

Abstract
From 2013 onwards, the spread of novel H5N6 highly pathogenic avian influenza (HPAI) viruses in China has posed great threats to not only poultry industry but also human health. Since late-2016 in particular, frequent outbreaks of clade 2.3.4.4 H5N6 HPAI viruses among wild birds have promoted viral dissemination in South Korea, Japan, and European countries. In response to those trends, we conducted molecular genetic analysis of global clade 2.3.4.4 H5N6 viruses in order to characterize spatio-temporal patterns of viral diffusion and genetic diversity among wild birds and poultry. The clade 2.3.4.4 H5N6 viruses were classified into three groups (Group B, C, and D). During the cocirculation of Group C/D H5N6 viruses from 2013 to 2017, viral movements occurred between close or adjacent regions of China, Vietnam, South Korea, and Japan. In addition, viral migration rates from Guangdong and Hunan to multiple adjacent provinces seemed to have been highly supported by transmission routes (Bayes factors >100), suggesting that southern China was an epicenter for the spread of H5N6 viruses in poultry during that period. Since the introduction of H5N6 viruses originating in wild birds in late-2016, evolving H5N6 viruses have lost most previous genotypes (e.g. G1, G2, and G1.2), whereas some prevailing genotypes (e.g. G1.1, G1.1.b, and G3) in aquatic birds have been dominated, and in particular, the effective population size of H5N6 originating in wild birds dramatically increased; however, the population size of poultry-origin H5N6 viruses declined during the same period, indicating that wild bird migration might accelerate the genetic diversity of H5N6 viruses. Phylogeographic approaches revealed that two independent paths of H5N6 viruses into South Korea and Japan from 2016 to 2018 and provided evidence of Group B and Group C H5N6 viruses were originated from Europe and China, respectively, as regions located in the East Asia–Australian migration flyway, which accelerated the genetic variability and dissemination. Altogether, our study provides insights to examine time of origin, evolutionary rate, diversification patterns, and phylogeographical approach of global clade 2.3.4.4 H5N6 HPAI viruses for assessing their evolutionary process and dissemination pathways.
Keywords: influenza, highly pathogenic H5N6 viruses, phylogeography, genetic diversity, dissemination
1. Introduction
First identified in 1996 in Guangdong province of China, H5N1 highly pathogenic avian influenza (HPAI) viruses have subsequently spread to multiple continents (Xu et al. 1999) and sparked global concerns due to their high pathogenicity in poultry and high fatality rate in humans. As of August 2020, a total of 863 human infection with 455 deaths, for a case fatality rate (CFR) of approximately 53% (https://iris.wpro.who.int/handle/10665.1/14460). The global cocirculation of the H5 subtype viruses has promoted the emergence of hemagglutinin (HA) gene variants, including ten distinct clades (0–9), with some of these clades further divided into as many as four-digit subclades (Smith et al. 2015). In 2014, a new clade of H5Nx viruses (2.3.4.4) emerged, this clade viruses had acquired an unprecedented number of neuraminidase (NA) subtypes, including N2, N3, N6, and N8 (Smith et al. 2015; Yamaji et al. 2020), which were detected in domestic and wild birds in Asia, North America, and Europe. In addition, as H5N8 HPAI viruses had been spread to Europe and North America via migratory birds (Hill et al. 2015; Lee et al. 2015; Verhagen et al. 2015) in early 2014, multiple outbreaks of novel reassortant H5N2, H5N6, and H5N8 HPAI viruses of clade 2.3.4.4 in East Asia had been reported (Lee et al. 2014; Wu et al. 2014; Xu et al. 2015; Jiao et al. 2016).
Novel reassortant H5N6 viruses were reported as the dominant subtype in domestic birds in mainland China during 2013 to 2016 (Bi et al. 2016a). These H5N6 HPAI viruses contained the HA gene of H5Nx clade 2.3.4.4, the internal genes of H5N1 clade 2.3.2, and the NA gene from the H6N6 AIV (Yang et al. 2017). Besides, the internal genes of H5N6 HPAI viruses had continued to undergo complex reassortment with low pathogenic avian influenza (LPAI) H3, H6, H7, H9, and H10 subtype viruses circulating locally (Bi et al. 2016a; Du et al. 2017; Zhang et al. 2018), which triggered genetic diversity of H5N6 viruses in China. In April 2014, the first fatal human case of H5N6 HPAI virus infection was reported in Sichuan province of China (Pan et al. 2016). As of July 2020, a total of 24 human infections with H5N6 HPAI viruses had been reported in China, including seven deaths, for a CFR of ∼30% (https://iris.wpro.who.int/handle/10665.1/14460), posing public health threat.
By genetic classification, clade 2.3.4.4 H5 subtype viruses had evolved into four groups, including Group A, B, C, and D (Lee et al. 2016). The H5N6 HPAI viruses in China, Vietnam, and Laos during 2014–16 appeared into Group C and Group D (Wong et al. 2015; Bi et al. 2016a; Chu et al. 2016). Group A H5 subtype viruses comprised H5N1, H5N2, and H5N8 HPAI viruses, which were identified among poultry and wild birds in China, Korea, Japan, the United States, and some European countries during 2014–15 (Jeong et al. 2014; Lee et al. 2014, 2016). Novel reassortant Group B H5 subtype viruses had been detected in domestic ducks and wild birds in China since 2013. In early-2014, outbreaks of Group B H5N8 viruses were reported in South Korea (Fan et al. 2014; Lee et al. 2014, 2015), and subsequently, these Group B H5N8 viruses were disseminated by migratory birds across the globe to Europe, North America, and Africa beginning in late October 2016 (Global Consortium for H5N8 and Related Influenza Viruses 2016). Interestingly, there had been no reports of Group B H5N6 viruses before 2017; however, during the 2017–18 wintering season, the novel reassortant Group B H5N6 HPAI viruses were detected in South Korea (Kwon et al. 2018b). Subsequently, those novel H5N6 variants disseminated to Switzerland, Iran, Finland, Sweden, Germany, the United Kingdom, and Denmark (Poen et al. 2019; Yamaji et al. 2020). Throughout their spread, the internal genes of poultry- and wild birds-origin H5N6 HPAI viruses have further reassorted with Eurasian LPAIV gene pool (Beerens et al. 2019; Poen et al. 2019; Pohlmann et al. 2019), which suggested that the genetic dynamics and global transmission of H5N6 HPAI viruses pose grave concerns for public health. Besides, several studies had documented that enhanced virulence of H5N6 viruses in experimentally infected mammals (Kim et al. 2018; Kwon et al. 2018a; Marchenko et al. 2018; Peng et al. 2018), and many mammalian adaption markers were observed in H5 subtype viruses (Herfst et al. 2012; Zhao et al. 2017; Peng et al. 2018; Sun et al. 2020), thus implying their potential threat to human health.
The direction and speed of the spread of AIV is determined by several interdependent factors, including wild bird migration (Kwon et al. 2018c; Liu et al. 2018; Fusaro et al. 2019; Poen et al. 2019), the live poultry trade (Su et al. 2017; Li et al. 2018; Yang et al. 2020), climate (Fusaro et al. 2019), and human population density (Herrick, Huettmann, and Lindgren 2013; Bui et al. 2018). In previous study, we demonstrated that the dissemination of H5 and H7 subtype influenza viruses in China among the domestic poultry is likely associated with the intensity of the live poultry trade (Yang et al. 2020). However, within their regional circulation, H5N6 HPAI viruses have expanded along distinct geographical and evolutionary pathways, likely via long-distance migratory bird dispersal onto several continents, and further evolved by reassorting with local viruses (Takemae et al. 2017; Lee et al. 2018; Liu et al. 2018; Mellor et al. 2018; Xu 2018; Beerens et al. 2019; Poen et al. 2019; Pohlmann et al. 2019). Notably, in 2017, a human death in Fujian province of China caused by an H5N6 virus derived from Group B clade 2.3.4.4 H5N8 was confirmed (Chen et al. 2019). Therefore, investigating the evolutionary dynamics and dissemination pathways of H5N6 HPAI viruses among poultry and wild birds is urgently needed. To that purpose, we sought to unravel the molecular evolution of HPAI H5N6 viruses from 2013 to 2019 with phylogenic analysis examining time of origin, evolutionary rate, and diversification patterns. To examine the potential dissemination routes of global H5N6 HPAI viruses among wild birds and poultry, we followed a phylogeographic approach as well.
