, TRPV1, and TRPM8 inhibitors on the respective channel activity (Extended Data Figure 4-2). TRP channel expression and calcitonin gene-related peptide release (Extended Data Figure 4-3). sEPSC in spinal dorsal horn lamina II neurons (Extended Data Figure 4-4) TRPV1 and TRPM8 activation through LPC 16:0. M, Representative Ca2+ response in primary sensory neurons of wild-type and TRPV1 knock-out mice as well as TRPM8 knock-out mice after stimulation with 5 μm LPC 18:1 for 60 s. N, Percentage of neurons responding to LPC 18:1 in wild-type and TRPV1 knock-out mice as well as TRPM8 knock-out mice. Data represent the mean SEM of n = 22–46 measurements; **p < 0.01; two-way ANOVA and Sidak's post hoc test. O, Schematic overview of LPC 18:1 action.
Functional testing of the effects of TRPA1, TRPV1, and TRPM8 inhibitors on the respective channel activity. A, C, E, Representative Ca2+ influx in primary sensory neurons. B, D, F, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by TRP channel agonist. Primary sensory neurons were stimulated two times with 100 µm AITC (TRPA1 agonist), 100 nm capsaicin (cap; TRPV1 agonist), or 100 µm menthol (TRPM8 agonist). Then, either preincubated for 2 min prior to the second stimulus with the corresponding TRP channel inhibitors (TRPA1, 20 µm HC 030031; TRPV1, 1 µM AMG 9810; TRPM8, 1 µM AMTB hydrochloride) or only with the corresponding vehicles of the TRP channel inhibitors (DMSO and H2O). Neurons were identified by stimulating the primary DRG culture with 50 mM KCl for 60 s. Data represent the mean SEM of n = 10–25 neurons/condition; **p < 0.01; ***p < 0.001; ##p < 0.01; ###p < 0.001; one-way ANOVA and Sidak's post hoc test. Download Figure 4-1, TIF file (509.3KB, tif) .
TRP channel expression and CGRP release. A, Relative expression levels of TRPV1, TRPA1, and TRPM8 in DRGs of oxaliplatin-treated animals after 24 h. Data represent the mean ± SEM from DRGs of five mice per group and transcript; one way-ANOVA and Dunnett's multiple comparison test. B, Concentration of CGRP release in picograms per milliliter after stimulating the primary sensory neurons with vehicle (DMSO+EtOH), 400 nm capsaicin (cap), 5 µm LPC 18:1, and 400 nm capsaicin + 5 µm LPC 18:1. Data represent the mean ± SEM from DRGs of four pooled mice per condition; *p < 0.05; **p < 0.01; one-way ANOVA and Sidak's post hoc test. Download Figure 4-2, TIF file (154.8KB, tif) .
sEPSC in spinal dorsal horn lamina II neurons. A, sEPSC frequency in hertz. B, sEPSC amplitude in picoamperes. Frequency and amplitude were recorded prior to and after incubating spinal dorsal horn lamina II neurons with 10 µm LPC 18:1. Data represent the mean ± SEM of n = 8 neurons from four mice; two-tailed Student's t test. Download Figure 4-3, TIF file (3.1MB, tif) .
LPC 16:0 activates TRPV1 and TRPM8. A, D, Representative Ca2+ response in primary sensory neurons after stimulation with 5 µm LPC 16:1 for 45 s prior to and after TRPM8 or TRPV1 and TRPM8 channel inhibition. B, E, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 16:0 after TRPM8 or TRPV1 and TRPM8 channel inhibition. Data represent the mean SEM from n = 10–14 neurons/condition; **p < 0.01; ***p < 0.001; #p < 0.05; ###p < 0.001; one-way ANOVA and Sidak's post hoc test. C, F, Percentage of responding neurons to LPC 16:0, after TRPM8 or TRPV1 and TRPM8 channel inhibition. Data represent the mean SEM of n = 7–13 measurements; *p < 0.05; ***p < 0.001; two-tailed unpaired t test with Welch's correction. Download Figure 4-4, TIF file (295.4KB, tif) .
We further investigated the effects of LPC 18:1 on spinal cord synaptic transmission by patch-clamp recordings in spinal cord slices. We measured the frequency and amplitude of spontaneous EPSCs (sEPSCs) in lamina II neurons before and after LPC 18:1 (10 μm) perfusion (n = 8 neurons). However, we could not observe any changes in frequency and amplitude of sEPSCs after LPC 18:1 treatment (Extended Data Fig. 4-3A,B). These findings are in line with our previous results, showing that a distinct neuronal population responds to LPC 18:1. In summary, these results show a central role of LPC 18:1 in nociceptive transmission via activation of TRPV1- and TRPM8-positive neurons.
