

Summary
Aging and endocrine transition states can significantly impact inflammation across organ systems. Neuroinflammation is well documented in Alzheimer disease (AD). Herein, we investigated neuroinflammation that emerges during mid-life aging, chronological and endocrinological, in the female brain as an early initiating mechanism driving AD risk later in life. Analyses were conducted in a translational rodent model of mid-life chronological and endocrinological aging followed by validation in transcriptomic profiles from women versus age-matched men. In the translational model, the neuroinflammatory profile of mid-life aging in females was endocrine and chronological state specific, dynamic, anatomically distributed, and persistent. Microarray dataset analyses of aging human hippocampus indicated a sex difference in neuroinflammatory profile in which women exhibited a profile comparable to the pattern discovered in our translational rodent model, whereas age-matched men exhibited a profile consistent with low neuroimmune activation. Translationally, these findings have implications for therapeutic interventions during mid-life to decrease late-onset AD risk.
Subject Areas: Neuroscience, Immunology, Endocrinology, Transcriptomics
Graphical Abstract
Highlights
-
•
Neuroimmune profile was dynamic across aging transitions in female brain
-
•
Microglial reactivity was spatially dependent and most evident in white matter
-
•
Neuroimmune signature of perimenopause parallels neurodegenerative profile
-
•
Discovery outcomes profiles human hippocampal immune microarray profile in women
Neuroscience; Immunology; Endocrinology; Transcriptomics
Introduction
Neuroinflammatory processes are at the core of the dysregulated pathophysiology observed in Alzheimer's disease (AD) (Akiyama et al., 2000; Eikelenboom et al., 1994; Griffin, 2006; Heneka et al., 2015; Itagaki et al., 1989; Mattiace et al., 1990; McGeer et al., 1987, 1989; Van Eldik et al., 2016). Yet, the inflammatory processes by which pathophysiology of AD is promoted and their contribution to the prodromal phase of late-onset AD remains to be fully understood.
The systems biology of immune response is dynamic and changes in concert with disease progression (Mishra and Brinton, 2018). Inflammation increases with age, in the brain and periphery (Chung et al., 2019; Lynch, 2010; Sanada et al., 2018; Simen et al., 2011). Although the increase in inflammation is evident in both females and males, the trajectories undertaken are different (Da Silva, 1995; Gubbels Bupp et al., 2018; Klein and Flanagan, 2016; Márquez et al., 2020; McCruden and Stimson, 1991; Roved et al., 2017; Ruggieri et al., 2018). Sex differences in immunity during aging have been well documented in peripheral immune cells (Klein and Flanagan, 2016; Márquez et al., 2020). Females experience an increase in inflammation during the perimenopausal transition, which is marked by an increase in peripheral CD4/CD8 T cell ratio, number of CD4 T cells and B cells, and cytokine levels—interferon (IFN)-γ, interleukin (IL)-6, and IL-8 (Klein and Flanagan, 2016; Mishra and Brinton, 2018; Straub, 2007). Much like in the periphery, menopause is associated with an increase in inflammation in brain (Mishra and Brinton, 2018; Sárvári et al., 2012, 2014; Sohrabji, 2007; Yin et al., 2015). Loss of ovarian hormones causes increased expression of microglial reactivity markers linked to interaction with T cells in the hippocampus and frontal cortex (Sárvári et al., 2012, 2014).
Sex-specific transitions in brain metabolism are apparent early in mid-life aging, which are coincident with endocrine transition of peri-to-post menopause (Brinton et al., 2015), and can be a tipping point in neurological aging (Brinton et al., 2015). The perimenopausal transition in females is marked by a bioenergetic deficit characterized by reduced glucose metabolism in brain as detected by 2-deoxy-2-[18F]fluoro-d-glucose positron emission tomography (18F-FDG-PET) and concomitant downregulation of glucose transporter 3 (GLUT3), pyruvate dehydrogenase 1 (PDH1), and oxidative phosphorylation (Ding et al., 2013a, 2013b; Yao and Brinton, 2012; Yao et al., 2012; Yin et al., 2015). Sex-differences in the bioenergetic trajectory of aging are apparent in both mechanistic and clinical analyses (Mosconi et al., 2017b, 2018; Yin et al., 2015; Zhao et al., 2016). Metabolic decline in the brain is an early indicator of the prodromal phase of AD and can be an initiator of chronic inflammation, which is involved in the pathophysiology of the disease (Wang et al., 2020; Yin et al., 2015, 2016). The greater prevalence of AD in women is often attributed to the 4.5 years of greater life span in women (Association, 2018; Nebel et al., 2018; Niu et al., 2017); however, midlife deficits in glucose metabolism and activation of inflammatory processes could be early indicators of prodromal AD contributing to the sex difference in AD prevalence.
To address the role of the neuroimmune system in mechanisms underlying AD in the female, we hypothesized that the mid-life endocrine aging transition in the female initiates neuroinflammation in the brain. To investigate the effect of each chronological and endocrinological aging phase on the neuroinflammatory phenotype, we conducted analyses in the perimenopausal animal model that mimics the perimenopausal transition in its emergence of reproductive irregularity followed by acyclicity (Yin et al., 2015; Bacon et al., 2019; Wang et al., 2020).
Using bulk RNA-Seq of the hippocampus, we investigated inflammatory pathways that are regulated during chronological and endocrine aging windows. Outcomes of these analyses indicated a neuroinflammatory phenotype that dynamically changes during female midlife aging involving a spectrum of glial phenotypes. Spatial mapping of major histocompatibility complex class II (MHC-II), a microglial reactivity marker, revealed increased immunoreactivity in white matter tracts: cingulum, corpus callosum, and fimbria during mid-endocrine aging phase. In parallel, microglial cells exhibited decreased phagocytic capacity. Dynamic changes in the immune profile were paralleled by glial-cell-specific alterations in mitochondrial function. Microglia exhibited a shift toward non-mitochondrial respiration, whereas astrocytes exhibited an increase in mitochondrial spare and maximal respiration. Estradiol administered immediately following ovariectomy prevented transition to an inflammatory profile, whereas estradiol administered weeks following ovariectomy was partially effective.