2. Materials and methods
2.1 Maximum likelihood phylogenies of the influenza A (H5N6) viruses
All the available full genomic sequences with the complete coding regions of influenza A (H5N6) viruses were downloaded from GISAID (http://www.gisaid.org/). The full genome sequences data set (sequences alignment was available on request) was then created. The downloaded gene sequences were aligned using the MAFFT (version 7.149) program (Katoh et al. 2002). Maximum likelihood (ML) phylogenies for the codon alignment of the full gene segments were estimated using the GTRGAMMA nucleotide substitution model in the RAxML (version 8.2) program (Stamatakis 2014). Node support was determined by non-parametric bootstrapping with 1,000 replicates. The phylogenetic tree was visualized in the FigTree (version 1.4.3) program (http://tree.bio.ed.ac.uk/software/figtree/). The genotypes of the influenza A(H5N6) viruses were nominated according to the combinations of lineages in segment trees and designated in line with Bi and colleagues’ research (Bi et al. 2016a).
2.2 Bayesian maximum clade credibility phylogeny and evolutionary dynamics analysis of the influenza A (H5N6) viruses
BEAST package (version 1.8.2) was used to estimate the time to the most recent common ancestor (tMRCA) and evolutionary rates. We estimated rates of evolutionary change (nucleotide substitution) in the HA and NA gene segments of the influenza A (H5N6) viruses. For efficiency of analysis, we focused on the influenza A (H5N6) viruses sampled at different times and locations, which were representative of phylogenetic diversity of the influenza A (H5N6) viruses. To ensure that these had sufficient temporal structure in HA and NA alignment for reliable rate estimation, we first performed a regression of root-to-tip genetic distances on the ML tree against exact sampling dates using the TempEst (Rambaut et al. 2016), and anomalous sequences were removed based on the estimated root-to-tip distance. To obtain a more robust rate estimate, we used the Bayesian Markov chain Monte Carlo (MCMC) method implemented in the BEAST package (version 1.8.2), employing the SRD06 nucleotide substitution model (Drummond et al. 2006), an uncorrected lognormal (UCLN) relaxed molecular clock model (Minin, Bloomquist, and Suchard 2008), and the Gaussian Markov random field (GMRF) Bayesian skyride coalescent tree prior (Minin, Bloomquist, and Suchard 2008). Multiple runs of MCMC method were combined using LogCombiner (version 1.8.3), utilizing 10,000,000,000 total steps for each set, with sampling every 10,000 steps. Convergence (i.e., effective sample sizes > 200) of relevant parameters was assessed using Tracer (version 1.6) (http://beast.bio.ed.ac.uk/). The posterior distribution of trees obtained from BEAST analysis (with 10% of runs removed as burn-in) was also used to obtain the Bayesian maximum clade credibility (MCC) tree for the HA and NA gene segments (Drummond et al. 2012). The BEAST package was used for the calculation of tMRCAs of each group.
Next, to estimate the population dynamics of H5N6 viruses in wild birds and poultry, we inferred H5N6 virus demographic history using the GMRF Bayesian Skyride plots. First, we performed a regression of root-to-tip genetic distances on the ML tree against exact sampling dates using the TempEst (Rambaut et al. 2016), which showed strong temporal signal (Supplementary Fig. S3). Then, we performed BEAST analysis on the 285 HA gene segments of H5N6 viruses in poultry, and 131 HA gene segments of H5N6 viruses in wild birds, with same parameters to estimate relative genetic diversity by excepting GMRF Bayesian skyride analysis. All phylogenetic trees visualized in Figtree with ascending mode order were provided in Supplementary Figs S9 and S10. We also estimate the substitution rates of the HA and NA genes of Group B, Group C, and Group D H5N6 viruses. We performed a regression of root-to-tip genetic distances on the ML tree against exact sampling dates using the TempEst (Rambaut et al. 2016), which showed strong temporal signal (Supplementary Figs S4 and S5). Next, we performed BEAST analysis on the HA and NA genes of Group B, Group C, and Group D H5N6 viruses, with same parameters to access the substitution rate. All phylogenetic trees visualized in Figtree with ascending mode order were provided in Supplementary Figs S11–S16.
2.3 Sequence data of phylogeographic analysis
We accessed all available HA genomic sequences of influenza A (H5N6) viruses during 2013–19 from GISAID Database. Duplicate entries and recombinant strains identified using RDP4 were removed (Martin et al. 2015). For efficiency of analysis, we focused on the influenza A (H5N6) viruses sampled at different times and locations. In order to focus on the viral dissemination in poultry and wild birds, it is important to exclude the genomic sequences of the H5N6 viruses derived from mammalian species, including human, cat, ferret, and swine. Identical sequences within the same year and region were removed and the resulting sequences were clustered using CD-HIT v4.6 (Li and Godzik 2006). To infer ancestor location and migration events, we found that clade 2.3.4.4 H5N6 viruses have evolved into three relatively groups, including Group B, C, and D. Group B H5N6 viruses emerged from clade 2.3.4.4 Group B H5N8 viruses. Hence, we conducted a separate analysis of Group B H5 subtype (H5N6 and H5N8) viruses and Group C/D H5N6 viruses, respectively. In order to ameliorate the potential sampling biases; subsequently, we randomly subsampled the database in a stratified manner to create a more equitable spatio-temporal distribution of the HA genome sequences of three group viruses. In detail, Group B H5N6 and H5N8 viruses during 2016–20 were subsampled to 216 sequences (1,704 nt), and Group C and D H5N6 viruses during 2013–20 were subsampled to 356 sequences (1,704 nt). Sequences from over-sampled years and provinces or countries were removed randomly so that there was at most six sequences per year and per locations, which can increase the evenness of sampling whilst retaining a wide range of sampling dates and locations. The discrete sampling locations of Group B H5 subtype viruses in this study include Bangladesh, China, Egypt, Germany, Italy, Japan, South Korea, Netherlands, Poland, Russia, and South Africa. The discrete sampling locations of Group C/D H5N6 viruses include China (including Chongqing, Fujian, Guangdong, Guangxi, Hubei, Hunan, Jiangsu, Jiangxi, Ningxia, Sichuan, Yunnan, and Zhejiang provinces), Vietnam, South Korea, and Japan. Spatial and temporal distribution of HA sequences of H5N6 and H5N8 viruses from each group used in this study before/after down-sampling were provided in Supplementary Figs S17–S20. Detailed information of subsampled HA gene sequences of the clade 2.3.4.4 Group B, C, and D H5 subtype viruses used in this study were provide in Supplementary Tables S6 and S7. To ensure that the relationship between the distances of sampling locations and migrations rates, we repeated the phylogeographic analysis after randomizing the locations assigned to each sequence (Supplementary Fig. S22).
