

Summary
Tumors comprised a tightly surrounded tumor microenvironment, made up of non-cellular extracellular matrix (ECM) and stromal cells. Although treatment response is often attributed to tumor heterogeneity, progression and malignancy are profoundly influenced by tumor cell interactions with the surrounding ECM. Here, we used a tumor organoid model, consisting of hepatic stellate cells (HSCs) embedded in collagen type 1 (Col1) and colorectal cancer cell (HCT-116) spheroids, to determine the relationship between the ECM architecture, cancer cell malignancy, and chemoresistance. Exogenous transforming growth factor beta (TGF-β) used to activate the HSCs increased the remodeling and bundling of Col1 in the ECM around the cancer spheroid. A dense ECM architecture inhibited tumor cell growth, reversed their mesenchymal phenotype, preserved stem cell population, and reduced chemotherapy response. Overall, our results demonstrate that controlled biofabrication and manipulation of the ECM in tumor organoids results enables studying tumor cell-ECM interactions and better understand tumor cell response to chemotherapies.
Subject Areas: Biological Sciences, Cancer, Bioengineering
Graphical Abstract
Highlights
-
•
A 3D tumor organoid model with stromal cells and colorectal cancer cell spheroids
-
•
ECM architecture influences cancer cell phenotype, malignancy, and chemoresistance
-
•
Denser ECM reverses tumor cell mesenchymal phenotype and preserves stem cell population
-
•
Looser ECM architecture supports mesenchymal phenotype and increased chemosensitivity
Biological Sciences; Cancer; Bioengineering
Introduction
Colorectal cancer (CRC) is the third most deadly and fourth most commonly diagnosed cancer worldwide (Rawla et al., 2019). Decreased mortality rate in CRC has been attributed to early detection efforts that lead to the removal of pre-cancerous or un-metastasized polyps in the colon (Rawla et al., 2019). CRC arises when certain cells of the epithelium acquire a series of mutations that enable them to grow uncontrollably, ultimately leading to a hyper-proliferative cohort of cancer cells that eventually metastasis. Population-based studies have shown that 25–30% of patients with CRC will develop metastases in their liver, which ultimately results in death in two-thirds of these patients (van der Pool et al., 2012; Christina Hackl et al., 2014; Manfredi et al., 2006).
The colorectal liver metastatic tumor microenvironment (TME) consists mainly of cancer-associated fibroblasts (CAFs), hepatic stellate cells (HSCs), immune cells, and endothelial cells (Williamson et al., 2019). CAFs are a crucial component of the TME responsible for cytokine and growth factor secretion along with extracellular matrix (ECM) remodeling (Erdogan and Webb, 2017; Tao et al., 2017). CAFs can be derived and activated from multiple origins in response to radiation, chemical, or immune triggers (Mueller et al., 2007; Ishii et al., 2016). In the liver, however, HSCs represent the major cell source of activated CAFs (Affo et al., 2017). HSC activation occurs through the release of various factors including transforming growth factor beta (TGF-β) by immune cells, thus inducing ECM remodeling, collagen deposition, local stiffness changes, and ECM linearization by the activated HSCs (Song et al., 2016; Dewidar et al., 2015; Ho et al., 2014). The ECM component of the TME has been shown to have a great influence on cancer proliferation, migration, and apoptosis (Yuzhalin et al., 2018; Paolillo and Schinelli, 2019; Eble and Niland, 2019; Cox and Erler, 2011). These changes could be a result of an altered signaling pathway that is transduced by ECM specific integrin binding such as focal adhesion kinase (FAK) (Seong et al., 2013). It is also understood that under normal conditions, the ECM undergoes constant active remodeling and stimulates biochemical and biophysical cues that influence cell behavior. Abnormalities in the homeostatic control of ECM production and degradation can have substantial effects on surrounding cells (Cox and Erler, 2011; Eble and Niland, 2019; Emon et al., 2018; Paolillo and Schinelli, 2019). For example, collagen, the most significant ECM component, has been shown to be upregulated in patients with colorectal to liver metastasis (van Huizen et al., 2019). Collagen type 1 (Col1) is an important ECM protein that provides tissue strength and support. Structural organization and levels of Col1 can indirectly influence cancer phenotype and drug efficacy (Plou et al., 2018; Brabrand et al., 2015; Holle et al., 2016). The ECM architecture surrounding most solid tumors has been established to be stiffer and denser compared to healthy tissue (Paolillo and Schinelli, 2019; Baiocchini et al., 2016). A dense TME has thus been shown to enhance metastasis and change the phenotype of mammary carcinoma in patient samples; however, the exact role of collagen deposition and density in tumor progression and drug response is nuanced (Pickup et al., 2013; Walker et al., 2018).
Hepatic resection is the only treatment that offers an increased long-term survival with a 5-year survival rate of 40%; however, only 25% of patients are suitable for this invasive surgery (Kanas et al., 2012; Christina Hackl et al., 2014). Among the many that are not eligible for resection, adjuvant chemotherapy such as 5-fluorouracil (5-FU) combined with leucovorin, oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) is used for CRC and metastasis management (Chow and Chok, 2019). Unfortunately, drug resistance remains a major factor in low survival rates for patients with CRC. Recently, the impact of the TME, including the cellular stroma and the acellular ECM, has gained attention for CRC research due to its increasing role in drug resistance and cancer progression (Walker et al., 2018; Senthebane et al., 2018; Sun, 2016).
It is now well understood that interactions between tumor cells, microenvironment-specific stromal cells, and ECM are essential for tumor growth and significantly affect drug response (Senthebane et al., 2017, 2018). The TME is not easily modeled in previously developed two-dimensional (2D) in vitro techniques; therefore, the consideration of new methods for visualization and manipulation has been investigated. Conventional 2D techniques are advantageous for their high-throughput capabilities and low cost; however, they lack the potential to mimic the complexity of the TME and are relatively limited in studying cancer metastasis and drug resistance mechanisms (Devarasetty et al., 2018). Additionally, in vivo animal models are expensive and inefficient, influencing the use of three-dimensional (3D) culture systems, such as spheroids, organoids, or microfluidics, to study the TME effect on cancer progression and chemotherapy response (Neal et al., 2018; Skardal et al., 2015). Three-dimensional culture systems have also proven useful in studying cancer stem cells (CSCs) due to its ability to maintain ECM density, hypoxia, and low nutrients (Lee et al., 2020). Organoids are defined as clusters of cells that represent a fraction of a particular tissue environment and function (Nantasanti et al., 2016). Organoid and 3D culture systems have been increasingly popular in cancer research due to the ability to model some, but not all, aspects of the TME interactions with cancer cells (Buzzelli et al., 2018; Neal et al., 2018).
