

Abstract
The obligatory biotrophic oomycetes Plasmopara viticola is the causal agent of downy mildew, a destructive disease of grapevine worldwide. So far, chemical fungicides are widely employed to limit this pathogen, but their adverse effects are stimulating the quest for environmentally friendly alternative approaches. Here, we report on the search for mycoviruses that might be later developed as biocontrol agents for this pathogen. Symptomatic leaves were collected from various regions in Spain and Italy and mycelia associated to leaf lesions was harvested. Total RNA extractions were depleted of rRNA and metatranscriptomes were generated using a high-throughput sequencing approach. The virome associated to leaf lesions was then characterized through a bioinformatic pipeline relying on blast searches against current viral databases. Here, we present an inventory of 283 new RNA viruses: 222 positive strand RNA viruses, 29 negative strand RNA viruses, 27 double-stranded RNA viruses and 5 ORFan virus RdRP segments, which could not be reliably assigned to any existing group in the Riboviria. In addition to ORFan viruses, we found other surprising new evolutionary trajectories in this wide inventory of viruses. The most represented viruses in our collection are those in phylum Lenarviricota, and, among them, a group of mycovirus segments distantly related to narnaviruses, but characterized by a polymerase palm domain lacking subdomain C, with the putative GDD catalytic triad. We also provided evidence of a strict association between two RNA segments that form a new mycovirus clade of positive strand RNA in the phylum Kitrinoviricota, order Martellivirales. In the phylum Negarnaviricota, we report for the first time in the order Mononegavirales a clade of viruses that is ambisense, a feature that so far was present only in the order Bunyavirales. Furthermore, in the same phylum we detected the widespread occurrence and abundant accumulation in our libraries of a distinct mycovirus clade distantly related to the Muvirales and Goujanvirales orders, which so far include only viruses infecting invertebrates. Possible new oomycetes-specific virus clades are also described in the phylum Duplornaviricota. These data greatly expand the evolutionary history of mycoviruses adding new layers of diversity to the realm Riboviria.
Keywords: mycovirus, virome, Plasmopara, narnavirus
1. Introduction
The recent coronavirus outbreak has once again reminded of the powerful pathogenetic potential of viruses and their ability to cause deadly epidemics on a world-wide scale (Wu et al. 2020). Nevertheless, it is important not to forget that viruses are fundamental motors of evolution (Ryan 2009) and play fundamental ecological roles, for example, regulating the biological pump in the ocean, a process of great impact on carbon cycles (Suttle 2007) and possibly on climate change (Danovaro et al. 2011). Furthermore, many viruses have a long story of co-evolution with their host, resulting often in a neutral or even positive symbiotic relationship (Roossinck 2011). Viruses are also used as biotechnological tools, and nanotechnological platforms (Wen and Steinmetz 2016). From an anthropocentric point of view, viruses can be exploited as biocontrol agents (BCAs) of plant diseases and plant pests, although their pathogenic potential is currently not fully explored. Their use as BCA is limited to a few applications: phage therapy against bacterial diseases (Balogh et al. 2010; Vandersteegen et al. 2013; Wang et al. 2019), the treatment of the chestnut blight pathogen, Cryphonectria parasitica, by infection with the naturally occurring Cryphonectria hypovirus 1 (Turina and Rostagno 2007), and the use of baculovirus for the containment of some lepidopteran pests (Popham, Nusawardani, and Bonning 2016). In the framework of the European Union funded H2020 project VIROPLANT, we aim at finding new viruses to be used as BCA for plant pests and diseases by a non-selective whole RNA sequencing (RNAseq) approach. Among several case studies present in the project, in this article, we focus on downy mildew of grapevine (DMG), a pathogen affecting a valuable crop on a world-wide scale. In particular, the disease results in damaged leaves, fruits, and shoots, especially young tissues. In warm and humid climates, it can be particularly destructive (Gessler, Pertot, and Perazzolli 2011). At present, its containment relies on a number of chemical fungicide applications, which raise environmental and health concern in highly populated areas (Nicolopoulou-Stamati et al. 2016; Pertot et al. 2017; Brauer et al. 2019). In organic farming, alternative treatments are based on copper, which is also in the process of more stringent regulation in Europe (Dagostin et al. 2011; Lamichhane et al. 2018). DMG is caused by the oomycete Plasmopara viticola, which is an obligatory biotroph and therefore cannot be cultivated in axenic conditions in vitro. Its recently described genome (Dussert 2016; Yin et al. 2017) together with the fully annotated grapevine one, (Jaillon et al. 2007; Velasco et al. 2007) is a crucial step for studying this very interesting and challenging host-pathogen system through all the different -omics tools (Brilli et al. 2018).
Here, we report the characterization of the virome associated to the complex of microbes present on P. viticola lesions. Previously the same dataset was used to describe putative plant viruses present in the metatranscriptomic sample (Chiapello et al. 2020). Here, we describe a catalogue of new putative mycoviruses expanding our knowledge of virus evolution with more than 280 new species, some of which constitute completely new lineages of positive and negative sense RNA genomes.
2. Methods
2.1 Sample collection
Samples were collected during the summer of 2018 in Italy and Spain on several varieties of grapevine infected by P. viticola, a heterothallic oomycete causal agent of DMG. After collection, depending on the infection stage of the lesion, infected leaves where immediately processed, or stored at room temperature in a moist bag, to allow the fungus to grow and to develop fruiting bodies (sporangia). Mycelia were then harvested, and RNA extracted.
Italian samples were collected mainly in the north of Italy, spanning different longitudes, from Piedmont to Veneto regions. This also reflected a wide range of grapevine varieties: Marzemino, Rebo, Groppello, Barbera, Merlot, Cabernet, Chardonnay, Pinot nero, Nebbiolo, Dolcetto, Erbaluce, Platano, Croatina, Malvasia, Trebbiano, Moscato, and Riesling. A smaller proportion of the samples were also collected from central and southern Italy, to cover as much as possible the country latitudes. A total of 158 DMG samples were collected in five different regions of the country (Supplementary Fig. S1A).
In Spain, a total of 139 samples were collected (DMS; Supplementary Fig. S1B) in four different regions: La Rioja, Penedés, Jerez, and Ribera de Duero. The regions of sampling covered the country from north to south and from east to west, and included different grapevine cultivars as Palomino, Pedro Ximénez, and Tempranillo. Samples collection followed the same protocol described for the Italian ones.
A table listing the sample names, the RNAseq pool they belong to (see below), the country and region of sampling is provided as Table S1.
2.2 RNA extraction from DMG
Grapevine leaves with P. viticola lesions were cut in small fragments (only the lesioned area with fresh mycelia and sporangiophores), submerged in sterile water (20 ml) and the lower surfaces were gently brushed to remove the emerging fungal mycelia and spores. The water suspension containing the fungal matrix was centrifuged at 5,800 g for 10 min. The resulting pellet was washed with sterile water (1 ml) and centrifuged at 14,000 g for 1 min. The pellet was finally resuspended in lysis buffer (Spectrum Plant Total RNA kit from Sigma-Aldrich) and 0.5 ml of glass beads (0.1 mm) were added in the same tube (video protocol: youtube.com/watch? v = 9ZMrUGRzMvw&t=). Two cycles of beadbeater (FastPrep-24 MP Biomedicals) at maximum speed for 30 s alternated with 2 min pauses were used to disrupt the fungal tissues. After RNA extraction following the RNA kit instructions (Spectrum Plant Total RNA kit from Sigma-Aldrich), sample concentrations were measured using a spectrophotometer (Nanodrop 2000, Thermo Scientific). An arbitrary quantity threshold was set of 50 ng/µl and only 141 RNAs passed the check (74 from Italy and 67 from Spain). Selected samples were then pooled to have the same final concentration (7 ng/µl; Supplementary Table S1), resulting in seven pools from Italy and nine pools from Spain.
2.3 RNAseq and bioinformatics pipeline
RNA sample pools were sent to Macrogen Inc. (Seoul, Republic of Korea) for library preparation (Illumina TrueSeq Stranded) and sequencing. For each library more than 100 million of 150-bp paired-end reads were retrieved. All the raw reads have been deposited in the Sequence Read Archive (SRA): Bioproject PRJNA613358, Biosamples from SAMN14402112 to SAMN14402127, SRA runs from 13869930 to 13869945. This dataset of sequencing reads was previously used to infer putative-associated plant viruses (Chiapello et al. 2020). The bioinformatic pipeline to assemble and identify in silico the viruses was divided in four steps: 1, cleaning; 2, de novo assembly; 3, viral sequence identification; and 4, mapping. Read cleaning was performed using BBTools (Bushnell, Rood, and Singer 2017), by removing adapters, artifacts, short reads, and residual ribosomal sequences. The output of the cleaning step was used as input for the de novo assembly. For this step Trinity software (version 2.3.2; Grabherr et al. 2011) was used. The third step used Blast to identify viral sequences. First, a custom viral database was queried with the assembled contigs via NCBI blast toolkit (version 2.8). The results were manually inspected and reliable viral sequences, based on identity percentage, alignment length and query length, were selected for the following analysis steps. The candidate contigs were blasted against the NCBI nr database (release October 2018) using DIAMOND software (Buchfink, Xie, and Huson 2015). These second blast results were used to discriminate between real virus sequences and host sequences present in viral genomes (e.g. heath shock related proteins present in the family Closteroviridae) or already discovered viruses. Cleaned reads were mapped on selected virus sequences using bowtie2 (Langmead and Salzberg 2012) and visualized with Tablet software (Milne et al. 2013). The detailed script with the parameters is available as Supplementary File S1. For open reading frames (ORFs) prediction, ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) was used with default parameters (Rombel et al. 2002). Domain search on each ORF encoded by each virus was performed with ‘motif search’ available at genome.jp/tools/motif with default parameters. Only ORFs with a predicted molecular mass of 5 kDa were graphically reported in genome organizations. Although virus identification was performed separately for each library, to reduce redundancy of viruses present in different libraries, we compared the results of each library with the others to obtain a list of unique viruses (uni-virus). Each viral contig was blasted against the whole list of viral contigs and those with nucleotide identity over 90 per cent and length over 1,000 nucleotides (nt) were grouped and considered a single virus contig in our final list. In this way, we simplified the information about sequence variants present in our samples, which is beyond the purpose of our inquiry. We selected as leading (representative) virus contig the longest of the group, which is also the contig deposited in GenBank. We developed custom R scripts for this ‘uni-contig’ selection (Supplementary Files S2–S4).
2.4 Taxonomy analysis
In order to evaluate the taxonomic complexity of the meta-samples present in our libraries we used Kraken2 (Wood, Lu, and Langmead 2019) in combination with Pavian (Breitwieser and Salzberg 2019) and a custom R script to create the visualization (Supplementary File S5).
2.5 Validation of selected viruses
For a selection of the most interesting contigs, their presence in the samples was validated through qRT-PCR. For this purpose, each of the 141 RNA samples was retro-transcribed into cDNA using the high-capacity RNA-to-cDNATM Kit (Applied Biosystems). Quantitative reverse transcriptase PCR (qRT-PCR) was used not only to assess the real presence of the in silico identified viruses into the samples, but also to associate specific viruses to specific samples. In fact, the computational identification was made on pool of samples, while the qRT-PCRs were conducted on individual samples of each pool. The list of primers used is displayed in Supplementary Table S2.