To verify whether LPC 18:1 effectively activates the TRPV1 and the TRPM8 channels, we next inhibited TRPV1 and TRPM8, with respective inhibitors. We first stimulated the sensory neurons with LPC 18:1. Afterward, we incubated the neurons with the TRPV1 or TRPM8 inhibitor before the second LPC 18:1 stimulus (Fig. 4D,G). The inhibition of TRPV1 or TRPM8 could partly block the LPC 18:1-mediated Ca2+ influx (TRPV1: n = 21–18 neurons; control vs TRPV1 inhibitor 2 peak, p = 0.0173; TRPV1 inhibitor 1 peak vs 2 peak, p = 0.0018; TRPM8: n = 9–10 neurons; control vs TRPM-inhibitor 2 peak, p = 0.0212; TRPM8 inhibitor 1 peak vs 2 peak, p = 0.0075; one-way ANOVA and Sidak's post hoc test; Fig. 4E,H). Using TRPV1 and TRPM8 inhibitors, the number of neurons responding to LPC 18:1 could be reduced by ∼50% (TRPV1 inhibitor: n = 9–13 independent measurements, p = 0.0018; TRPM8 inhibitor, n = 7–8 independent measurements; p = 0.0453; two-tailed t test and Welch's correction; Fig. 4F,I). This again suggests that LPC 18:1 is likely to activate a subpopulation of TRPV1- and TRPM8-expressing neurons (Fig. 4O). To test whether TRPV1 and TRPM8 are affected by LPC 18:1, we next used a combination of both inhibitors (Fig. 4J). Inhibition of TRPV1 and TRPM8 showed a complete blockage of LPC 18:1-mediated Ca2+ influx (n = 10–11 neurons; Δ ratio control vs TRPV1+TRPM8-inhibitor 2 peak, p = 0.0002; TRPV1+TRPM8 inhibitor 1 peak vs 2 peak, p = 0.0036; one-way ANOVA and Sidak's post hoc test; n = 9 independent measurements; percentage of responding neurons, p = 0.0012; two-tailed t test and Welch's correction; Fig. 4K,L). Additionally, we investigated the LPC-induced calcium responses in TRPV1- and TRPM8-deficient mice (Fig. 4M). In the TRPV1-deficient animals, we observed that significantly fewer neurons responded to LPC 18:1 (n = 22–46 independent measurements; p = 0.0045; one-way ANOVA and Sidak's post hoc test; Fig. 4N). In the TRPM8-deficient mice, we found a tendency of reduced neuronal response to LPC 18:1 (Fig. 4N).
Likewise, we could observe that ∼50% of the sensory neurons reacted on LPC 16:0 using only the TRPM8 inhibitor (n = 10–13 neurons; Δ ratio control vs TRPM8-inhibitor 2 peak, p = 0.0462; TRPM8-inhibitor 1 peak vs 2 peak, p = 0.0063; one-way ANOVA and Sidak's post hoc test; n = 7–8 independent measurements; percentage of responding neurons, p = 0.0385; two-tailed t test and Welch's correction; Extended Data Fig. 4-4A–C). Using the TRPV1 and the TRPM8 inhibitors together, we saw a nearly complete blockage of the LPC 16:0-mediated Ca2+ influx (n = 13–14 neurons, Δ ratio control vs TRPV1+TRPM8 inhibitor 2 peak, p < 0.0001; TRPV1+TRPM8 inhibitor 1 peak vs 2 peak, p = 0.0002; one-way ANOVA and Sidak's post hoc test; n = 9–10 independent measurements; percentage of responding neurons, p < 0.0001; two-tailed t test and Welch's correction; Extended Data Fig. 4-4D–F).
LPC 18:1 induces mechanical hypersensitivity in vivo
To investigate whether treatment with LPC 18:1 has any effect in vivo, we subcutaneously injected 10 and 50 μm LPC 18:1 or its vehicle in the left hindpaw of wild-type mice and measured the paw withdrawal latency at 0.5, 1, 2, 3, 4, and 6 h after injection (n = 6–10 mice/group). Monitoring the mechanical threshold, we observed that mice that received LPC 18:1 had already developed a mechanical hypersensitivity after 0.5 h, which lasted for 2 h (10 μm LPC 18:1: 0.5 h, p = 0.0002; 1 h, p = 0.0031; 2 h, p = 0.0093; 50 μm LPC 18:1: 1 h, p = 0.0002; 2 h, p = 0.0056; two-way ANOVA and Bonferroni's post hoc test; Fig. 5). The contralateral and ipsilateral paws of vehicle-treated animals showed no changes in paw withdrawal latency, and the contralateral paw of LPC 18:1-treated animals was also unaffected (Extended Data Fig. 5-1A–C).
Figure 5.
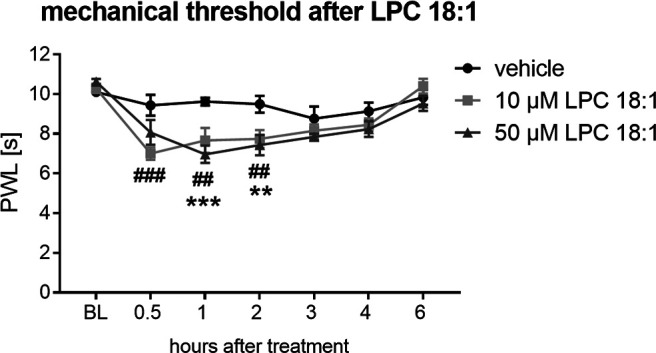
LPC 18:1 induces mechanical hypersensitivity. Paw withdrawal latency in seconds up to 6 h after subcutaneous injection of 10 and 50 μm LPC 18:1 and vehicle in the left hindpaw of wild-type mice. Data represent the mean SEM of 6–10 mice/group; LPC 18:1, 10 μm: ##p < 0.01; ###p < 0.001; LPC 18:1, 50 μm: **p < 0.01; ***p < 0.001; two-way ANOVA and Bonferroni's post hoc test. BL, Baseline (Extended Data Figure 5-1, control data).