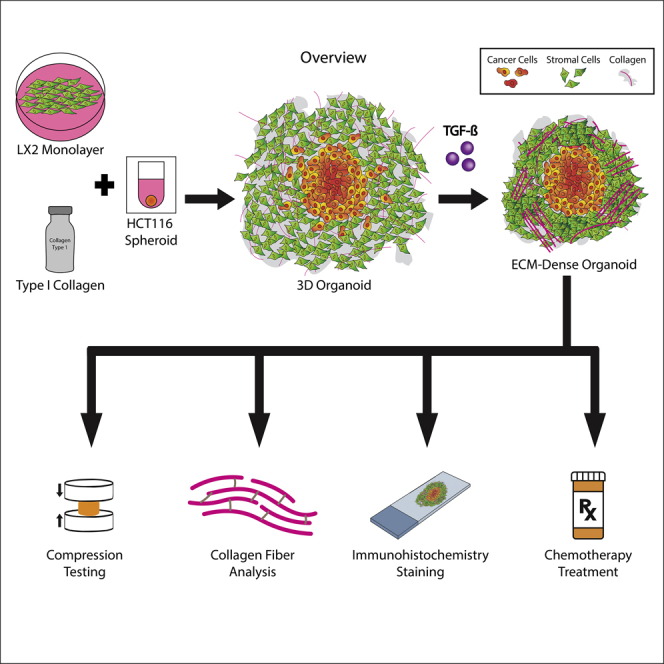
Our innovative approach uses advanced biofabrication methods that mimic in vivo conditions in order to create a microenvironment similar to that of a colorectal tumor that has metastasized to the liver. We have recently reported on the fabrication of CRC organoids by embedding tumor cell spheroids in Col1-suspended stromal cells (smooth muscle cells and fibroblasts) (Devarasetty et al., 2017). The stromal cells were able to remodel the Col1 gel, resulting in 3D organoids with well-structured stromal ECM that we implanted in mice. Herein, we describe the utilization of our tumor organoid platform to analyze the interactions between an HSC cell line of the liver and metastatic CRC cell lines. In this study, we expose cancer cells to various HSC-produced ECM densities using TGF-β and determine macroscopic characteristics of the collagen remodeling and its effect on embedded cells. Finally, our tumor organoid platform is capable of testing the effects of the tumor-stroma organization on tumor cell response to chemotherapy. The overall goal of this research is to determine how structural/mechanical changes in the TME, specifically the ECM, impact tumor cell phenotype and their response to chemotherapy.
Results
TGF-β Induces Myofibroblastic-like Phenotype of LX-2 Cells within the Organoids
HSCs are a major component of the liver mesenchymal cell population that react to injury or insult through transdifferentiation into highly proliferative and motile myofibroblasts. Various cytokines, including TGF-β, activate HSC to myofibroblasts that steadily remodel the liver ECM via deposition of new ECM components and structural remodeling of the preexisting ECM (Carloni et al., 2014). To model the effect of TGF-β-induced HSC activation, we constructed 3D tissue equivalents (organoids) consisting of HSC line (LX-2) suspended in Col1 hydrogel (Figure 2A). We then examined the expression of several fibroblastic markers in response to TGF-β by immunostaining organoids treated with TGF-β compared to control (Figure 1A). Staining for fibroblast activation protein (FAP), a protein overexpressed on HSC upon activation, revealed a greater number of FAP-expressing LX-2 cells in the presence of TGF-β compared to the control. Similar results were observed for the expression of α-smooth muscle actin (αSMA), a marker for HSC activation and liver fibrosis. Quantification of stained images using VisioPharm software confirmed that LX-2 cells in organoids cultured in the presence of TGF-β increased expression of FAP and αSMA by 2.07- and 2.56-fold, respectively (Figure 1B, p value = 0.045 and 0.019, respectively). Lastly, we measured the numbers of proliferating cells in the organoids by immunostaining for Ki-67. TGF-β increased LX-2 cell proliferation in the organoids by 2.02 times compared to control (p value of 0.0026). These results indicate that the TGF-β activation of LX-2 cells induces myofibroblastic-like differentiation.
Figure 2.

Establishing a Tumor Organoid Model with ECM Dense Stroma
(A) Culture molds were produced by additive printing a negative mold then deposited PDMS was cured to generate a 6-well plate microwell insert. The cell-collagen type I solution was deposited, with a suspended preformed HCT-116 spheroid, into microwells and allowed to gel. Culture media, with or without 10 ng/mL of TGF-β was added to the culture media after the organoid gelated and allowed to grow for up to 7 days before being harvested. Organoids cultured in TGF-β, right, underwent sizable contraction compared to organoids in control media, center, and collagen-only controls, left.
(B) Organoid diameter was measured over 96 hr in culture conditions. Graphs represent mean ± s.e.m (n = 4).
(C) Organoid stiffness was measured over 96 hr in culture using a rheometer to determine the young's modulus of the construct. Graphs represent mean ± s.e.m (n = 4) (∗∗p ≤ 0.01; ∗∗∗∗p ≤ 0.0001).
Figure 1.

TGF-β Induces Myofibroblastic-Like Phenotype of the Hepatic Stellate Cells (LX-2s) within the Organoid
(A) LX-2 organoids are grown for 7 days with or without 10 ng/mL TGF-β and IHC stained for α-SMA or FAP (green), DAPI (blue), and Ki-67 (red).
(B) LX-2 activation was determined by IHC quantification of FAP, a-SMA, and Ki-67 using VisioPharm software by calculating the percentage of positively expressed cells by the total nuclei present. Graphs represent mean ± s.e.m (n = 5) and generated using GraphPad prism.
TGF-β Induces Physical and Structural Changes to the ECM of Tumor Organoids
The results presented above demonstrated that TGF-β induced transdifferentiation of LX-2, which may have a subsequent effect on ECM remodeling in the organoids. Anatomically, the remodeled ECM yields a dense environment (Graphical Abstract). To further model the effect of ECM density and architecture on metastatic tumor growth, we embedded spheroids of CRC cells (HCT-116) in the LX-2/Col1 suspension. Macroscopic observations of organoids cultured in the presence of TGF-β revealed a significant difference compared with organoids grown in control media (Figures 2B and 2C). Organoids grown in 10 ng/mL TGF-β for 96 hr contracted 57% from the starting diameter of 7.5 mm whereas organoids grown in control media contracted 47% (Figure 2B). The amount of contraction comparing the two groups was statistically significant with a p value ≤ 0.01. In contrast, organoids lacking HSC and made with bare Col1 only did not contract over time in either of the media conditions (not shown). The significant change in LX-2-Col1 organoids' diameter in the presence of TGF-β was associated with a significant increase in material stiffness of the organoids in comparison to control (Figure 2C). Rheological data was converted to a fold increase from the t = 0 measurement of ∼2 kPa (Bare Col1) showing that organoids exposed to TGF-β achieved a fold increase of 16.4 in Young's modulus whereas organoids grown in control media only reached an increase of 4.9 times after 96 hr in culture (p value < 0.0001). Bare Col1 organoids did not change in stiffness over time in either media condition (not shown). These morphological results indicate that exposure to exogenous TGF-β results in a dramatic change in ECM density. There is evidence that physical and structural changes associated with collagen fiber in the TME can alter the phenotype of tumor cells (Brabrand et al., 2015).
To further study changes in the ECM in response to TGF-β stimulation, we characterized the collagen fiber structure using PS-Red staining on sections of paraffin-embedded organoids. PS-Red stain highlights collagen bundles in samples imaged under polarized light from highly bundled fibers in orange/red to naive fibers in green (Figure 3A). Organoids exposed to TGF-β exhibited a high amount of orange/red signal, suggesting active remodeling of Col1 by the LX-2 cells to yield a large number of collagen fiber bundling. Hue quantification of ROIs throughout both the control and TGF-β groups verified the degree of collagen bundling that can be detected from the staining pattern (Figure 3B). Organoids exposed to TGF-β comprised 89% red/orange pixels compared to 9% in the control group. Bare Col1 organoids displayed exclusively green signal, indicating unbundled collagen. Collagen fiber architecture was also analyzed to identify the length, width, and alignment of the individual fibers by CT-FIRE, a program designed to quantify the fiber characteristic by identifying edges through curvelet transformation and applying a fiber extraction algorithm (Bredfeldt et al., 2014; Candès et al., 2006). Fiber angles in both organoid groups produced a unimodal distribution whereas the collagen fibers in the control organoids had a wider angular distribution, indicating a less parallel assortment of fibers (Figure 3C). The average angle variance between the fibers from both groups was highly significant (p value < 0.0001), suggesting that TGF-β activated the LX-2 cells to successfully remodel and align collagen fibers as seen in ECM dense fibrotic tissues (Baiocchini et al., 2016; Lee and Friedman, 2011). Fiber length and width measurements showed that organoids exposed to TGF-β contained longer and wider fibers than control (Figures 3E and 3F; p value < 0.0001). These results build on the results shown in Figure 1 and indicate that TGF-β-mediated activation of LX-2 cells results in an ECM dense environment in the organoids.