2.6 Phylogenetic analysis
RNA-dependent RNA polymerase proteins (RdRps) from all identified viruses and closest homologues from NCBI were aligned using the online version of Clustal Omega software with default parameters (Sievers et al. 2011) at the EBI Web Services (Madeira et al. 2019). Retrieved alignments were then submitted to IQ-TREE software (Trifinopoulos et al. 2016) to produce accurate phylogenetic trees under maximum likelihood model (default parameters). Alternatively, the maximum likelihood methodology was run with RaXL at CIPRES (Miller, Pfeiffer, and Schwartz 2010) for a subset of trees. The accession numbers of the proteins and the corresponding virus names and acronyms are displayed on the trees.
2.7 ORFan contig detection
Assembled contigs from each library were submitted to a DIAMOND search of the NCBI non-redundant whole database (version: October 2019). All contigs with a homologue were discarded, whereas the remaining contigs that were over 1 kb in length and encoded a protein of at least 150 amino acids (∼15 kDa) were kept, defining a preliminary set of contigs coding for ‘ORFan’ proteins. Reads were mapped to those contigs considering their orientation that is, whether they mapped in sense or antisense orientation. All the contigs that showed only positive or only negative reads were discarded, since a typical feature of replicating viruses is the presence of a minus and plus sense genomic template for replication. Remaining contigs were further analyzed for protein homology among themselves with Clustal Omega.
2.8 Reads correlation between true fungi/oomycetes and viruses
Correlation between reads from two taxonomical classes: Dothideomycetes (belonging to true fungi) and Oomycetes and number of reads of a specific virus has been computed. Only viruses identified in more than nine pools have been used to compute the correlation. Positive correlation means that the presence of the virus is positively correlated with the presence of a Class of organisms; therefore, the virus is likely to be hosted by an organism belonging to either Dothideomycetes or Oomycetes.
2.9 Virus names
Viruses identified in this article have been named using the following criteria: I) the first part of the name is the source of the virus; II) the second part of the name identifies the virus taxonomical group; and III) the last part of the name is a progressive number. The contig DMG-C_TRINITY_DN27100_c0_g3_i1 has as first match Rhizoctonia solani negative-stranded virus 4 and we named it: (P. viticola lesion associated)[I part] (mycobunyavirales-like virus)[II] (1)[III part].
3. Results
3.1 The virome associated to DMG
Across the sixteen studied pools, ∼623 M reads in total and ∼38 M reads, on average, per library were identified, with 68 per cent of reads classified. Supplementary Fig. S2A and Table S3provided a bird’s-eye view on the relative abundance of several taxonomical ‘Class’ (reads above 100 K) in each sample, showing a representation of the complexity of organisms present in the samples considering fungal, arthropods, plant and bacterial components. In most Italian samples, the highest ranking (abundance) taxa (order level) was, as expected, Oomycetes, followed by true fungi (with Dothideomycetes and Sordariomycetes as the most represented orders). Samples from Spain showed a different scenario, and Oomycetes and true fungi were predominant ranking taxa in only a few samples.
An initial quality control of our pipeline was performed assigning the reads of each library to Vitis vinifera, Alternaria sp., and P. viticola. Alternaria spp. (Alternaria alternata and Alternaria arborescens) is a complex of endophytic fungi of V. vinifera and the most abundant true fungus identified in our analysis. For each pool, cleaned reads have been blasted against the NCBI nt database using Kraken2 software (Wood, Lu, and Langmead 2019). Supplementary Fig. S2B shows the results of the analysis, highlighting that in eleven out of sixteen pools the number of P. viticola reads were more or equally abundant than V. vinifera reads, and, among them, in some cases the ratio between oomycete- and plant-mapping reads was above fifty times.
Initially, each pool was analyzed as a separated unit and viruses were identified independently from the other pools, resulting in sixteen distinct lists of viral contigs. The unicontig selection reduced the number of identified viral contigs from 1,280 to 423. Manual inspection and BLAST analysis of such contigs allowed us to classify them in different groups (taxonomically based, when feasible) or as putative different segments of multi-segmented viral genomes. The initial list of unicontigs contains some previously characterized viruses and putative plant viruses. Such list of viruses (already detected in grapevine, or new for grapevine) has been recently published and discussed thoroughly (Chiapello et al. 2020). In this work, we concentrated only on sequences that showed as first hits, after BlastX searches against nr protein database, viruses with known fungal/oomycetes hosts or other unspecified hosts, but not plant hosts. The total number of contigs reported in this study is 315, corresponding to 283 distinct virus RdRps. The viruses which show novelties or define new genera or families, or new phylogenetic trajectories are reported and discussed in the main text, whereas the remaining ones are described in Supplementary Text S1.
3.2 Positive strand viruses
A total of 227 positive strand RNA viral unicontigs were identified. They correspond to 222 viruses, based on the identified RdRps. The extra segments belong to a Jingmen-like virus with homology to NS3 and NS5 (being NS5 the RdRp) of flavivirus and four segments belonging to putative new bipartite viruses. The viruses we identified belong to twelve groups: sobemo-like viruses (one), picorna-ifla-dicistro-like viruses (six), tombus-like viruses (two), tobamo-virga-alpha-like viruses (eight), fusari/hypo-like viruses (three), poty-like viruses (four), ambiguiviruses (two), Jingmen-like viruses (one), yadokariviruses (one), amalgaviruses (one), flavi-like viruses (one), mitoviruses (fifty-five), narnaviruses (forty-three), leviviruses (one), botourmiaviruses (ninety-one), and orfanplasmovirus (five). The most represented taxonomic groups are those in the Lenarviricota (levivirus, narnavirus, mitovirus, and botourmiavirus). Supplementary Table S4 reports all the identified single-stranded RNA (ssRNA) (+) viruses and some of their features, including the GenBank accession number.
3.2.1. Narna-like viruses
Recently, an effort to put all the RdRp-encoding viruses in a single phylogenetic tree resulted in the recognition that bacterial leviviruses, narnaviruses, mitoviruses, and botourmiaviruses are in a single branch at the root of the tree (Wolf et al. 2018 2019). Such effort has been criticized by some researchers due to the low overall conservation of the alignment (Holmes and Duchene 2019). Nevertheless, in this work, we will follow this attempt at unifying RNA virus phylogeny, since it provides a taxonomic framework (Koonin et al. 2020) that can be continuously updated whenever more comprehensive alignments are available, increasing the likelihood of getting a more reliable phylogenetic tree. All the viruses comprised in the basal branch of the five-branch tree of this new megataxonomic framework are now in the phylum Lenarviricota, which now includes the families Mitoviridae, Narnaviridae, Leviviridae, and Botourmiaviridae (Koonin et al. 2020). Apart from characterized leviviruses, which can have multiple ORFs encoded by a single RNA, all the other viruses belonging to Lenarviricota are also monosegmented but have a single ORF, with the exception of plant ourmiaviruses and, possibly, some narnaviruses infecting trypanosomatid hosts (Rastgou et al. 2009; Charon et al. 2019).
In our study, we identified fifty-five mito-like viruses (Supplementary Table S4 and Fig. S3), forty-three narna-like viruses (Supplementary Table S4 and Fig. S4), and ninety-one new members of botourmia-like viruses across Italy and Spain (Supplementary Table S4 and Fig. S5). The only levivirus identified (PVaLevi1) is present exclusively in one library in Italy (DMG-D). Leviviruses have a monopartite, linear ssRNA genome of about 3.5 kb (Supplementary Table S4 and Fig. S4), so far stably associated only to bacteria.
Although the majority of mitoviruses was concentrated in Italian regions, with few viruses present in Spain and only one exclusively in the Iberic peninsula (PVaMito52), narna-like viruses are present both in Italy and Spain. Among them, P. viticola lesion-associated narnavirus8 (PVaNarna8) was extremely abundant in all the libraries, reaching over 1 million reads in library DMG-G. Two other narnaviruses (PVaNarna7 and PVaNarna10) were also widespread in most libraries and are fairly abundant.
Among the identified narnaviruses, nine encode for two ORFs: one is the putative RdRp in the positive sense orientation, whereas a second ORF is present in the negative sense orientation (rORF; Fig. 1B). We did not find any degree of conservation aligning the rORF of the nine narnaviruses (PVaNarna15, 22, 23, 24, 25, 26, 41, 42, and 43) as already shown for other ambisense narnaviruses (Dinan et al. 2020). We deposited the additional ORFs in NCBI as ‘putative proteins’.
Figure 1.

(A) Lenarviricota phylogenetic tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: VT+F + I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −377870.722. At nodes the percentage bootstrap values. The tree focus is on narnaviruses. Red stars indicate viruses with missing GDD motif. Color dots correspond to established or proposed new genera. (B) Genomic organization of nine ambinarnaviruses with a second ORF in reverse genomic orientation. Top ruler indicates the length in kb. (C) Alignment of narna-like viruses without the GDD domain and some of the closest homologues with the GDD domain. In the consensus row an exclamation mark ‘!’ stands for a conserved amino acid, while an asterisk ‘*’ stands for positions in which there is a majority of sequences agreeing. Positions in which the sequences disagree are left blank in the consensus sequence. Color code indicates the aminoacidic properties. The two boxes show the GDD and the conserved MGDP/M or L conserved motifs.
All the fifty-five contigs that were assigned to mitoviruses are recognized as RdRps through a motif search software.
Some botourmia-like viruses are ubiquitously spread across the different sampling sites in Italy and Spain (among the most abundant PVaOurmia7, 29, 39, 40, and 70) and only a few are library specific (i.e. PVaOurmia3, 12, 46, and 47; Supplementary Fig. S5). All the ninety-one sequences of the RdRps have a number of conserved amino acids in the alignment, in specific around the GDD motif (data not shown). Nevertheless, motif-searches did not recognize a typical RdRp motif in nine of these sequences (PVaOurmia3, 49, 50, 58,59, 60, 61, 80, and 81).
All the identified viruses in the Lenarviricota phylum have been used to derive a phylogenetic tree together with representatives of sequences already present in public databases (273 RdRp sequences) to confirm the correct virus association and the presence of possible out-group viruses. The tree is displayed in three different figures to maximize the resolution (Supplementary Figs S6 and S7 and Fig. 1A). Botourmia-like viruses form a single clade in the phylogenetic tree, with all the RdRps included, confirming that all the ninety-one identified viruses have a strong support from the phylogenetic analysis to belong to the family Botourmiaviridae (Supplementary Fig. S6). Inside the recognized Botourmiaviridae, the three established genera Botoulivirus, Scleroulivirus, and Magoulivirus (Ayllón et al. 2020) can be extended to include some, but not all, the new botourmiaviruses characterized in this work. For this reason, new genera should be established, one of which could include so far sequences only from this study.
The mitovirus tree (Supplementary Fig. S7) shows that also this group of viruses includes a remarkably diverse array of viruses with a number of sub-clades. As shown before, plant mitoviruses all derived from a specific branch (Bruenn, Warner, and Yerramsetty 2015; Nibert et al. 2018; Nerva et al. 2019c).
The levivirus present in our virome study is clustering with other leviviruses and its association is strong (Fig. 1A); given the presence of some bacterial reads in library DMG-D, we suggest that this is probably a true bacteriophage (Supplementary Fig. S2).