To address the translational validity of these findings, microarray data from mid-life human female and male hippocampi were analyzed. A sex difference in neuroinflammatory profile was evident in which women exhibited a profile comparable to the pattern discovered in our translational rodent model, whereas age-matched men exhibited a profile consistent with low neuroimmune activation. As in the translational model, the neuroinflammatory profile in women was dynamic, anatomically distributed, and persistent. Collectively, these findings provide a foundation upon which to create a precision therapeutic strategy to prevent, delay, or treat neuroinflammation as an initiating and accelerating driver of Alzheimer risk in women.
Results
Hippocampal Neuroinflammatory Transcriptomic Profile Is Chronological Aging and Endocrinological Aging Specific
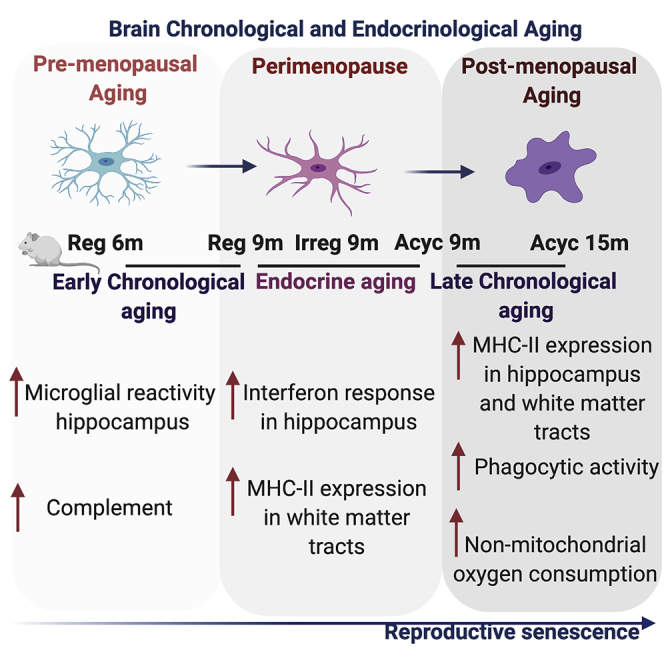
To interrogate a broad immune systems biology, an unbiased transcriptomic analysis was conducted. Transcriptomic profiling conducted by bulk RNA-Seq of the hippocampus during chronological and endocrinological aging revealed that genes involved in neuroinflammation were significantly affected during the course of chronological and endocrinological aging, in comparison to regular cycling 6-month-old (Reg 6m) (Figure 1 and Table S1). Further, each stage of the aging window exhibited a unique profile. Upregulation of Apoe, Trem2, Tyrobp, a gene expression signature that has recently been implicated with a disease-associated microglia (DAM) phenotype (Keren-Shaul et al., 2017), was evident in the chronological aging phase that precedes the onset of perimenopause, (Reg 9m versus Reg 6m) (Figure 1). Co-incident with the expression of DAM genes was an upregulation of complement genes, and microglial reactivity including Cd68, Aif1, Fscn1, Tgfa, and MHC-I and -II (Rt1-A1, Rt1-Dmb). The Cd200 gene that codes for a protein required for communication between microglia and neurons through Cd200r was significantly downregulated at onset of chronological aging (Reg 9m versus Reg 6m).The perimenopause (Irreg 9m versus Reg 6m) was uniquely characterized by downregulation of genes involved in transforming growth factor-β (TGF-β) signaling (Mapk3, Nog, Tgfb3), Apoe, Trem2, Tyrobp, and complement signaling (Figure 1). Interestingly, the perimenopause was characterized by upregulation in genes involved in lipid metabolism (Abca1, Vegfa, Nos3, Ide, Pla2g10) and type I and type II interferon response (B2m, Irf4, Itgb7, Ifnar2, Txnip, Usp18) (Figure 1). The significant rise in the perimenopausal gene expression profile appeared to be initiated during the preceding chronological aging as evidenced by upregulation of B2m, Irf1, Irf2, and Irf7 (Figure 1).
Figure 1.

Transcriptomic Profiling of Endocrine and Chronological Aging (Early and Late) Female Rat Hippocampus
Heatmap visualization of hippocampal differential gene expression analysis of Reg 9m, Irreg 9m, Acyc 9m, and Acyc 15m with respect to Reg 6m, pathway analysis focused on microglial reactivity, complement, lipid metabolism, transforming growth factor β (TGF-β) signaling, major histocompatibility complex (MHC) class I and class II, tumor necrosis factor (TNF) signaling, type I and type II interferon signaling, and T cell markers and signaling. ∗p < 0.05 for differential gene expression analysis of the biological groups with respect to Reg 6m; calculated using student t-test. Detailed p-values and adjusted p values<0.05 for the differential gene expression analysis with respect to Reg 6m are listed in Table S1).
Transition into acyclicity and reproductive senescence (Acyc 9m) was marked by downregulation of type I and type II interferon responses and lipid metabolism (Figure 1). The Acyc 9m group was also characterized by an upregulation of gene expression of phospholipase Pla2g4b, lipid transport Abca7, and microglial lysosomal marker Cd68. Late-chronological aging post-menopause (Acyc 15m) was characterized by an upregulation of the MHC-II genes (Rt1-Ba, Rt1-Bb, Rt1-Da, Rt1-Db1, and Rt1-Db2) (Figure 1). Upregulation of genes involved in myelin breakdown, (Pla2g4b), lipid transport (Abca7), and lysosomal microglial marker (Cd68) was sustained into late chronological aging. Co-incident with this expression profile was an upregulation in genes involved in tumor necrosis factor (TNF) signaling and T helper cell marker Cd4. Increased in Cd4 expression was accompanied by increased T cell activation marker expression, Cd69, and adhesive molecule Icam-1 (Figure 1). Validation was conducted by real-time quantitative PCR (RT-PCR). Consistent with RNAseq result, MHC-II expression was significantly increased in the Acyc 15m group (Rt1-Ba fold change = 1.38, Rt1-Da fold change = 6.27, and Rt1-Db fold change = 9.56, with respect to Reg 6m control).