2.4 Phylogeographic inference
Time-measured phylogenies were inferred using Bayesian discrete phylogeographic approach implemented in BEAST package (version 1.8.2) (Drummond et al. 2012). We first performed a regression of root-to-tip genetic distances on the ML tree against exact sampling dates using the TempEst (Rambaut et al. 2016), which showed strong temporal signal (Supplementary Fig. S2). Then, we used an UCLN relaxed molecular clock model (Drummond et al. 2006), the SRD06 nucleotide substitution have the highest negative log likelihood scores (Minin, Bloomquist, and Suchard 2008). Besides, a Bayesian stochastic search variable selection (BSSVS) model with asymmetric substitution was also used. For each independent dataset, multiple runs of MCMC method were combined using LogCombiner (version 1.8.3), utilizing 10,000,000,000 total steps for each set, with sampling every 10,000 steps. All phylogenetic trees visualized in Figtree with ascending mode order were provided in Supplementary Figs S7 and S8. Subsequently, we used SpreaD3 v0.9.6 to develop interactive visualizations of the dispersal process through time and to compute a Bayes factors (BFs) test to assess the support for significant individual transitions between distinct geographic locations. SpreaD3 takes a rate matrix file for location states generated under the BEAST analysis using the BSSVS procedure (Bielejec et al. 2016). The BF values >100 indicated robust statistical support. BF values >30 and ≤100 indicated very strong statistical support. BF values >10 and ≤30 indicated strong statistical support. BF values >3 and ≤10 indicated substantial statistical support. BF values <3 indicated poor statistical support (Lemey et al. 2009). We used R (https://www.r-project.org) and ArcGIS Desktop 10.4 software (http://www.esri.com/software/arcgis/arcgis-for-desktop/) to create plots showing results of BF tests.
3. Results
3.1 Distribution of H5N6 viruses
To better understand the evolution and prevalence of global clade 2.3.4.4 H5N6 HPAI viruses, 1225 HA gene sequences of H5N6 viruses from GISAID’s Database sampled at different times and locations together with other clade 2.3.4.4 H5 subtype viruses were studied by performing multiple sequence alignment and phylogenetic analysis (Katoh et al. 2002; Stamatakis 2014). In the ML tree of the HA gene, clade 2.3.4.4 H5 subtype viruses had evolved into four distinct genetic subclades. Following a previous study (Yamaji et al. 2020), four subclades were designated with Group A (a group represented by A/gyrfalcon/Washington/41088-6/2014), Group B (a group represented by A/Fujian-Sanyuan/21099/2017), Group C (a group represented by A/Hubei/29578/2016), and Group D (a group represented by A/Sichuan/26221/2014). Nearly all H5N6 viruses derived from clade 2.3.4.4 H5Nx and could be classified into Group B, Group C, and Group D (Fig. 1A). Although all H5N6 viruses in Group D were from Vietnam, Laos, and mainland China, Group C included the H5N6 gene sequences from those countries along with South Korea and Japan. A total of 28 human-origin H5N6 isolates were observed in Group C, whereas only one human-origin virus from Sichuan province of China emerged in Group D (Fig. 1A). HPAI H5N6 viruses were first identified from wild birds in late-2016; and subsequently, multiple outbreaks of H5N6 viruses among wild and domestic birds emerged in more than eleven European countries (Fig. 1B and C). Since mid-2017, H5N6 viruses in Group B had emerged in Asia (i.e. China, Bangladesh, South Korea, and Japan) and Europe (Greece, England, Switzerland, Italy, the Netherlands, Germany, Denmark, and et al.) (Fig. 1B and C). Only one human-origin Group B H5N6 virus designating A/Fujian-Shanyuan/21099/2017 was isolated from Fujian, China in 2017. All of the Group B H5N6 viruses from Bangladesh, South Korea, and Japan were isolated from domestic poultry, including chickens, ducks, and geese, whereas 46% of them were derived from wild birds (Fig. 1A; Supplementary Table S9). Besides, we also found that Group B H5N6 viruses from some Asian and European countries grouped together with European H5N8 viruses (Fig. 1A), which indicated that the cocirculation of H5N6 and H5N8 viruses occurred among both poultry and wild birds.
Figure 1.

Evolutionary history of clade 2.3.4.4 HAPI H5N6 viruses. (A) ML tree of HA gene of H5N6 viruses. Different subtypes, hosts and years are denoted by different markers. All branch lengths are scaled according to the numbers of substitutions per site (subs/site). The tree is rooted using clade 2.3.2 A/duck/Vietnam/LBM360c1-4-1/2013(H5N6), which was collected on 6 February 2013. (B) Global distribution of H5N6 viruses occurring in each country according to the OIE (https://www.oie.int). (C) The timeline of H5N6 viruses from 2013 to 2019. [1] represents Greece, Myanmar, Netherlands, and Philippines. [2] represents Denmark, Finland, Germany, Ireland, Netherlands, Sweden, Switzerland, United Kingdom.
3.2 Evolutionary dynamics of H5N6 viruses
Using a root-to-tip regression, our analysis of temporal structure revealed aspects of the clock-like structure of HA (n = 397, correlation coefficient = 0.85; R2 = 0.72) and NA (Group B, correlation coefficient = 0.92, R2 = 0.84; Group C, correlation coefficient = 0.92, R2 = 0.84; Group D, correlation coefficient = 0.73, R2 = 0.53) (Fig. 2A; Supplementary Fig. S5). The epidemic H5N6 HPAI viruses were classified into three groups. We estimated the times of origin of H5N6 viruses in each group with 95% highest probability density (HPD) as follows: Group B (November 2015–August 2016), Group C (January 2012–June 2013), and Group D (August 2012–May 2013) for HA; and Group B (March 2012–March 2014), Group C (December 2012–September 2013), and Group D (August 2011–April 2013) for NA (Fig. 2B; Table 1; Supplementary Figs S6 and S7). Those findings suggested that the tMRCAs of Group C (i.e. November 2012 for HA and May 2013 for NA) and Group D (i.e. January 2013 for HA and July 2012 for NA) were earlier than their counterparts in Group B (i.e. April 2016 for HA and March 2014 for NA) (Table 1), as well as that two parallel evolutionary pathways (i.e. Group C and D) of H5N6 viruses occurred in China. We also found that H5N6 viruses originating in wild birds mainly exhibited traits of Group B-W and Group C-W (in which “W” represents wild birds) (Fig. 2B). Such viruses in South Korea and Japan belonged to Group B-W and Group C-W, while Group B-W H5N6 viruses dominant in some European countries, South Korea, and Japan. The tMRCAs of the HA and NA of Group B-W were December 2016 (95% HPD, October 2016–February 2017) and January 2016 (95% HPD, November 2014–September 2016), respectively, whereas those of the HA and NA of Group C-W are March 2016 (95% HPD, December 2015–May 2016) and April 2016 (95% HPD, January 2016–July 2016), also respectively (Table 1; Fig. 2B; Supplementary Figs S6 and S14–S16). These results imply that the introduction of H5N6 viruses originating in wild birds might have occurred at least twice in South Korea and Japan.
Figure 2.

Time-scaled evolution of HA gene of H5N6 viruses. (A) Analysis of root-to-tip divergence against sampling date for HA gene segment (n = 397). (B) A MCC tree of the HA sequence of H7N9 viruses sampled in China (n = 397) is shown, H5N6 viruses from different countries are denoted by different colors. Shaded bars represent the 95% highest probability distribution for the age of each node.