More complex is the narna-like phylogenetic tree (Fig. 1A). The narnaviruses are indeed classified in a single well supported clade, but several subgroups could be established, either genera or families given the phylogenetic distances and deep branches that support some of the clades. In specific, there is a new group of narnaviruses that lacks both a typical palm domain and the GDD triplet in their RdRps: PVaNarna1, 2, 3, 5, 6, 7 (very abundant in most libraries), 19, and 20. In particular, PVaNarna20 stems from a very deep branch at the root of this group and shows very limited amino-acid conservation in the alignment (Fig. 1C). This group of viruses should probably represent a new taxon (possibly a new family or higher-ranking taxa) for which we propose the name GDDminusnarnaviridae. Another new group of narnavirus maintains the GDD triplet but has also a conserved M-G-D-[MLP] motif in common with the above mentioned group (PVaNarna11-12-13-14-29-30-32-33-34-35-36-37; Fig. 1C). Other authors have recently shown that existing narnavirus should at least comprise two genera, (Alphanarnavirus and Betanarnavirus; Dinan et al. 2020), but the clades described above are distinct from those already observed, so we think higher new ranking taxa should be assigned. The new narnaviruses with ambisense genomes that we present in this study can be grouped in one phylogenetic homogenous clade and we propose for this clade the name mycoambinarnaviruses.
3.2.2. Tobamo/virga-like viruses
One alphavirus-like virus has been identified in our study (PvLaAlpha-LV1) in Italy with very low estimated accumulation (Supplementary Fig. S8). Its genome organization shows two ORFs: ORF1 (2007 amino acids) contains an RNA helicase and an RdRp motif, while the ORF2 (184 amino acids) has no conserved domains and matches with a hypothetical protein (QGW08826.1) of Fusarium graminearum alphavirus-like virus 1 (Fig. 2A).
Figure 2.
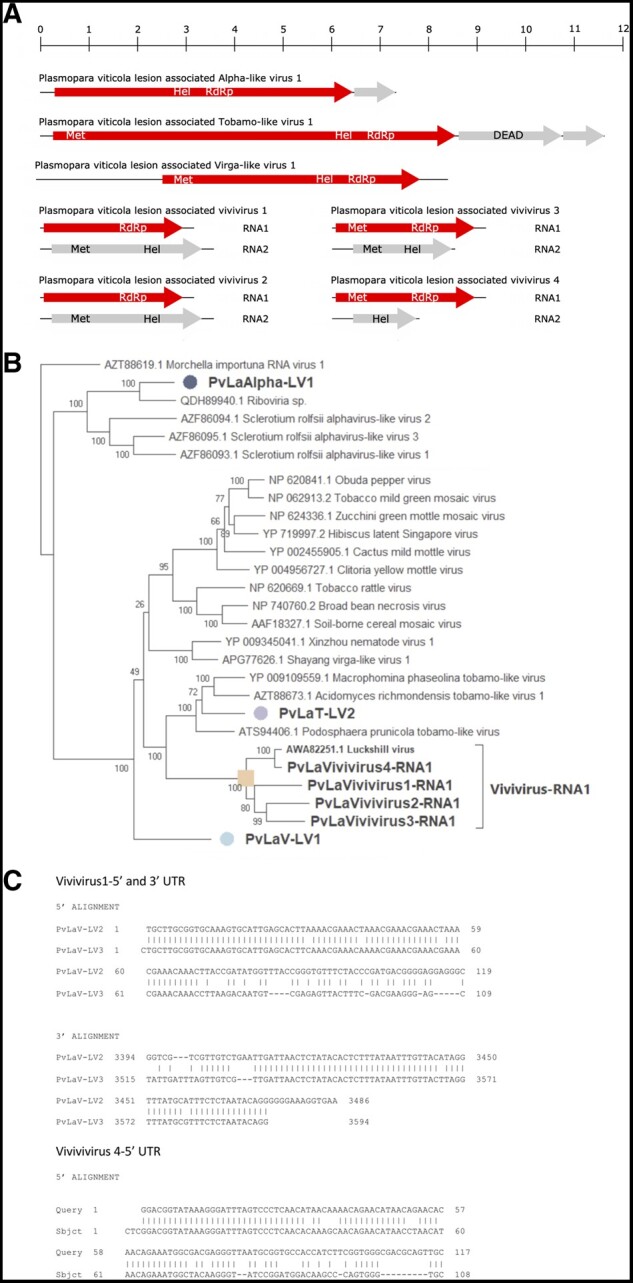
(A) Tobamo-virga-alpha genome organizations. Top ruler indicates the length in kb. (B) Tobamo-virga-alpha virus tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: Blosum64+F-I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −120571.621. Purple label indicates the tobamovirus, orange labels indicate viviviruses in different sub-clades, light blue label indicates virga-like virus, and the blue one indicates the alphavirus. At nodes the percentage bootstrap values. (C) Sequence alignment of vivivirus 5′ and 3′ UTRs.
A single tobamo-like virus has been identified (PvLaT-LV2) in three samples from Italy (Supplementary Fig. S8). Its genome organization comprises three ORFs, one encoding a putative protein carrying the methyltransferase, helicase, and RdRp domains, whereas the second ORF carries a DEAD motif, but is not conserved with any ORFs in public databases. Finally, the third ORF-encoded protein has no conserved motif, and does not have any homologue in public databases (Fig. 2A).
Nine virga-like virus fragments have been identified. PvLaV-LV1 has a coding region of over 8 kb and encodes a single ORF with methyltransferase, helicase and RdRp domains. The remaining eight segments have a length between 2 and 3.5 kb: four of them carry the RdRp domain and the other four carry the helicase domain, whereas the methyltransferase domains are present or absent depending on the specific viral segment (Fig 2A). These two groups of four sequences have as first blast hit two putative viral segments: one carrying the RdRp motif (Luckshill virus fragments) and one carrying the helicase motif (Cyrill virus fragments; Medd et al. 2018). Strict association of read abundance and presence in the same libraries of corresponding Luckshill- and Cyrill-like fragments allow us to hypothesize that each of the segment is part of a bipartite virus, which we name Plasmopara viticola lesion-associated vivivirus 1–4, since four of these strict associations are present in our libraries (Supplementary Fig. S8). Another element that support the strict association of these two segments in a single virus genome is the conservation of the 5′ and 3′ terminal nt sequences, which are specifically recognized by the RdRp during replication: in facts, PvLaVivivirus1 RNA1 and RNA2 share high conservation over the first 60 nt both at the 5′ and 3′ ends, while PvLaVivivirus4 RNA1 and RNA2 share conservation only at the 5′ end (Fig. 2C).
Sequence alignment, and phylogenetic analysis (Fig. 2B) shows that the four RdRp-encoding virga-like fragments are in a clade together with Lukeshill virus characterized in the Drosophila suzukii virome (Medd et al. 2018). The alpha-like virus (PvLaAlpha-LV1) belongs to a clade that groups a viral fragment from an environmental sample and three other mycoviruses. The tobamo-like virus (PvLaT-LV2) also belongs to a statistically well-supported clade of mycoviruses, which should probably constitute a new viral family.
3.3. Negative strand RNA virus
Twenty-nine negative-stranded RNA viruses have been identified (Supplementary Table S5). Those twenty-nine viruses have been assigned to four major taxonomical groups of negative strand viruses: eleven viruses to the order Bunyavirales, twelve to the order Mononegavirales, three to the Serpentovirales, and three to yue-like virus family/order.
3.3.1. Bunyavirales
The Bunyavirales order includes several families of viruses infecting vertebrates, invertebrates, plants; only recently specific clades infecting fungi were also characterized (Lin et al. 2019; Nerva et al. 2019b; Velasco et al. 2019). The members of this order are linear negative-sense, single-stranded segmented viruses. Their genome size and organization are family specific, but at least bi-segmented. Other RdRp-encoding mycobunya-like sequences have also been previously characterized (Donaire, Pagán, and Ayllón 2016; Marzano and Domier 2016) but associated segments encoding for at least the nucleocapsid (Nc) protein are missing. The bunya-like viruses identified in this work are distributed across Italy and Spain. The mapped reads showed a general low accumulation of the identified viruses, apart from a new arena-like virus which is abundant in most Italian libraries (Supplementary Fig. S9) and we provisionally named it Plasmopara viticola lesion-associated bunyaarena-like virus 1 (Fig. 3A). The ten new tentative members of the Bunyavirales have been deposited in NCBI and named PVaBunyavirales1 to PVaBunyavirales10 (Supplementary Fig. S9).
Figure 3.
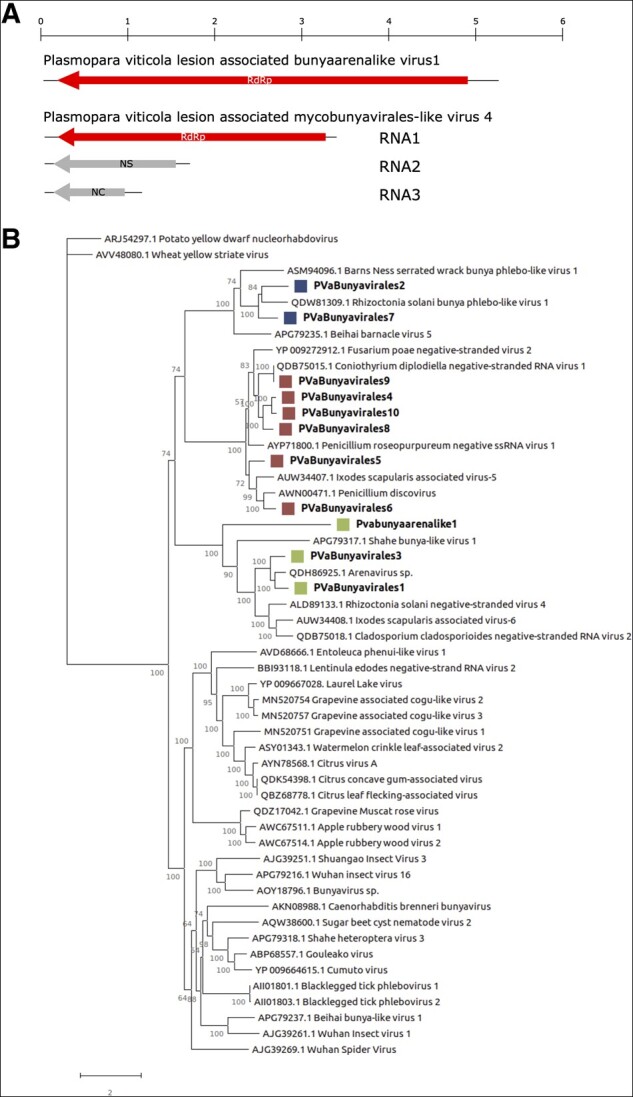
(A) Genome organization of a representative mycovbunyavirus and a bunya–arena virus. Top rule indicates the base pair in kb. (B) Bunyavirales phylogenetic tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: rtREV+F + I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −220431.575864. At nodes the percentage bootstrap values. Different colors indicate different subgroups.