Figure 3.

TGF-β-Activated Organoids Display a Dense ECM Architecture
(A) Control and TGF-β-treated liver organoids without (left) and with (right) an HCT-116 colorectal cancer spheroid was sectioned and stained with PSR to highlight naive (green) and remodeled/bundled (red/orange) collagen fibers under polarized light.
(B) Hue signal of PSR images was quantified using MATLAB software to compare groups.
(C–F) Collagen fiber architecture was analyzed with segmentation software, CT-FIRE, to generate distributions of fiber angle (C), fiber angle variance (D), and box and whisker plots of fiber length (E), and width (F). MATLAB was used to generate graphs from extracted data. Box and whisker plots with Tukey formatting of pooled fibers performed in triplicate, representing 500–5000 fibers in total. Error bars indicate ± s.e.m (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001).
Dense ECM Reverses Mesenchymal Properties of CRC Cells in the Organoids
After establishing a model of TGF-β-induced HSC transdifferentiation that resulted in a dense ECM architecture, we investigated the responsive behavior of the CRC cell spheroids within the organoids to changes in ECM (Figure 4A). We utilized four membrane stains, E-cadherin, N-cadherin, β-catenin, and CD44 to determine the phenotypic change of the tumor cells in each degree of ECM architecture (Figures 4B and S1). E-cadherin immunostaining demonstrated that HCT-116 cells along the spheroid periphery in the LX-2-Col1 organoids that were exposed to TGF-β expressed a higher level of membrane-bound E-cadherin, yet not statistically significant, compared to HCT-116 cells in organoids cultured in control media (Figure 4B). HCT-116 cells within bare Col1 organoids expressed a similar level of E-cadherin as LX-2-Col1 organoids in control media. N-cadherin immunostaining results demonstrated a higher N-cadherin expression in HCT-116 cells within LX-2-Col1 organoids in control media compared with nearly absent N-cadherin in organoids exposed to TGF-β (Figure 4B). The expression of N-cadherin in HCT-116 cells was even higher in HCT-116 spheroids within bare Col1 organoids. Similar results were observed for CD44 staining, with higher CD44 expression in HCT-116 cells within LX-2-Col1 organoids in control media compared with HCT116 cells in organoids exposed to TGF-β (Figure S1). The expression of CD44 in HCT-116 cells was even higher in HCT-116 spheroids within bare Col1 organoids (Figure S1). We then analyzed β-catenin expression and localization in HCT-116 spheroids inside the organoids. Immunostaining results of β-catenin demonstrated that exposure to TGF-β led to an increase in membrane-associated β-catenin in HCT-116 cells within LX-2-Col1 organoids compared to control media and within bare collagen organoids (Figure 4B). Lastly, we examined the presence of a CSC population within the HCT116 spheroids by staining for CD133 (Figure 4B-bottom). The number of CD133-positive cells was significantly higher in organoids exposed to TGF-β compared with organoids in control. Only a few CD133-positive cells were observed in bare Col1 organoids. The staining results were further quantified using VisioPharm software to determine the significance in staining results for the different EMT markers described above (Figures 4C and S3). In general, HCT-116 spheroids inside LX-2-Col1 organoids that were exposed to TGF-β demonstrated a significant decrease in N-cadherin and CD44 expression compared with organoids in control media. In parallel, exposure to TGF-β induced a significant increase in membranal β-catenin compared with control media. Next, we analyzed organoids for cell proliferation and expression of stem cell marker CD133 in order to determine the effects of ECM architecture tumor cell stemness. HCT-116 spheroids within bare Col1 organoids demonstrated higher proliferation compared with LX-2-Col1 organoids either in the presence or the absence of TGF-β (Figure 4C). In parallel, CD133 expression was significantly increased in bare Col1 organoids compared to LX-2-Col1 organoids and in organoids treated with TGF-β compared to control media (Figures 4C and S1B). Together, the results shown in Figures 2 and 4 suggest that HCT-116 cells inside organoids with ECM dense environment (organoids exposed to TGF-β) demonstrated a reduced EMT phenotype and cell proliferation compared with a more malignant phenotype of cancer cells in organoids with a loose ECM environment. In contrast, the ECM dense environment preserved a higher number of CD133-positive CSC population.
Figure 4.

Organoid with Dense ECM Reduces EMT Marker Expression of Tumor Cells
(A) A brightfield image of an H&E stained organoid section reveals the orientation of future IHC images with HCT-116 cells (CRC) in a preformed spheroid on the top right of the cropped image with LX-2 (hepatic stellate cells) or only type I collagen on the bottom left.
(B) IHC staining of organoid sections for cell adhesion markers (red), Ki-67 (green) and DAPI (blue) at 40 x magnification. All images use the same scale bar = 50 μm. White box on top right of each individual frame is a 2 x zoom of the white box within image to show the red signal within individual cells.
(C) IHC quantification of HCT-116 cells was determined using VisioPharm software by calculating the percentage of positively expressed cells by the total nuclei present. TGF-β inhibitor addition was at 24hr. Graphs represent mean ± s.e.m (n = 5) and generated using GraphPad prism.
ECM composition and architecture have been shown to affect the initiation of metastasis and cancer cell growth (Paolillo and Schinelli, 2019). However, our model described above using HCT-116 spheroids embedded in LX-2-Col1 suspension better represent the growth of established liver metastases rather than an initial seeding in the liver. To better model the latter, we used a homogeneous mixture of HCT-116 and LX-2 in Col1 and allowed the organoids to form and grow for 7 days in culture media. This technique allowed us to analyze foci formation from a single cell within the organoids with the foundation that larger foci translates to highl malignancy and to test how different ECM densities impact tumor foci growth. Identification of HCT-116 cells inside the LX-2-Col1 organoids was achieved by immunohistochemistry (IHC) with high levels of E-cadherin antibody that doesn't stain LX-2 cells (red, Figure 5A). We then analyzed the numbers and size of HCT-116 cell foci within HCT-116 cells only and co-culture Col1 organoids and determined the effects of TGF-β exposure and ECM architecture (Figure 5). Smaller HCT-116 foci were observed in the co-culture organoids (Figure 5A-upper left) compared to the larger multicellular foci in the HCT-116 only organoids (Figure 5A-upper right). Exposure of the co-culture organoids to TGF-β further reduced the size of HCT-116 foci, especially close to the center of the organoids (Figure 5A-lower left); however, TGF-β did not produce an apparent difference in the size of HCT-116 foci in the HCT-116 only organoids (Figure 5A-lower right). These results were further quantified using a custom image analysis MATLAB script (Figure 5B). It is important to note that the organoids contract due to Col1 remodeling, resulting in a “cupped” construct, with LX-2 cells on one side of the construct. When sectioning the “cup” from the top or bottom, the center of the section is the center of the cup, where most of the remodeling occurs. We see the smallest HCT-116 foci at this location, the center of the section. This area is not necessarily the center of the 3D organoid, rather the center of the section, which is selected in order to show areas with varying levels of remodeling at the same plane.
Figure 5.

Microspheroid Cluster Formation is Hindered in Organoids with Dense ECM
(A) Co-cultured (HCT-116 & LX-2) (I & III) or monocultured (HCT-116) (II & IV) organoids in type I collagen are grown for 7 days with (III & IV) or without (I & II) 10 ng/mL TGF-β and IHC stained for HCT-116 cells (E-cadherin, red), DAPI (blue), and Ki-67 (green). Arrows indicate where the zoomed in image are spatially located in the organoid section.
(B) VisioPharm software was used to identify the mean HCT-116 cluster size in mono/co-culture with/without TGF-β. Graphs represent mean ± s.e.m (n = 6) and generated using GraphPad prism.