Table 1.
tMRCA estimates of HA and NA genes of clade 2.3.4.4 H5N6 viruses in different groups.
| Group | HA gene |
NA gene |
||||
|---|---|---|---|---|---|---|
| Median tMRCA | Low 95% HPD | Upper 95% HPD | Median tMRCA | Low 95% HPD | Upper 95% HPD | |
| Group B |
2,016.256 (2 April 2016) |
2,015.8707 (14 November 2015) |
2,016.6652 (30 August 2016) |
2,013.0982 (6 February 2013) |
2,012.2069 (14 March 2012) |
2,014.1753 (3 March 2014) |
| Group C |
2012.8354 (1 November 2012) |
2012.0675 (25 January 2012) |
2013.4603 (16 June 2013) |
2013.3722 (14 May 2013) |
2012.9281 (4 December 2012) |
2013.7434 (14 September 2013) |
| Group D |
2,013.0392 (15 January 2013) |
2,012.6316 (18 August 2012) |
2,013.3811 (18 May 2013) |
2,012.5806 (29 July 2012) |
2,011.6435 (22 August 2011) |
2,013.3083 (21 April 2013) |
| Group B-W |
2,016.9598 (16 December 2016) |
2,016.7818 (12 October 2016) |
2,017.1104 (10 February 2017) |
2,016.0561 (21 January 2016) |
2,014.8840 (19 November 2014) |
2,016.6888 (8 September 2016) |
| Group C-W |
2,016.2034 (14 March 2016) |
2,015.9949 (29 December 2015) |
2,016.4078 (27 May 2016) |
2,016.2954 (17 April 2016) |
2,016.0510 (19 January 2016) |
2,016.5032 (1 July 2016) |
tMRCA, the time to the most recent common ancestor; HPD, highest probability density.
In addition, we estimated that evolutionary rates in Group B were 7.376 × 10−3 (95% HPD, 5.001 × 10−3 to 9.981 × 10−3 substitutions/site/year) for HA and 7.292 × 10−3 (95% HPD, 5.57 × 10−3 to 9.096 × 10−2 substitutions/site/year) for NA, which were higher than that of the HA and NA genes of Group C and D H5N6 viruses (Supplementary Table S8; Figs S6 and S14–S16) and within the expected range for RNA viruses (Duffy et al. 2008). To estimate the population dynamics of H5N6 viruses in wild birds and poultry, we inferred H5N6 virus demographic history using the GMRF Bayesian Skyride plots, which revealed the changes in genetic diversity of H5N6 viruses (Supplementary Figs S3 and S9 and S10). In poultry, the effective population size fluctuated slightly from early-2013 to mid-2013, and from mid-2013 to early-2014, genetic diversity dramatically increased and stably maintained (Fig. 3B). However, a sharp decrease of the effective population size occurred during early-2015 to mid-2015, while from mid-2015 to 2017, the genetic diversity decreased slowly, and after 2017, the effective population size remained constant (Fig. 3B). In wild birds, the genetic diversity increased sharply from early-2017 to mid-2017 and declined steadily until 2019 (Fig. 3C), which was different from that of the poultry-origin H5N6 viruses during the same period.
Figure 3.

Evolutionary dynamics of H5N6 viruses during 2013–19. (A) Diversity of the genotypes of H5N6 viruses during 2013–19. There are 46 genotypes in the H5N6 viruses across the globe. GMRF Bayesian Skyride plot of HA genes of clade 2.3.4.4 H5Nx viruses from poultry and wild birds. A GMRF Bayesian Skyride analysis of HA gene of clade 2.3.4.4 H5N6 viruses from poultry (B) and wild birds (C) to display changes in the effective population size over time. The solid red line indicates the median value, and the shaded red area represents the 95% highest posterior density of genetic diversity estimates.
3.3 Genotypic analyses of H5N6 viruses
We conducted phylogenic analysis of internal genes and classified them into different lineages according to the nucleotide similarity of less than 98% and bootstrap values exceeding 80%. In line with Bi’s classification, we then designated 46 distinct genotypes (Supplementary Figs S1 and S23; Table S1). From 2013 to 2014, 39.32% and 40.17% of the H5N6 viruses belonged to G1 and G2, respectively, whereas only 9.87% of them were classified into G1.1. However, a sharp increase in the number of genotypes (n = 326) occurred in 2015 (Fig. 3A; Supplementary Table S4), and G1.1 (n = 326) became the dominant genotype during that period, followed by G1 (n = 79), G2 (n = 33), and G1.2 (n = 79) (Fig. 3A; Supplementary Table S4). From 2016 to 2017, the evolving H5N6 viruses lost most their previous genotypes, including G1, G2, and G1.1, and a new G1.1.b (n = 222) became the dominant genotype from 2016 to 2018 in China (Fig. 4). After the outbreaks of H5N6 viruses among wild birds, G1.1.b and G3 genotypes were widely prevalent in wild birds populations from 2017 to 2019 (Supplementary Table S1; Fig. 3A). In particular, G1.1, G1.1.b, and G1.1.c genotypes H5N6 viruses originating in wild birds had cocirculated in Japan in late-2016, and the G3 genotype had circulated there since late-2017 (Figs 3A and 4), which suggested that two independent genotypes of H5N6 viruses originating in wild birds were introduced into Asia. Together, those findings indicated that the viral exchange occurred between H5N6 viruses originating in domestic versus wild birds and that some genotypes, including G1.1, G1.1.b, and G3, might have gained greater viral fitness in birds. In addition, we also found that the main hosts of H5N6 viruses were the aquatic poultry and wild birds (Supplementary Table S5; Fig. S21). The aquatic poultry, especially ducks, exhibited the highest number of genotypes of H5N6 viruses compared with other hosts during 2015, which indicated that aquatic poultry might contributed to the reassortment of H5N6 viruses. After 2016, the number of genotypes of H5N6 viruses in chicken decreased; however, the number of genotypes of wild birds-origin H5N6 viruses increased (Supplementary Table S5; Fig. S21), which indicated that wild bird migration accelerates the genetic diversity of H5N6 viruses.
Figure 4.

Timeline of emergence of different genotypes of global H5N6 viruses during 2013–19. Green bars represent duration of circulation of H5N6 viruses during 2013–19.
3.4 The spread of H5N6 viruses
We reconstructed the worldwide spatial dispersal networks of H5N6 HPAI viruses in domestic and wild birds. Analyzing transmission routes with BF values exceeding 3 were selected for analysis (Lemey et al. 2009). Since Group B clade 2.3.4.4 H5N6 viruses emerged from clade 2.3.4.4 H5N8 viruses, we conducted separate phylogeography analysis of Group B clade 2.3.4.4 H5N6 viruses together with Group B H5N8 viruses during 2016–20 and Group C/D H5N6 viruses during 2013–18 (Supplementary Figs S17–S20). There were eleven and fifteen discrete sampling locations in Group B H5 subtype viruses and Group C/D H5N6 viruses, respectively. Among the results, seven and twenty-four significant transmission routes were observed in Group B H5N6 viruses and Group C/D H5N6 viruses (Supplementary Table S2 and S3), respectively. The lineage movements of Group C/D H5N6 viruses occurred between adjacent or close provinces of China or countries, and the lineage transition rates between different locations were inversely related to the distance (Figs 5 and 7A), indicating that virus movements occur over shorter distances; however, some long-distance movements of Group B H5N6 viruses occurred across borders from 2016 to 2019 (Figs 6 and 7B).
Figure 5.