Phylogenetic analysis (Fig. 3B) shows that all the identified viruses are indeed in the order Bunyavirales. At least three distinct clades are obvious, and most members of these clades are infecting fungi. The clade that includes PVaBunyavirales4, 5, 6, 8, 9, and 10 include also Penicillium discovirus, Penicillium-roseopurpureum-negative-ssRNA-virus-1, and Coniothyrium diplodiella negative-stranded RNA virus 1. These three viruses are confirmed mycoviruses and have three identified genomic segments, coding for an RdRp, a Nc, and a non-structural protein (NSs) (Nerva et al. 2019b,c). Their Nc and NSs proteins were used to search our downy mildew libraries to look for homologous proteins. Indeed, many Nc and NSs were retrieved and assigned to specific RdRp encoding segments on the base of phylogenetic and read/abundance correlations. We could therefore derive some likely full-length genomes (PVaBunyavirales 4, 8, and 9), although final confirmation should come from characterization of RNA associated to purified Nc or purified particles. The in silico association was checked and confirmed by qRT-PCR on the original sample RNA extracts. In fact, some specific samples inside the pools were positive for all three segments associated to the putative PVaBunyavirales4, 8, and 9 (Table 1).
Table 1.
Presence and titer of viral contigs belonging to tripartite Bunyavirales determined by qRT-PCR using as template RNA extracted from specific downy mildew lesion samples
| Sample | Primer | Ct (Min) | Virus | Domain |
|---|---|---|---|---|
| DMG37 | T31-107 | 32.95 | Bunyavirale9 | NSs |
| DMG37 | T31-108 | 31.44 | Bunyavirale9 | NC |
| DMG37 | T31-27 | 30.21 | Bunyavirale9 | RdRp |
| DMG71 | T31-111 | 30.2 | Bunyavirale8 | NSs |
| DMG71 | T31-112 | 27.51 | Bunyavirale8 | NC |
| DMG71 | T31-26 | 30.43 | Bunyavirale8 | RdRp |
| PB-DM14 | T31-113 | 26.2 | Bunyavirale4 | NC |
| PB-DM14 | T31-114 | 25.64 | Bunyavirale4 | NSs |
| PB-DM14 | T31-24 | 28.04 | Bunyavirale4 | RdRp |
| PB-DM15 | T31-113 | 26.48 | Bunyavirale4 | NC |
| PB-DM15 | T31-114 | 26.14 | Bunyavirale4 | NSs |
| PB-DM15 | T31-24 | 27.4 | Bunyavirale4 | RdRp |
PVaBunyavirales2 and 7 belong to a clade that includes a confirmed mycovirus, Rhizoctonia solani bunya phlebo-like virus 1 (Picarelli et al. 2019), which so far has no specific Nc or NSs associated.
Also PVaBunyavirales1 and 3 belong to a clade that includes confirmed mycoviruses that is, R. solani negative-stranded virus 4 (Marzano et al. 2016) and Cladosporium cladosporioides negative-stranded RNA virus 2 (Nerva et al. 2019c; Fig. 3B). Also for this group of viruses only the RdRp is known. This clade also includes our Pvabunyaarenalike1, a very distant arena-like virus, which has instead as first hit a putative plant virus of the cogu-like clade (Citrus concave gum-associated virus).
3.3.2. Mononegavirales
The order Mononegavirales includes negative-stranded RNA viruses mostly monopartite, with multiple ORFs, generally all in the same orientation. In our libraries, we found a number of viruses putatively belonging to this order showing different genomic organization (Fig. 4A). Specifically, viruses PvLamononegaambiV1, 2, 3, 4, 5, 6, 7, and 8 had as first hit in Blast searches Kiln Barn virus (Medd et al. 2018), and the recently reported Penicillium glabrum negative-stranded RNA virus 1 and Penicillium adametzioides negative-stranded RNA virus 1 (Nerva et al. 2019c). Kiln Barn virus does not seem to be complete, based on the alignment with other virus sequences with homologous RdRps, which are longer than 6.5 kb and have the potential to encode a small ORFs also in the sense orientation (compared with the antisense genomic orientation of the RdRp). Such small ORF (around 200 amino acids long), found at the 5′ of PvLamononegaambiV2, 4, and 6 genomes (Fig. 4A), is partly conserved, both in position and in some specific amino acid motifs highlighted in the alignment (Fig. 4C); The ambisense nature of these contigs was confirmed through RT-PCR across the junction (not shown). Since the ORF is absent in our shorter virus contigs (PvLamononegaambiV1, 3, 5, 7, and 8) and because of the low coverage of the genome (Supplementary Fig. S10), we can hypothesize that they are not complete. On the basis of these direct and indirect pieces of evidence, we suggest that this second ORF in sense orientation is indeed a feature of a new viral taxon inside the mononegavirales, for which we propose the name mononegaambivirus.
Figure 4.

(A): Mononegavirales genome organizations. Top ruler indicates the length in kb. (B) Mononegavirales phylogenetic tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: LG+F + I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −124341.266. At nodes the percentage bootstrap values. Different colors indicate different subgroups. (C) Alignment of PvLamononegaambi2, 4, and 6 ORF2 with other Nc sequences from PvLamymonaV1. In the consensus row an exclamation mark ‘!’ stands for a conserved amino acid, while an asterisk ‘*’ stands for positions in which there is a majority of sequences agreeing. Positions in which the sequences disagree are left blank in the consensus sequence.
Plasmopara viticola lesion-associated mononega virus 1 (PvLamononegaV1) is abundant only in one library (DMG-B) and encodes two ORFs (Supplementary Fig. S10 and Fig. 4A). It has low homology with an invertebrate rhabdo-like virus, Hubei rhabdo-like virus 2 (Shi et al. 2016), which codes for four ORFs. Plasmopara viticola lesion-associated mymonavirus 1 (PvLamymonaV1) and Plasmopara viticola lesion-associated mymonavirus 2 (PvLamymonaV2) have respectively three and four ORFs and are typical mymonaviruses (Jiang et al. 2019; Fig. 4A).
Mononegavirales are more present in Italy than in Spain, both in terms of identified viruses and in term of inferred virus load. Seven out of twelve were exclusively identified in Italy and none of the five identified in Spain was uniquely present in that country (Supplementary Fig. S10).
Phylogenetic analysis (Fig. 4B) shows that PvLamymonaV1 and 2 are closely related to the recently characterized Sclerotimonavirus genus (Jiang et al. 2019). The new proposed group of monoambinegavirus also belongs to a well-defined and distinct clade of mostly fungal viruses, even though Kiln Barn Virus comes from a Drosophila suzukii metagenome study (Medd et al. 2018). PvLamononegaV1 belongs to a clade grouping already well characterized rhabdoviruses, even though its branch length indicates a very basal relationship. The closest sequences to PvLamononegaV1 are associated to invertebrates.
3.3.3. Yue-like mycoviruses
Our bionformatic pipeline retrieved three distinct segments encoding three putative RdRps having as first BLASTx hits members of the Yuevirus and Qinvirus groups (Shi et al. 2016). These segments are 3.6–4 kb in length and encoded a single ORF, coding for the putative RdRp. Yueviruses and Qinviruses belong to Goujianvirales and Muvirales orders respectively in the current ICTV taxonomy (Koonin et al. 2020). The three viral contigs, PVaYuevirus1, 2, and 3, are highly represented in all the libraries in both countries, in some cases with over one million mapping reads (Supplementary Fig. S11). This could suggest a virus integration in some host genomes common to all the metagenomic samples. Consequently, we checked for their presence in either vitis or plasmopara genomes, but we did not retrieve any positive hit. We also checked evidence of DNA corresponding to the three segments through qPCR and demonstrated that the viruses are not integrated (data not shown). Therefore, they can be considered bona fide replicating RNA segments.
Phylogenetic analysis (Fig. 5A) shows that these viruses constitute a well-defined sister clade of the two Yuevirus so far characterized. The conserved putative catalytic domain of this new fungal yue-like viruses are IDD, similarly to the one of qin-like viruses so far characterized, but differently from the one of Yueviruses which is SDD (Fig. 5B). Both the Yueviruses and Qinviruses characterized from invertebrates have a second genomic segment coding for a protein of unknown function, conserved among each of the two groups of viruses (Shi et al. 2016); our attempts to find associated segments with different approaches were so far unsuccessful.
Figure 5.

A: Yue-like virus phylogenetic tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: LG+F + I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −72054.223. At nodes the percentage bootstrap values. Red labels indicate viruses identified in this study. (B) Alignment of discovered yue-like viruses (PVaYuevirus1, 2, and 3) with some of the closest homologues. The RdRp domain is highlighted in red; 50 amino acids across the GDD/SDD/IDD domains are shown.
3.3.4. Serpentovirales-like viruses
Typically, ophioviruses are plant viruses belonging to the order Serpentovirales, family Aspiviridae, and are segmented negative-stranded RNA linear genomes, which can contain up to four segments. We identified three ophio-like viral RdRps, encoded by RNA segments ranging from 7.7 to 8.4 kb. Two of these segments (PVaophio4 and 5) carry also a small ORF at the 3′ terminus in the antisense orientation. PVaophio5 is almost identical (82% at nt level) to Fusarium poae negative-stranded virus 1 (Osaki et al. 2016), the first mycoophiovirus identified. Such segments are mostly restricted to Italian libraries, and PVaophio1 is particularly abundant with more than 200,000 reads mapping on the segment, whereas PVaophio4 and 5 are present with a very limited number of reads (Supplementary Fig. S12). Phylogenetic analysis (Supplementary Fig. S13) reveals that they indeed belong to a well-defined clade that includes a number of mycoviruses recently characterized, for which we propose a family taxon named mycoaspiviridae. The likely presence of extra genomic segments associated to them was not yet revealed for any of these phylogenetically distant viruses.
3.4. Double-stranded RNA virus
The double-stranded RNA (dsRNA) viruses have been classified in seven sub-groups: bipartite (a proposed new family not yet accepted by ICTV described in Nerva et al. 2016), botybirna, partiti, partiti-like, polymycovirus, totivirus, and toti-like virus. The identified viruses have been mapped against all the Italian and Spanish libraries in order to display the coverage and highlight the presence of the viruses in the different pools (Supplementary Table S6 and Fig. S14).
For the multi-partite viruses, when possible, the read count/library has also been used as a guide to identify segments belonging to the same virus; when this approach resulted in ambiguous associations, the qRT-PCR approach to find strict associations in specific samples in the pools has been used (data not shown).
In the recent effort to include all RNA viruses in a single phylogenetic tree, dsRNA viruses have been shown to have originated at least twice independently (Wolf et al. 2018). They are present as a distinct lineage in Branch 2 of the picornavirus supergroup, which now corresponds to the Phylum Pisuviricota, Class Duplopiviricetes (polymyco-, ‘bipartite’, partiti, and partiti-like viruses; Koonin et al. 2020), while toti, toti-like, and botybirna viruses belong to the second lineage that includes the whole Branch 4 (Wolf et al. 2018), which now corresponds to the phylum Duplornaviricota (Koonin et al. 2020). We will follow this subdivision in describing the dsRNA viruses found in our library.
3.4.1. Group 1
Group 1 contains polymyco, ‘bipartite’, partiti and partiti-like viruses. Polymycoviruses have four to eight dsRNA genomic segments, four of which show some degree of conservation among species. The overall size of the genome ranges from 7.5 to 12.5 kb. We identified five polymycoviruses (PvaPolymyco1–5) in both Italian and Spanish sites (Supplementary Fig. S14): for all of them we found the RdRp, and for three of them we were able to identify also additional segments. Polymycoviruses (also previously known as tetramycoviruses) have been recently classified by the ICTV under the Polymycoviridae family, a floating family inside the Realm Riboviria (Koonin et al. 2020).