(∗∗p ≤ 0.01; ∗∗∗∗p ≤ 0.0001) (C) Serial sections were stained for either E-cadherin (HCT-116, red), DAPI (blue), and Ki-67 (green) shown in the first column or PSR in the second column. Arrows indicate the location of the HCT-116 cluster from the first column on the serial section in the second column stained with PSR to identify the presence of absence of collagen remodeling.
We subsequently analyzed the spatial association of HCT-116 foci size and its relationship to the ECM architecture by staining serial sections for high levels of E-cadherin (Figure 5C-left) and PS-Red (Figure 5C-right). This visual representation demonstrated that larger HCT-116 foci were mostly surrounded by unbundled/lamellar Col1 (stained green in PS-Red), whereas smaller foci were embedded inside highly remodeled and bundled Col1 (stained red/orange in PS-Red). This spatial relationship of foci size and ECM architecture is apparent among the different experimental conditions and furthermore within a single condition. Co-culture organoids exposed to TGF-β, demonstrated a heterogeneous ECM architecture, with areas of highly bundled Col1 fibers (dense orange staining in PS-Red) and areas of less bundled Col1 fibers (sparse orange staining in PS-Red). In these organoids, smaller HCT-116 foci were present in the areas of highly bundled Col1 fibers (Figure 5C-fourth row) compared with slightly larger foci in the areas of less bundled Col1 fibers (Figure 5C-fifth row). The proliferation of HCT-116 cells within the cell foci was determined by Ki-67 staining. Overall, there were more proliferating HCT-116 cells in foci within unbundled/lamellar Col1 (Figure 5C-first and third rows) compared with foci within highly bundled Col1 (Figure 5C-fourth and fifth rows). Altogether, ECM dense environments prevented the formation of large tumor cell foci with a high proliferation rate compared with the loose ECM environment that supported tumor cell foci growth and cell proliferation.
Manipulation of WNT and FAK Signaling in Organoids can Modulate HCT-116 Proliferation
Cells are known to transduce signals from the ECM through integrin binding which can initiate signaling pathways through FAK which can drive metastasis, survival, and regulate downstream pathways such as WNT. In order to determine if Col1 fiber organization can alter FAK expression, we stained sections of organoids consisting of HCT-116 spheroids with different stroma conditions for FAK (green) and Ki-67 (red). In Col1 organoids, there was prominent FAK staining present in most cancer cells. In LX-2 containing organoids, cytoplasmic FAK staining decreased and is predominantly in the membrane on the cells (Figure 6A-bottom). Similar FAK staining was observed in LX-2 containing organoids treated with TGF-β, in spite of the significant difference in ECM architecture (PS-Red staining). However, in organoids treated with TGF-β and its inhibitor at 24hr, FAK staining was localized back into the cytoplasm at a higher signal. Additionally, we were able to observe that concomitant FAK expression was associated with higher rates of proliferation in cancer cells, as shown by immunostaining of HCT-116 cells for Ki-67 expression (Figure 6B). Quantification of Ki-67 positive ratios corroborated our visual observations that Col1 organoids showed significantly higher levels of HCT-116 proliferation compared with Col1+LX-2 organoids. In order to examine the role of FAK in tumor cell proliferation in the organoids, Col1 organoids were treated with an FAK-phosphorylation inhibitor, Defactinib. In the presence of the FAK inhibitor, we observed that FAK staining was localized more to the cell membrane, as seen in LX-2 containing organoids (Figure 6A-right), as well as a significant reduction in the number of Ki-67+ proliferating HCT-116 cells compared with untreated Col1 organoids (Figure 6B). These results suggest that inhibition of FAK-phosphorylation, and possibly FAK downstream signaling, could control HCT-116 proliferation in organoids with unbundled/lamellar Col1, that were shown to favor tumor cell proliferation.
Figure 6.

The Effects of ECM Architecture on FAK Expression and Localization
(A) Organoids consisting of HCT-116 spheroids with the indicated stroma were treated with TGF-β, TGF-β+SB 431542 (Inhibitor) at 24hr or Defactinib (100nM) at 0h, an FAK-Phosphorylation inhibitor, as indicated. Sections of the organoids were stained with PS-Red (top), FAK/Ki-67/DAPI (middle), and FAK (bottom).
(B) Quantification of Ki-67 positive HCT-116 cells were determined using VisioPharm software by calculating the positively expressed cells relative to the control. Graphs represent mean ± s.e.m (n = 5) and generated using GraphPad prism.
TGF-β Inhibition Alters ECM Remodeling and Tumor Cell Chemosensitivity
The results presented above link ECM architecture to tumor cells phenotype and proliferation inside the tumor organoids. We found that TGF-β significantly induced the formation of a dense ECM environment and subsequently reduced tumor cell proliferation and a change to a less malignant phenotype. As such, inhibition of structural changes to the ECM could serve as a therapeutic target. Specifically, SB 431542 is a potent small-molecule TGF-β receptor inhibitor of TGF-β intracellular signaling (Figure S2) (Inman et al., 2002). We first analyzed LX-2-Col1 organoid contraction, stiffness, and collagen fiber characteristics in the presence of TGF-β and the inhibitor at 24hr (Figure 7). Organoid contraction was significantly repressed when the TGF-β inhibitor was added simultaneously with the exogenous TGF-β (Figure 7A, p value < 0.001). Inclusion of the TGF-β inhibitor at different times after the addition of TGF-β demonstrated a time-dependent inhibition of organoid stiffness (Figure 7B) Addition of the TGF-β inhibitor up to 24 hr post-exposure of the organoids to exogenous TGF-β completely inhibited TGF-β-induced organoid stiffness after 96 hr, in spite of an initial stiffness increase when the inhibitor was added 8 and 24 hr post-exposure to TGF-β. However, when the inhibitor was added 48- and 72-hr post-exposure to TGF-β, the stiffness measured after 96 hr was significantly higher compared with the addition of the inhibitor at the earlier time points. Interestingly, the addition of the inhibitor at these later time points (48 and 72 hr) still had significant inhibition of organoids' stiffness compared to organoids incubated with TGF-β in the absence of the inhibitor. Collagen fiber analysis using CT-FIRE supported the organoid's contraction and stiffness results and show that the TGF-β inhibitor at 24hr significantly blocked TGF-β induction of collagen fiber alignment (Figure 7C), its corresponding variance (Figure 7D) and fiber lengthening (Figure 7E) and widening (Figure 7F) (p value < 0.0001). The addition of the inhibitor 24 hr post-exposure of TGF-β resulted in an epithelial phenotype quantified between the control group and the TGF-β group, as expected (Figure 4C). Altogether, these results showed that TGF-β exerts a progressive induction of ECM remodeling in the organoids and that our organoid system can serve as a good model to determine the time of intervention when targeting the ECM architecture remodeling process.
Figure 7.

Inhibiting the TGF-β Modulates the Degree of ECM Density and Fiber Organization
(A) HCT-116-containing LX-2 organoid diameter was measured over 96 hr in culture. A TGF-β inhibitor (SB 431542, 10 μM) was added to the media containing TGF-β (10 ng/mL) at the indicated time points. Graphs represent mean ± s.e.m (n = 4).
(B) Organoid stiffness was measured over 96 hr in culture conditions using a rheometer to determine the young's modulus of the construct. Graphs represent mean ± s.e.m (n = 4).
