

Abstract
Post-translational modifications (PTMs) impact the pathological aggregation of α-synuclein (αS), a hallmark of Parkinson’s Disease (PD). Here, we synthesize αS phosphorylated at tyrosine 39 (pY39) through a novel route using in vitro enzymatic phosphorylation of a fragment followed by ligation to form the full-length protein. We can execute this synthesis in combination with unnatural amino acid mutagenesis to include two fluorescent labels for Förster resonance energy transfer (FRET) studies. We determine the effect of pY39 on the aggregation of αS, and compare our authentically phosphorylated material to the corresponding glutamate 39 “phosphomimetic.” Intriguingly, we find that αS-pY39 can either accelerate or decelerate aggregation, depending on the fraction of phosphorylated protein. The αS-E39 mutant can qualitatively reproduce some, but not all, of these effects. FRET measurements and analysis of existing structures of αS help to provide an explanation for this phenomenon. Our results have important implications for treatment of PD patients with tyrosine kinase inhibitors and highlight the importance of validating phosphomimetics through studies of authentic PTMs.
Graphical Abstract
The 140 amino acid α-synuclein (αS) protein is found at the presynaptic termini of neurons and is involved in the pathogenesis of several neurodegenerative disorders, of which Parkinson’s Disease (PD) is most prominent.1 A natively disordered monomer, αS forms amyloid fibrils that deposit in Lewy Bodies, which characterize PD. Recent structures of αS, both monomer ensembles based on NMR, FRET and other data, as well as fibril structures obtained by solid state NMR (ssNMR) or cryo-electron microscopy (cryo-EM) help to shed light on its folding and aggregation.2–6
Various factors are responsible for changes in αS’s propensity to aggregate, including post-translational modifications (PTMs).7–13 Recently, the presence of αS phosphorylated at Tyr39 (pY39), resulting from c-Abl kinase activity, has been identified in vivo14 and PD patients display elevated levels of c-Abl compared to healthy individuals.14 However, reports are in conflict as to the functional impact of αS phosphorylation. On one hand, co-expression of c-Abl with αS in mice was found to increase aggregation and neurotoxicity induced by αS-pY39.15 On the other hand, in vitro aggregation assays have demonstrated that αS-pY39 slows fibrilization.16
Several approaches have been used to study tyrosine phosphorylation in proteins. The most common approach is mutation of the Tyr residue to Glu/Asp, a so-called phosphomimetic; although straightforward to introduce, Glu/Asp does not provide a good representation of the steric bulk or dianionic charge of Tyr phosphorylation. Two studies have reported the incorporation of pY (or an analog) using amber codon suppression, but these methods are not easily compatible with introducing multiple synthetic modifications.17, 18 Three other approaches have been used to study αS-pY39 itself: (1) direct treatment of full-length wild type (WT) αS with c-Abl kinase in vitro, (2) mutation of all other Tyr to Phe followed by in vitro enzymatic phosphorylation of the protein to improve specificity on a near-WT construct; and (3) native chemical ligation (NCL) to effect a total synthesis of the authentic, site-specifically phosphorylated protein.15, 16 The first approach has imperfect site-selectivity, and the second approach, while selective, is non-ideal as it requires a number of careful control experiments to show that the Phe mutations do not produce confounding effects. Total synthesis is appealing as it allows great freedom in introducing other modifications. However, we sought to improve upon the previous methods to enable straightforward introduction of not only pY39, but also as many as two fluorophores for Förster resonance energy transfer (FRET) experiments by combining phosphorylation methods with unnatural amino acid mutagenesis and NCL, used previously by ouselves and others for double labeling.19
RESULTS AND DISCUSSION
Comparison of strategies for generating αS phosphorylated at Tyr39.
To generate phosphorylated αS in a fashion that would ultimately be compatible with double labeling for FRET, we first investigated direct phosphorylation of WT αS with recombinant, purified c-Abl. Active c-Abl kinase was expressed, purified, and incubated with αS in vitro with ATP and MgCl2. While we found that we were able to phosphorylate Tyr39, we were not able to drive this reaction to completion without phosphorylating other Tyr residues as well (Fig. S29 in SI). Previous studies using this approach also report an inability to obtain homogeneous protein phosphorylated only at Tyr39.14 Although separation of the mixture of phosphorylated products was in theory possible by high performance liquid chromatography (HPLC), we were concerned about introducing an additional purification step that would ultimately need to be performed in combination with FRET labeling.
We then compared three semi-synthetic strategies to produce site-specifically phosphorylated protein: synthetic peptide ligation, expressed protein ligation, and chemoenzymatic phosphorylation followed by ligation. While αS bears four native tyrosines (at positions 39, 125, 133, and 136), semi-synthesis permits the site-specific introduction of phosphorylation at Tyr39. In our first semi-synthetic approach, the incorporation of pY39 was carried out through NCL20 of two synthetic peptides followed by ligation to a recombinant protein fragment, similar to previous reports by Lashuel and coworkers.16 The N-terminal peptide αS1–29 was generated by solid phase peptide synthesis (SPPS) with an acyl hydrazide (8% yield) for use in NCL.21 The peptide αS30–55-C30pY39, bearing the phosphotyrosine at position 39 and a cysteine for ligation, was also made by SPPS with an acyl hydrazide (15% yield). Following ligation of the two peptides, the intermediate (αS1–55-C30pY39) was ligated with recombinant fragment αS56–140-C56 (4.2 mg per L of culture), and the product was desulfurized to convert Cys ligation sites to Ala and produce αS-pY39 in 11% yield. Although there was sufficient material for preliminary measurements, improvements in yield were necessary for full biochemical studies.