A ‘bipartite’-like virus, with both its segments, was detected in all the Italian libraries at very low accumulation, whereas in Spain it is present in four pools with the highest concentration in DMS6 (Supplementary Fig. S14). PVaBipar1 clusters with other unclassified dsRNA viruses with small genomes (Supplementary Fig. S15), which include the mutualistic Curvularia thermal tolerance virus (Marquez et al. 2007).
Partitiviruses are the most represented taxon in our dataset (ten distinct species), and two of them are ubiquitously distributed in Italian and Spanish libraries (PVaPartit6 and PVaPartit7) accumulating abundantly in each library, based on read counts. Conversely, PVaPartit5 is present only in one sample (DMG-G). Some of the partitiviruses seem to be country specific, namely PVaPartit9, PVaPartit4, PVaPartit5, PVaPartit2 for Italy and PVaPartit8 for Spain. The Partitiviridae is a family of bisegmented (occasionally tri-segmented) dsRNA viruses with genomes of 3–4.8 kb. The two genome segments are individually encapsidated. The family includes five genera with characteristic hosts for members of each genus.
PVaPartLike1 has been detected at high level in all samples and PVaPartLike2 is present only in one Italian sample and two Spanish samples.
Phylogenetic analysis (Supplementary Fig. S15) confirms that all the polymycoviruses we identified belong to a statistically well supported clade with bootstrap values of 100 per cent. Partitiviruses are spread across three sub-groups. PVaBipar1 is indeed in a clade well sustained by a high bootstrap value with other ‘bipartite’-like viruses. The two partiti-like viruses belong to a well-supported clade that should be recognized as a new virus genus inside the family Partitiviridae, but PVaPartLike1 stems from a separated deep branch compared with the other partiti-like viruses.
3.4.2. Group 2
Group 2 contains toti, toti-like, botybirna viruses, and an unclassified dsRNA virus (Supplementary Fig. S14). Four bona fide totiviruses have been identified: three exclusively present in Spain and one exclusively present in northern Italy. There is a specific site in Spain (DMS6—Penedés) where three of these viruses are present at a high accumulation. Totiviruses consist in a single molecule of linear dsRNA, 4.6–7.0 kb in size. The genome usually contains two overlapping ORFs, coding for CP and RdRp. Phylogenetic tree shows totiviruses clustering in a single clade (Fig. 6B), even though one of them (PVaToti3) stems from a branch separated from the other totiviruses characterized in this study.
Figure 6.
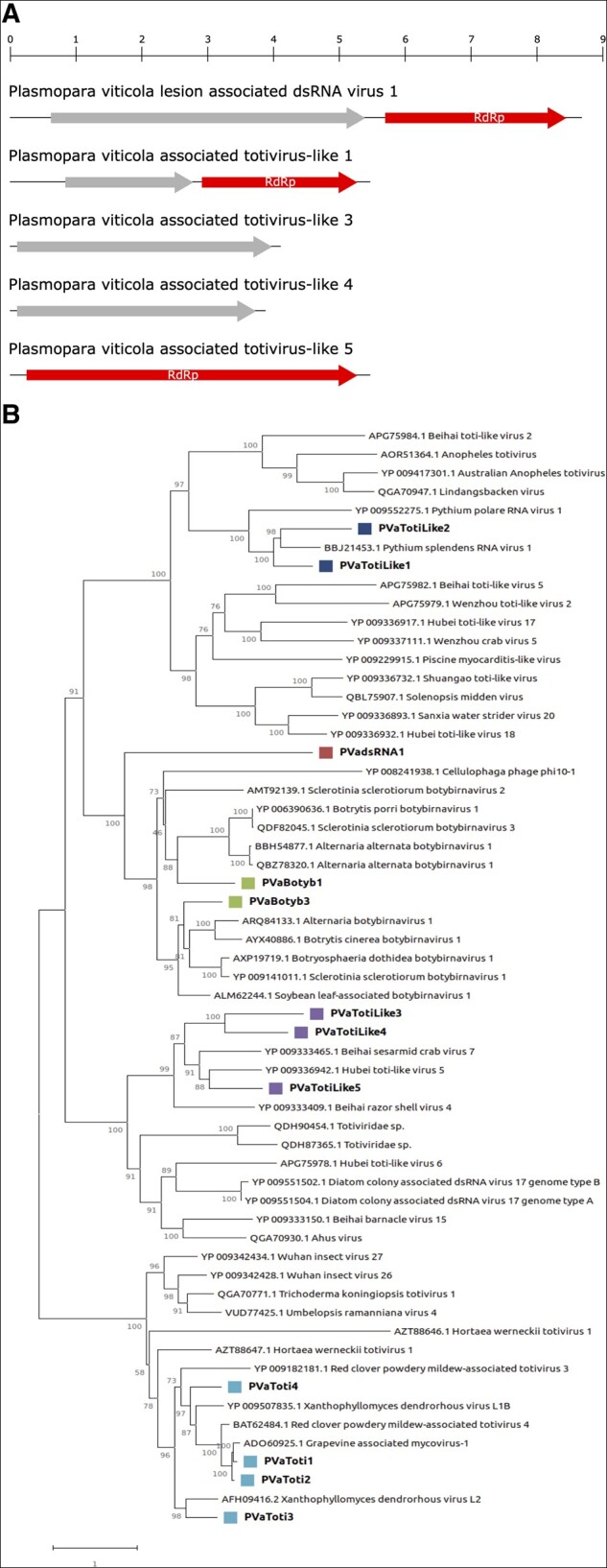
(A) genome organization of representative viruses from dsRNA group2. Top rule indicates the length in kb. (B) dsRNA group2 phylogenetic tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: VT+F + I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −132016.713. At nodes the percentage bootstrap values. Different colors indicate different subgroups.
The identified toti-like viruses are ubiquitous in all regions of both countries, with a very high number of reads, with the exception of PVaTotilike2, which was not detected in four libraries from Spain. The five toti-like virus segments showed different genome organizations (Fig. 6A). PVaTotilike1 and 2 codes for two ORFs: one is the RdRp and the other has no conserved domains; therefore, these can be considered two distinct self-sufficient viruses. The other three segments encode a single ORF: PVaTotilike5 codes for a putative RdRp while in the other two virus contigs (PVaTotilike3 and 4) no conserved domain was found. The phylogenetic analysis shows that toti-like viruses are divided into two groups and the identified viruses are spread across them (Fig. 6B): PVaTotilike1 and 2 are in a clade with other mycoviruses infecting oomycetes, whereas PVaTotilike3, 4, and 5 are in a clade with viruses from invertebrates.
One new dsRNA virus is present in all the libraries of both countries (Supplementary Fig. S14). Its genome (∼8.6 kb) contains two ORFs carrying an RdRp and a second ORF with no conserved domains (Fig. 6A) and a distant homology with another dsRNA discovered in another member of Peronosporaceae, Bremia lactucae (GenBank accession: QIP68007.1, unpublished). Interestingly the virus does not cluster with any other virus in the tree (Fig. 6B), even if it seems to be distantly related to botybirnaviruses.
Botybirnaviruses have a bipartite genome, with two types of structural proteins (p85/80, p70) and an RdRp. They are currently classified in a floating Genus—Botybirnavirus—in the Orthornavirae Kingdom (Koonin et al. 2020). Two botybirnaviruses were identified in this study: one is present in all Italian samples and three Spanish samples, whereas the other one is only present in Spain (Supplementary Fig. S12). Both Plasmopara viticola lesion-associated botybirnaviruses cluster with known botybirnaviruses in the phylogenetic tree (Fig. 6B).
3.5. Orfans of likely viral origin
Our pipeline designed to identify new viruses in our dataset is based on homology to virus proteins already present in NCBI databases. This approach has the inherent limitation of missing new virus clades or virus segments that encode proteins that are not sufficiently conserved to be retrieved through Blast searches. We applied a different pipeline to identify ‘ORFan’ protein-encoding contigs (putative viral) and the initial search with DIAMOND identified 333 unicontigs. When manually inspecting the number of positive-and negative-stranded reads mapping to each of them, we found hundreds of relatively abundant new putative virus segments (based on the abundance of both plus and minus strand mapping reads in the library). A further analysis aiming at revealing clusters of homologous proteins within them (and therefore new clusters of conserved proteins) highlighted a number of putative groups. For one of these clusters of sequences we were able to detect some distant relationship to existing virus RdRps (not detected by our Blast search), which revealed a new lineage of RNA viruses. In particular, we discovered five sequences, named PvLaORFPlV1–5, that form a new clade inside (or closely related to) the Lernaviricota phylum that comprises abundant virus segments widespread in many libraries both from Italy and Spain (Supplementary Fig. S8). Phylogenetic analysis clusters these sequences as a sister clade of characterized leviviruses (Fig. 7A); an alignment with members of the Lenarviricota shows conservation of the A, B, and C palm subdomains (te Velthuis 2014; Fig. 7B).
Figure 7.

(A) Lenarviricota phylogenetic tree computed by IQ-TREE stochastic algorithm to infer phylogenetic trees by maximum likelihood. Model of substitution: VT+F + I+G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −385880.540585. At nodes the percentage bootstrap values. The tree focus is on a new clade of putative mycoviruses closely related to leviviruses and named orfaplasmovirus. Colored squares correspond to the proposed new family of viruses. (B) Alignments of the new clade of viruses with some levivirus and two new narnaviruses: only residues without gaps were maintained. The A, B, C, and D subdomains are highlighted with boxes.
4. Discussion
4.1. The metagenomic nature of our samples and uncertainties in virus–host assignment
The purpose of this study was to find new virus-based BCAs to use against DMG. A first step in this quest would be an inventory of viruses infecting P. viticola, irrespective of their pathogenic potential. Currently, to the best of our knowledge, there are no specific mycovirus associated to P. viticola, but a number of viruses have been associated to other Peronosporales (Yokoi et al. 1999; Yokoi, Yamashita, and Hibi 2003; Heller-Dohmen et al. 2011; Grasse et al. 2013; Grasse and Spring 2015). Many well-studied virus–host interactions are reported for viruses of Phytophtora spp. (Cai and Hillman 2013; Cai et al. 2013 2019a,b), and the study of the Globisporangium genus (Pythiaceae family) provided the characterization of some mycoviruses (Gillings, Tesoriero, and Gunn 1993; Sasai et al. 2018; Shiba et al. 2018, 2019). A summary of the viruses so far studied in oomycetes was recently reviewed (Sutela, Poimala, and Vainio 2019). Some of the viruses found were specific to this taxonomic group of protists. In our study we found viruses, such as PVaNarna 8 and 10, closely related to Phytophthora infestans virus 4 (Cai et al. 2012), and PVaTotilike1 and 2, related to those recently characterized in Pythium spp. (Shiba et al. 2019) and in Phytophthora cactorum (Poimala and Vainio 2020); both are therefore good candidates to have P. viticola as host.
Given the complexity of the metatranscriptomics samples, assigning all the viruses we found to the target host is a difficult task and other approaches, besides the in silico analyses, need to be planned. Reproducing infection in vivo, using a set of inocula from the field, and qRT-PCR on single sporangia for specific viruses might be interesting complementary approaches to define a stricter association between at least some of the viruses we found and their putative host.