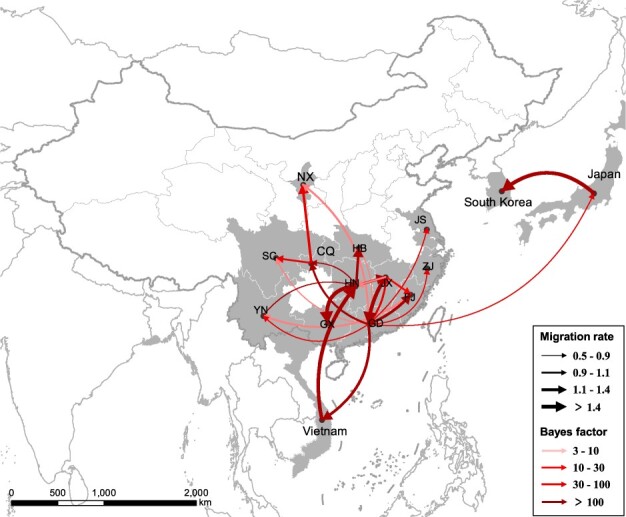
Spatiotemporal dissemination of clade 2.3.4.4 Group C/D H5N6 viruses among domestic poultry and wild birds, which was determined by Bayesian phylogeography inference of HA gene sequences. Curves show the among-province virus lineage transitions statistically supported with BF >3 for H5N6 viruses. Curve widths represent transition rate values; curve colors represent corresponding statistical support (BF value) for each transition rate. Abbreviations: NX, Ningxia; SC, Sichuan; CQ, Chongqing; YN, Yunnan; GX, Guangxi; GD, Guangdong, HN, Hunan; HB, Hubei; JX, Jiangxi; FJ, Fujian; ZJ, Zhejiang; JS, Jiangsu.
Figure 7.

Level of BF support for each transmission routes. Among-province virus lineage transition rates (supported with BF >3) decrease with geodesic distance between provinces for clade 2.3.4.4 Group C/D H5N6 viruses (A) and Group B H5N6/H5N8 viruses (B), respectively. The left and right panels display the level of BF support for each of the transmission routes considered for clade 2.3.4.4 Group C/D H5N6 analyses (C) and Group B H5N6/H5N8 analyses (D), respectively. The x-axis represents the origin location and subtypes, and the y-axis represents the destination and subtypes.
Figure 6.

Spatiotemporal dissemination of clade 2.3.4.4 Group B H5N6/H5N8 viruses among domestic poultry and wild birds, which was determined by Bayesian phylogeography inference of HA gene sequences. Curves show the among-province virus lineage transitions statistically supported with BF >3 for H5N6 viruses. Curve widths represent transition rate values; curve colors represent corresponding statistical support (BF value) for each transition rate.
Considering the continuous process of geographic dissemination of H5N6 HPAI viruses, we sought to pinpoint the dissemination pathways of H5N6 HPAI viruses. For Group C and D H5N6 viruses, Guangdong, Hunan, and Jiangxi provinces of China acted as the epicenters for the spread of H5N6 viruses in domestic birds. Guangdong was linked with eleven locations from 2013 to 2018, including Chongqing (BF = 3436), Fujian (BF = 2110), Vietnam (BF = 293), Zhejiang (BF = 223), Jiangxi (BF = 47), Jiangsu (BF = 42), Yunnan (BF = 36), Japan (BF = 10), Hubei (BF = 9), Sichuan (BF = 3), and Ningxia (BF = 3) (Fig. 7C; Supplementary Table S2). Hunan was linked with five locations during the same period, including Guangxi (BF = 385), Chongqing (BF = 315), Hubei (BF = 178), Jiangxi (BF = 124), and Vietnam (BF = 109) (Fig. 7C; Supplementary Table S2). In addition, we observed that the higher viral migration rates occurred between closer provinces or countries, including from Hunan to Hubei (migration rate = 1.54), from Guangdong to Vietnam (migration rate = 1.39), from Chongqing to Ningxia (migration rate = 1.19), and from Japan to South Korea (migration rate = 1.90) (Fig. 7A; Supplementary Table S2). Remarkably, a special case got our attention. We observed that a strong signal of viral dissemination (migration rate = 2.86) from Hunan to Vietnam occurred in 2015, even though the two places are approximately 1,500 km apart, which suggests that the H5N6 viruses in Vietnam arose within China, as consistent with the results of Du’s phylogenetic analysis (Du et al. 2017).
For Group B H5N6 viruses, the transmission routes of H5N6 differed starkly from the Group C/D H5N6 viruses and extended onto multiple continents. In Fig. 7, we found that South Korea played important roles in the worldwide dissemination of H5N6 viruses. Using phylogeographical analysis, Group B H5N6 viruses from South Korea was linked with Group B H5N8 viruses from Netherlands (BF = 12) and Germany (BF = 4) (Figs 6 and 7D), suggesting that H5N6 viruses from South Korea were originated from H5N8 viruses in Europe. In addition, we also found that Group B H5N6 viruses from Bangladesh during 2018 was linked with wild birds-origin H5N8 viruses in Egypt (BF = 33) and China (BF = 4) (Figs 6 and 7D; Supplementary Tables S2 and S3), indicating that transmission routes of these H5N6 viruses from Bangladesh differed from that of H5N6 viruses in South Korea. Remarkably, using phylogeopraphical approach, we also found that the cocirculation of Group B H5N6 and H5N8 viruses occurred in European countries, indicative of the occurrence of continuous reassortment events of H5 subtype viruses. Due to poor statistical support for viral migration, phylogenetic analysis precluded inferring, we cannot infer the dissemination pathways of H5N6 viruses in those regions. However, a strong statistical support of viral migration of Group B H5N6 viruses was observed from South Korea to Japan (Fig. 6; Supplementary Table S3).
4. Discussion
Our study revealed significant aspects of the molecular evolution of clade 2.3.4.4 HPAI H5N6 viruses from 2013 to 2019 following a phylogenetic analysis that examined time of origin, evolutionary rate, diversification patterns, and potential dissemination routes guided by a phylogeographic approach. Our findings suggested from 2013 to 2016, the cocirculation of Group C/D H5N6 viruses occurred in China and Vietnam and that viral movements occurred between adjacent or close provinces of China. The transition rates between locations were inversely related to the distances, thereby indicating that viral movements largely occurred over shorter distances. Considering evidence that H5N1 and H7N9 viruses were likely associated with the live poultry trade (Bui et al. 2018; Li et al. 2018; Yang et al. 2020), in this study, based on the phylogeographic approach, we found that H5N6 viruses from Guangdong and Hunan provinces were more likely to have originated from, and migrated to, at least eleven locations and four locations, respectively, which implies that H5N6 viruses endemic in southern China might have jointly resulted from the gene flow through circular poultry movements. Livestock breeding is extensive in southern China, with many breeding poultry farms, particularly “free range” ducks that are raised in large amount of small backyard farms, and live poultry markets (Li et al. 2018). Most of the poultry products are sold to adjacent regions, which might accelerate the dissemination of H5N6 viruses.
Not only an epidemic in China, H5N6 viruses have expanded in distinct geographical and evolutionary pathways, likely via long-distance migratory bird dispersal into several countries. We observed that Group B and C reassortant H5N6 viruses were widely prevalent in wild birds in South Korea and Japan. Two independent paths of H5N6 viruses into South Korea from 2016 to 2018 provided evidence of Group B reassortant H5N6 viruses in South Korea were originated from Group B H5N8 viruses in Europe, and Group C H5N6 viruses were originated from Group C H5N6 viruses in Guangdong, China. Notably, we also found that Group B H5N6 viruses from Bangladesh was linked with two locations, including Qinghai Lake of China and Egypt, since these regions here lie along major migratory bird pathways–the central Asian, east Africa-west Asian, and Black Sea-Mediterranean routes (Verhagen et al. 2015). H5N6 HPAI viruses in wild birds have been reported simultaneously with poultry outbreaks in multiple countries (Yamaji et al. 2020). Moreover, the migration of wild birds makes several stopovers along the migratory flyways where they can transmit influenza virus to local domestic aquatic poultry, environment, and mammals (Hinshaw et al. 1981; Bi et al. 2016b; Domanska-Blicharz et al. 2010). Direct and indirect contacts with wild birds, not farm-to-farm transmission, have frequently been as the most probable cause of viral introduction into domestic poultry (Beerens et al. 2018; Ndumu et al. 2018; Wade et al. 2018; Abolnik et al. 2019), which can subsequently cause outbreaks of influenza viruses among domestic birds due to low biosecurity systems of poultry farms (Ssematimba et al. 2013). Prevention and controls efforts should be implemented in live poultry markets and poultry farms to improve biosecurity measures.