Given that the amount of peptide produced by SPPS is limited as peptide length increases, we explored an alternative ligation site at Val37 which permitted a shorter central fragment and expression of the 1–36 fragment as a C-terminal intein fusion (9.2 mg per L of culture) that was converted to a thioester, αS1–36-SR, for NCL.22 The partner peptide, αS37–55-V*37pY39 (V* is penicillamine), was synthesized with a C-terminal acyl hydrazide as before (19% yield). The use of penicillamine as the ligation handle allowed the recovery of the native valine by desulfurization after ligation.23 This new NCL reaction sequence was also followed by ligation with the recombinant fragment αS56–140-C56 and desulfurization. In this case, we obtained αS-pY39 in 14% yield. Although the yield for the NCL steps was only slightly higher than the previous synthesis, this route was deemed superior because we could scale up production of the N-terminal fragment without HPLC purification simply by increasing culture size.
In order to further capitalize on the scalability of protein expression, we investigated a third semi-synthetic approach that takes advantage of enzyme chemistry on only a fragment of the protein, bypassing SPPS to allow for increased production of αS-pY39. In this chemoenzymatic approach, we ligated an expressed and phosphorylated αS1–55 fragment thioester (produced at 8.0 mg per L of culture by the intein method used for αS1–36-SR above) to the expressed αS56–140-C56 fragment. Phosphorylation at Tyr39 was achieved enzymatically on the αS1–55 fragment prior to ligation, without modifying the other tyrosines which are all found in the αS56–140-C56 fragment. Using the purified c-Abl construct from our attempts to phosphorylate full-length αS (above), we were able to form phosphorylated αS1–55-C30pY39-MES in 80% yield following HPLC purification.
Many biophysical studies require the presence of one or two fluorescent labels on the αS constructs, presenting a further synthetic challenge. For singly fluorescently labeled αS-pY39, we made an S9C mutation to attach Alexa Fluor 488 (AF488) via maleimide chemistry. For doubly labeled protein, the unnatural amino acid propargyl-tyrosine (π) was synthesized and incorporated at various positions in the αS56–140-C56 fragment via amber codon suppression.24 Alexa Fluor 594 (AF594) azide was attached at the π site using copper-catalyzed cycloaddition. Thus, we were able to use our chemoenzymatic semi-synthesis strategy in combination with these two orthogonal reactions to produce triply modified protein, including a PTM, in 63% yield. Figure 1 depicts the synthesis of αS-C4889pY39π594136 as an example. This strategy provides a convergent synthesis, where although some HPLC purification is necessary, it can be performed on more tractable fragments. Although this combination of enzymatic modification, unnatural amino acid mutagenesis, and NCL is rarely used, in some cases it can be the most efficient way of triply labeling a protein.19, 24, 25
Figure 1.
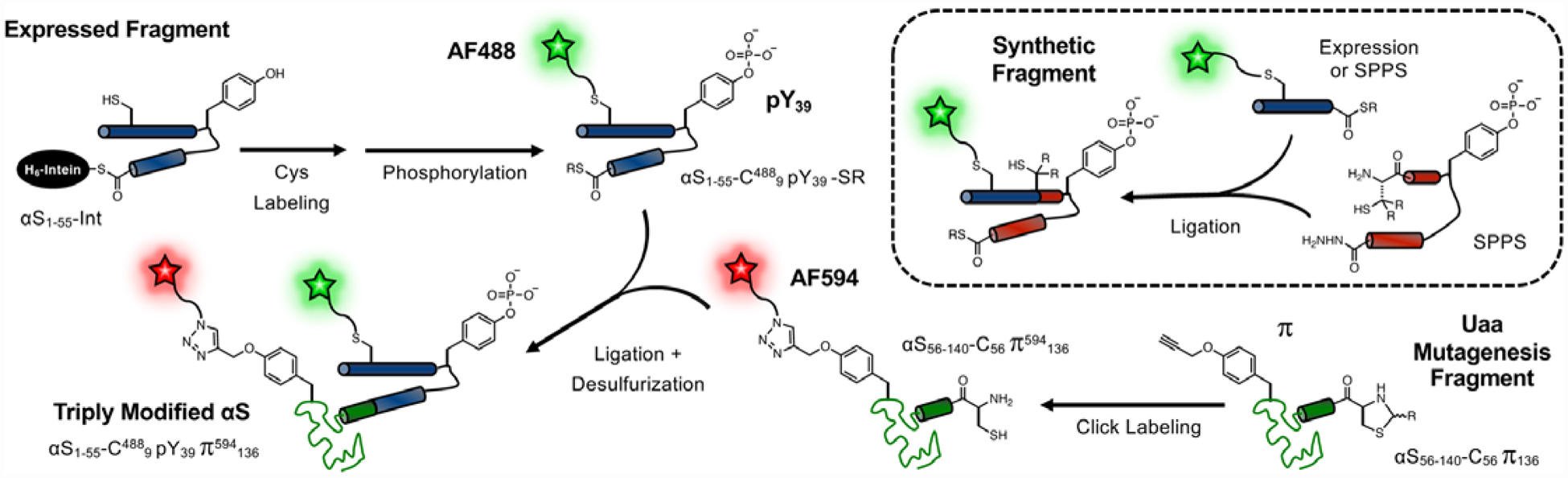
Chemoenzymatic Semi-Synthesis Scheme. Expressed Fragment: αS1–55 is expressed as an intein fusion, labeled with AF488 maleimide, and enzymatically phosphorylated with c-Abl kinase. Uaa Mutagenesis Fragment: αS56–140 is expressed with π incorporated by unnatural amino acid mutagenesis and labeled with AF594 azide. Triply Modified αS: The αS1–55 and αS56–140 fragments are ligated and desulfurized to give phosphorylated protein for FRET experiments. Synthetic Fragment: AF488-labeled, phosphorylated αS1–55 produced by ligation of a synthesized (1–29) or expressed (1–36) N-terminus and a synthetic central fragment (30–55 or 37–55).