(C–F) Collagen fiber architecture in organoids treated with TGF- β inhibitor 24hr after TGF-β addition was analyzed with segmentation software, CT-FIRE, to generate distributions of fiber angle (C), fiber angle variance (D), and box and whisker plots of fiber length (E), and width (F). MATLAB was used to generate graphs from extracted data. Box and whisker plots with Tukey formatting of pooled fibers performed in triplicate, representing 500–5000 fibers in total. (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001)
As stated above, the process of ECM remodeling around the tumor could present a novel target for anti-cancer therapy. To test this hypothesis in our tumor organoid model, we treated HCT-116 spheroids within LX-2 and bare Col1 organoids with TGF-β and its inhibitor at 24hr after TGF-β addition and measured tumor cell response to three 5-FU-based chemotherapy combination (FOLFIRI, FOLFOX, and 5-FU alone) (Figure 8). Organoids were harvested 72 hr post-chemotherapy treatment and histological sections of the organoids in the different treatment groups were analyzed by immunostaining with antibodies against cleaved caspase-3 (green), Ki-67 (red), and with DAPI (blue) (Figure 8A). Expression of caspase-3 and Ki-67 in HCT-116 cells (within the white dashed line) was analyzed using VisioPharm software (Figure S3). Caspase-3 to Ki-67 ratio for each drug was graphed as a measure for chemosensitivity (Figures 8B–8E).
Figure 8.

Organoid with Dense ECM Reduces Tumor Cell Response to Specific Fluorouracil-Based Chemotherapy
(A) Organoids were treated with FOLFIRI (5-FU, 1mM; Leucovorin, 20 μM; Irinotecan, 50 μM), FOLFOX (5-FU, 1mM; Leucovorin, 20 μM; Oxaliplatin, 50 μM), 5-FU (1mM) or Control (DMSO) for 72hr after organoid was allowed to self-organize for 96hr. Paraffin sections were stained with DAPI (blue), Ki-67 (red), and Caspase-3 (green) where the HCT-116 cells can be seen within the spheroid (white dashed circle) and the LX-2s are surrounding.
(B–E) Immunofluorescent images of treated organoids were analyzed using VisioPharm to determine total HCT-116 (dash gray line) caspase-3 to Ki-67 ratio in each drug condition. TGF-β inhibitor was giving at 24hr post TGF-β addition. Graphs represent mean ± s.e.m (n = 12) and generated using GraphPad prism. (∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p ≤ 0.0001).
Organoids in the four experimental conditions (Col1, LX-2-Col1, LX-2-Col1 & Inhibitor, and LX-2-Col1 & TGF-β) exhibited the lowest cell apoptosis-to-proliferation (Cas-3:Ki-67) rates when grown in the presence of vehicle control media (Figure 8A). Treatment of LX-2-Col1 organoids with FOLFIRI showed no significant difference in Cas-3:Ki-67 ratio compared to vehicle control media (Figure 8B). However, we observed a drastic increase in Cas-3:Ki-67 ratio in TGF-β-treated LX-2-Col1 organoids (Figure 8B). This was most likely an anomaly of the HCT-116 cell line due to its low plasticity, but further investigation was not done. FOLFOX and 5-FU treatment of Organoids in the four experimental conditions revealed a trend that aligned with our initial hypothesis (Figures 8D and 8E). As the ECM density increased, chemotherapy response, in terms of apoptosis-to-proliferation (Cas-3:Ki-67) ratio, decreased. We also noticed that organoids incubated with the TGF-β inhibitor 24hr after initial activation with TGF-β exhibited similar Cas-3:Ki-67 values as organoids treated just with TGF-β. This response to chemotherapy could be a combined effect ECM density and also HSC activation, which was allowed to happen for the first 24hr before the inhibitor was added. Together, these results suggest a correlation between tumor cell phenotype, proliferation, and 5-FU-based chemotherapy sensitivity to ECM remodeling and stromal cell activation.
Discussion
A tumor mass consists of more than simply cancerous cells in isolation but also the tightly surrounding TME, made up of both the non-cellular ECM and various types of stromal cells. Although chemotherapy resistance is often attributed to tumor heterogeneity, cancer progression and malignancy is also profoundly influenced by its interaction with the surrounding TME. Recently, there has been a move to incorporate stromal cells and TME components in experimental testing in vitro and understanding of liver fibrosis and disease as well as the development of chemotherapeutic agents (van Grunsven, 2017; Skardal et al., 2015; Khawar et al., 2018). It is clear that the TME is a major influencer in surrounding cell fate; however, there remain unanswered questions on the exact function the TME plays in cancer progression.
Residing HSCs in the liver are considered a major component of a liver TME since they transdifferentiate into highly proliferative and motile myofibroblasts that are associated with desmoplasia and tumor growth. HSC activation can occur in response to various cytokines, such as TGF-β, from either a neighboring cell in response to an injury or from cancerous cells within the tissue. Myofibroblastic HSCs are responsible for the major changes that transpire in the liver ECM associated with an aggressive TME via two mechanisms, the deposition of new ECM components and the physical remodeling of the preexisting ECM. Our advanced approach uses biofabrication methods that mimic in vivo conditions in order to create a microenvironment similar to that of a colorectal tumor that has metastasized to the liver. Herein, we described the utilization of our tumor organoid platform to analyze the interactions between an HSC cell line of the liver and metastatic CRC cell line. In this study, we exposed cancer cells to various HSC-produced ECM densities and determined macroscopic characteristics of the collagen remodeling and its effect on embedded cancer cells. Our tumor organoid platform was capable of determining the effects of the tumor-stroma organization on tumor cell response to chemotherapy. The 3D organoid technology allows for full control over the structural and cellular properties of the TME, including stromal fibroblast cells, to better mimic the physiological nature of an in vivo tumor.
The goal of this research was to test the relationship between the TME architecture and cancer cell malignancy, as well as chemoresistance by developing a Col1 based 3D organoid model that can be manipulated around a cancer spheroid. We classify our 3D constructs as organoids based on its ability to self-remodel the surrounding collagen in a matter of days that mimics the long, ongoing process that occurs in an aging liver. The LX-2 cell line responsible for the collagen remodeling remains genetically similar to primary HSC and responds to TGF-β by increasing expression of a-SMA, Col1, and other profibrotic markers (Fabre et al., 2014; Xu et al., 2005; Yuan et al., 2019). Our organoid constructs consisting of LX-2s displays a rapid increase in contraction and stiffness when treated with exogenous TGF-β, suggesting that the LX-2s are actively remodeling the surrounding Col1 3-D hydrogel. Individual collagen fiber analysis confirms that the LX-2s are performing normal collagen bundling and remodeling in control cell culture conditions and excessive remodeling when exposed to TGF-β. We identified that the Col1 fibers in organoids treated with TGF-β are more aligned, longer, and wider, consistent with HSC activation and fibrotic tissue (Friedman, 2008; Bonnans et al., 2014). The collagen fiber characteristics of the different groups indicate the ability to generate a range of fibrotic environments consisting of dense, aligned collagen ECM. Since activation of HSCs is thought to be progenitors to αSMA-positive myofibroblasts, a marker for CAFS, we stained organoid sections with αSMA and FAP antibodies confirming TGF-β induces a myofibroblastic-like phenotype in the LX-2s within the Col1 hydrogel (Friedman, 2008; Biffi et al., 2019). Altogether, the stromal stiffening seen in the organoids upon exposure to TGF-β is a result of the increased matrix deposition, crosslinking, and bundling by the LX-2s, consistent with CAF behavior in the liver that can change cancer cell characteristics and create chemoresistance (Kumar and Weaver, 2009; Rasanen and Vaheri, 2010).