Collectively, these data indicate that chronological pre-menopausal aging phase was characterized by upregulation of DAM genes, whereas the endocrine peri-menopausal phase by type I and type II interferon response and the post-menopausal phase was marked by upregulation of MHC-II genes, indicating a dynamic shift in microglial reactivity. Collectively, these transcriptomic data are consistent with a dynamic trajectory in patterns of lipid metabolism and microglial gene expression predictive of a shift in neuroimmune transcriptome and functional phenotype.
Microglial Reactivity during Endocrine Aging Selectively Targeted White Matter
On the basis of transcriptomic evidence, the distribution of microglial reactivity was mapped in brain using the microglial marker, ionized calcium binding adaptor molecule (IBA-I, red), and MHC-II (green) microglial reactivity marker (Figures 2A–2E).
Figure 2.

Spatial Mapping of Microglial Reactivity in the Female Aging Rat Brain
(A–D) Several regions in the brain were mapped for microglial reactivity by staining for microglial marker IBA-I (red) and reactivity marker, MHC-II (green), and DAPI (blue). (A) Representative images demonstrating the colocalization of MHC-II with IBA-I and the spatial distribution and differences in extent of MHC-II upregulation in the corpus callosum between Reg 9m and Irreg9m groups (scale bars, 100 μm). Quantification of the colocalization signals between IBA-I and MHC-II using Pearson's correlation in the (B) corpus callosum, (C) fimbria, and (D) hippocampus across five biological groups. (E) Representative images demonstrating the spatial mapping of IBA-I and MHC-II in the corpus callosum, fimbria, and hippocampus across the 5 biological groups (scale bars, 100 μm). Data are represented as mean ± SEM. ∗p ≤ 0.05, ∗∗p ≤ 0.01; calculated using unpaired-student t test.
Spatial mapping of microglial reactivity across the brain revealed that white matter areas, corpus callosum, cingulum, and fimbria, exhibited increased microglial reactivity evidenced by elevated MHC-II positive microglia in early endocrine aging (Irreg 9m) (Figures 2A–2C and 2E) and in late chronological aging (Acyc 15m) (Figures 2B, 2C, and 2E). The hippocampus proper exhibited an increase in microglial reactivity in Irreg 9-month group, although to a lesser extent relative to the white matter (Figures 2D and 2E). Increased presence of microglial reactivity in white matter tracts is consistent with earlier findings (Klosinski et al., 2015) and indicate the potential for myelin catabolism as a source of ketone bodies to fuel energy demand of the brain (Klosinski et al., 2015).
Dynamics of Microglial Oxidative Stress, Phagocytosis, and Reactivity across Chronological and Endocrine Aging
During endocrine aging transitions upregulation of type I and type II interferon responses were coincident with increased microglial expression of MHC-II, a marker of reactivity in white matter. Based on this evidence, we hypothesized that microglial phagocytic function and associated free radical generation would be affected in the transition to reproductive senescence. To investigate this issue, single cell suspensions of pooled dissociated cortices, hippocampi, fimbria, and corpus callosum from each group were generated followed by flow cytometry detection of microglial mitochondrial free radical production, phagocytic capacity, and reactivity marker expression. Microglial cells were gated first for total cells (Figure 3A), followed by singlets (Figure 3B), followed by gating for CD11b (FITC) high and CD45 (VioBlue) intermediate (Figure 3C).
Figure 3.

Microglial Oxidative Stress, Function, and Reactivity Across Chronological and Endocrinological Aging
(A–F) Figures A–F are representative figures of the gating strategy utilized for the assessment. Microglia were selected from the single cell suspension by first gating for (A) total cells from the suspension, then (B) singlets, and (C) then gating for microglia using CD11b (FITC) high and CD45 (Vio-Blue) intermediate as a gating strategy. Microglial cells expressing CD11b (FITC) were used for gating for (D) mitochondrial oxidative stress (PE) (E) output of phagocytic capacity (PE) and (F) MHC-II expression (PE-Vio770).
(G–I) (G) Quantification of mitochondrial oxidative stress. (H) Quantification of phagocytic capacity. (I) Quantification of MHC-II expression. Data are represented as mean ± SEM. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001 calculated using one-way ANOVA.
Microglial mitochondrial generation of reactive oxygen species (ROS) was assessed by measurement of MitoSOX positive microglial cells (Figure 3D). The uptake of MitoSOX was highest in the mid-endocrine aging (Irreg 9m) group (Figure 3G). Coincident with the increase in microglial cells contributing to mitochondrial ROS production was a reduction in microglia participating in phagocytosis (Figures 3E and 3H) evidenced by reduction in percentage of cells phagocytosing pHrodo Staphylococcus aureus Bioparticle conjugates (Figures 3E and 3H). Phagocytic capacity increased and reached maximum after late chronological aging (Acyc 12m group). Microglial reactivity marker—MHC-II expression—increased after late chronological aging in the Acyc 12m group, whereas the mid-aging group (9 months) trended to show an increase in the reactivity marker (Figures 3F and 3I). These data suggest that microglial mitochondrial oxidative stress and phagocytic capacity are inversely associated during endocrine aging. Dynamic shifts in microglial cells contributing to oxidative stress and phagocytic activity in the brain indicate a shift in the neural milieu during endocrine aging and the activation of an acute microglial response.
Astrocytic and Microglial Mitochondrial Function Are Differentially Expressed across Chronological and Endocrinological Aging
Evidence thus far suggested that chronological and endocrinological aging impact microglial reactivity, free radical generation, and phagocytic capacity. We hypothesized that microglial and astrocytic metabolic status would reflect the transitions in the neuroimmune phenotype. To test this hypothesis, metabolic status of glial cells, pooled from cortex, hippocampus, and white matter regions was investigated by measuring microglial and astrocytic mitochondrial respiratory capacity in primary cultures of microglia and astrocytes derived from 6-month-old and 12-month-old animals.
Microglial maximal respiration and ATP production, although not significantly affected, trended higher in younger 6-month-old animals (Figures 4A and 4B). However, microglia from the 12-month-old female brain exhibited significantly higher non-mitochondrial oxygen consumption.
Figure 4.

Mitochondrial Respiratory Capacity in Glial Cells across Age
Seahorse XFe24 analyzer was used to conduct metabolic flux assays on astrocytes and microglia derived from 6-month to 12-month-old female brains.