Aggregation of αS phosphorylated at Tyr39.
We used our semi-synthetic proteins to investigate the impact of pY39 on the aggregation of αS. Unlabeled αS-pY39 was mixed at various percentages with WT αS at 100 μM total protein concentration and shaken at 37 °C for 48 h to form fibrils. Congo Red aggregation assays (Fig. 2, Left) showed that αS-pY39 has an unusual bidirectional effect on aggregation. It accelerates aggregation at low doses, where 1%−5% of the starting material consists of αS-pY39. However, αS-pY39 decelerates aggregation at higher doses, with a 2.6-fold slowing effect at 100% αS-pY39, similar to the observation of Dikiy et al.16 A comparison was made to the commonly used phosphomimetic Glu at position 39 (E39). Aggregation studies of αS-E39 display the acceleration/deceleration effect in a qualitatively similar fashion, but with a turn-around dose of between 10%−25%, in contrast to the 5%−10% turn-around for authentic αS-pY39. After the final aggregation time point, the amount of protein incorporated into the fibrils was quantified (Fig. 2, Right). The authentic αS-pY39 resulted in >84% incorporation (relative to WT) of the monomeric starting material into fibrils, irrespective of the fraction of αS-pY39. In contrast, the αS-E39 mimic caused total fibril incorporation to decrease by half at high percentages of αS-E39/WT. Thus, although the E39 mutation qualitatively reproduces some of the effects of the pY39 PTM, there are clear differences, and the use of the E39 mutation as a phosphomimetic must be regarded with some caution.
Figure 2.
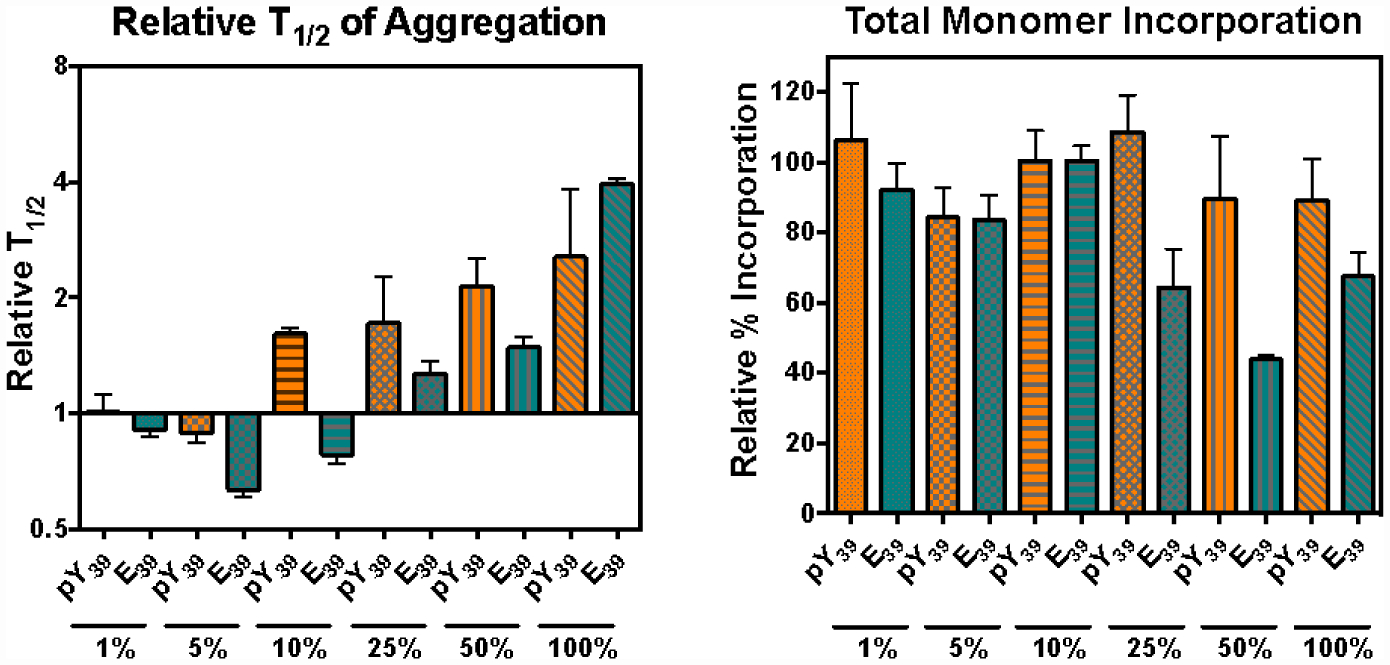
pY39 has a Bidirectional Effect on αS Aggregation. Left: Addition of αS-pY39 (orange bars) or αS-E39 (teal bars) increases aggregation rates at low percentages, but slows aggregation at high percentages. Right: The amount of total monomer incorporated into fibrils is changed more by αS-E39 than by αS-pY39. Experimental details and primary data given in SI.