LPC 18:1 induces mechanical hypersensitivity. Paw withdrawal latency (PWL) in seconds up to 6 hours after subcutaneous injection of 10 and 50 µm LPC 18:1 and vehicle in the left hindpaw (ipsilateral paw) of wild-type mice. A, PWL of the contralateral and ipsilateral paws of control animals. B, PWL of the contralateral paw of mice treated with vehicle, 10 µm or 50 m LPC 18:1. C, PWL of the contralateral and ipsilateral paw of mice treated with 10 and 50 µm LPC 18:1. Data represent the mean SEM of 6–10 mice/group; 10 µm LPC 18:1: ##p < 0.01; ###p < 0.001; ####p < 0.0001; 50 µm LPC 18:1: **p < 0.01; ***p < 0.001; ****p < 0.0001; two-way ANOVA and Bonferroni's post hoc test. BL, Baseline. Download Figure 5-1, TIF file (255.9KB, tif) .
Discussion
In summary, we observed that the synthesis of various lipids was significantly altered in nervous system tissue after oxaliplatin treatment. The LC-MS/MS measurements revealed a significant reduction of prostanoid concentrations in DRGs and an increase in 9,10-EpOME concentrations. The latter lipid has previously been shown to sensitize the TRPV1 channel in sensory neurons and to cause mechanical and thermal hypersensitivity in vivo (Sisignano et al., 2016). Furthermore, the untargeted lipidomics analysis showed increased signals of lysophosphatidylcholines, particularly LPC 18:1 in DRGs and sciatic nerves in oxaliplatin-treated mice. Characterizing LPC 18:1, we observed that the lipid causes Ca2+ transients in TRPV1- and TRPM8-positive sensory neurons.
In the present study, we did not observe changes in levels of chemokines, growth factors, and cytokines as well as in astrocyte activation in the dorsal spinal cord, which indicates that classical inflammatory processes and related mediators appear to have a minor contribution to oxaliplatin-induced acute pain. Analyzing the mRNA expression of ROS-producing enzymes in DRGs of oxaliplatin-treated animals with qPCR, no significant increase in expression could be detected. In several studies, astrocyte activation as well as increased proinflammatory cytokines, and ROS levels were found that may contribute to CIPN (Ledeboer et al., 2007; Kiguchi et al., 2008a,b; Mihara et al., 2011; Doyle et al., 2012; Ji et al., 2013; Sisignano et al., 2014; Janes et al., 2015). However, the authors of these studies mainly investigated later timepoints of CIPN (days or weeks after initial treatment). Our results indicate that classical inflammatory pathways and related mediators are not as critically involved in the onset of oxaliplatin-induced pain as in other chronic pain states and may thus be secondary responses that are initiated in later phases of oxaliplatin-induced neuropathy. Moreover, although a difference in lipid concentrations between male and female mice could be observed, the degree of mechanical hypersensitivity after oxaliplatin treatment is similar in male and female mice, suggesting at least no direct effect of these concentration differences on mechanical hypersensitivity following oxaliplatin treatment in different sexes.
The untargeted lipidomic measurement showed a significant increase of LPC 18:1 in sciatic nerve and DRGs as well as of LPC 16:0 in spinal cord of oxaliplatin-treated animals. Previous studies could report that LPC species can in general modulate the open probability of the TRPM8 channel in transfected HEK293 cells (Abeele et al., 2006). Andersson et al. (2007) could observe the same effect with LPC 16:0 in TRPM8-transfected CHO cells. Our results are consistent with these two studies but add the important observation that LPC 18:1 and LPC 16:0 act on both TRPM8 and TRPV1 channels in DRG neurons. In contrast to our study, Nieto-Posadas et al. (2012) and Canul-Sánchez et al. (2018) could not detect a TRPV1 activation in HEK293 cells caused by LPC by performing inside-out patches and measurement of single-channel currents, which indicates that the calcium transients we observed in the heterologous expression system are caused by an indirect activation of TRPV1 by LPC 18:1.
Furthermore, our in vivo data showed that LPC 18:1 induces mechanical hypersensitivity. Hence, it could be possible that the increased LPC 18:1 level that was observed in sciatic nerve and DRGs may directly contribute to oxaliplatin-induced acute peripheral pain. In addition, we see a significant increase of the linoleic acid-derived metabolite 9,10-EpOME in DRGs of oxaliplatin-treated animals after 24 h. Previous studies showed that 9,10-EpOME synthesized by CYP2J6 not only has a direct and sensitizing effect on the TRPV1 channel but also that it induces mechanical hypersensitivity and thermal pain in vivo (Sisignano et al., 2016). Consistent with the increased levels of LPC and free fatty acids, we could show that the cPLA2 and iPLA2 pathways seem to be activated in DRGs of oxaliplatin-treated animals. These phospholipases are responsible for the liberation of free fatty acids (cPLA2) and lysophospholipids (iPLA2) from membrane phospholipids (Dennis et al., 2011). A study targeting the iPLA2 pathway with the specific iPLA2 inhibitor bromoenol lactone (BEL) could demonstrate that a TRPM8 activation in DRGs through cold and icilin (TRPM8 agonist) could be blocked (Andersson et al., 2007). Animal studies with BEL showed that LPC 16:0-induced cold hypersensitivity could not be prevented by the inhibition of iPLA2 in vivo (Gentry et al., 2010). Moreover, iPLA2 is involved in the production of prostaglandins and the inhibition of iPLA2 through BEL leads to reduced prostaglandin concentrations (Tsuchida et al., 2015). Our data suggest that targeting the iPLA2 pathway and reducing 9,10-EpOME synthesis by inhibiting CYP2J6 may ameliorate oxaliplatin-induced acute pain. However, there are currently no selective iPLA2 antagonists that could be used as clinical candidates and more research is required to investigate potential physiological roles of iPLA2 that may cause unwanted effects in treated patients.