In an attempt to associate the viruses we found with a possible host, the complex nature of our samples was analyzed. Some samples have a lower overall quality of reads and some bacterial contaminations, but in most libraries plant, bacterial and animal components are limited. Nevertheless, we were initially surprised to find a strong fungal component in most samples, quantitatively comparable to that of the oomycete P. viticola. A closer inspection of the fungal component showed that it was fairly constant among the different libraries, and the most represented fungi were those of the Alternaria spp. complex (A. alternata and A. arborescens). Occurrence of such fungi as endophytes (and here probably also as microorganisms of the phyllosphere) in grapevine is common and was previously studied (Polizzotto et al. 2012; Tao et al. 2014; Elena et al. 2019). More interestingly Alternaria species from grapevine were also isolated from P. viticola-infected leaves for their potential as BCAs (Musetti et al. 2006) possibly through the secretion of a specific fungal toxin (Musetti et al. 2007). Regulation of fungal toxin production by mycoviruses was recently shown (Nerva et al. 2019a), and this work could indeed suggest a further level of complexity in the production of such toxin, if also in Alternaria spp. the mycovirus presence alters its expression.
Also, to be as accurate as possible in assigning a putative host in silico, we took advantage of a correlation analysis between reads abundance for a specific virus and that of the non-virus components of the metagenomic sample. In fact, given that the various libraries we analysed have a different percentage of reads mapping to fungi at large (true fungi and oomycetes), it is possible to correlate abundance of a specific virus in each sample to the relative abundance of their class of hosts. Applying this principle, it is easy to infer that most of the viruses we described here are indeed mycoviruses (having true fungi or oomycetes as hosts). As an example, the new clade of yue-like viral RdRps that we identified correlates well with the presence of fungal reads, being higher in some of the libraries with the highest oomycetous-fungal content, and much lower in libraries with a high contamination of plant or bacterial reads (Supplementary Fig. S16).
Authors have argued in favor or against the inclusion of viruses from metagenomic sample in taxonomy without a certain host assignment (Simmonds et al. 2017; Kaefer et al. 2019). Taking indeed for granted the added value of establishing with certainty the host for a number of our most representative viruses, the effort to annotate 283 new viruses from a fairly defined metagenomic sample sheds new lights on virus biodiversity with the discovery of new virus lineages that are worth further investigations. In the next paragraphs we will point out only the most relevant that expand our knowledge of RNA virus biodiversity in general.
4.2. New narnavirus-like evolutionary trajectories
Narnaviruses were first discovered in Baker’s yeast, and thoroughly studied in this model system and for a long time they were thought to infect only fungi and oomycetes (Kadowaki and Halvorson 1971; Wesolowski and Wickner 1984; Hillman and Cai 2013). Their role remains mostly cryptic, but an interesting recent paper shows they can affect the biology of their fungal host in a four-way symbiosis that involves also an endobacterium (Espino-Vázquez et al. 2020). The vast majority of the members of the phylum Lenarviricota (Koonin et al. 2020; comprising levivirus, mitovirus, botourmiavirus, and narnavirus clades) encodes only the RdRp, but recent convincing indirect evidence suggests that some narnaviruses might also carry a functional ORF in their negative strand (rORF; DeRisi et al. 2019; Dinan et al. 2020). Narnavirus (intended here as the members of the newly established Class Amabiliviricetes, Order Wolframvirales) natural hosts are fungi (including oomycetes; Cai et al. 2012; Hillman and Cai 2013), and protists (Akopyants et al. 2016; Charon et al. 2019), but recently evidence that a narnavirus is infecting Caenorhabditis elegans was provided (Richaud et al. 2019). In addition, narnaviruses seem to infect also insects, particularly mosquitos (Chandler, Liu, and Bennett 2015; Goertz et al. 2019), opening to the possibility that also some viruses characterized in a previous invertebrate meta-transcriptomic virome characterization study, can indeed be viruses of arthropods or of other invertebrate groups (Shi et al. 2016). Our study adds further layers of biodiversity to this group of viruses; in specific we characterized the genomes of new viruses forming at least three new clades in the Lernaviricota that potentially open new perspectives in the evolutionary history of this branch of viruses. The first clade (likely a new genus in the family Narnaviridae) is the mycoambinarnaviruses. In our study, we describe a rather uniform clade that includes putative complete or incomplete genomic fragments of narnaviruses that encode a putative protein in the negative strand orientation. Such genomic organization has been described at length in a recent work where the authors also searched transcriptomes available in repositories for transcripts of viral origin having this ambisense genomic organization (Dinan et al. 2020). The viruses with the same genome organization identified in our work constitute a well-defined clade separated from those already described before, with the exclusion of PVaNarna41, which belongs to the insect-infecting clade cited before. A second clade (likely a new family or a new order, given its diversity in the new to be established order Wolframvirales) is the one we provisionally named GDDminusviruses (or Gammanarnaviridae, following previously proposed nomenclature; Dinan et al. 2020). We here provide a number of new sequences forming a statistically well supported clade of viruses together with some virus sequences that had already been characterized as bona fide fungal viruses in axenic fungal cultures: Fusarium poae narnavirus2 (Osaki et al. 2016), two narnavirus from Aspergillus fumigatus (Aspergillus fumigatus narnavirus1 and 2; Zoll , Verweij, and Melchers 2018) and three more from a study of grapevine endophytes that is, Neufusicoccum parvum narnavirus2, Cladosporium tenuissimum narnavirus 1 (CtNV1), and Alternaria tenuissima narnavirus 1 (AtNV1; Nerva et al. 2019c). In all previous studies, the authors classified such viruses as narnavirus segments encoding RdRps, but did not notice that the sequence alignment shows that the central domain carrying the GDD signature triplet present in the palm domain is missing, raising the questions if these are indeed functional RdRp encoding segments. On the contrary, such lack of GDD motif was recently noticed in a Magnaporte oryzae narna-like sequence (Lin et al. 2020). In the case of AtNV1 and CtNV1, the isolates that harbor them do not have any other RdRp coinfecting them (Nerva et al. 2019c), which therefore seems to exclude the occurrence of a typical RdRp (with a conserved palm domain) that replicates them in trans. Given that all these isolates cluster phylogenetically, and some of them are fairly abundant in their libraries, we would exclude the possibility that they all are assembled contigs truncated at the 3′ end. Interestingly, some of these newly characterized viruses accumulate a much higher amount of minus strand RNA than plus strand (Supplementary Fig. S17). A further possibility is that this incomplete RdRp-encoded protein is functionally complemented by another viral-encoded protein not conserved enough to be detected by our pipeline.
Finally, the third clade of narnavirus-like sequences that we newly characterized derives from our initial inspection of ORFan-encoding sequences of likely viral origin; therefore, we name this new group of viruses orfanplasmovirus. We identified this GDD carrying clade through the analysis of ORFans proteins encoded by contigs that had abundant reads mapping both in plus sense and minus sense RNA. Surprisingly, phylogenetic analysis assigns this clade of new viral segments to a basal deep branch that stems at the origin of leviviruses. This group of virus segments is widespread and abundant in our collection, with a correlation to the oomycetes-fungal prevalence. In this case, our alignment with distantly related levi-like and some distantly related narna-like viruses shows some confident alignments of the A, B, and C palm subdomains, with the D, G and GDD conserved residues in each of the three subdomain, respectively (te Velthuis 2014).
Regarding the closely related Botourmiaviridae, our addition of more than ninety putative new species suggests some new genera outside the three genera currently recognized (Ayllón et al. 2020). Furthermore, our tree suggests that plant ourmiaviruses are derived from invertebrate ourmia-like viruses, and not from fungal ourmiaviruses as previously hypothesized (Rastgou et al. 2009).
4.3. New plus-stranded genomic associations
Other plus-stranded RNA viruses identified in our collection constitute interesting new previously undescribed virus lineages in the virga/tobamo/alpha-like group of viruses (corresponding to the new Order Martellivirales). They are, in fact, sufficiently abundant and with a high read coverage to infer their existence as mycoviruses. In specific, we propose the name vivivirus (from virga-virga virus) for an at least bi-segmented virus group, which has one segment containing the methyltransferase and helicase, and the other the RdRp domain and sometimes a second methyltransferase domain. This association is also confirmed by analyzing 5′ and 3′ untranslated region (UTR) sequences between the two putative viral segments (in case of putative full-length sequences). In fact, PvLaVivivirus1 RNA1 and RNA2 share high conservation over the first 60 nt both at the 5′ and 3′ ends, whereas PvLaVivivirus4 RNA1 and RNA2 share conservation only at the 5′ end (Fig. 2C). The lack of conservation observed in PvLaVivivirus2 and 3 is likely due to their genomic sequence incompleteness, given the low virus coverage.
The other interesting case we want to highlight is the virga-like virus PvLaV-LV1, a long contig encoding a single ORF that stems from a very deep branch in the phylogenetic tree, without a clear assignment to a specific cluster. This virus is the most abundant and prevalent virga-like virus among the different libraries and is indeed a good candidate for a possible infectious clone to be used to verify its biological properties, because virga-like viruses are easily manageable through reverse genetic approaches.
4.4. Further biodiversity of negative-stranded mycoviruses
Negative strand viruses are a relatively recent discovery in fungi, and only a few of them have been so far characterized through virus purification and whole genome characterization (Liu et al. 2014). Our work allowed us to add new viruses to different orders of this viral group and to highlight some unique features that are at the base of the proposal for new taxa. In the Bunyavirales order, we characterize a new RdRp (Pvabunyaarenalike1) that is highly prevalent and abundant in all the libraries and stems from a very deep basal branch, which should likely warrant a new family status, as soon as more members of this family are unveiled. In the Mononegavirales, we added two putative mycoviruses close to those already characterized in the family Mymonaviridae (Jiang et al. 2019) and a rhabdo-like putative mycovirus distantly related to mostly invertebrate viruses. The most surprising result in the Mononegavirales was the characterization of the genome organization of some complete and putatively incomplete genomes coding for two ORFs. The largest ORF codes for a conserved RdRp, whereas the smallest probably codes for the Nc protein, since it aligns with Ncs of other mymonaviruses (Fig. 4C). Interestingly, and rather uniquely in the Mononegavirales, the two ORFs are in opposite sense orientation with a short intergenic region of 50 nt. Since homologues of these viruses are present in filamentous fungi axenically isolated from grapevine (Nerva et al. 2019c), we are confident that this is a new clade of negative strand mycoviruses awaiting taxonomic classification, for which we propose the name mononegaambivirus.
Finally, we reported here for the first time the abundant presence of yue/qin-like mycoviruses. Such groups of viruses have been recently described in invertebrates only (Shi et al. 2016) and have been taxonomically placed in two distinct orders: the Muvirales and the Goujianvirales. They are likely accompanied by a putative Nc encoded by a second RNA segment, but in our case, we could not reliably retrieve such segments even if a number of possible candidates are present in the list of ORFan virus-like contigs.
4.5. New mycovirus in the dsRNA virus groups
PVaTotilike1 and 2 are rather abundant in most libraries and are most closely related to viruses recently characterized in Phythium polare and Pythium splendens (Shiba et al. 2019), with conserved genomic features; therefore these two viruses are likely true P. viticola full-length mycoviruses. PVaTotilike3, 4, and 5 share the characteristic of having a much higher number of reads mapping to the negative strands compared with the positive strand, contrary to all the other dsRNA genomes characterized in our study (Supplementary Fig. S14). They align with genomes of recently characterized dsRNA viruses from invertebrates, such as Beihai searmid crab virus 7 and Hubei toti-like virus 5 (Shi et al. 2016), but only PVaTotilike5 carries the RdRp motif. PVaTotilike3 and 4 align to each other and with a third protein encoded by a viral segment from a still unpublished B. lactucae work (GenBank accession: QIP68022.1). We here hypothesize that either PVaTotilike3 or 4 contigs could be associated to PVaTotilike5 and another RdRp-carrying contig is missing; our attempt to confirm the association in single samples of the three contigs, was inconclusive, since the three segments are present in all the samples we tested.