Analysis of impact of Tyr39 phosphorylation on αS fibril structures.
Analysis of ssNMR and cryo-EM fibril structures suggests a mechanism for these effects. In all of the available structures, Tyr39 stacks directly upon Tyr39 in the next monomer of the fibril (Fig. 3 and Fig. S27 in SI). Thus, at high percentages of either αS-pY39 or αS-E39, electrostatic repulsion of these charged residues could destabilize fibril formation. If this is the basis for slowing of aggregation by Tyr39 phosphorylation, then it is not surprising that the effect is larger for the phosphorylated dianion (pY second pKa is 5.526) than it is for singly-charged Glu. What is less clear is why the negatively charged residues would increase aggregation rates at low percentages. We hypothesized that this could be due either to stabilizing effects once incorporated in fibrils that become unfavorable only at high percentages of αS-pY39, or to destabilizing effects on monomer conformation, making αS-pY39 more prone to adopt aggregating conformations.
Figure 3.
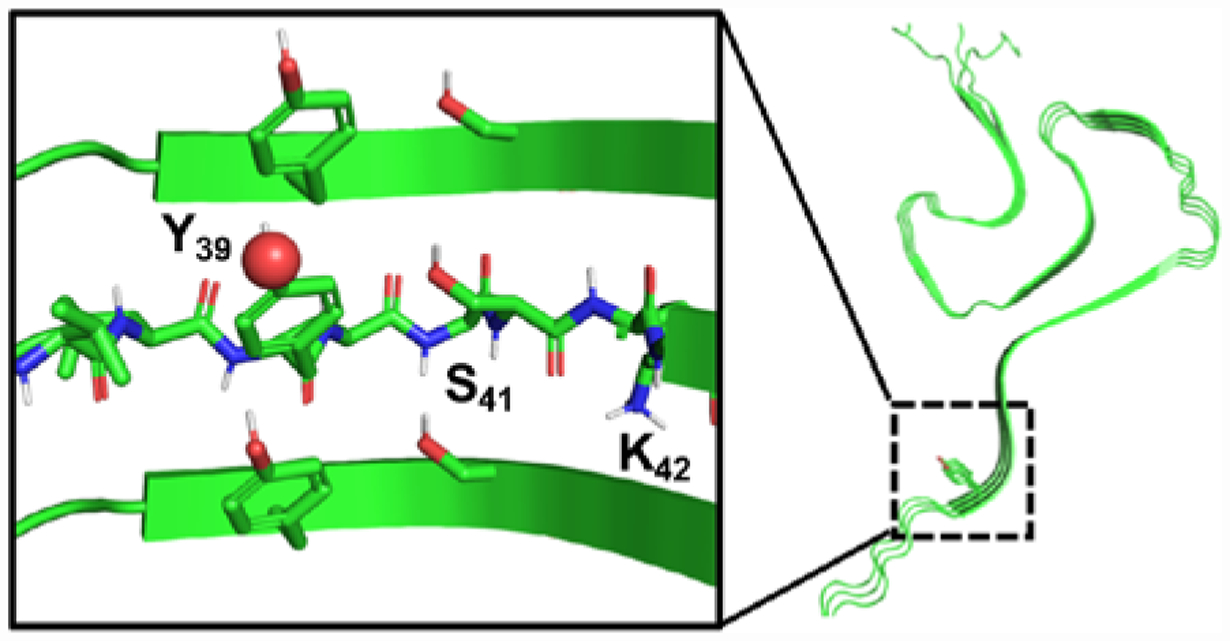
Potential Interactions of pY39 in Fibrils. ssNMR structure (PDB ID: 2N0A) showing Y39 viewed down fibril axis or side-on (inset).6 Views of Y39 in other fibril structures in SI.
FRET studies of effects of Tyr39 phosphorylation on αS monomer.
To test the hypothesis that Tyr39 phosphorylation accelerates αS aggregation by destabilizing the monomer, we studied the monomer conformation by single molecule FRET, as in previous studies of αS membrane interactions and aggregation-prone states.27, 28 In FRET, the efficiency of energy transfer (ETeff) depends on the distance between the fluorophores and can be used to examine a biomolecule’s conformation.29 αS-pY39 constructs labeled with AF488 and AF594 at residues 9 and 72 (αS-C4889pY39π59472) or 9 and 136 (αS-C4889pY39π594136) were used to probe the N-terminal half of αS or the entire protein, respectively (Fig. 4). For 9–72, a small shift in the peak of the ETeff histogram reflects compaction of αS-pY39 relative to WT αS, with a broadening of the ETeff distribution indicative of increased conformational sampling or slower dynamics. For 9–136, a small shift in the peak of the ETeff histogram to lower ETeff was observed, reflecting an expansion of αS-pY39. These results are consistent with NMR measurements on αS with pY39 in a construct in which all other Tyr had been mutated to Phe.16 Our findings imply that pY39 primarily alters aggregation through effects in the fibrils, although we cannot rule out some contribution from altered monomer conformational ensembles or intrachain dynamics as we have observed with tyrosine nitration in αS.30
Figure 4.