(A–D) (A) Microglia: oxygen consumption rate (OCR) normalized to protein in microglia. (B) Microglial mitochondrial function parameters. (C) Astrocytes: oxygen consumption rate (OCR) normalized to protein in astrocytes. (D) Astrocytic mitochondrial function parameters. NMOC - Non-Mitochondrial Oxygen Consumption. Data are represented as mean ± SEM. ∗p ≤ 0.05, ∗∗∗∗p ≤ 0.0001; calculated by using two-way ANOVA and Sidak multiple test correction.
Astrocytes derived from 12-month-old female brain exhibited a distinct spare respiratory phenotype. Consistent with increased β-oxidation (Jiang and Cadenas, 2014), astrocytic maximal respiration and spare respiratory capacity was significantly increased (Figures 4C and 4D), whereas basal respiration, proton leak, and ATP production were not impacted.
These data suggest that microglia and astrocytes react differently to chronological aging. The mechanisms to compensate for increased energy demand due to increased inflammation had a cell-specific difference, addressing their broader role in initiating, amplifying, and regulating inflammatory signals.
Estradiol Modulates Neuroinflammation in the Aging Female Brain
To investigate if the hippocampal transcriptome neuroinflammatory profile observed during chronological and endocrinological aging was due to depletion in steroidal hormones, 6-month-old female rats were ovariectomized (OVX). To determine if estradiol would mitigate neuroinflammation and the reactive phenotype of glial cells, two interventions were introduced: an estradiol prevention versus an estradiol treatment paradigm. The prevention paradigm was initiated 24 h post-OVX and was administered over a period of 5 weeks, whereas estradiol treatment paradigm was initiated 2 weeks following OVX and was 3 weeks in duration. Experimental paradigm for both groups equaled 5 weeks.
Bulk RNA-Seq of the hippocampus revealed that ovariectomy induced an upregulation of genes involved in type I and type II interferon response (B2m, Irf7, Ifngr1, Fcer1g) (Figure 5A and Table S2), a gene expression pattern previously observed during endocrine aging (Irreg 9m). Ovariectomy also induced an upregulation of genes involved in the complement system and Trem2 (Figure 5B and Table S2), which was previously observed in the chronological aging phases preceding the perimenopause (Figure 1).
Figure 5.

Estradiol Modulates Neuroinflammation in the Aging Female Brain
(A) Heatmap analysis of interferon response genes in the hippocampus, showing ovariectomy causes an upregulation.
(B) Heatmap analysis of genes contributing to inflammation, myelin metabolism, and neuronal markers, indicating ovariectomy impacts the systems. All genes shown in heatmap are p < 0.05 for OVX versus SHAM. ∗p < 0.05 for differential gene expression analysis with respect to Sham;calculated using student t-test. Detailed p values<0.05 are listed in Table S2.
Estradiol prevention paradigm induced robust suppression of genes involved in the interferon response and the complement system (Figures 5A and 5B). In contrast, the estradiol treatment paradigm partially mitigated the effect of ovariectomy with a decrease in B2m, Casp4, Irf7, and Fcer1g expression, indicating a decrease in interferon response. However, multiple genes involved in the interferon response (Ifit3, Ifngr1, MHC: Rt1-A2), C1s and Trem2, were not suppressed and remained unaffected in the estradiol treatment paradigm (Figures 5A and 5B).
These data suggest that estradiol depletion by ovariectomy induced upregulation of type I and type II interferon responses, which was prevented by introducing estradiol 24 h after ovariectomy. In contrast, delay of estradiol treatment induced a partial, but not complete, suppression of type I and II interferon responses.
Human Validation of Neuroinflammatory Gene Expression in Hippocampus across Aging in Women and Evaluation of Sex Differences
To determine the translational validity of the inflammatory gene expression profile from the chronological and endocrinological midlife aging in perimenopausal animal model, we conducted a proof-of-concept analysis of hippocampal gene expression analysis using a dataset listed uner Gene Expression Omnibus (GEO): GSE11882 (Berchtold et al., 2008) and GEO2R tool of gene expression microarray data collected in human samples. Three age groups, 20–34 years, 35–59 years, and 60–75 years (accession numbers are listed in Tables S3 and S4), designed to capture pre-menopausal/young-adult, peri-to-menopausal/mid-life adult, and post-menopausal/late-life adult, respectively were investigated for differentially expressed genes. Gene expression profiles of age-matched men were analyzed in parallel. In women, multiple immune system related genes were within the top significantly different 250 genes across the age groups. Consistent with our preclinical findings, expression of MHC-I and -II, increased with age (Figure 6). MHC-II expression especially, HLA-DMA, HLA-DMB, HLA-DQA1, HLA-DRB1, HLA-DRB6, was increased during mid-life (35–59 years) relative to 20–34 years—young-adult group. Late-life phase in women was associated with an increased expression of MHC-II genes—HLA-DMA, HLA-DQB1, HLA-DRA, HLA-DRB6—by 2- to 10-fold (Figure 6A) relative to the early aging group (20–34 years).
Figure 6.

Translational Validation of Neuroinflammatory Gene Expression Profile
(A–D) Hippocampal gene expression analysis was conducted on Gene Expression Omnibus (GEO):GSE11882 on stratified age groups in females and males. Fold changes computed by GEO2R tool are plotted. MHC-II expression stratified by age in (A) females and (B) males. Markers for microglial and neuron communication stratified by age in (C) females and (D) males. Plotted as fold change with respect to 20–34 years as control. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001 calculated by GEO2R tool.
In age-matched men, a similar pattern of MHC-II gene expression occurred with increasing age. However, the magnitude of increase, 1- to 3-fold, was lower than the 2- to 10-fold increase in age-matched women. The number of significantly affected genes in men was also less than women. Unlike women, a significant increase in men in MHC-II was only evident by HLA-DRB1 gene expression during late life between 60 and 75 years (Figure 6B).