Since stromal components are known to play a key role in the process of tumor progression, we analyzed the effect a fibrotic, ECM dense organoid has on HCT-116 CRC spheroids that are metastatic in nature. IHC staining of common epithelial markers, E-cadherin and β-catenin, and mesenchymal marker, N-cadherin and CD44, reveals that embedded cancer cells in bare Col1 exhibit a highly proliferative, mesenchymal phenotype whereas cells within an increasingly ECM-dense environment begin to only express epithelial markers. N-cadherin is a cell membrane protein that is usually expressed at low levels in normal epithelial cells, whereas E-cadherin is a well-known tumor-suppressor membrane protein and loss of expression is associated with the EMT, as we see in HCT-116 cells along the periphery of the spheroid embedded in bare Col1 (Kalluri and Weinberg, 2009). CD44 is highly expressed in primary and metastatic colon cancer compared to low levels in normal tissues and upregulation of CD44 expression induces EMT and the acquisition of invasive and metastatic properties (Cho et al., 2012). E-cadherin expression by HCT-116 cells in the periphery of spheroids in organoids with denser ECM is high relative to other organoid types but not significantly. β-catenin is a component of the adherens junction complex helping anchor E-cadherin to intracellular actin on the cell membrane of many epithelial cells which is seen in high stiffened organoids. In contrast, upon EMT and Wnt activation in many cancer cells, β-catenin is mostly found inside the cell cytoplasm/nucleus and is associated with malignant transformation as we have seen in HCT-116 embedded in bare Col1 (Kalluri and Weinberg, 2009). In parallel, CD44 has been shown to inhibit the membrane-associated E-cadherin- β-catenin complex, which results in nuclear translocation of β-catenin (Xu et al., 2015). This phenomenon associates high levels of CD44 with proliferation and a mesenchymal phenotype, a characteristic we observed in bare Col1 organoids. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion (Cho et al., 2012).
In order to determine the use of this tumor organoid model to simulate tumor cell seeding and initial metastatic growth, we use a 3D co-culture system using single cell suspension of LX-2 and HCT116. This technique enables us to analyze foci development from a single-cell within the differing ECM architectural organoids with the foundation that larger foci translate to high malignancy. Through IHC staining, we identified that an ECM-dense environment created by activated LX-2s inhibited HCT-116 microspheroid foci formation compared to a lesser dense ECM. We then looked at serial sections of the same organoid to locate cancer foci via IHC in relationship to their surrounding collagen fiber architecture using PS-Red staining. We can visually see that even within the same organoid, larger foci are adjacent to naive Col1, whereas smaller foci are neighboring highly remodeled architecture. These findings are evidence that both activated myofibroblastic-like cells and ECM architecture play a role in inhibiting cancer initiation and can act as possible tumor suppressors.
We then test a potential transduction pathway of ECM architecture through FAK – a known oncogenic pathway (Basu et al., 2016; Kamposioras et al., 2013). We had identified the variable activity of the FAK pathway between differentially organized matrices. Treatment of Col1 organoids with Defactinib, an inhibitor of FAK phosphorylation, changed FAK staining localization from the cytoplasm to the membrane, as seen in LX-2 containing organoids, and significantly reduced HCT-116 cell proliferation. This result suggests that FAK activation may have a role in supporting tumor cell proliferation in Col1 organoids that lack well-structured stromal ECM. In fact, FAK activity is well documented in CRC, and higher expression is associated with aggressive cancers (Gao et al., 2015; Baker et al., 2013; Sulzmaier et al., 2014). This sheds light on a possible signaling pathway that may be involved ECM-tumor cell interactions and could influence tumor cell phenotype and proliferation.
Next, we employed a TGF-β receptor inhibitor, SB 431542, that can completely eliminate exogenous TGF-β when given simultaneously. We used this inhibitor at different time points to attempt to modulate the effects of TGF-β in the organoid. In the normal setting, stromal cells express TGF-β inhibitors and are thought to reduce the inflammatory response and effect of the surrounding fibroblasts (Costanza et al., 2017). Subsequently, low levels of TGF-β inhibitors, like asporin, can be correlated with poor survival. Rescuing the ECM from remodeling within our organoid model leads to a controllable fibrotic environment that surrounds the cancer cells. We see that when given the organoids TGF-β inhibitor 24hr after TGF-β addition, the organoids revert to a less stiff (Figure 7B), less remodeled (Figures 7C–7F) collagen architecture by 96hr. Conversely, the addition of TGF-β inhibitor at 48hr or after does not completely revert to the original stiffness without any TGF-β addition (Figure 7B). These results suggest that there is a time threshold before remodeling can be stopped and that the remodeling activity during the first 24hr with TGF-β may be irreversible.
After determining a dissimilarity in HCT-116 proliferation and phenotype in differing ECM density conditions, we tested chemotherapy efficacy of common fluoropyrimidine-based regimens. Fluoropyrimidine drugs, such as 5-FU, are widely used in the treatment of multiple cancers including CRC; however, 5-FU has a response rate of about 10–30% for metastatic CRC and differs depending on the stage of cancer (Longley et al., 2003; Siegel et al., 2017; Christina Hackl et al., 2014). 5-FU achieves its therapeutic efficacy by the conversion to metabolites upon entering the cell, disrupting DNA synthesis through misincorporation, along with inhibiting thymidylate synthase activity halting cell division of a rapidly dividing cell. In current clinical practices, 5-FU-based chemotherapy shows the greatest impact against CRC when combined with other therapeutic agents like leucovorin, irinotecan (FOLFIRI), and oxaliplatin (FOLFOX) (Christina Hackl et al., 2014). After treating spheroids embedded in organoids, we found that HCT-116 cells are less susceptible to most 5-FU-based chemotherapies tested when surrounded by a dense, remodeled ECM architecture produced by activated HSC. Conversely, HCT-116 spheroids in bare collagen, which are more proliferative, are more susceptible to 5-FU and FOLFOX. Due to the mechanism of action of 5-FU, it is not surprising that the difference in proliferation seen in the various conditions correlates with chemotherapy sensitivity. The high Cas-3:Ki-67 seen by FOLFIRI induction of apoptosis and halting proliferation in a highly dense ECM is most likely an anomaly in part due to the use of a low malleable cell line or an insight on how irinotecan functions in ECM dense environments. Altogether, we show that ECM density created by activated stromal cells can later chemosensitize nearby tumor cells.
CSCs are known to display a heightened resistance to chemotherapy, similar to the therapy resistance exhibited by recurrent tumors in patients with cancer [35]. Contrary to the literature, we see a significant decrease in HCT-116 proliferation and mesenchymal phenotype in organoids with denser ECM, indicating a potential influence on chemosensitivity. We have asked ourselves if the lower chemotherapy response in organoids with denser ECM (Figure 8) may be related to the preservation of stem cell populations of HCT116 in organoids treated with TGF-β. The cell surface marker CD133 has been identified as a CSC marker in CRC and high CD133 expression is thought to contribute to tumor progression and therapy resistance (Ke Wang et al., 2012). CD133 staining, reveals a significantly larger number of CD133-positive cells, presumably CRC, in organoids with denser ECM (Figure 4). This leads us to suggest that the organoids with denser ECM enable the CSC population in the HCT-116 cell line to remain dormant rather than differentiate and proliferate, as seen in the bare Col1 organoids. Tumor dormancy is an important mechanism underlying the ability for cancer cells to evade chemotherapy, maintain residual disease, and metastasize slowly over time (Barkan et al., 2010; Jahanban-Esfahlan et al., 2019; Tauriello et al., 2017). Future studies will focus on the effects of tumor ECM density on tumor dormancy and if changes in the ECM architecture may release tumors from a dormant stage.
The cellular and non-cellular aspects of the TME has been recently attributed to how tumors metastasize, proliferate, evade immune systems, and resist chemotherapy (Erdogan and Webb, 2017; Paolillo and Schinelli, 2019; Lu et al., 2012; Senthebane et al., 2017, 2018; Hoye and Erler, 2016). However, a lack of in vitro models that accurately replicate changes in the ECM architecture has made it difficult to pinpoint fibrotic effects on cancer that has metastasized to the liver, among many others. Our CRC organoid platform enables us to study the interactions between tumor cells and the controllable surrounding ECM. We are able to identify changes in a low plasticity cell line, HCT-116, when changes in the ECM architecture occur that result in differing chemotherapy resistance and sensitivities. This system will allow us to begin investigating the effect ECM density has on tumor samples directly from patients to help better understand the underlying mechanisms of chemoresistance through precision medicine.