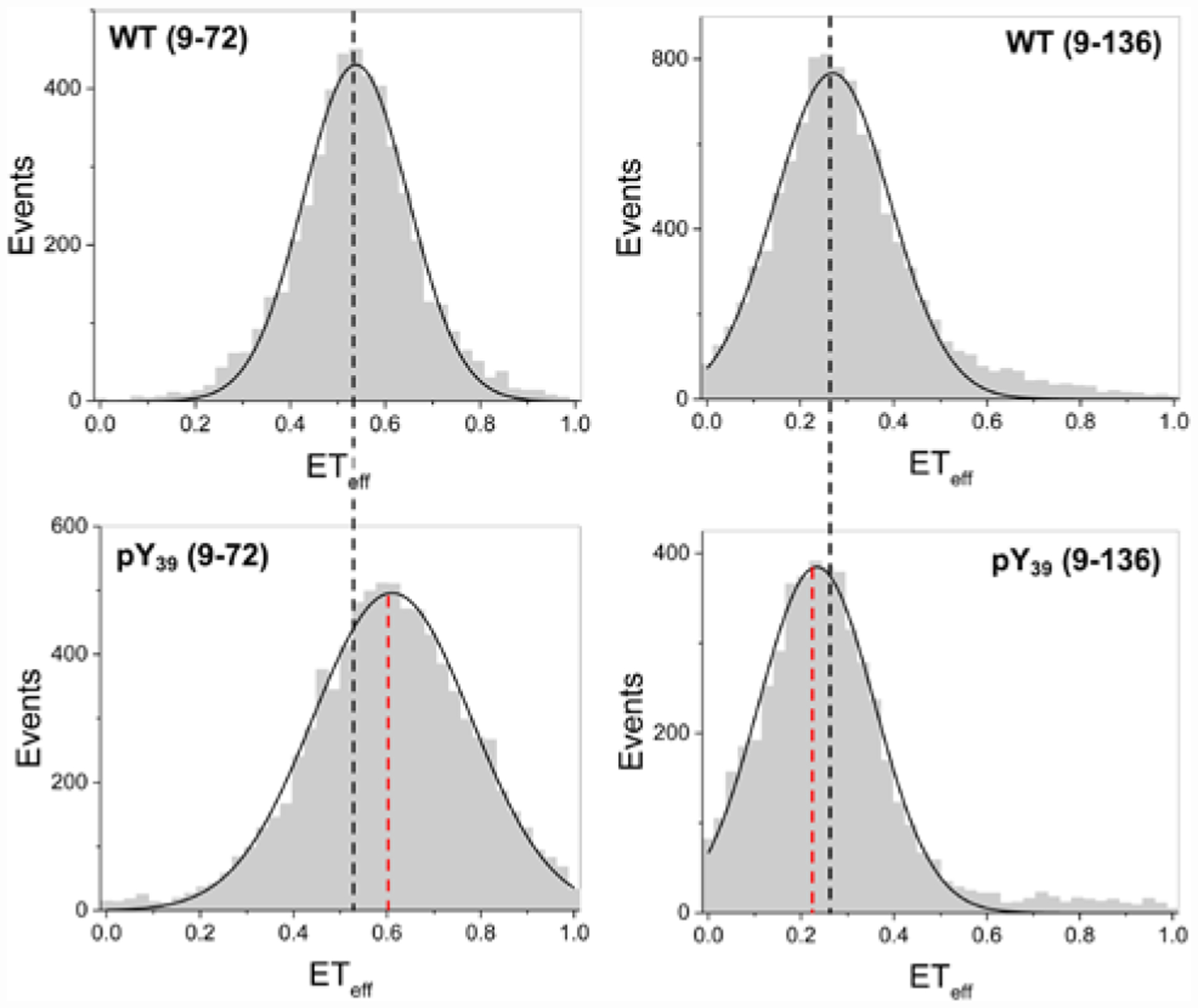
Conformational Changes in αS-pY39 Monomer. Single molecule FRET histograms showing compaction between positions 9 and 72 (ETeff for αS-C4889π59472 vs. αS-C4889pY39π59472) with expansion between positions 9 and 136 (αS-C4889π594136 vs. αS-C4889pY39π594136). Details of data fitting are given in SI.
Development of a chemoenzymatic semi-synthesis strategy allowed us to access mg quantities of αS-pY39 as well as doubly fluorescently labeled protein for FRET studies. Aggregation assays with αS-pY39 demonstrated that it has a dose-dependent, bidirectional effect on aggregation. We hypothesized that this unusual phenomenon results from interactions in the fibrils that switch from being stabilizing to destabilizing at percentages of αS-pY39 higher than 10%. Analysis of existing fibril structures supports this hypothesis, where pY39 can participate in ionic or hydrogen-bonding interactions with Y39, S41 and K42 (in some structures) in adjacent strands (Fig. 3 and Fig. S27 in SI). While the E39 mutant behaved in a generally similar way, there were significant mismatches in the dose-dependence; at 10% modified protein αS-E39 stabilizes fibrils while αS-pY39 destabilizes fibrils. This result underscores the necessity for installation of authentic phosphotyrosine in studying the effect of this PTM in αS.
While the electrostatic explanation for fibril destabilization is consistent with our results, other explanations are also worth considering. Since we do not see a dramatic change in αS-pY39 fibril amounts based on the gel analysis in Figure 2, the effect of αS-pY39 may be to destabilize a protofibril intermediate, resulting in a larger effect on kinetics than on endpoints. It should also be noted that other charged residues in αS would also necessarily participate in similar short-range interactions upon fibril formation, and a dose-dependent bidirectional phenomenon has not, to our knowledge, been previously been observed with charge-bearing mutants. Therefore, this phenomenon may be specific to the 39 site. It is also possible that the introduction of Tyr39 phosphorylation alters the fibril conformation more dramatically. Transmission electron microscopy (TEM) imaging of fibrils composed of 25% αS-pY39 do not appear to adopt a significantly different morphology than WT αS fibrils (Fig. S28 in SI). Ultimately, high resolution structural analysis through ssNMR or cryo-EM may be necessary to understand the structural basis for our observations.