PVadsRNA1 is also a very abundant virus widespread in all the libraries we have sequenced. It stems from a long branch basal to a group of dsRNA viruses that includes the botybirnaviruses. This virus is likely hosted by P. viticola since, the closest hit in the database is a still unpublished virus present in B. lactucae (QIP68007.1) and, the number of reads mapping on the contig is comparable to the one mapping to oomycetes sequences (Supplementary Fig. S16), adding this virus to those that can be confidently assigned to P. viticola.
5. Conclusions
In summary, this work is, to our knowledge, the largest collection of putative mycoviruses so far characterized in a single study and greatly expands the view on mycovirus biodiversity and their contribution to evolution of RNA viruses. Given the vast biodiversity of viruses characterized here and, despite our searches for conserved representative Rep proteins, it was surprising not to find any virus related to the ssDNA mycoviruses recently described in fungi (Yu et al. 2010).
Supplementary data
Supplementary data are available at Virus Evolution online.
Supplementary Material
Acknowledgements
The authors thank Simona Abbà and Marta Vallino for valuable discussions and comments on the manuscript. Particular thanks to Vara y Pulgar C. B. (Jerez) and wineries Recaredo (Penedés), Pradorey-Real Sitio de Ventosilla (Ribera del Duero), and Bodegas Roda (La Rioja) for providing grapevine samples used in this study.
Funding
This work was fully supported financially by VIROPLANT, a project that received funding from the European Union’s Horizon H2020 Research and Innovation Program, grant agreement number 773567.
Conflict of interest: None declared.
Contributor Information
M Chiapello, Institute for Sustainable Plant Protection, CNR, Strada delle Cacce 73, Torino 10135, Italy.
J Rodríguez-Romero, Centro de Biotecnología y Genómica de Plantas, Universidad Politécnica de Madrid (UPM)-Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Campus de Montegancedo, Pozuelo de Alarcón, Madrid 28223, Spain; Departamento de Biotecnología-Biología Vegetal, Escuela Técnica Superior de Ingeniería Agronómica, Alimentaria y de Biosistemas, Universidad Politécnica de Madrid (UPM), Madrid 28040, Spain.
M A Ayllón, Centro de Biotecnología y Genómica de Plantas, Universidad Politécnica de Madrid (UPM)-Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Campus de Montegancedo, Pozuelo de Alarcón, Madrid 28223, Spain; Departamento de Biotecnología-Biología Vegetal, Escuela Técnica Superior de Ingeniería Agronómica, Alimentaria y de Biosistemas, Universidad Politécnica de Madrid (UPM), Madrid 28040, Spain.
M Turina, Institute for Sustainable Plant Protection, CNR, Strada delle Cacce 73, Torino 10135, Italy.
References
- Akopyants N. S. et al. (2016) ‘A Narnavirus in the Trypanosomatid Protist Plant Pathogen Phytomonas serpens’, Genome Announcements, 4: e00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayllón M. A. et al. (2020) ‘ICTV Virus Taxonomy Profile: Botourmiaviridae’, Journal of General Virology, 101: 454–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh B. et al. (2010) ‘Phage Therapy for Plant Disease Control’, Current Pharmaceutical Biotechnology, 11: 48–57. [DOI] [PubMed] [Google Scholar]
- Brauer V. S. et al. (2019) ‘Antifungal Agents in Agriculture: Friends and Foes of Public Health’, Biomolecules, 9: 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser F. P., Salzberg S. L. (2019) ‘Pavian: Interactive Analysis of Metagenomics Data for Microbiome Studies and Pathogen Identification’, Bioinformatics, 36: 1303–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brilli M. et al. (2018) ‘A Multi-Omics Study of the Grapevine-Downy Mildew (Plasmopara viticola) Pathosystem Unveils a Complex Protein Coding- and Noncoding-Based Arms Race during Infection’, Scientific Reports, 8: 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruenn J. A., Warner B. E., Yerramsetty P. (2015) ‘Widespread Mitovirus Sequences in Plant Genomes’, PeerJournal, 3: e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Bushnell B., Rood J., Singer E. (2017) ‘BBMerge–Accurate Paired Shotgun Read Merging via Overlap’, PLoS One, 12: e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G. H., Hillman B. I. (2013), ‘Phytophthora Viruses’, Advances in Virus Research, 86: pp. 327–50. [DOI] [PubMed] [Google Scholar]
- Cai G. H. et al. (2013) ‘A New Virus from the Plant Pathogenic Oomycete Phytophthora infestans with an 8 kb dsRNA Genome: The Sixth Member of a Proposed New Virus Genus’, Virology, 435: 341–9. [DOI] [PubMed] [Google Scholar]
- Cai G. et al. (2019. a) ‘PiRV-2 Stimulates Sporulation in Phytophthora infestans’, Virus Research, 271: 197674. [DOI] [PubMed] [Google Scholar]
- Cai G. et al. (2012) ‘A Member of the Virus Family Narnaviridae from the Plant Pathogenic Oomycete Phytophthora infestans’, Archives of Virology, 157: 165–9. [DOI] [PubMed] [Google Scholar]
- Cai G. et al. (2019. b) ‘Phytophthora infestans RNA Virus 2, a Novel RNA Virus from Phytophthora infestans, Does Not Belong to Any Known Virus Group’, Archives of Virology, 164: 567–72. [DOI] [PubMed] [Google Scholar]
- Chandler J. A., Liu R. M., Bennett S. N. (2015) ‘RNA Shotgun Metagenomic Sequencing of Northern California (USA) Mosquitoes Uncovers Viruses, Bacteria, and Fungi’, Frontiers in Microbiology, 06: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon J. et al. (2019) ‘Novel RNA Viruses Associated with Plasmodium vivax in Human Malaria and Leucocytozoon parasites in Avian Disease’, PLoS Pathogens, 15: e1008216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiapello M. et al. (2020) ‘Putative New Plant Viruses Associated with Plasmopara viticola-Infected Grapevine Samples’, Annals of Applied Biology, 176: 180–91. [Google Scholar]
- Dagostin S. et al. (2011) ‘Are There Alternatives to Copper for Controlling Grapevine Downy Mildew in Organic Viticulture?’, Crop Protection, 30: 776–88. [Google Scholar]
- Danovaro R. et al. (2011) ‘Marine Viruses and Global Climate Change’, FEMS Microbiology Reviews, 35: 993–1034. [DOI] [PubMed] [Google Scholar]
- DeRisi J. L. et al. (2019) ‘An Exploration of Ambigrammatic Sequences in Narnaviruses’, Scientific Reports, 9: 17982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan A. M. et al. (2020) ‘A Case for a Negative-Strand Coding Sequence in a Group of Positive-Sense RNA Viruses’, Virus Evolution, 6: veaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaire L., Pagán I., Ayllón M. A. (2016) ‘Characterization of Botrytis Cinerea Negative-Stranded RNA Virus 1, a New Mycovirus Related to Plant Viruses, and a Reconstruction of Host Pattern Evolution in Negative-Sense ssRNA Viruses’, Virology, 499: 212–8. [DOI] [PubMed] [Google Scholar]
- Dussert Y. (2016) ‘Draft Genome Sequence of Plasmopara viticola, the Grapevine Downy Mildew Pathogen’, Microbiology Resource Announcements, 4: e00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena G. et al. (2019) ‘Microbiota of Grapevine Woody Tissues with or without Esca-Foliar Symptoms in Northeast Spain’, Phytopathologia Mediterranea, 57: 425–38. [Google Scholar]
- Espino-Vázquez A. N. et al. (2020) ‘Narnaviruses: Novel Players in Fungal–Bacterial Symbioses’, The ISME Journal, 14: 1743–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessler C., Pertot I., Perazzolli M. (2011) ‘Plasmopara viticola: A Review of Knowledge on Downy Mildew of Grapevine and Effective Disease Management’, Phytopathologia Mediterranea, 50: 3–44. [Google Scholar]
- Gillings M. R., Tesoriero L. A., Gunn L. V. (1993) ‘Detection of Double-Stranded-RNA and Virus-like Particles in Australian Isolates of Pythium irregulare’, Plant Pathology, 42: 6–15. [Google Scholar]
- Goertz G. P. et al. (2019) ‘Mosquito Small RNA Responses to West Nile and Insect-Specific Virus Infections in Aedes and Culex Mosquito Cells’, Viruses-Basel, 11: 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G. et al. (2011) ‘Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome’, Nature Biotechnology, 29: 644–U130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasse W., Spring O. (2015) ‘Occurrence and Genetic Diversity of the Plasmopara halstedii Virus in Sunflower Downy Mildew Populations of the World’, Fungal Biology, 119: 170–8. [DOI] [PubMed] [Google Scholar]
- Grasse W. et al. (2013) ‘Plasmopara halstedii Virus Causes Hypovirulence in Plasmopara halstedii, the Downy Mildew Pathogen of the Sunflower’, Fungal Genetics and Biology, 57: 42–7. [DOI] [PubMed] [Google Scholar]
- Heller-Dohmen M. et al. (2011) ‘The Nucleotide Sequence and Genome Organization of Plasmopara halstedii Virus’, Virology Journal, 8: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman B. I., Cai G. (2013) ‘The Family Narnaviridae: Simplest of RNA Viruses’, Advances in Virus Research, 86: 149–76. [DOI] [PubMed] [Google Scholar]
- Holmes E. C., Duchene S. (2019) ‘Can Sequence Phylogenies Safely Infer the Origin of the Global Virome?’, mBio, 10: e00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O. et al. (2007) ‘The Grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla’, Nature, 449: 463–U5. [DOI] [PubMed] [Google Scholar]
- Jiang D. et al. (2019) ‘ICTV Virus Taxonomy Profile: Mymonaviridae’, Journal of General Virology, 100: 1343–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki K., Halvorson H. O. (1971) ‘Appearance of a New Species of Ribonucleic Acid during Sporulation in Saccharomyces cerevisiae’, Journal of Bacteriology, 105: 826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaefer S. et al. (2019) ‘Re-Assessing the Diversity of Negative Strand RNA Viruses in Insects’, PLoS Pathogens, 15: e1008224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2020) ‘Global Organization and Proposed Megataxonomy of the Virus World’, Microbiology and Molecular Biology Reviews, 84: e00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane J. R. et al. (2018) ‘Thirteen Decades of Antimicrobial Copper Compounds Applied in Agriculture’, Agronomy for Sustainable Development, 28. [Google Scholar]
- Langmead B., Salzberg S. L. (2012) ‘Fast Gapped-Read Alignment with Bowtie 2’, Nature Methods, 9: 357–U54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. H. et al. (2019) ‘Two Novel Fungal Negative-Strand RNA Viruses Related to Mymonaviruses and Phenuiviruses in the Shiitake Mushroom (Lentinula edodes)’, Virology, 533: 125–36. [DOI] [PubMed] [Google Scholar]
- Lin Y. et al. (2020) ‘A Novel Narnavirus from the Plant-Pathogenic Fungus Magnaporthe oryzae’, Archives of Virology, 165: 1235–40. [DOI] [PubMed] [Google Scholar]
- Liu L. et al. (2014) ‘Fungal Negative-Stranded RNA Virus That is Related to Bornaviruses and Nyaviruses’, Proceedings of the National Academy of Sciences of Sciences of the United States of America, 111: 12205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F. et al. (2019) ‘The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019’, Nucleic Acids Research, 47: W636–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez L. M. et al. (2007) ‘A Virus in a Fungus in a Plant: Three-Way Symbiosis Required for Thermal Tolerance’, Science, 315: 513–5. [DOI] [PubMed] [Google Scholar]
- Marzano S.-Y. L., Domier L. L. (2016) ‘Novel Mycoviruses Discovered from Metatranscriptomics Survey of Soybean Phyllosphere Phytobiomes’, Virus Research, 213: 332–42. [DOI] [PubMed] [Google Scholar]
- Marzano S.-Y. L. et al. (2016) ‘Identification of Diverse Mycoviruses through Metatranscriptomics Characterization of the Viromes of Five Major Fungal Plant Pathogens’, Journal of Virology, 90: 6846–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medd N. C. et al. (2018) ‘The Virome of Drosophila Suzukii, an Invasive Pest of Soft Fruit’, Virus Evolution, 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A., Pfeiffer W., Schwartz T. (2010), ‘Creating the CIPRES Science Gateway for inference of large phylogenetic’, 2010 Gateway Computing Environments Workshop (GCE), pp. 1–8. New Orleans, LA: IEEE.