Microglial and neuronal communication genes that changed in the perimenopausal animal model were affected during aging in females and males. Microglial phagocytic receptor TREM2 and TYROBP increased with aging in both females and males, although to a greater extent in females (Figures 6C and 6D). Interestingly, the microglial marker CX3CR1, which is involved in microglial checkpoint inhibition and homeostatic control (Deczkowska et al., 2018; Lee et al., 2010), was significantly upregulated during mid-life (35–59) years only in females (Figure 6C). The ligand for CX3CR1 expressed on neurons, CX3CL1, showed a similar pattern in expression as CX3CR1 in both females and males (Figures 6C and 6D). The CD200 neuronal communication marker expression was not significantly affected but followed a similar pattern of expression in female and male aging, where it increased during mid-aging and decreased during late-aging phases (Figures 6C and 6D).
These data indicate that the rodent model recapitulates multiple aspects of neuroimmune aging in women. The magnitude of MHC-II upregulation was much higher in females than males. Gene expression of microglial markers and checkpoint genes was also consistent with preclinical findings. These data highlight that aging with respect to inflammation in the hippocampus is dynamic and greater in magnitude in females relative to males.
Discussion
Using an animal model that closely resembles the human perimenopausal transition, we investigated unique contributions of chronological and endocrinological female aging to neuroimmune profiles in brain. This study is the first to characterize chronological and endocrinological effect of aging of the neuroimmune system in brain with specific emphasis on brain regions most impacted in AD.
Hippocampal transcriptomic profiling indicated that inflammation was not a linear continuum in female aging brain. Each aging window was typified by a distinctive neuroimmune program. Chronological aging preceding the endocrinological perimenopause was marked by distinct upregulation in a subset of genes associated with disease-associated microglial phenotype (Apoe, Trem2, Tyrobp, B2m) and microglial reactivity markers such as Aif1, C3, and Cd68. This early chronological aging window was associated with a sustained downregulation of Cd200, a neuronal marker that participates in checkpoint inhibition of microglial phagocytic responses (Deczkowska et al., 2018). Subsequent to early phase chronological aging, the endocrinological aging phase was typified by upregulation of type I and II interferon response genes (B2m, Irf1, Irf4, Ifnar2, adhesion molecule Vcam-1) and downregulation of TGF-β signaling, Apoe, Trem2, Tyrobp, and microglial reactivity markers. The onset of reproductive senescence induced downregulation of type I and II interferon response genes and MHC-II genes. Late-phase chronological aging of post-menopause was specifically marked by upregulation of MHC-II genes, TNF signaling, phospholipases, lysosomal marker Cd68, and T cell marker Cd4. The gene expression profile is consistent with the onset of adaptive immune responses and white matter catabolism.
Recent characterization of models of familial AD and neurodegeneration (5xFAD, CK-p25) revealed a distinct phenotype of microglia, which is disease state specific and involves the participation of type I and type II interferon response genes, MHC, Apoe, Trem2, Tyrobp, and TGF-β signaling (Keren-Shaul et al., 2017; Mathys et al., 2017). Herein, we reported that female aging and specifically the perimenopausal transition is associated with a molecular signature of microglia associated with neurodegeneration. Of note, the upregulation of type I and type II interferon response genes during the perimenopausal phase followed by the subsequent upregulation of MHC-II genes in the post-menopausal aged animals matches the “late response microglia” in the CK-p25 model (Mathys et al., 2017). Consistent with the expression of “late stage microglia” in neurodegeneration, the menopausal transition indicates that neurodegenerative programs can be activated early during midlife aging, possibly indicating the onset of a prodromal state.
Mapping of microglial reactivity revealed distinct localization of microglia to multiple white matter tracts that was coincident with early endocrine aging. Increased expression of MHC-II was evident in the corpus callosum, cingulum, and fimbria. Reduction in microglial phagocytosis was associated with age-related increase in myelin debris (Safaiyan et al., 2016), suggesting that the increase in MHC-II expression in white matter tracts observed during early endocrine aging is likely due to an increase in myelin debris. Microglial isolation from gray and white matter areas and subsequent functional profiling documented decline in phagocytosis during perimenopause. This microglial phenotype coupled with microglial mitochondrial respiratory phenotype and white matter track localization parallels earlier analyses of white matter catabolism for ketone body generation in brain during endocrine aging (Klosinski et al., 2015).
Microglial and astrocytic mitochondrial function paralleled immune phenotypic shifts during chronological and endocrinological aging. Microglia initiate the inflammatory response, whereas astrocytes amplify the inflammatory signals while meeting neuronal metabolic demands (Yin et al., 2016). Herein, we show that astrocytic maximal respiration and spare respiratory capacity and microglial non-mitochondrial oxygen consumption increased with age. The disparity between the cellular mitochondrial response to aging can be addressed by their broader function in the brain. Microglia respond to aging by increasing non-mitochondrial oxygen consumption, which possibly indicates increased anaerobic glycolysis and free radical production (Ghosh et al., 2018). Consistent with peripheral immune cells, microglia also react to inflammatory challenges by increasing anaerobic glycolysis and reduced mitochondrial respiration (Holland et al., 2018). In contrast, astrocytes increased spare respiratory capacity and maximal respiration, which is consistent with increased β-oxidation or response to energy demand of increased inflammation or both (Jiang and Cadenas, 2014).
Decline in estrogen during mid-life endocrine aging in the female is associated with a decline in glucose metabolism and activation of increased reliance on lipid-derived ketone bodies that can be supplied by white matter (Klosinski et al., 2015). Those analyses demonstrated a phased trajectory of white matter degeneration, lipid metabolism, and ketone body generation during endocrine aging (Klosinski et al., 2015). Consistent with increased mitochondrial oxidative stress reported herein, Klosinski et al. observed an increase in hydrogen peroxide production during early endocrine aging that activated the arachidonic acid and phospholipase A2 signaling pathways to activate sphingomyelinase metabolism of sphingomyelin followed by a staged activation of ceramidase and thiolase to generate fatty acid lipids that ultimately results in ketone body generation (Klosinski et al., 2015). The increase in maximal respiration and spare respiratory capacity in astrocytes with age is consistent with increased age-related inflammation (Jiang and Cadenas, 2014) and increased β-oxidation to generate ketone bodies (Klosinski et al., 2015). These findings in translational rodent models are consistent with human brain imaging, indicating that endocrine aging in women of perimenopausal to postmenopausal age exhibit decline in white matter volume and a rise in β-amyloid plague deposition (Mosconi et al., 2017a, 2017b, 2018).