Genotypic and genetic analysis of H5N6 viruses have demonstrated that H5N6 HPAI viruses constituted an epidemic in China from 2013 to 2015, one marked by the increasing genetic diversity of H5N6 viruses in 2014 (Gao et al. 2017). In particular, the G1 and G2 H5N6 viruses were widely prevalent during that period, and the HA genes of G1 and G2 H5N6 viruses belonged to clade 2.3.4.4 Group C and D H5N6 viruses, respectively. It is interesting to note that no substantial significant difference surfaced in the tMRCAs and evolutionary rates between Group C and D, which indicated that two parallel evolutionary pathways of H5N6 viruses arose in China from 2013 to 2014. However, a sharp increase in the number of genotypes of H5N6 viruses occurred in 2015, and G1.1 became the dominant genotype during that period. G1.1 H5N6 viruses bear the PB2 derived from duck-origin H6 subtype viruses and other internal genes from clade 2.3.2 H5N1 (Bi et al. 2016a), and subsequently, new G1.1.b H5N6 viruses bearing PB2 derived from duck-origin H6 subtype viruses and PB1 originating from H3 subtype Eurasian ‘gene pool’ (Zhang et al. 2017, 2018), so to speak, became the dominant genotype in domestic birds and humans from 2016 to 2018. In our previous study, G1.1.b H5N6 viruses were found to circulate in Guangxi province of China, and to genetically closely related to H5N6 viruses originating in wild birds from South Korea and Japan from late 2016 to early 2017 (Zhang et al. 2018), which further indicated that the outbreak of H5N6 viruses originating from wild birds during late-2016 to mid-2017 emerged from China. The emergence of G3 H5N6 viruses prevalent in most European countries, South Korea, and Bangladesh from 2018 to 2019 (Beerens et al. 2018; Yamaji et al. 2020) were distinct from H5N6 viruses from China. In addition, we found that the aquatic birds, including ducks, geese, and wild birds could accelerated the reassortment of HPAI H5N6 viruses, which exhibited the great genetic diversity of H5N6 viruses (Supplementary Table S5). The H6 subtype viruses were among the most frequently encountered in both wild and domestic aquatic birds in southern China (Huang et al. 2010). The reassortant H5N6 viruses spread to South Korea and Japan all harbored H6 origin segment, and the number of genotypes of H5N6 viruses from aquatic birds were far exceed than H5N6 viruses from chicken, thus indicating that H5N6 viruses were more adapted to aquatic birds.
We also observed that after the introduction of H5N6 viruses originating in wild birds since late-2016, the effective population size obviously increased; however, the effective population size of poultry-origin H5N6 viruses declined during the same period, which indicated that genetic diversity and increasing geographic distributions of H5N6 viruses might have promoted by wild bird migration. Wild birds migrate between wintering and breeding sites, which accelerated the dissemination of influenza viruses. The evidence showed that the increasing genetic diversity of H5N6 viruses occurred during the 2017 breeding season and the following winter (Fig. 3C), which suggested that H5N6 viruses among wild birds had resulted the global distribution. The HPAI H5N6 viruses had spread by migratory birds to multiple European countries. Thus, a key concern is whether these novel reassortant viruses could posed an elevated risk of causing human infection. Previous study showed that the human risk following exposure to the European reassortant HPAI H5N6 viruses remains low (Thornton et al. 2019). Although some mammalian adaption molecular markers were found in the H5N6 viruses (Peng et al. 2018; Sun et al. 2020), the HPAI H5N6 viruses could not be efficiently transmitted among ferrets (Herfst et al. 2018), which suggested that, despite of the global transmission of H5N6 viruses, it could not cause a pandemic among humans.
Vaccination has long been a major strategy for controlling and preventing H5 subtype avian influenza in China. The HA gene of the vaccine strains used in China since 2004 has been updated several times to ensure an antigenic match between the prevalent H5 subtype strains and vaccine strains. Since its emergence in 2013, clade 2.3.4.4 H5N6 has replaced H5N1 as a dominant avian influenza virus subtype in southern China (Bi et al. 2016a), and the current vaccine strains, Re-8 and rD8, which bear the HA gene derived from clade 2.3.4.4 representative of H5 subtype viruses, have been widely used in chicken populations since 2016 in China. Although vaccines can largely thwart avian influenza outbreaks, they can also accelerate the generation of novel variants, resulting in some positively selected mutations. These positively selected mutations of H5N6 viruses may primarily related to so-called ‘immune escape’ under vaccine selection as well as the antigenic changes of re-emerging HPAI H7N9 viruses in China (Zhang et al. 2020). Effective vaccines are only available for a few clades of H5 and H7 subtype viruses, and have been produced by only a few countries; hence, in addition to vaccination, the enhancement of the biosafety in poultry farms and companies could further reduce the spread of H5N6 viruses.
This study has several limitations. First, sampling bias may have affected the Bayesian phylogenic reconstruction, inference of the transmission networks, and association between the distance and migration rates. Second, the spatio-temporal datasets of HA gene sequences may not have accurately represented the times and locations of H5N6 viruses originating in wild birds. In most countries, domestic poultry are sampled more intensively than wild birds, and only few sampling sites of wild birds are concentrated. Taken together, our findings offer novel insights into the evolutionary dynamics and dissemination pathways of global H5N6 viruses. With the continuous evolution and increasing geographic distributions of H5N6 HPAI viruses, comprehensive surveillance and the enhancement of biosecurity precautions should be taken immediately performed to prevent the H5N6 virus epidemic from morphing into a pandemic.
Data availability
All the data needed to generate the conclusions made in the article are present in the article itself and/or the Supplementary data. Additional data related to this article may be requested from the authors.
Supplementary data
Supplementary data are available at Virus Evolution online.
Supplementary Material
Acknowledgements
We acknowledge the authors, originating and submitting laboratories of the sequences from GISAID’s EpiFlu Database and GenBank (http://ncbi.nlm.nih.gov/genbank/) Database on which this research is based. All submitters of data may be contacted directly through the GISAID website (www.gisaid.org).
Funding
This work was supported by the Key Research and Development Program of Guangdong Province (2019B020218004), National Natural Science Foundation of China (31672586, 31830097), the Earmarked Found for China Agriculture Research System [CARS-41-G16], The Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2018, Wenbao Qi), and Young Scholars of Yangtze River Scholar Professor Program (2019, Wenbao Qi).
Conflict of interest: None declared.
Contributor Information
Jiahao Zhang, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission; Ministry of Agricultural and Rural Affairs, Key Laboratory of Zoonoses; Key Laboratory of Zoonoses Prevention and Control of Guangdong Province, Wushan Rd, Tianhe District, Guangzhou, Guangdong 510642, P.R. China.
Yiqun Chen, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission.
Nan Shan, Nanjing Institute of Environmental Sciences, Ministry of Ecology and Environment of the People’s Republic of China; Jiangsu Center for Collaborative Innovation in Geographical Information Resource Development and Application, Nanjing, Jiangsu 210023, P.R. China.