Our findings show that misregulation of a PTM may play a complex role in protein dysfunction in vivo. While no study to date has precisely quantified pY39 levels, they are likely to lie in the 1–20% range where small changes in kinase and phosphatase activity will redirect the entire αS population to aggregate either faster or slower. Thus, our results are highly relevant to ongoing clinical studies of Abl Tyr kinase inhibitors in PD, which will shift pY39 levels.31 Such PTM variations may also be the aspect of the cellular “milieu” that have been attributed to differences in αS pathology in PD and related synucleinopathies.32 Additionally, a very recent study has shown that Tyr39 phosphorylation alters interactions with chaperone proteins.33 In the broader context, our chemoenzymatic semi-synthesis strategy could be employed for other proteins and other PTMs, and we have shown how access to proteins with authentic PTMs can be used to identify the importance of subtle differences in PTM levels on biological processes.
METHODS
Solid phase peptide synthesis (SPPS) of peptides.
Peptides were made via solid phase peptide synthesis (SPPS) following standard procedures34 using 2-chloro-trityl resin derivatized with Fmoc-hydrazine. Amino acid coupling was carried out by adding 5 equiv Fmoc-protected amino acid, 10 equiv DIPEA, and 5 equiv HBTU to the deprotected peptidyl resin with stirring for 30 min. Fmoc deprotection was done by stirring for 20 min with 20% (v/v) piperidine in DMF. Peptide cleavage from the resin was performed by adding cleavage cocktail (95% TFA, 2.5% TIPS, 2.5% H2O) and incubating with agitation for 1.5 h at room temperature. The peptidyl hydrazides αS30–55-C30pY39-NHNH2 and αS37–55-V*37pY39-NHNH2 were synthesized using phosphotyrosine (Fmoc-Tyr(PO(OBzl)OH)-OH) at position 39. To label αS1–29-C9, the peptide was dissolved in 20 mM Tris pH 8 and reacted with 2 equiv AF488-maleimide at 37 °C for 1–4 h. All peptides were purified by RP-HPLC using a C18 preparatory column.
In vitro phosphorylation of full-length αS and αS fragments.
WT αS, αS1–55, or the αS1–55-C9488 fragment was dissolved in buffer (50 mM Tris, 150 mM NaCl, pH 7.4) to a final concentration of ~70 μM. The sample was incubated with up to 0.1 equiv c-Abl enzyme, 2mM Mg-ATP, and 5mM MgCl2 in a 30 °C water bath for several h. The reaction was monitored by MALDI-MS and supplemented with additional Mg-ATP and MgCl2 as necessary. Phosphorylated αS1–55 or αS1–55-C9488 fragment was purified by RP-HPLC over a C18 column.
Native chemical ligation (NCL).
Peptide-acyl-hydrazide was dissolved in low pH NCL buffer (6 M GdnHCl, 200 mM Na2PO4, pH 3) for a final concentration of 2 mM and chilled to −15°C in an ice-salt bath. Hydrazide to azide conversion35 was achieved by adding 10 equiv NaNO2 and stirring for 15 min at −15°C. The partner peptide, pre-dissolved in NCL buffer pH 7.0 along with 40 equiv MPAA, was added to the reaction. The mixture was warmed to room temperature, and the pH was adjusted to 7.0. Reaction was incubated at 37 °C with agitation and supplemented with TCEP as necessary.
For ligation of expressed N-terminal protein fragment, the intein of αS fragment-MxeHis6 was cleaved with 200 mM MESNa to generate a thioester.22 To carry out ligation, the reacting partners were dissolved in NCL buffer (6 M GdnHCl, 200 mM Na2PO4, 30 mM TCEP, 50 mM MPAA, pH 7.0) and incubated at 37 °C with agitation for several hours to overnight. Following RP-HPLC purification of ligation products, the cysteines and penicillamines used in ligation were converted to the respective native alanines and valines by incubating the protein with 50 mM radical initiator VA-044 and 10% (v/v) t-BuSH in an argon-purged tube at 37 °C overnight. The full-length αS products were then purified by RP-HPLC.
Protein aggregation kinetics and percentage incorporation into fibrils.
Protein samples (100 μM total concentration in monomer units) in buffer (20 mM Tris, 100 mM NaCl, pH 7.5) were prepared in triplicate in Eppendorf tubes. Samples were aggregated by shaking at 1300 rpm at 37 °C. At each time point, an aliquot of each sample was added to Congo Red solution (20 μM in 20 mM Tris, 100 mM NaCl, pH 7.5), and the extent of aggregation was determined based on the ratio of Congo Red absorbance at 540 nm/480 nm. After the final time point, samples were spun down at maximum speed on a tabletop centrifuge for 90 min. The supernatant was removed, and pellet was resuspended in the original volume of buffer. To determine the percentage of monomer incorporation into fibrils, the resuspended pellets were supplemented with 20 mM SDS, boiled for 20 min, and chilled on ice prior to analysis by SDS-PAGE. Monomeric samples for calibration were prepared by 2-fold serial dilutions in water.
Single molecule Förster resonance energy transfer (smFRET).