Estrogen is a master regulator controlling neuronal and glial bioenergetics, neurogenesis, microglial inflammation, and glucose metabolism (Rettberg et al., 2014). Evidence for regulation of the inflammatory phenotype by estradiol is evident in the hippocampal transcriptomic profiling of ovariectomized animals. Ovariectomizing animals prior to onset of endocrine aging induced gene expression patterns that were a combination of chronological and endocrinological aging. Ovariectomy induced a transcriptomic profile indicative of accelerated aging in which early chronological and endocrinological aging (perimenopause) and late endocrinological aging are compressed into a single immune state (upregulation of C4b, Trem2, Clec7a, type I and II interferon response, MHC-I genes). Interestingly, ovariectomy did not cause significant changes in MHC-II expression, indicating that upregulation of MHC-II expression in aged reproductive senescent animals was in part caused by aging. Although ovariectomy mechanistically validates that estradiol regulates the neuroinflammatory phenotype, it also indicates that eliminating natural endocrine aging by ovariectomy causes an overt inflammatory reaction that combines inflammatory programs of chronological and endocrinological aging. The translational implication of accelerated immune reactivity is consistent with earlier clinical studies, indicating increased risk of Alzheimer in women oophorectomized prior to natural menopause (Rocca et al., 2008, 2014). The estradiol prevention treatment paradigm prevented ovariectomy-induced immune profile, whereas the estradiol treatment paradigm failed to restore the pre-ovariectomy immune profile. Mitigation of inflammation and especially interferon response genes by an estradiol prevention treatment paradigm could be effective for disease-state-specific targeting of inflammation in prodromal states of AD. Epidemiological data provide support for reduced risk of AD in women treated with estrogen or hormone therapy for menopausal symptoms (Brinton, 2008), whereas treatment of postmenopausal women in the absence of symptoms has no benefit and depending on the hormone formulation can increase risk in women aged 65 years or older (Brinton, 2008).
Microglial activation of T cells mediated by type I and type II interferon is also at the core of pathophysiology of multiple sclerosis, an autoimmune disorder more prevalent in women than men (Schetters et al., 2018) and a worsening in the clinical trajectory following menopause.
Clinical gene expression analysis of differentially expressed genes in females during aging (20–34 years: early-life, 35–59 years: mid-life; and 60–75 years: late-life and reproductively senescent) was consistent with our rodent brain analyses. As endocrine aging status of the clinical samples was unavailable, stratification of groups was based on average age of perimenopausal to menopausal transition. Incremental upregulation of MHC-II and interferon response genes with age in females is indicative of immune activation in the brain after menopause. These findings suggest that the interferon and MHC signaling are likely linked to estrogen decline in the female brain (Wang et al., 2020).
Outcomes of analyses reported herein demonstrate dynamic shifts in the neuroinflammatory phenotype across chronological and endocrine aging in the female brain. Coincident with dysregulation of glucose metabolism during the perimenopausal transition in female brain (Ding et al., 2013a, 2013b; Klosinski et al., 2015; Wang et al., 2020; Yin et al., 2015) is the upregulation of interferon response and MHC-II expression in the hippocampus and white matter tracts. The inflammatory phenotype and upregulation of interferon response and MHC-II genes observed in the perimenopausal model was validated in ovariectomized animal models and human transcriptome data. Glial redox status, phagocytic capacity, and respiratory capacity also paralleled aging transitions. These findings highlight that the perimenopausal transition causes a significant shift in neuroinflammation, affecting glial function and metabolism.
Herein we report the emergence of neuroimmune responses in brain that emerge during midlife chronological and endocrinological aging transitions in the female brain. The profile of neuroimmune responses were dynamic, localized to white matter and unique to the stage of aging. The data supporting this complex biology are consistent across investigational methods, relevant to earlier fundamental and clinical science reports and have translational validity to the human. From a therapeutic perspective, these findings provide a foundation upon which to create a precision therapeutic strategy to prevent, delay, or treat neuroinflammation as an initiating and accelerating driver of Alzheimer risk in women.
Limitations of the Study
Although we provide evidence of upregulation of MHC-II in menopausal and post-menopausal aged women in a human micro-array dataset, these findings are not controlled for confounding variables. These analyses were conducted as a proof-of-concept evaluation and require replication in larger datasets. Furthermore, causal relationships between the upregulation of interferon response with metabolic dysfunction in brain requires further investigation. Notably, the role of microglial and astrocytic crosstalk in female brain aging needs to be further characterized. Interventional studies of MHC-II and interferon signaling therapeutics to investigate impact on white matter integrity, inflammation, and cognition could validate the role of interferon signaling in the aging female brain. Aging transitions that lead to the development of autoimmune profile such as multiple sclerosis and neurodegenerative diseases such as Alzheimer's require further exploration.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Roberta Diaz Brinton (rbrinton@arizona.edu)
Data Availability
The accession number for the hippocampal RNA-Seq data from the perimenopausal animal model analyzed in this paper is GEO: GSE161142. The accession number for the hippocampal RNA-Seq datas from the ovariectomy, estradiol treatment, and prevention experiment analyzed in this paper is GEO: GSE161233.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by NIH: National Institute on Aging, United States grants P01-AG026572 to R.D.B.; Project 1 to R.D.B. and Analytic Core to F.Y., R37AG053589 and, 1R01AG057931 to R.D.B.
Author Contributions
Conceptualization, A.M. and R.D.B; Methodology, A.M., Y.S, Y.W., and E.B.; Formal analysis, A.M, Y.S., and F.Y.; Investigation, A.M.; Writing—Original Draft, A.M. and R.D.B.; Writing—Review and Editing, A.M. and R.D.B; Funding acquisition, R.D.B. and F.Y.
Declaration of Interest
The authors declare no competing financial interest.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101829.