Apart from lipid-signaling pathways, our results show that targeting TRP channels may be beneficial in preventing oxaliplatin-induced acute peripheral pain. Other studies also demonstrated that at later time points the TRPM8 as well as the TRPV1 transcript are increased in DRGs after oxaliplatin treatment and may contribute to cold allodynia (Kawashiri et al., 2012; Chukyo et al., 2018). Furthermore, it was reported that the inhibition of the TRPV1 and TRPM8 channels by capsazepine, but not inhibition of merely the TRPV1 channel, with 5´-iodo resiniferatoxin reduced oxaliplatin-induced cold allodynia in vivo (Gauchan et al., 2009). However, there is currently no TRP channel antagonist available for clinical use. Moreover, previous early clinical trials with TRPV1 antagonists failed because of hyperthermia in patients, which indicates a central role of TRPV1 in maintaining body temperature (Wong and Gavva, 2009) and in the hampering of clinical development in safe TRPV1 antagonists.
In summary, our study highlights the importance of early changes in neuronal lipid synthesis and metabolism after oxaliplatin treatment and identifies a crucial role of signaling lipids of the LPC-EpOME–TRPV1–TRPM8 pathway in oxaliplatin-induced acute pain in vivo.
Footnotes
This work was supported by Grants SFB1039 A09, Z01, and A08 of the Deutsche Forschungsgemeinschaft (German Research Foundation) and from the State of Hessen to the LOEWE Research Center for Translational Medicine and Pharmacology as well as the Fraunhofer Foundation Project: Neuropathic Pain and the Fraunhofer Cluster of Excellence for Immune-Mediated Diseases (CIMD).
The authors declare no competing financial interests.
References
- Abeele FV, Zholos A, Bidaux G, Shuba Y, Thebault S, Beck B, Flourakis M, Panchin Y, Skryma R, Prevarskaya N (2006) Ca2+-independent phospholipase A(2)-dependent Gating of TRPM8 by lysophospholipids. J Biol Chem 281:40174–40182. 10.1074/jbc.M605779200 [DOI] [PubMed] [Google Scholar]
- Anand U, Otto WR, Anand P (2010) Sensitization of capsaicin and icilin responses in oxaliplatin treated adult rat DRG neurons. Mol Pain 6:82 10.1186/1744-8069-6-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DA, Nash M, Bevan S (2007) Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J Neurosci 27:3347–3355. 10.1523/JNEUROSCI.4846-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NA, Latremoliere A, Basbaum AI, Mogil JS, Porreca F, Rice ASC, Woolf CJ, Currie GL, Dworkin RH, Eisenach JC, Evans S, Gewandter JS, Gover TD, Handwerker H, Huang W, Iyengar S, Jensen MP, Kennedy JD, Lee N, Levine J, et al. (2016) Ensuring transparency and minimization of methodologic bias in preclinical pain research: PPRECISE considerations. Pain 157:901–909. 10.1097/j.pain.0000000000000458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyriou AA, Cavaletti G, Briani C, Velasco R, Bruna J, Campagnolo M, Alberti P, Bergamo F, Cortinovis D, Cazzaniga M, Santos C, Papadimitriou K, Kalofonos HP (2013) Clinical pattern and associations of oxaliplatin acute neurotoxicity. Cancer 119:438–444. 10.1002/cncr.27732 [DOI] [PubMed] [Google Scholar]
- Basso L, Altier C (2017) Transient receptor potential channels in neuropathic pain. Curr Opin Pharmacol 32:9–15. 10.1016/j.coph.2016.10.002 [DOI] [PubMed] [Google Scholar]
- Brenneis C, Sisignano M, Coste O, Altenrath K, Fischer MJ, Angioni C, Fleming I, Brandes RP, Reeh PW, Woolf CJ, Geisslinger G, Scholich K (2011) Soluble epoxide hydrolase limits mechanical hyperalgesia during inflammation. Mol Pain 7:78. 10.1186/1744-8069-7-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JR, Morrison G, Dolan ME, Fleming GF (2016) Chemotherapy-induced peripheral neuropathy: current status and progress. Gynecol Oncol 140:176–183. 10.1016/j.ygyno.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canul-Sánchez JA, Hernández-Araiza I, Hernández-García E, Llorente I, Morales-Lázaro SL, Islas LD, Rosenbaum T (2018) Different agonists induce distinct single-channel conductance states in TRPV1 channels. J Gen Physiol 150:1735–1746. 10.1085/jgp.201812141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Xie RG, Gao YJ, Xu ZZ, Zhao LX, Bang SS, Berta T, Park CK, Lay M, Chen W, Ji RR (2016) beta-arrestin-2 regulates NMDA receptor function in spinal lamina II neurons and duration of persistent pain. Nat Commun 7:12531. 10.1038/ncomms12531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yang G, Grosser T (2013) Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat 104-105:58–66. 10.1016/j.prostaglandins.2012.08.006 [DOI] [PubMed] [Google Scholar]
- Chukyo A, Chiba T, Kambe T, Yamamoto K, Kawakami K, Taguchi K, Abe K (2018) Oxaliplatin-induced changes in expression of transient receptor potential channels in the dorsal root ganglion as a neuropathic mechanism for cold hypersensitivity. Neuropeptides 67:95–101. 10.1016/j.npep.2017.12.002 [DOI] [PubMed] [Google Scholar]
- Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G (2011) Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev 111:6130–6185. 10.1021/cr200085w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, Couette B, Busserolles J, Courteix C, Noel J, Lazdunski M, Eschalier A, Authier N, Bourinet E (2011) Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med 3:266–278. 10.1002/emmm.201100134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle T, Chen ZM, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, Dagostino C, Ryerse J, Rausaria S, Kamadulski A, Neumann WL, Salvemini D (2012) Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci 32:6149–6160. 10.1523/JNEUROSCI.6343-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid SR, Crown ED, Moore EL, Liang HYA, Choong KC, Dima S, Henze DA, Kane SA, Urban MO (2008) HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain 4:48. 10.1186/1744-8069-4-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatters SJL, Bennett GJ (2004) Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain 109:150–161. 10.1016/j.pain.2004.01.029 [DOI] [PubMed] [Google Scholar]
- Gadient RA, Cron KC, Otten U (1990) Interleukin-1-β and tumor necrosis factor-α synergistically stimulate nerve growth-factor (Ngf) release from cultured rat astrocytes. Neurosci Lett 117:335–340. 10.1016/0304-3940(90)90687-5 [DOI] [PubMed] [Google Scholar]
- Gauchan P, Andoh T, Kato A, Kuraishi Y (2009) Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin-induced cold allodynia in mice. Neurosci Lett 458:93–95. 10.1016/j.neulet.2009.04.029 [DOI] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu YS, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJS (2005) AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther 313:474–484. 10.1124/jpet.104.079855 [DOI] [PubMed] [Google Scholar]
- Gentry C, Stoakley N, Andersson DA, Bevan S (2010) The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol Pain 6:4. 10.1186/1744-8069-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahnefeld L, Gurke R, Thomas D, Schreiber Y, Schäfer SMG, Trautmann S, Snodgrass IF, Kratz D, Geisslinger G, Ferreirós N (2020) Implementation of lipidomics in clinical routine: can fluoride/citrate blood sampling tubes improve preanalytical stability? Talanta 209:120593. 10.1016/j.talanta.2019.120593 [DOI] [PubMed] [Google Scholar]
- Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Brück J, Teske A, Valtcheva N, Fuchs K, Kneilling M, Park JH, Kim KH, Kim KW, Hoffmann P, Krenn C, Hai T, Ghoreschi K, Biederman T, Röcken M (2011) ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med 18:128–134. 10.1038/nm.2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann SW, Angioni C, Tunaru S, Lee S, Woolf CJ, Offermanns S, Geisslinger G, Scholich K, Sisignano M (2017) The G2A receptor (GPR132) contributes to oxaliplatin-induced mechanical pain hypersensitivity. Sci Rep 7:446. 10.1038/s41598-017-00591-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Hammock BD (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8:794–805. 10.1038/nrd2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K, Wahlman C, Little JW, Doyle T, Tosh DK, Jacobson KA, Salvemini D (2015) Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A(3) adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun 44:91–99. 10.1016/j.bbi.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Wang X, Lo EH (2009) Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol Sci 30:336–340. 10.1016/j.tips.2009.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Gao YJ (2014) Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov 13:533–548. 10.1038/nrd4334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji XT, Qian NS, Zhang T, Li JM, Li XK, Wang P, Zhao DS, Huang G, Zhang L, Fei Z, Jia D, Niu L (2013) Spinal astrocytic activation contributes to mechanical allodynia in a rat chemotherapy-induced neuropathic pain model. Plos One 8:e60733. 10.1371/journal.pone.0060733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D. (2013) TRP Channels and Pain. Annu Rev Cell Dev Biol 29, 29:355–384. 10.1146/annurev-cellbio-101011-155833 [DOI] [PubMed] [Google Scholar]
- Kawashiri T, Egashira N, Kurobe K, Tsutsumi K, Yamashita Y, Ushio S, Yano T, Oishi R (2012) L type Ca2+ channel blockers prevent oxaliplatin-induced cold hyperalgesia and TRPM8 overexpression in rats. Mol Pain 8:7 10.1186/1744-8069-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Maeda T, Kobayashi Y, Kishioka S (2008a) Up-regulation of tumor necrosis factor-alpha in spinal cord contributes to vincristine-induced mechanical allodynia in mice. Neurosci Lett 445:140–143. 10.1016/j.neulet.2008.09.009 [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Maeda T, Kobayashi Y, Kondo T, Ozaki M, Kishioka S (2008b) The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. Eur J Pharmacol 592:87–92. 10.1016/j.ejphar.2008.07.008 [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010) Improving bioscience research reporting: the arrive guidelines for reporting animal research. PLoS Biol 8:e1000412. 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashinger ESR, Steiginga MS, Hieble JP, Leon LA, Gardner SD, Nagilla R, Davenport EA, Hoffman BE, Laping NJ, Su X (2008) AMTB, a TRPM8 channel blocker: evidence in rats for activity in overactive bladder and painful bladder syndrome. Am J Physiol Renal Physiol 295:F803–F810. 10.1152/ajprenal.90269.2008 [DOI] [PubMed] [Google Scholar]