All smFRET measurements were made on a MicroTime 200 inverse time- resolved confocal microscope (PicoQuant). Eight-chambered Nunc coverslips (Thermo Fisher Scientific) were plasma cleaned and coated with poly(ethylene glycol) poly(L-lysine) overnight. Each smFRET measurement was taken using 30 pM αS labeled with AF488 and AF594 in buffer (20 mM Tris, 100 mM NaCl, pH 7.4). 485 nm and 560 nm lasers pulsed at 40 MHz were adjusted to 30 μW before entering the microscope. Fluorescence was collected through the objective and passed through a 100 μm pinhole. Excitation and emission were discriminated by passing the photons through a HQ585LP dichroic in combination with ET525/50M and HQ600LP filters. Signal was detected by photodiodes. Photon traces were collected in 1-ms time bins for an hour. A threshold of 30 counts/ms total in the donor and acceptor channels was used to discriminate events from noise. For each event, the energy transfer efficiency between donor and acceptor fluorophore (ETeff) was calculated in the SymPhoTime 64 software and the resulting histograms were fit using Origin (OriginLab Corp, Northampton, MA) to Gaussian distributions as described in SI.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by the National Institutes of Health (NIH NS103873 and NS102435 to E.J.P.; NS079955 to E.R.). Instruments supported by the National Science Foundation include a matrix-assisted laser desorption mass spectrometer (NSF MRI-0820996). B.P. thanks the University of Pennsylvania for support through a Dissertation Completion Fellowship.
Footnotes
The Supporting Information (SI) is available free of charge on the ACS Publications website. Detailed procedures for peptide synthesis, protein expression, ligations procedures and purification, as well as aggregation and FRET data. (PDF)
REFERENCES
- [1].Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, and Goedert M (1997) Alpha-Synuclein in Lewy Bodies, Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- [2].Li BS, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye SL, Eisenberg DS, Zhou ZH, and Jiang L (2018) Cryo-Em of Full-Length Alpha-Synuclein Reveals Fibril Polymorphs with a Common Structural Kernel, Nat. Commun 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li YW, Zhao CY, Luo F, Liu ZY, Gui XR, Luo ZP, Zhang X, Li D, Liu C, and Li XM (2018) Amyloid Fibril Structure of Alpha-Synuclein Determined by Cryoelectron Microscopy, Cell Res. 28, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lv GH, Kumar A, Huang Y, and Eliezer D (2018) A Protofilament-Protofilament Interface in the Structure of Mouse Alpha-Synuclein Fibrils, Biophys. J 114, 2811–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guerrero-Ferreira R, Taylor NMI, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, and Stahlberg H (2018) Cryo-Em Structure of Alpha-Synuclein Fibrils, eLife 7, e36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, and Rienstra CM (2016) Solid-State Nmr Structure of a Pathogenic Fibril of Full-Length Human Alpha-Synuclein, Nat. Struct. Mol. Biol 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, Arnold DB, Langen R, and Pratt MR (2015) O-Glcnac Modification Blocks the Aggregation and Toxicity of the Protein Alpha-Synuclein Associated with Parkinson’s Disease, Nat. Chem 7, 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang JL, Han XM, Leu NA, Sterling S, Kurosaka S, Fina M, Lee VM, Dong DW, Yates JR, and Kashina A (2017) Protein Arginylation Targets Alpha Synuclein, Facilitates Normal Brain Health, and Prevents Neurodegeneration, Sci Rep 7, 11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Giasson BI, Duda JE, Murray IVJ, Chen QP, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, and Lee VMY (2000) Oxidative Damage Linked to Neurodegeneration by Selective Alpha-Synuclein Nitration in Synucleinopathy Lesions, Science 290, 985–989. [DOI] [PubMed] [Google Scholar]
- [10].Hara S, Arawaka S, Sato H, Machiya Y, Cui C, Sasaki A, Koyama S, and Kato T (2013) Serine 129 Phosphorylation of Membrane-Associated Alpha-Synuclein Modulates Dopamine Transporter Function in a G Protein-Coupled Receptor Kinase-Dependent Manner, Mol. Biol. Cell 24, 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, Chiappe D, Moniatte M, Schneider BL, Aebischer P, Eliezer D, Zweckstetter M, Masliah E, and Lashuel HA (2010) Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits Alpha-Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions, J. Neurosci 30, 3184–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hejjaoui M, Butterfield S, Fauvet B, Vercruysse F, Cui J, Dikiy I, Prudent M, Olschewski D, Zhang Y, Eliezer D, and Lashuel HA (2012) Elucidating the Role of C-Terminal Post-Translational Modifications Using Protein Semisynthesis Strategies: Alpha-Synuclein Phosphorylation at Tyrosine 125, J. Am. Chem. Soc 134, 5196–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schmid AW, Fauvet B, Moniatte M, and Lashuel HA (2013) Alpha-Synuclein Post-Translational Modifications as Potential Biomarkers for Parkinson Disease and Other Synucleinopathies, Mol. Cell. Proteom 12, 3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mahul-Mellier AL, Fauvet B, Gysbers A, Dikiy I, Oueslati A, Georgeon S, Lamontanara