Limitations of the Study
Several study limitations are acknowledged in this work. Our findings are restricted by the use of only established cell lines, HCT-116 (CRC), and LX-2 (HSC). Future work using primary tumor cells from human CRC biopsies is needed to validate these studies. Our tumor ECM is composed only of Col1. Although Col1 is the major component of the tumor ECM, other ECM proteins are found in the tumor-stroma and may undergo different remodeling than Col1 and may interact differently with the tumor cells. Further studies using a variety of ECM proteins will have a more complete recapitulation of the tumor ECM and its role in tumor progression. Finally, this study tested the tumor organoids only under in vitro conditions, which are far different than the in vivo environment and may not take into account normal physiological circulation and immune/inflammatory system.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shay Soker (ssoker@wakehealth.edu).
Materials Availability
This study did no generate new unique reagents. There are restrictions on the availability of LX-2 cell line since they were gifted from Dr. Scott Friedman at Icahn School of Medicine.
Data and Code Availability
The data sets generated in this study is available upon request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Scott Friedman from the Icahn School of Medicine at Mount Sinai, New York, for providing LX2 cells. The authors acknowledge funding through the NIH NCI grant R33CA202822. The authors wish to acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Tumor Tissue and Pathology Shared Resource supported by the National Cancer Institute's Cancer Center Support Grant award number P30CA012197.
Authors Contribution
Conceptualization, A.D., M.D., and S.S.; Methodology, A.D., M.D., and S.S.; Software, A.D. and M.D.; Validation, A.D. and S.S.; Formal Analysis, A.D.; Writing – Original Draft, A.D., and S.S.; Writing – Review & Editing, A.D., M.D., and S.S. Supervision, A.D., M.D., and S.S.
Declaration of Interests
The authors declare no competing interest.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101851.
Supplemental Information
References
- Affo S., Yu L.X., Schwabe R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017;12:153–186. doi: 10.1146/annurev-pathol-052016-100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baiocchini A., Montaldo C., Conigliaro A., Grimaldi A., Correani V., Mura F., Ciccosanti F., Rotiroti N., Brenna A., Montalbano M. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS One. 2016;11:e0151736. doi: 10.1371/journal.pone.0151736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A.M., Bird D., Lang G., Cox T.R., Erler J.T. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene. 2013;32:1863–1868. doi: 10.1038/onc.2012.202. [DOI] [PubMed] [Google Scholar]
- Barkan D., El Touny L.H., Michalowski A.M., Smith J.A., Chu I., Davis A.S., Webster J.D., Hoover S., Simpson R.M., Gauldie J. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010;70:5706–5716. doi: 10.1158/0008-5472.CAN-09-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Haase G., Ben-Ze'ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Res. 2016;5:F1000. doi: 10.12688/f1000research.7579.1. Faculty Rev-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G., Oni T.E., Spielman B., Hao Y., Elyada E., Park Y. IL1-Induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301. doi: 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C., Chou J., Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabrand A., Kariuki, Engstrom M.J., Haugen O.A., Dyrnes L.A., Asvold B.O., Lilledahl M.B., Bofin A.M. Alterations in collagen fibre patterns in breast cancer. A premise for tumour invasiveness? APMIS. 2015;123:1–8. doi: 10.1111/apm.12298. [DOI] [PubMed] [Google Scholar]
- Bredfeldt J.S., Liu Y., Pehlke C.A., Conklin M.W., Szulczewski J.M., Inman D.R., Keely P.J., Nowak R.D., Mackie T.R., Eliceiri K.W. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J. Biomed. Opt. 2014;19:16007. doi: 10.1117/1.JBO.19.1.016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzelli J.N., Ouaret D., Brown G., Allen P.D., Muschel R.J. Colorectal cancer liver metastases organoids retain characteristics of original tumor and acquire chemotherapy resistance. Stem Cell Res. 2018;27:109–120. doi: 10.1016/j.scr.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candès E., Demanet L., Donoho D., Ying L. Fast discrete curvelet transforms. Multiscale Model. Simulat. 2006;5:861–899. [Google Scholar]
- Carloni V., Luong T.V., Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34:834–843. doi: 10.1111/liv.12465. [DOI] [PubMed] [Google Scholar]
- Cho S.H., Park Y.S., Kim H.J., Kim C.H., Lim S.W., Huh J.W., Lee J.H., Kim H.R. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int. J. Oncol. 2012;41:211–218. doi: 10.3892/ijo.2012.1453. [DOI] [PubMed] [Google Scholar]
- Chow F.C., Chok K.S. Colorectal liver metastases: an update on multidisciplinary approach. World J. Hepatol. 2019;11:150–172. doi: 10.4254/wjh.v11.i2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christina H., Neumann P., Gerken M., Loss M., Klinkhammer-Schalke M., Schlitt H.J. Treatment of colorectal liver metastases in Germany: a ten-year population-based analysis of 5772 cases of primary colorectal adenocarcinoma. BMC Cancer. 2014;14:810. doi: 10.1186/1471-2407-14-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanza B., Umelo I.A., Bellier J., Castronovo V., Turtoi A. Stromal modulators of TGF-beta in cancer. J. Clin. Med. 2017;6:7. doi: 10.3390/jcm6010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T.R., Erler J.T. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarasetty M., Skardal A., Cowdrick K., Marini F., Soker S. Bioengineered submucosal organoids for in vitro modeling of colorectal cancer. Tissue Eng. A. 2017;23:1026–1041. doi: 10.1089/ten.tea.2017.0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarasetty M., Mazzocchi A.R., Skardal A. Applications of bioengineered 3D tissue and tumor organoids in drug development and precision medicine: current and future. BioDrugs. 2018;32:53–68. doi: 10.1007/s40259-017-0258-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewidar B., Soukupova J., Fabregat I., Dooley S. TGF-β in hepatic stellate cell activation and liver fibrogenesis: updated. Curr. Pathobiol. Rep. 2015;3:291–305. [Google Scholar]
- Eble J.A., Niland S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis. 2019;36:171–198. doi: 10.1007/s10585-019-09966-1. [DOI] [PubMed] [Google Scholar]
- Emon B., Bauer J., Jain Y., Jung B., Saif T. Biophysics of tumor microenvironment and cancer metastasis - a mini review. Comput. Struct. Biotechnol. J. 2018;16:279–287. doi: 10.1016/j.csbj.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogan B., Webb D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017;45:229–236. doi: 10.1042/BST20160387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre T., Kared H., Friedman S.L., Shoukry N.H. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-beta receptor on hepatic stellate cells in a JNK-dependent manner. J. Immunol. 2014;193:3925–3933. doi: 10.4049/jimmunol.1400861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C., Chen G., Kuan S.F., Zhang D.H., Schlaepfer D.D., Hu J. FAK/PYK2 promotes the Wnt/beta-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3beta. Elife. 2015;4:e10072. doi: 10.7554/eLife.10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Grunsven L.A. 3D in vitro models of liver fibrosis. Adv. Drug Deliv. Rev. 2017;121:133–146. doi: 10.1016/j.addr.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Ho M.T., Kim Y.M., Yu D.-Y., Lee D.H., Cho M., Hyun C. TGF-β secreted from activated hepatic stellate cells may induce the transdifferentiation of hepatocytes into hepatocarcinoma in HBx-expressing livers. J. Korean Soc. Appl. Biol. Chem. 2014;57:529–538. [Google Scholar]
- Holle A.W., Young J.L., Spatz J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016;97:270–279. doi: 10.1016/j.addr.2015.10.007. [DOI] [PubMed] [Google Scholar]
- Hoye A.M., Erler J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016;310:C955–C967. doi: 10.1152/ajpcell.00326.2015. [DOI] [PubMed] [Google Scholar]