- Law SH, Chan ML, Marathe GK, Parveen F, Chen CH, Ke LY (2019) An updated review of lysophosphatidylcholine metabolism in human diseases. Int J Mol Sci 20:1149 10.3390/ijms20051149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR (2007) Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun 21:686–698. 10.1016/j.bbi.2006.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr M, Griessbach K (2000) Involvement of different protein kinases and phospholipases A2 in phorbol ester (TPA)-induced arachidonic acid liberation in bovine platelets. Mediators Inflamm 9:31–34. 10.1080/09629350050024357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PP, Zhu W, Chen C, Yan B, Zhu L, Chen X, Peng C (2020) The mechanisms of lysophosphatidylcholine in the development of diseases. Life Sci 247:117443. 10.1016/j.lfs.2020.117443 [DOI] [PubMed] [Google Scholar]
- Liu XJ, Tonello R, Ling YJ, Gao YJ, Berta T (2019) Paclitaxel-activated astrocytes produce mechanical allodynia in mice by releasing tumor necrosis factor-alpha and stromal-derived cell factor 1. J Neuroinflammation 16:209. 10.1186/s12974-019-1619-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Makker PGS, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O, Park SB, Goldstein D, Moalem-Taylor G (2017) Characterisation of immune and neuroinflammatory changes associated with chemotherapy-induced peripheral neuropathy. PLoS One 12:e0170814. 10.1371/journal.pone.0170814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmiroli P, Scuteri A, Cornblath DR, Cavaletti G (2017) Pain in chemotherapy-induced peripheral neurotoxicity. J Peripher Nerv Syst 22:156–161. 10.1111/jns.12226 [DOI] [PubMed] [Google Scholar]
- Mihara Y, Egashira N, Sada H, Kawashiri T, Ushio S, Yano T, Ikesue H, Oishi R (2011) Involvement of spinal NR2B-containing NMDA receptors in oxaliplatin-induced mechanical allodynia in rats. Mol Pain 7:8. 10.1186/1744-8069-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassini R, Gees M, Harrison S, De Siena G, Materazzi S, Moretto N, Failli P, Preti D, Marchetti N, Cavazzini A, Mancini F, Pedretti P, Nilius B, Patacchini R, Geppetti P (2011) Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 152:1621–1631. 10.1016/j.pain.2011.02.051 [DOI] [PubMed] [Google Scholar]
- Nieto-Posadas A, Picazo-Juárez G, Llorente I, Jara-Oseguera A, Morales-Lázaro S, Escalante-Alcalde D, Islas LD, Rosenbaum T (2012) Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nat Chem Biol 8:737 10.1038/nchembio0812-737c [DOI] [PubMed] [Google Scholar]
- Pachman DR, Qin R, Seisler DK, Smith EML, Beutler AS, Ta LE, Lafky JM, Wagner-Johnston ND, Ruddy KJ, Dakhil S, Staff NP, Grothey A, Loprinzi CL (2015) Clinical course of oxaliplatin-induced neuropathy: results from the randomized phase III trial N08CB (alliance). J Clin Oncol 33:3416–3422. 10.1200/JCO.2014.58.8533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SB, Goldstein D, Krishnan AV, Lin CS, Friedlander ML, Cassidy J, Koltzenburg M, Kiernan MC (2013) Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J Clin 63:419–437. 10.3322/caac.21204 [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Akopian AN, Ruparel NB, Diogenes A, Weintraub ST, Uhlson C, Murphy RC, Hargreaves KM (2010) Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J Clin Invest 120:1617–1626. 10.1172/JCI41678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Sasso O (2014) Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci 17:164–174. 10.1038/nn.3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden CE, Domenichiello AF, Yuan ZX, Sapio MR, Keyes GS, Mishra SK, Gross JR, Majchrzak-Hong S, Zamora D, Horowitz MS, Davis JM, Sorokin AV, Dey A, LaPaglia DM, Wheeler JJ, Vasko MR, Mehta NN, Mannes AJ, Iadarola MJ (2017) A systems approach for discovering linoleic acid derivatives that potentially mediate pain and itch. Sci Signal 10:eaal5241 10.1126/scisignal.aal5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CR, Zhang H, Dougherty PM (2014) Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience 274:308–317. 10.1016/j.neuroscience.2014.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Little JW, Doyle T, Neumann WL (2011) Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med 51:951–966. 10.1016/j.freeradbiomed.2011.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisignano M, Park CK, Angioni C, Zhang DD, von Hehn C, Cobos EJ, Ghasemlou N, Xu ZZ, Kumaran V, Lu R, Grant A, Fischer MJ, Schmidtko A, Reeh P, Ji RR, Woolf CJ, Geisslinger G, Scholich K, Brenneis C (2012) 5,6-EET is released upon neuronal activity and induces mechanical pain hypersensitivity via TRPA1 on central afferent terminals. J Neurosci 32:6364–6372. 10.1523/JNEUROSCI.5793-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisignano M, Baron R, Scholich K, Geisslinger G (2014) Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat Rev Neurol 10:694–707. 10.1038/nrneurol.2014.211 [DOI] [PubMed] [Google Scholar]
- Sisignano M, Angioni C, Park CK, Meyer Dos Santos S, Jordan H, Kuzikov M, Liu D, Zinn S, Hohman SW, Schreiber Y, Zimmer B, Schmidt M, Lu R, Suo J, Zhang DD, Schafer SM, Hofmann M, Yekkirala AS, de Bruin N, Parnham MJ, et al. (2016) Targeting CYP2J to reduce paclitaxel-induced peripheral neuropathic pain. Proc Natl Acad Sci U S A 113:12544–12549. 10.1073/pnas.1613246113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Weir GA, Schiavo G (2016) A simple, step-by-step dissection protocol for the rapid isolation of mouse dorsal root ganglia. BMC Res Notes 9:82. 10.1186/s13104-016-1915-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockstill K, Doyle TM, Yan XS, Chen ZM, Janes K, Little JW, Braden K, Lauro F, Giancotti LA, Harada CM, Yadav R, Xiao WH, Lionberger JM, Neumann WL, Bennett GJ, Weng HR, Spiegel S, Salvemini D (2018) Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain. J Exp Med 215:1301–1313. 10.1084/jem.20170584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarighi N, Menger D, Pierre S, Kornstädt L, Thomas D, Ferreirós N, Nüsing RM, Geisslinger G, Scholich K (2019) Thromboxane-induced α-CGRP release from peripheral neurons is an essential positive feedback loop in capsaicin-induced neurogenic inflammation. J Invest Dermatol 139:656–664. 10.1016/j.jid.2018.10.011 [DOI] [PubMed] [Google Scholar]