- Milne I. et al. (2013) ‘Using Tablet for Visual Exploration of Second-Generation Sequencing Data’, Briefings in Bioinformatics, 14: 193–202. [DOI] [PubMed] [Google Scholar]
- Musetti R. et al. (2007) ‘Antifungal Activity of Diketopiperazines Extracted from Alternaria alternata against Plasmopara viticola: An Ultrastructural Study’, Micron, 38: 643–50. [DOI] [PubMed] [Google Scholar]
- Musetti R. et al. (2006) ‘Inhibition of Sporulation and Ultrastructural Alterations of Grapevine Downy Mildew by the Endophytic Fungus Alternaria alternata’, Phytopathology®, 96: 689–98. [DOI] [PubMed] [Google Scholar]
- Nerva L. et al. (2016) ‘Multiple Approaches for the Detection and Characterization of Viral and Plasmid Symbionts from a Collection of Marine Fungi’, Virus Research, 219: 22–38. [DOI] [PubMed] [Google Scholar]
- Nerva L. et al. (2019. a) ‘Mycoviruses Mediate Mycotoxin Regulation in Aspergillus ochraceus’, Environmental Microbiology, 21: 1957–68. [DOI] [PubMed] [Google Scholar]
- Nerva L. et al. (2019. b) ‘Biological and Molecular Characterization of Chenopodium quinoa Mitovirus 1 Reveals a Distinct Small RNA Response Compared to Those of Cytoplasmic RNA Viruses’, Journal of Virology, 93: e01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerva L. et al. (2019. c) ‘Isolation, Molecular Characterization and Virome Analysis of Culturable Wood Fungal Endophytes in Esca Symptomatic and Asymptomatic Grapevine Plants’, Environmental Microbiology, 21: 2886–904. [DOI] [PubMed] [Google Scholar]
- Nibert M. L. et al. (2018) ‘Evidence for Contemporary Plant Mitoviruses’, Virology, 518: 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolopoulou-Stamati P. et al. (2016) ‘Chemical Pesticides and Human Health: The Urgent Need for a New Concept in Agriculture’, Frontiers in Public Health, 4: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaki H. et al. (2016) ‘Multiple Virus Infection in a Single Strain of Fusarium poae Shown by Deep Sequencing’, Virus Genes, 52: 835–47. [DOI] [PubMed] [Google Scholar]
- Pertot I. et al. (2017) ‘A Critical Review of Plant Protection Tools for Reducing Pesticide Use on Grapevine and New Perspectives for the Implementation of IPM in Viticulture’, Crop Protection, 97: 70–84. [Google Scholar]
- Picarelli M. A. S. C. et al. (2019) ‘Extreme Diversity of Mycoviruses Present in Isolates of Rhizoctonia solani AG2-2 LP from Zoysia japonica from Brazil’, Frontiers in Cellular and Infection Microbiology, 9: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poimala A., Vainio E. J. (2020) ‘Complete Genome Sequence of a Novel Toti-like Virus from the Plant-Pathogenic Oomycete Phytophthora cactorum’, Archives of Virology, 165: 1679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polizzotto R. et al. (2012) ‘A Polyphasic Approach for the Characterization of Endophytic Alternaria Strains Isolated from Grapevines’, Journal of Microbiological Methods, 88: 162–71. [DOI] [PubMed] [Google Scholar]
- Popham H. J. R., Nusawardani T., Bonning B. C. (2016), ‘Introduction to the Use of Baculoviruses as Biological Insecticides’, in Murhammer D. (ed.) Baculovirus and Insect Cell Expression Protocols, 3rd edn, New York, NY, Vol. 1350, pp. 383–92. [DOI] [PubMed] [Google Scholar]
- Rastgou M. et al. (2009) ‘Molecular Characterization of the Plant Virus Genus Ourmiavirus and Evidence of Inter-Kingdom Reassortment of Viral Genome Segments as Its Possible Route of Origin’, Journal of General Virology, 90: 2525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richaud A. et al. (2019) ‘Vertical Transmission in Caenorhabditis Nematodes of RNA Molecules Encoding a Viral RNA-Dependent RNA Polymerase’, Proceedings of the National Academy of Sciences of the United States of America, 116: 24738–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombel I. T. et al. (2002) ‘ORF-FINDER: A Vector for High-Throughput Gene Identification’, Gene, 282: 33–41. [DOI] [PubMed] [Google Scholar]
- Roossinck M. J. (2011) ‘The Good Viruses: Viral Mutualistic Symbioses’, Nature Reviews Microbiology, 9: 99–108. [DOI] [PubMed] [Google Scholar]
- Ryan F. P. (2009) ‘An Alternative Approach to Medical Genetics Based on Modern Evolutionary Biology. Part 1: Mutation and Symbiogenesis’, Journal of the Royal Society of Medicine, 102: 272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai S. et al. (2018) ‘A Novel Non-Segmented Double-Stranded RNA Virus from an Arctic Isolate of Pythium polare ’, Virology, 522: 234–43. [DOI] [PubMed] [Google Scholar]
- Shi M. et al. (2016) ‘Redefining the Invertebrate RNA Virosphere’, Nature, 540: 539–43. [DOI] [PubMed] [Google Scholar]
- Shiba K. et al. (2018) ‘Genome Sequence of a Novel Partitivirus Identified from the Oomycete Pythium nunn’, Archives of Virology, 163: 2561–3. [DOI] [PubMed] [Google Scholar]
- Shiba K. et al. (2019) ‘A Novel Toti-like Virus from a Plant Pathogenic Oomycete Globisporangium splendens’, Virology, 537: 165–71. [DOI] [PubMed] [Google Scholar]
- Sievers F. et al. (2011) ‘Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega’, Molecular Systems Biology, 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. et al. (2017) ‘Virus Taxonomy in the Age of Metagenomics’, Nature Reviews Microbiology, 15: 161–8. [DOI] [PubMed] [Google Scholar]
- Sutela S., Poimala A., Vainio E. J. (2019) ‘Viruses of Fungi and Oomycetes in the Soil Environment’, FEMS Microbiology Ecology, 95: fiz119. [DOI] [PubMed] [Google Scholar]
- Suttle C. A. (2007) ‘Marine Viruses - Major Players in the Global Ecosystem’, Nature Reviews Microbiology, 5: 801–12. [DOI] [PubMed] [Google Scholar]
- Tao W. C. et al. (2014) ‘A New Alternaria Species from Grapevine in China’, Mycological Progress, 13: 1119–25. [Google Scholar]
- te Velthuis A. J. W. (2014) ‘Common and Unique Features of Viral RNA-Dependent Polymerases’, Cellular and Molecular Life Sciences, 71: 4403–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifinopoulos J. et al. (2016) ‘W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis’, Nucleic Acids Research, 44: W232–W35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turina M., Rostagno L. (2007) ‘Virus-Induced Hypovirulence in Cryphonectria parasitica: Still an Unresolved Conundrum’, Journal of Plant Pathology, 89: 165–78. [Google Scholar]
- Vandersteegen K. et al. (2013) ‘Romulus and Remus, Two Phage Isolates Representing a Distinct Clade within the Twortlikevirus Genus, Display Suitable Properties for Phage Therapy Applications’, Journal of Virology, 87: 3237–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco R. et al. (2007) ‘A High Quality Draft Consensus Sequence of the Genome of a Heterozygous Grapevine Variety’, PLoS One, 2: e1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco L. et al. (2019) ‘Viromes in Xylariaceae fungi infecting avocado in Spain’, Virology, 532: 11–21. [DOI] [PubMed] [Google Scholar]
- Wang X. et al. (2019) ‘Phage Combination Therapies for Bacterial Wilt Disease in Tomato’, Nature Biotechnology, 37: 1513–20. [DOI] [PubMed] [Google Scholar]
- Wen A. M., Steinmetz N. F. (2016) ‘Design of Virus-Based Nanomaterials for Medicine, Biotechnology, and Energy’, Chemical Society Reviews, 45: 4074–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowski M., Wickner R. B. (1984) ‘2 New Double-Stranded-RNA Molecules Showing Nonmendelian Inheritance and Heat Inducibility in Saccharomyces cerevisiae’, Molecular and Cellular Biology, 4: 181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y. I. et al. (2018) ‘Origins and Evolution of the Global RNA Virome’, mBio, 9: e02329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y. I. et al. (2019) ‘Reply to Holmes and Duchene, “Can Sequence Phylogenies Safely Infer the Origin of the Global Virome?”: Deep Phylogenetic Analysis of RNA Viruses is Highly Challenging but Not Meaningless’, mBio, 10: e00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D. E., Lu J., Langmead B. (2019) ‘Improved Metagenomic Analysis with Kraken ’, Genome Biology, 20: 2'. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L. et al. (2017) ‘Genome Sequence of Plasmopara viticola and Insight into the Pathogenic Mechanism’, Scientific Reports, 7: 46553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi T., Yamashita S., Hibi T. (2003) ‘The Nucleotide Sequence and Genome Organization of Sclerophthora macrospora Virus A’, Virology, 311: 394–9. [DOI] [PubMed] [Google Scholar]
- Yokoi T. et al. (1999) ‘The Nucleotide Sequence and Genome Organization of Sclerophthora macrospora Virus B’, Virology, 264: 344–9. [DOI] [PubMed] [Google Scholar]
- Yu X. et al. (2010) ‘A Geminivirus-Related DNA Mycovirus That Confers Hypovirulence to a Plant Pathogenic Fungus’, Proceedings of the National Academy of Sciences of Sciences of the United States of America, 107: 8387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoll J., Verweij P. E., Melchers W. J. G. (2018) ‘Discovery and Characterization of Novel Aspergillus fumigatus Mycoviruses’, PLoS One, 13: e0200511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.