Xiaomin Wang, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission.
Shuxia Lin, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission.
Kaixiong Ma, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission.
Bo Li, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission.
Huanan Li, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission.
Ming Liao, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission; Ministry of Agricultural and Rural Affairs, Key Laboratory of Zoonoses; Key Laboratory of Zoonoses Prevention and Control of Guangdong Province, Wushan Rd, Tianhe District, Guangzhou, Guangdong 510642, P.R. China; Guangdong Laboratory for Lingnan Modern Agriculture, Wushan Rd, Tianhe District, Guangzhou, Guangdong 510642, P.R. China.
Wenbao Qi, College of Veterinary Medicine, South China Agricultural University; National Avian Influenza Para-Reference Laboratory; National and Regional Joint Engineering Laboratory for Medicament of Zoonoses Prevention and Control, National Development and Reform Commission; Ministry of Agricultural and Rural Affairs, Key Laboratory of Zoonoses; Key Laboratory of Zoonoses Prevention and Control of Guangdong Province, Wushan Rd, Tianhe District, Guangzhou, Guangdong 510642, P.R. China; Guangdong Laboratory for Lingnan Modern Agriculture, Wushan Rd, Tianhe District, Guangzhou, Guangdong 510642, P.R. China.
References
- Abolnik C. et al. (2019) ‘The Incursion and Spread of Highly Pathogenic Avian Influenza H5N8 Clade 2.3.4.4 within South Africa’, Avian Diseases, 63: 149–56. [DOI] [PubMed] [Google Scholar]
- Beerens N. et al. (2018) ‘Novel Highly Pathogenic Avian Influenza A(H5N6) Virus in The Netherlands, December 2017’, Emerging Infectious Diseases, 24: 770–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N. et al. (2019) ‘Genetic Relationship between Poultry and Wild Bird Viruses during the Highly Pathogenic Avian Influenza H5N6 Epidemic in The Netherlands, 2017–2018’, Transboundary and Emerging Diseases, 66: 1370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Y. et al. (2016. a) ‘Genesis, Evolution and Prevalence of H5N6 Avian Influenza Viruses in China’, Cell Host & Microbe, 20: 810–21. [DOI] [PubMed] [Google Scholar]
- Bi Y. et al. (2016. b) ‘Novel Avian Influenza A (H5N6) Viruses Isolated in Migratory Waterfowl before the First Human Case Reported in China, 2014’, Science Reports, 6: 29888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielejec F. et al. (2016) ‘SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes’, Molecular Biology and Evolution, 33: 2167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui C. M. et al. (2018) ‘Characterising Routes of H5N1 and H7N9 Spread in China Using Bayesian Phylogeographical Analysis’, Emerging Microbes & Infections, 7: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. et al. (2019) ‘A Study of the Relationship between Human Infection with Avian Influenza a (H5N6) and Environmental Avian Influenza Viruses in Fujian’, BMC Infectious Diseases, 19: 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D. H. et al. (2016) ‘Genetic and Antigenic Characterization of H5, H6 and H9 Avian Influenza Viruses Circulating in Live Bird Markets with Intervention in the Center Part of Vietnam’, Veterinary Microbiology, 192: 194–203. [DOI] [PubMed] [Google Scholar]
- Domanska-Blicharz K. et al. (2010) ‘H5N1 High Pathogenicity Avian Influenza Virus Survival in Different Types of Water’, Avian Diseases, 54: 734–7. [DOI] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2006) ‘Relaxed Phylogenetics and Dating with Confidence’, PLoS Biology, 4: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2012) ‘Bayesian Phylogenetics with BEAUti and the BEAST 1.7’, Molecular Biology and Evolution, 29: 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y. et al. (2017) ‘Molecular Evolution and Emergence of H5N6 Avian Influenza Virus in Central China’, Journal of Virology, 91: e00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S. et al. (2008) ‘Rates of Evolutionary Change in Viruses: Patterns and Determinants’, Nature Reviews Genetics, 9: 267–76. [DOI] [PubMed] [Google Scholar]
- Fan S. et al. (2014) ‘A Novel Highly Pathogenic H5N8 Avian Influenza Virus Isolated from a Wild Duck in China’, Influenza and Other Respiratory Viruses, 8: 646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro A. et al. (2019) ‘Disentangling the Role of Africa in the Global Spread of H5 Highly Pathogenic Avian Influenza’, Nature Communications, 10: 5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. et al. (2017) ‘Increasing Genetic Diversity of H5N6 Avian Influenza Virus in China: A Serious Threat to Persistence and Dissemination in Guangdong Province’, Journal of Infection, 75: 586–90. [DOI] [PubMed] [Google Scholar]
- Global Consortium for H5N8 and Related Influenza Viruses. (2016) ‘Role for Migratory Wild Birds in the Global Spread of Avian Influenza H5N8’, Science, 354: 213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herfst S. et al. (2012) ‘Airborne Transmission of Influenza a/H5N1 Virus between Ferrets’, Science, 336: 1534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herfst S. et al. (2018) ‘Human Clade 2.3.4.4 a/H5N6 Influenza Virus Lacks Mammalian Adaptation Markers and Does Not Transmit via the Airborne Route between Ferrets’, mSphere, 3: e00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick K. A., Huettmann F., Lindgren M. A. (2013) ‘A Global Model of Avian Influenza Prediction in Wild Birds: The Importance of Northern Regions’, Veterinary Research, 44: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S. C. et al. (2015) ‘Wild Waterfowl Migration and Domestic Duck Density Shape the Epidemiology of Highly Pathogenic H5N8 Influenza in the Republic Of Korea’, Infection, Genetics, and Evolution, 34: 267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw V. S. et al. (1981) ‘Replication of Avian Influenza a Viruses in Mammals’, Infection and Immunity, 34: 354–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K. et al. (2010) ‘Establishment of an H6N2 Influenza Virus Lineage in Domestic Ducks in Southern China’, Journal of Virology, 84: 6978–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J. et al. (2014) ‘Highly Pathogenic Avian Influenza Virus (H5N8) in Domestic Poultry and Its Relationship with Migratory Birds in South Korea during 2014’, Veterinary Microbiology, 173: 249–57. [DOI] [PubMed] [Google Scholar]
- Jiao P. et al. (2016) ‘New Reassortant H5N6 Highly Pathogenic Avian Influenza Viruses in Southern China, 2014’, Frontiers in Microbiology, 7: 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. et al. (2002) ‘MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform’, Nucleic Acids Research, 30: 3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. I. et al. (2018) ‘Pathogenicity and Genetic Characterisation of a Novel Reassortant, Highly Pathogenic Avian Influenza (HPAI) H5N6 Virus Isolated in Korea, 2017’, Euro Surveillance, 23: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H. I. et al. (2018. a) ‘Comparison of the Pathogenic Potential of Highly Pathogenic Avian Influenza (HPAI) H5N6, and H5N8 Viruses Isolated in South Korea during the 2016-2017 Winter Season’, Emerging Microbes & Infections, 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J. H. et al. (2018. b) ‘New Reassortant Clade 2.3.4.4b Avian Influenza A(H5N6) Virus in Wild Birds, South Korea, 2017–18’, Emerging Infectious Diseases, 24: 1953–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. H. et al. (2015) ‘Intercontinental Spread of Asian-Origin H5N8 to North America through Beringia by Migratory Birds’, Journal of Virology, 89: 6521–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. H. et al. (2016) ‘Highly Pathogenic Avian Influenza Viruses and Generation of Novel Reassortants, United States, 2014–2015’, Emerging Infectious Diseases, 22: 1283–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. K. et al. (2018) ‘Characterization of a Novel Reassortant H5N6 Highly Pathogenic Avian Influenza Virus Clade 2.3.4.4 in Korea, 2017’, Emerging Microbes & Infections, 7: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. J. et al. (2014) ‘Novel Reassortant Influenza A(H5N8) Viruses, South Korea, 2014’, Emerging Infectious Diseases, 20: 1087–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P. et al. (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R. et al. (2018) ‘Live Poultry Trading Drives China's H7N9 Viral Evolution and Geographical Network Propagation’, Front Public Health, 6: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A. (2006) ‘Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences’, Bioinformatics, 22: 1658–9. [DOI] [PubMed] [Google Scholar]