- Tsuchida K, Ibuki T, Matsumura K (2015) Bromoenol lactone, an inhibitor of calcium-independent phospholipase A(2), suppresses carrageenan-induced prostaglandin production and hyperalgesia in rat hind paw. Mediators Inflamm 2015:605727. 10.1155/2015/605727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windebank AJ, Grisold WG (2008) Chemotherapy-induced neuropathy. J Peripher Nerv Syst 13:27–46. 10.1111/j.1529-8027.2008.00156.x [DOI] [PubMed] [Google Scholar]
- Wong GY, Gavva NR (2009) Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: recent advances and setbacks. Brain Res Rev 60:267–277. 10.1016/j.brainresrev.2008.12.006 [DOI] [PubMed] [Google Scholar]
- Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM (2011) Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain 152:844–852. 10.1016/j.pain.2010.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Isami K, Nakamura S, Shirakawa H, Nakagawa T, Kaneko S (2012) Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol Pain 8:55. 10.1186/1744-8069-8-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer B, Angioni C, Osthues T, Toewe A, Thomas D, Pierre SC, Geisslinger G, Scholich K, Sisignano M (2018) The oxidized linoleic acid metabolite 12,13-DiHOME mediates thermal hyperalgesia during inflammatory pain. Biochim Biophys Acta Mol Cell Biol Lipids 1863:669–678. 10.1016/j.bbalip.2018.03.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cold allodynia in oxaliplatin-induced acute peripheral pain. A, Cold-score time course after intraperitoneal injection of oxaliplatin (3 mg/kg) and vehicle. B, Paw withdrawal latency in seconds up to 48 h after intraperitoneal injection of oxaliplatin (3 mg/kg) and vehicle. Data represent the mean SEM of 6 mice/group; two-way ANOVA and Bonferroni's post hoc test. Download Figure 1-1, TIF file (148.7KB, tif) .
Involvement of chemokines, growth factors, and cytokines during oxaliplatin-induced acute peripheral pain. A–O, Measurement of different cytokines and chemokines in DRGs and spinal cords 24 h after oxaliplatin (3 mg/kg, i.p.) treatment. Granulocyte macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-2, TNF-α, and macrophage inflammatory protein 2 (MIP-2) were measured but not detectable. Data represent the mean SEM from DRGs and spinal cord of three to six mice per group; one-way ANOVA and Sidak's post hoc test. P, Relative expression levels of NGF, NOX2, NOX4, XDH, iNOS, ATF3, and MMP9 transcripts in DRGs of oxaliplatin-treated animals after 24 h. The transcripts TNF-α, IFN-γ, and MMP3 were measured but could not be detected. Data represent the mean ± SEM from DRGs of four to nine mice per group and transcript; one-way ANOVA and Dunnett's multiple comparison test. IL, Interleukin. Download Figure 1-2, TIF file (707KB, tif) .
Immunohistochemistry of dorsal spinal cord astrocytes with GFAP 24 h after oxaliplatin or vehicle treatment. A, Representative GFAP immunohistochemistry image of the dorsal spinal cord captured with 50× magnification. B, Representative GFAP immunohistochemistry image of the marked dotted square area in A with 100× magnification. C, Representative GFAP immunohistochemistry image of the marked dotted square area in B with 200× magnification. Data represent a representative image of spinal cord slices. Download Figure 1-3, TIF file (4.1MB, tif) .
Lipid syntheses of prostanoids and linoleic acid metabolites in oxaliplatin-induced acute peripheral pain after 24 h. A, B, Concentration in picograms per milligram of the prostanoids PGF2α and 6-keto PGF1α in sciatic nerve, DRGs, and spinal cord 24 h after intraperitoneal injection of vehicle and oxaliplatin (3 mg/kg) in male and female mice. C–F, Concentrations of linoleic acid-derived metabolites (12,13-EpOME, 12,13-DiHOME, 13-HODE, and 9-HODE) in sciatic nerve, DRGs, and spinal cord 24 h after intraperitoneal injection of vehicle and oxaliplatin in male and female mice. Data represent the mean SEM from DRGs, sciatic nerve, and spinal cord of 4–6 mice/group; *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA and Tukey's post hoc test. Download Figure 1-4, TIF file (511.5KB, tif) .
Lipid syntheses of prostanoids in oxaliplatin-induced acute peripheral pain after 72 h. A–F, Concentration in picograms per milligram of the prostanoids PGE2, PGD2, TXB2, PGF2α, and 6-keto PGF1α in sciatic nerve, DRGs, and spinal cord 72 h after intraperitoneal injection of vehicle and oxaliplatin (3 mg/kg) in male and female mice. Data represent the mean SEM from DRGs, sciatic nerve, and spinal cord of three to five mice per group; *p < 0.05; ***p < 0.001; one-way ANOVA and Tukey's post hoc test. Download Figure 1-5, TIF file (289.7KB, tif) .