- van Huizen N.A., Coebergh van den Braak R.R.J., Doukas M., Dekker L.J.M., JNM I.J., Luider T.M. Up-regulation of collagen proteins in colorectal liver metastasis compared with normal liver tissue. J. Biol. Chem. 2019;294:281–289. doi: 10.1074/jbc.RA118.005087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman G.J., Nicolás F.J., Callahan J.F., Harling J.D., Gaster L.M., Reith A.D., Laping N.J., Hill C.S. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Ishii G., Ochiai A., Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016;99(Pt B):186–196. doi: 10.1016/j.addr.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Jahanban-Esfahlan R., Seidi K., Manjili M.H., Jahanban-Esfahlan A., Javaheri T., Zare P. Tumor cell dormancy: threat or opportunity in the fight against cancer. Cancers (Basel) 2019;11:1207. doi: 10.3390/cancers11081207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamposioras K., Konstantara A., Kotoula V., Lakis S., Kouvatseas G., Akriviadis E., Vrettou E., Dionysopoulos D., Krikelis D., Papadopoulou K. The prognostic significance of WNT pathway in surgically-treated colorectal cancer: beta-catenin expression predicts for disease-free survival. Anticancer Res. 2013;33:4573–4584. [PubMed] [Google Scholar]
- Kanas G.P., Taylor A., Primrose J.N., Langeberg W.J., Kelsh M.A., Mowat F.S., Alexander D.D., Choti M.A., Poston G. Survival after liver resection in metastatic colorectal cancer: review and meta-analysis of prognostic factors. Clin. Epidemiol. 2012;4:283–301. doi: 10.2147/CLEP.S34285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Wang J.X., Zhang Junshu, Huang Jian. Prognostic role of CD133 expression in colorectal cancer- a meta-analysis. BMC Cancer. 2012;12:573. doi: 10.1186/1471-2407-12-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawar I.A., Park J.K., Jung E.S., Lee M.A., Chang S., Kuh H.J. Three dimensional mixed-cell spheroids mimic stroma-mediated chemoresistance and invasive migration in hepatocellular carcinoma. Neoplasia. 2018;20:800–812. doi: 10.1016/j.neo.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Weaver V.M. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee U.E., Friedman S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Reed-Newman T., Anant S., Ramasamy T.S. Regulatory role of quiescence in the biological function of cancer stem cells. Stem Cell Rev. Rep. 2020;16:1185–1207. doi: 10.1007/s12015-020-10031-8. [DOI] [PubMed] [Google Scholar]
- Longley D.B., Harkin D.P., Johnston P.G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Lu P., Weaver V.M., Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi S., Lepage C., Hatem C., Coatmeur O., Faivre J., Bouvier A.M. Epidemiology and management of liver metastases from colorectal cancer. Ann. Surg. 2006;244:254–259. doi: 10.1097/01.sla.0000217629.94941.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller L., Goumas F.A., Affeldt M., Sandtner S., Gehling U.M., Brilloff S., Walter J., Karnatz N., Lamszus K., Rogiers X. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 2007;171:1608–1618. doi: 10.2353/ajpath.2007.060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nantasanti S., de Bruin A., Rothuizen J., Penning L.C., Schotanus B.A. Concise review: organoids are a powerful tool for the study of liver disease and personalized treatment design in humans and animals. Stem Cells Transl. Med. 2016;5:325–330. doi: 10.5966/sctm.2015-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal J.T., Li X., Zhu J., Giangarra V., Grzeskowiak C.L., Ju J., Liu I.H., Chiou S.H., Salahudeen A.A., Smith A.R. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175:1972–1988 e16. doi: 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolillo M., Schinelli S. Extracellular matrix alterations in metastatic processes. Int. J. Mol. Sci. 2019;20:4947. doi: 10.3390/ijms20194947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup M.W., Laklai H., Acerbi I., Owens P., Gorska A.E., Chytil A., Aakre M., Weaver V.M., Moses H.L. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res. 2013;73:5336–5346. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plou J., Juste-Lanas Y., Olivares V., Del Amo C., Borau C., Garcia-Aznar J.M. From individual to collective 3D cancer dissemination: roles of collagen concentration and TGF-beta. Sci. Rep. 2018;8:12723. doi: 10.1038/s41598-018-30683-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pool A.E., Damhuis R.A., Ijzermans J.N., de Wilt J.H., Eggermont A.M., Kranse R., Verhoef C. Trends in incidence, treatment and survival of patients with stage IV colorectal cancer: a population-based series. Colorectal Dis. 2012;14:56–61. doi: 10.1111/j.1463-1318.2010.02539.x. [DOI] [PubMed] [Google Scholar]
- Rasanen K., Vaheri A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010;316:2713–2722. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- Rawla P., Sunkara T., Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14:89–103. doi: 10.5114/pg.2018.81072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senthebane D.A., Rowe A., Thomford N.E., Shipanga H., Munro D., Mazeedi M., Almazyadi H.A.M., Kallmeyer K., Dandara C., Pepper M.S. The role of tumor microenvironment in chemoresistance: to survive, keep your enemies closer. Int. J. Mol. Sci. 2017;18:1586. doi: 10.3390/ijms18071586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senthebane D.A., Jonker T., Rowe A., Thomford N.E., Munro D., Dandara C., Wonkam A., Govender D., Calder B., Soares N.C. The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. Int. J. Mol. Sci. 2018;19:2861. doi: 10.3390/ijms19102861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J., Tajik A., Sun J., Guan J.L., Humphries M.J., Craig S.E., Shekaran A., García A.J., Lu S., Lin M.Z. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. U S A. 2013;110:19372–19377. doi: 10.1073/pnas.1307405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Fedewa S.A., Ahnen D.J., Meester R.G.S., Barzi A., Jemal A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017;67:177–193. doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- Skardal A., Devarasetty M., Rodman C., Atala A., Soker S. Liver-tumor hybrid organoids for modeling tumor growth and drug response in vitro. Ann. Biomed. Eng. 2015;43:2361–2373. doi: 10.1007/s10439-015-1298-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Kim S.H., Kim K.M., Choi E.K., Kim J., Seo H.R. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci. Rep. 2016;6:36750. doi: 10.1038/srep36750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzmaier F.J., Jean C., Schlaepfer D.D. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016;380:205–215. doi: 10.1016/j.canlet.2015.07.044. [DOI] [PubMed] [Google Scholar]
- Tao L., Huang G., Song H., Chen Y., Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol. Lett. 2017;14:2611–2620. doi: 10.3892/ol.2017.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello D.V., Calon A., Lonardo E., Batlle E. Determinants of metastatic competency in colorectal cancer. Mol. Oncol. 2017;11:97–119. doi: 10.1002/1878-0261.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C., Mojares E., Del Rio Hernandez A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018;19:3028. doi: 10.3390/ijms19103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson T., Sultanpuram N., Sendi H. The role of liver microenvironment in hepatic metastasis. Clin. Transl Med. 2019;8:21. doi: 10.1186/s40169-019-0237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Hui A.Y., Albanis E., Arthur M.J., O'Byrne S.M., Blaner W.S., Mukherjee P., Friedman S.L., Eng F.J. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Tian Y., Yuan X., Wu H., Liu Q., Pestell R.G., Wu K. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. 2015;8:3783–3792. doi: 10.2147/OTT.S95470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B., Chen Y., Wu Z., Zhang L., Zhuang Y., Zhao X., Niu H., Cheng J.C., Zeng Z. Proteomic profiling of human hepatic stellate cell line LX2 responses to irradiation and TGF-beta1. J. Proteome Res. 2019;18:508–521. doi: 10.1021/acs.jproteome.8b00814. [DOI] [PubMed] [Google Scholar]
- Yuzhalin A.E., Lim S.Y., Kutikhin A.G., Gordon-Weeks A.N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta Rev. Cancer. 2018;1870:207–228. doi: 10.1016/j.bbcan.2018.09.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated in this study is available upon request.