Supplemental Information
References
- Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G.M., Cooper N.R., Eikelenboom P., Emmerling M., Fiebich B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association A.s. 2018 Alzheimer's disease facts and figures. Alzheimer's Demen. 2018;14:367–429. [Google Scholar]
- Bacon E.R., Mishra A., Wang Y., Desai M.K., Yin F., Brinton R.D. Neuroendocrine aging precedes perimenopause and is regulated by DNA methylation. Neurobiology of aging. 2019;74:213–224. doi: 10.1016/j.neurobiolaging.2018.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold N.C., Cribbs D.H., Coleman P.D., Rogers J., Head E., Kim R., Beach T., Miller C., Troncoso J., Trojanowski J.Q. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. 2008;105:15605. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton R.D. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosciences. 2008;31:529–537. doi: 10.1016/j.tins.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton R.D., Yao J., Yin F., Mack W.J., Cadenas E. Perimenopause as a neurological transition state. Nat. Rev. Endocrinol. 2015;11:393–405. doi: 10.1038/nrendo.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.Y., Kim D.H., Lee E.K., Chung K.W., Chung S., Lee B., Seo A.Y., Chung J.H., Jung Y.S., Im E. Redefining chronic inflammation in aging and age-related diseases: proposal of the senoinflammation concept. Aging Dis. 2019;10:367–382. doi: 10.14336/AD.2018.0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva J.A. Sex hormones, glucocorticoids and autoimmunity: facts and hypotheses. Ann. Rheum. Dis. 1995;54:6–16. doi: 10.1136/ard.54.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A., Amit I., Schwartz M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 2018;21:779–786. doi: 10.1038/s41593-018-0145-x. [DOI] [PubMed] [Google Scholar]
- Ding F., Yao J., Rettberg J.R., Chen S., Brinton R.D. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PLoS One. 2013;8:e79977. doi: 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F., Yao J., Zhao L., Mao Z., Chen S., Brinton R.D. Ovariectomy induces a shift in fuel availability and metabolism in the hippocampus of the female transgenic model of familial Alzheimer's. PLoS One. 2013;8:e59825. doi: 10.1371/journal.pone.0059825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P., Zhan S.-S., van Gool W.A., Allsop D. Inflammatory mechanisms in Alzheimer's disease. Trends Pharmacol. Sci. 1994;15:447–450. doi: 10.1016/0165-6147(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Castillo E., Frias E.S., Swanson R.A. Bioenergetic regulation of microglia. Glia. 2018;66:1200–1212. doi: 10.1002/glia.23271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin W.S.T. Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 2006;83:470S–474S. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- Gubbels Bupp M.R., Potluri T., Fink A.L., Klein S.L. The confluence of sex hormones and aging on immunity. Front. Immunol. 2018;9:1269. doi: 10.3389/fimmu.2018.01269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M.T., Carson M.J., El Khoury J., Landreth G.E., Brosseron F., Feinstein D.L., Jacobs A.H., Wyss-Coray T., Vitorica J., Ransohoff R.M. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland R., McIntosh A.L., Finucane O.M., Mela V., Rubio-Araiz A., Timmons G., McCarthy S.A., Gun'ko Y.K., Lynch M.A. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav. Immun. 2018;68:183–196. doi: 10.1016/j.bbi.2017.10.017. [DOI] [PubMed] [Google Scholar]
- Itagaki S., McGeer P.L., Akiyama H., Zhu S., Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- Jiang T., Cadenas E. Astrocytic metabolic and inflammatory changes as a function of age. Aging cell. 2014;13:1059–1067. doi: 10.1111/acel.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B. A unique microglia type Associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e1217. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Klein S.L., Flanagan K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016;16:626. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- Klosinski L.P., Yao J., Yin F., Fonteh A.N., Harrington M.G., Christensen T.A., Trushina E., Brinton R.D. White matter lipids as a ketogenic fuel supply in aging female brain: implications for alzheimer's disease. EBioMedicine. 2015;2:1888–1904. doi: 10.1016/j.ebiom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Varvel N.H., Konerth M.E., Xu G., Cardona A.E., Ransohoff R.M., Lamb B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am. J. Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. Age-related neuroinflammatory changes negatively impact on neuronal function. Front. Aging Neurosci. 2010;1:6. doi: 10.3389/neuro.24.006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez E.J., Chung C.-h., Marches R., Rossi R.J., Nehar-Belaid D., Eroglu A., Mellert D.J., Kuchel G.A., Banchereau J., Ucar D. Sexual-dimorphism in human immune system aging. Nat. Commun. 2020;11:751. doi: 10.1038/s41467-020-14396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H., Adaikkan C., Gao F., Young J.Z., Manet E., Hemberg M., De Jager P.L., Ransohoff R.M., Regev A., Tsai L.-H. Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 2017;21:366–380. doi: 10.1016/j.celrep.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiace L.A., Davies P., Yen S.H., Dickson D.W. Microglia in cerebellar plaques in Alzheimer's disease. Acta Neuropathologica. 1990;80:493–498. doi: 10.1007/BF00294609. [DOI] [PubMed] [Google Scholar]
- McCruden A.B., Stimson W.H. Sex hormones and immune function. In: Psychoneuroimmunology R. Ader, D.L. Felten, Cohen N., editors. Academic Press; 1991. pp. 475–493. [Google Scholar]
- McGeer P.L., Akiyama H., Itagaki S., McGeer E.G. Immune system response in alzheimer's disease. Can. J. Neurol. Sci. 1989;16:516–527. doi: 10.1017/s0317167100029863. [DOI] [PubMed] [Google Scholar]