LPC 16:0 causes calcium transients in sensory neurons. A, Representative Ca2+ influx in primary sensory neurons after stimulation with LPC 16:0 (1, 5, and 10 µM; 45 s). Neurons were identified by stimulating the primary DRG culture with 50 mM KCl for 60 s. B, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 16:0 (1, 5, and 10 µM; 45 s) as well as their vehicle (EtOH). Data represent the mean SEM from n = 18–34 neurons/condition; **p < 0.01; ***p < 0.001; one-way ANOVA and Sidak's post hoc test. C, Percentage of neurons responding to LPC 16:0 (1, 5, and 10 µM; 45 s) as well as their corresponding vehicle (EtOH). Data represent the mean SEM of n = 22–44 measurements/condition; **p < 0.01; one-way ANOVA and Sidak's post hoc test. Download Figure 3-1, TIF file (135.9KB, tif) .
Transfection control of (h)TRPA1-, (h)TRPV1-, and (r)TRPM8-transfected, as well as empty vector-transfected HEK293 cells. Representative images of empty vector-, (h)TRPA1-, (h)TRPV1-, and (r)TRPM8-transfected HEK293 cells. The left panels show the phase contrast of transfected-HEK293 cells. The middle panels show the GFP signal of transfected HEK293 cells. The right panels show the merged images of the GFP signal and the phase of transfected HEK293 cells. The images were captured with 50× magnification. Download Figure 3-2, TIF file (2.2MB, tif) .
Functional testing of the effects of TRPA1, TRPV1, and TRPM8 inhibitors on the respective channel activity. A, C, E, Representative Ca2+ influx in primary sensory neurons. B, D, F, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by TRP channel agonist. Primary sensory neurons were stimulated two times with 100 µm AITC (TRPA1 agonist), 100 nm capsaicin (cap; TRPV1 agonist), or 100 µm menthol (TRPM8 agonist). Then, either preincubated for 2 min prior to the second stimulus with the corresponding TRP channel inhibitors (TRPA1, 20 µm HC 030031; TRPV1, 1 µM AMG 9810; TRPM8, 1 µM AMTB hydrochloride) or only with the corresponding vehicles of the TRP channel inhibitors (DMSO and H2O). Neurons were identified by stimulating the primary DRG culture with 50 mM KCl for 60 s. Data represent the mean SEM of n = 10–25 neurons/condition; **p < 0.01; ***p < 0.001; ##p < 0.01; ###p < 0.001; one-way ANOVA and Sidak's post hoc test. Download Figure 4-1, TIF file (509.3KB, tif) .
TRP channel expression and CGRP release. A, Relative expression levels of TRPV1, TRPA1, and TRPM8 in DRGs of oxaliplatin-treated animals after 24 h. Data represent the mean ± SEM from DRGs of five mice per group and transcript; one way-ANOVA and Dunnett's multiple comparison test. B, Concentration of CGRP release in picograms per milliliter after stimulating the primary sensory neurons with vehicle (DMSO+EtOH), 400 nm capsaicin (cap), 5 µm LPC 18:1, and 400 nm capsaicin + 5 µm LPC 18:1. Data represent the mean ± SEM from DRGs of four pooled mice per condition; *p < 0.05; **p < 0.01; one-way ANOVA and Sidak's post hoc test. Download Figure 4-2, TIF file (154.8KB, tif) .
sEPSC in spinal dorsal horn lamina II neurons. A, sEPSC frequency in hertz. B, sEPSC amplitude in picoamperes. Frequency and amplitude were recorded prior to and after incubating spinal dorsal horn lamina II neurons with 10 µm LPC 18:1. Data represent the mean ± SEM of n = 8 neurons from four mice; two-tailed Student's t test. Download Figure 4-3, TIF file (3.1MB, tif) .
LPC 16:0 activates TRPV1 and TRPM8. A, D, Representative Ca2+ response in primary sensory neurons after stimulation with 5 µm LPC 16:1 for 45 s prior to and after TRPM8 or TRPV1 and TRPM8 channel inhibition. B, E, Δratio F340/F380 of the amplitude after a transient Ca2+ response caused by LPC 16:0 after TRPM8 or TRPV1 and TRPM8 channel inhibition. Data represent the mean SEM from n = 10–14 neurons/condition; **p < 0.01; ***p < 0.001; #p < 0.05; ###p < 0.001; one-way ANOVA and Sidak's post hoc test. C, F, Percentage of responding neurons to LPC 16:0, after TRPM8 or TRPV1 and TRPM8 channel inhibition. Data represent the mean SEM of n = 7–13 measurements; *p < 0.05; ***p < 0.001; two-tailed unpaired t test with Welch's correction. Download Figure 4-4, TIF file (295.4KB, tif) .
LPC 18:1 induces mechanical hypersensitivity. Paw withdrawal latency (PWL) in seconds up to 6 hours after subcutaneous injection of 10 and 50 µm LPC 18:1 and vehicle in the left hindpaw (ipsilateral paw) of wild-type mice. A, PWL of the contralateral and ipsilateral paws of control animals. B, PWL of the contralateral paw of mice treated with vehicle, 10 µm or 50 m LPC 18:1. C, PWL of the contralateral and ipsilateral paw of mice treated with 10 and 50 µm LPC 18:1. Data represent the mean SEM of 6–10 mice/group; 10 µm LPC 18:1: ##p < 0.01; ###p < 0.001; ####p < 0.0001; 50 µm LPC 18:1: **p < 0.01; ***p < 0.001; ****p < 0.0001; two-way ANOVA and Bonferroni's post hoc test. BL, Baseline. Download Figure 5-1, TIF file (255.9KB, tif) .