- Liu Y. P. et al. (2018) ‘Detection of Reassortant H5N6 Clade 2.3.4.4 Highly Pathogenic Avian Influenza Virus in a Black-Faced Spoonbill (Platalea Minor) Found Dead, Taiwan, 2017’, Infection, Genetics, and Evolution, 62: 275–8. [DOI] [PubMed] [Google Scholar]
- Marchenko V. et al. (2018) ‘Isolation and Characterization of H5Nx Highly Pathogenic Avian Influenza Viruses of Clade 2.3.4.4 in Russia’, Virology, 525: 216–23. [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor K. C. et al. (2018) ‘Comparative Epidemiology of Highly Pathogenic Avian Influenza Virus H5N1 and H5N6 in Vietnamese Live Bird Markets: Spatiotemporal Patterns of Distribution and Risk Factors’, Frontiers in Veterinary Science, 5: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V. N., Bloomquist E. W., Suchard M. A. (2008) ‘Smooth Skyride through a Rough Skyline: Bayesian Coalescent-Based Inference of Population Dynamics’, Molecular Biology and Evolution, 25: 1459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndumu D. et al. (2018) ‘Highly Pathogenic Avian Influenza H5N8 Clade 2.3.4.4B Virus in Uganda, 2017’, Infection, Genetics, and Evolution, 66: 269–71. [DOI] [PubMed] [Google Scholar]
- Pan M. et al. (2016) ‘Human Infection with a Novel, Highly Pathogenic Avian Influenza A (H5N6) Virus: Virological and Clinical Findings’, Journal of Infection, 72: 52–9. [DOI] [PubMed] [Google Scholar]
- Peng X. et al. (2018) ‘Amino Acid Substitutions HA A150V, PA A343T, and PB2 E627K Increase the Virulence of H5N6 Influenza Virus in Mice’, Frontiers in Microbiology, 9: 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poen M. J. et al. (2019) ‘Co-Circulation of Genetically Distinct Highly Pathogenic Avian Influenza a Clade 2.3.4.4 (H5N6) Viruses in Wild Waterfowl and Poultry in Europe and East Asia, 2017-18’, Virus Evol, 5: vez004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlmann A. et al. (2019) ‘Genetic Characterization and Zoonotic Potential of Highly Pathogenic Avian Influenza Virus A(H5N6/H5N5), Germany, 2017–2018’, Emerging Infectious Diseases, 25: 1973–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. J. et al. (2015) ‘Nomenclature Updates Resulting from the Evolution of Avian Influenza A(H5) Virus Clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013–2014’, Influenza and Other Respiratory Viruses, 9: 271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ssematimba A. et al. (2013) ‘Avian Influenza Transmission Risks: Analysis of Biosecurity Measures and Contact Structure in Dutch Poultry Farming’, Preventive Veterinary Medicine, 109: 106–15. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies’, Bioinformatics, 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. et al. (2017) ‘Epidemiology, Evolution, and Pathogenesis of H7N9 Influenza Viruses in Five Epidemic Waves since 2013 in China’, Trends in Microbiology, 25: 713–28. [DOI] [PubMed] [Google Scholar]
- Sun Y. et al. (2020) ‘An R195K Mutation in the PA-X Protein Increases the Virulence and Transmission of Influenza a Virus in Mammalian Hosts’, Journal of Virology, 94: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemae N. et al. (2017) ‘Five Distinct Reassortants of H5N6 Highly Pathogenic Avian Influenza a Viruses Affected Japan during the Winter of 2016–2017’, Virology, 512: 8–20. [DOI] [PubMed] [Google Scholar]
- Thornton A. C. et al. (2019) ‘Human Exposures to H5N6 Avian Influenza, England, 2018’, The Journal of Infectious Diseases, 220: 20–2. [DOI] [PubMed] [Google Scholar]
- Verhagen J. H. et al. (2015) ‘Wild Bird Surveillance around Outbreaks of Highly Pathogenic Avian Influenza A(H5N8) Virus in The Netherlands, 2014, within the Context of Global Flyways’, Eurosurveillance, 20: 21069. [DOI] [PubMed] [Google Scholar]
- Wade A. et al. (2018) ‘Highly Pathogenic Avian Influenza A(H5N8) Virus, Cameroon, 2017’, Emerging Infectious Diseases, 24: 1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F. Y. et al. (2015) ‘Reassortant Highly Pathogenic Influenza A(H5N6) Virus in Laos’, Emerging Infectious Diseases, 21: 511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. et al. (2014) ‘Novel Reassortant Influenza A(H5N8) Viruses in Domestic Ducks, Eastern China’, Emerging Infectious Diseases, 20: 1315–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. et al. (2015) ‘Genomic and Phylogenetic Characterization of Novel, Recombinant H5N2 Avian Influenza Virus Strains Isolated from Vaccinated Chickens with Clinical Symptoms in China’, Viruses, 7: 887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. et al. (1999) ‘Genetic Characterization of the Pathogenic Influenza a/Goose/Guangdong/1/96 (H5N1) Virus: Similarity of Its Hemagglutinin Gene to Those of H5N1 Viruses from the 1997 Outbreaks in Hong Kong’, Virology, 261: 15–9. [DOI] [PubMed] [Google Scholar]
- Xu Y. F. (2018) ‘Novel Highly Pathogenic Avian Influenza A(H5N6) Viruses in The Netherlands’, Emerging Infectious Diseases, 24: 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji R. et al. (2020) ‘Pandemic Potential of Highly Pathogenic Avian Influenza Clade 2.3.4.4 A(H5) Viruses’, Reviews in Medical Virology, 30: e2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. et al. (2017) ‘Genesis and Dissemination of Highly Pathogenic H5N6 Avian Influenza Viruses’, Journal of Virology, 91: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q. et al. (2020) ‘Assessing the Role of Live Poultry Trade in Community-Structured Transmission of Avian Influenza in China’, Proceedings of the National Academy of Sciences of the United States of America, 117: 5949–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. et al. (2018) ‘Genetic Diversity and Dissemination Pathways of Highly Pathogenic H5N6 Avian Influenza Viruses from Birds in Southwestern China along the East Asian-Australian Migration Flyway’, Journal of Infection, 76: 418–422. [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. (2020) ‘Evolution and Antigenic Drift of Influenza A (H7N9) Viruses, China, 2017-2019’, Emerging Infectious Diseases, 26: 1906–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2017) ‘Human Infections with Novel Reassortant H5N6 Avian Influenza Viruses in China’, Emerging Microbes & Infections, 6: e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D. et al. (2017) ‘Glycosylation of the Hemagglutinin Protein of H5N1 Influenza Virus Increases Its Virulence in Mice by Exacerbating the Host Immune Response’, Journal of Virology, 91: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data needed to generate the conclusions made in the article are present in the article itself and/or the Supplementary data. Additional data related to this article may be requested from the authors.