- McGeer P.L., Itagaki S., Tago H., McGeer E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- Mishra A., Brinton R.D. Inflammation: bridging age, menopause and APOEε4 genotype to Alzheimer’s disease. Front. Aging Neurosci. 2018;10:312. doi: 10.3389/fnagi.2018.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Berti V., Quinn C., McHugh P., Petrongolo G., Osorio R.S., Connaughty C., Pupi A., Vallabhajosula S., Isaacson R.S. Perimenopause and emergence of an Alzheimer's bioenergetic phenotype in brain and periphery. PLoS One. 2017;12:e0185926. doi: 10.1371/journal.pone.0185926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Berti V., Quinn C., McHugh P., Petrongolo G., Varsavsky I., Osorio R.S., Pupi A., Vallabhajosula S., Isaacson R.S. Sex differences in Alzheimer risk: brain imaging of endocrine vs chronologic aging. Neurology. 2017;89:1382–1390. doi: 10.1212/WNL.0000000000004425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L., Rahman A., Diaz I., Wu X., Scheyer O., Hristov H.W., Vallabhajosula S., Isaacson R.S., de Leon M.J., Brinton R.D. Increased Alzheimer's risk during the menopause transition: a 3-year longitudinal brain imaging study. PLoS One. 2018;13:e0207885. doi: 10.1371/journal.pone.0207885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebel R.A., Aggarwal N.T., Barnes L.L., Gallagher A., Goldstein J.M., Kantarci K., Mallampalli M.P., Mormino E.C., Scott L., Yu W.H. Understanding the impact of sex and gender in Alzheimer's disease: a call to action. Alzheimer's Demen. 2018;14:1171–1183. doi: 10.1016/j.jalz.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H., Álvarez-Álvarez I., Guillén-Grima F., Aguinaga-Ontoso I. Prevalence and incidence of Alzheimer's disease in Europe: a meta-analysis. Neurología (English Edition) 2017;32:523–532. doi: 10.1016/j.nrl.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Rettberg J.R., Yao J., Brinton R.D. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front. neuroendocrinology. 2014;35:8–30. doi: 10.1016/j.yfrne.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca W.A., Grossardt B.R., Maraganore D.M. The long-term effects of oophorectomy on cognitive and motor aging are age dependent. Neuro-degenerative Dis. 2008;5:257–260. doi: 10.1159/000113718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca W.A., Grossardt B.R., Shuster L.T. Oophorectomy, estrogen, and dementia: a 2014 update. Mol. Cell. Endocrinol. 2014;389:7–12. doi: 10.1016/j.mce.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roved J., Westerdahl H., Hasselquist D. Sex differences in immune responses: hormonal effects, antagonistic selection, and evolutionary consequences. Horm. Behav. 2017;88:95–105. doi: 10.1016/j.yhbeh.2016.11.017. [DOI] [PubMed] [Google Scholar]
- Ruggieri A., Gagliardi M.C., Anticoli S. Sex-dependent outcome of hepatitis B and C viruses infections: synergy of sex hormones and immune responses? Front. Immunol. 2018;9:2302. doi: 10.3389/fimmu.2018.02302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaiyan S., Kannaiyan N., Snaidero N., Brioschi S., Biber K., Yona S., Edinger A.L., Jung S., Rossner M.J., Simons M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016;19:995–998. doi: 10.1038/nn.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada F., Taniyama Y., Muratsu J., Otsu R., Shimizu H., Rakugi H., Morishita R. Source of chronic inflammation in aging. Front. Cardiovasc. Med. 2018;5:12. doi: 10.3389/fcvm.2018.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sárvári M., Hrabovszky E., Kalló I., Solymosi N., Likó I., Berchtold N., Cotman C., Liposits Z. Menopause leads to elevated expression of macrophage-associated genes in the aging frontal cortex: rat and human studies identify strikingly similar changes. J. Neuroinflammation. 2012;9:264. doi: 10.1186/1742-2094-9-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sárvári M., Kalló I., Hrabovszky E., Solymosi N., Liposits Z. Ovariectomy and subsequent treatment with estrogen receptor agonists tune the innate immune system of the Hippocampus in middle-aged female rats. PLoS One. 2014;9:e88540. doi: 10.1371/journal.pone.0088540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schetters S.T.T., Gomez-Nicola D., Garcia-Vallejo J.J., Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front. Immunol. 2018;8:1905. doi: 10.3389/fimmu.2017.01905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simen A.A., Bordner K.A., Martin M.P., Moy L.A., Barry L.C. Cognitive dysfunction with aging and the role of inflammation. Ther. Adv. Chronic Dis. 2011;2:175–195. doi: 10.1177/2040622311399145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrabji F. Guarding the blood-brain barrier: a role for estrogen in the etiology of neurodegenerative disease. Gene Expr. 2007;13:311–319. doi: 10.3727/000000006781510723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007;28:521–574. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- Van Eldik L.J., Carrillo M.C., Cole P.E., Feuerbach D., Greenberg B.D., Hendrix J.A., Kennedy M., Kozauer N., Margolin R.A., Molinuevo J.L. The roles of inflammation and immune mechanisms in Alzheimer's disease. Alzheimer's Dementia (N Y) 2016;2:99–109. doi: 10.1016/j.trci.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Mishra A., Brinton R.D. Vol. 9. 2020. Transitions in Metabolic and Immune Systems from Pre-menopause to Post-menopause: Implications for Age-Associated Neurodegenerative Diseases; p. F1000Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Shang Y., Mishra A., Bacon E., Yin F., Brinton R. Midlife Chronological and Endocrinological Transitions in Brain Metabolism: System Biology Basis for Increased Alzheimer’s Risk in Female Brain. Scientific Reports. 2020;10:8528. doi: 10.1038/s41598-020-65402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Brinton R.D. Estrogen regulation of mitochondrial bioenergetics: implications for prevention of Alzheimer's disease. Adv. Pharmacol. 2012;64:327–371. doi: 10.1016/B978-0-12-394816-8.00010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Irwin R., Chen S., Hamilton R., Cadenas E., Brinton R.D. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiol. Aging. 2012;33:1507–1521. doi: 10.1016/j.neurobiolaging.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F., Sancheti H., Patil I., Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radic. Biol. Med. 2016;100:108–122. doi: 10.1016/j.freeradbiomed.2016.04.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F., Yao J., Sancheti H., Feng T., Melcangi R.C., Morgan T.E., Finch C.E., Pike C.J., Mack W.J., Cadenas E. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging. 2015;36:2282–2295. doi: 10.1016/j.neurobiolaging.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L., Mao Z., Woody S.K., Brinton R.D. Sex differences in metabolic aging of the brain: insights into female susceptibility to Alzheimer's disease. Neurobiol. Aging. 2016;42:69–79. doi: 10.1016/j.neurobiolaging.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the hippocampal RNA-Seq data from the perimenopausal animal model analyzed in this paper is GEO: GSE161142. The accession number for the hippocampal RNA-Seq datas from the ovariectomy, estradiol treatment, and prevention experiment analyzed in this paper is GEO: GSE161233.