AJ, Bisquertt A, Eliezer D, Masliah E, Halliday G, Hantschel O, and Lashuel HA (2014) C-Abl Phosphorylates Alpha-Synuclein and Regulates Its Degradation: Implication for Alpha-Synuclein Clearance and Contribution to the Pathogenesis of Parkinson’s Disease, Hum. Mol. Genet 23, 2858–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brahmachari S, Ge P, Lee SH, Kim D, Karuppagounder SS, Kumar M, Mao XB, Shin JH, Lee Y, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, and Ko HS (2016) Activation of Tyrosine Kinase C-Abl Contributes to Alpha-Synuclein-Induced Neurodegeneration, J. Clin. Invest 126, 2970–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dikiy I, Fauvet B, Jovicic A, Mahul-Mellier AL, Desobry C, El-Turk F, Gitler AD, Lashuel HA, and Eliezer D (2016) Semisynthetic and in Vitro Phosphorylation of Alpha-Synuclein at Y39 Promotes Functional Partly Helical Membrane-Bound States Resembling Those Induced by Pd Mutations, ACS Chem. Biol 16, 2428–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fan CG, Ip K, and Soll D (2016) Expanding the Genetic Code of Escherichia Coli with Phosphotyrosine, FEBS Lett. 590, 3040–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luo XZ, Fu GS, Wang RSE, Zhu XY, Zambaldo C, Liu RH, Liu T, Lyu XX, Du JT, Xuan WM, Yao AZ, Reed SA, Kang MC, Zhang YH, Guo H, Huang CH, Yang PY, Wilson IA, Schultz PG, and Wang F (2017) Genetically Encoding Phosphotyrosine and Its Nonhydrolyzable Analog in Bacteria, Nat. Chem. Biol 13, 845–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haney CM, Wissner RF, and Petersson EJ (2015) Multiply Labeling Proteins for Studies of Folding and Stability, Curr. Opin. Chem. Biol 28, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dawson PE, Muir TW, Clarklewis I, and Kent SBH (1994) Synthesis of Proteins by Native Chemical Ligation, Science 266, 776–779. [DOI] [PubMed] [Google Scholar]
- [21].Zheng JS, Tang S, Qi YK, Wang ZP, and Liu L (2013) Chemical Synthesis of Proteins Using Peptide Hydrazides as Thioester Surrogates, Nat. Protoc 8, 2483–2495. [DOI] [PubMed] [Google Scholar]
- [22].Muir TW, Sondhi D, and Cole PA (1998) Expressed Protein Ligation: A General Method for Protein Engineering, Proc. Natl. Acad. Sci. U. S. A 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen J, Wan Q, Yuan Y, Zhu J, and Danishefsky SJ (2008) Native Chemical Ligation at Valine: A Contribution to Peptide and Glycopeptide Synthesis, Angew. Chem.-Int. Edit 47, 8521–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Haney CM, Wissner RF, Warner JB, Wang YXJ, Ferrie JJ, Covell DJ, Karpowicz RJ, Lee VMY, and Petersson EJ (2016) Comparison of Strategies for Non-Perturbing Labeling of Alpha-Synuclein to Study Amyloidogenesis, Org. Biomol. Chem 14, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tanaka T, Wagner AM, Warner JB, Wang YJ, and Petersson EJ (2013) Expressed Protein Ligation at Methionine: N-Terminal Attachment of Homocysteine, Ligation, and Masking, Angew. Chem.-Int. Edit 52, 6210–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wojciechowski M, Grycuk T, Antosiewicz JM, and Lesyng B (2003) Prediction of Secondary Ionization of the Phosphate Group in Phosphotyrosine Peptides, Biophys. J 84, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Trexler AJ, and Rhoades E (2010) Single Molecule Characterization of A-Synuclein in Aggregation-Prone States, Biophys. J 99, 3048–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Trexler AJ, and Rhoades E (2009) A-Synuclein Binds Large Unilamellar Vesicles as an Extended Helix, Biochemistry 48, 2304–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Forster T (1948) *Zwischenmolekulare Energiewanderung Und Fluoreszenz, Ann. Phys.-Berlin 2, 55–75. [Google Scholar]
- [30].Sevcsik E, Trexler AJ, Dunn JM, and Rhoades E (2011) Allostery in a Disordered Protein: Oxidative Modifications to A-Synuclein Act Distally to Regulate Membrane Binding, J. Am. Chem. Soc 133, 7152–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pagan FL, Hebron ML, Wilmarth B, Torres-Yaghi Y, Lawler A, Mundel EE, Yusuf N, Starr NJ, Arellano J, Howard HH, Peyton M, Matar S, Liu X, Fowler AJ, Schwartz SL, Ahn J, and Moussa C (2019) Pharmacokinetics and Pharmacodynamics of a Single Dose Nilotinib in Individuals with Parkinson’s Disease, Pharmacol. Res. Perspect 7, e00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, Trojanowski JQ, and Lee VMY (2018) Cellular Milieu Imparts Distinct Pathological Alpha-Synuclein Strains in Alpha-Synucleinopathies, Nature 557, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Burmann BM, Gerez JA, Matečko-Burmann I, Campioni S, Kumari P, Ghosh D, Mazur A, Aspholm EE, Šulskis D, Wawrzyniuk M, Bock T, Schmidt A, Rüdiger SGD, Riek R, and Hiller S (2020) Regulation of A-Synuclein by Chaperones in Mammalian Cells, Nature 577, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Merrifield RB (1963) Solid Phase Peptide Synthesis .1. Synthesis of a Tetrapeptide, J. Am. Chem. Soc 85, 2149–2154. [Google Scholar]
- [35].Fang GM, Li YM, Shen F, Huang YC, Li JB, Lin Y, Cui HK, and Liu L (2011) Protein Chemical Synthesis by Ligation of Peptide Hydrazides, Angew. Chem.-Int. Edit 50, 7645–7649. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.