

Abstract
Adoptive T cell therapy has emerged as a promising treatment for cancer. However, it is unknown whether adoptively transferred anti-tumor T cells can form immunological memory and provide continuous protection against cancer metastasis. Herein, we used TCR transgenic Pmel-1 CD8+ T cells as a model to investigate whether early transferred Pmel-1 CD8+ T cells can generate immunological memory to prevent later melanoma metastasis. Upon stimulation with the cognate melanoma-associated hgp100 antigen, in vitro cultured Pmel-1 CD8+ T cells developed into effector T (Teff) cells that exhibited potent cytotoxic activity against B16F10 melanoma cells. Next, B16F10 melanoma cells were intravenously injected into C57BL/6 (B6) mice to establish experimental lung metastasis. In vitro generated Pmel-1 Teff cells were adoptively transferred into the mice on the same day of or three weeks prior to B16F10 cell inoculation. We found that adoptive Pmel-1 Teff cell therapy significantly inhibited the B16F10 lung metastasis and prolonged the animal survival. Importantly, Pmel-1 Teff cells transferred three weeks prior to tumor inoculation were as potent as the Pmel-1 Teff cells transferred on the same day in inhibiting melanoma metastasis. Hence, our results suggest that adoptive CD8+ Teff cell therapy generates immunological memory that continuously protect against melanoma metastasis.
Keywords: Pmel-1, CD8+ T cell, adoptive cell therapy, B16 melanoma, cancer metastasis, IVIS
Introduction
In recent years, T cell-based cancer immunotherapy has become a new pillar in cancer treatment. The major breakthroughs include the immune checkpoint blockade therapy and adoptive T-cell therapy (e.g. chimeric antigen receptor T (CAR-T) cells) [1-4]. The immune checkpoint blockades enhance the activity of endogenous anti-cancer T cells in patients, whereas in CAR-T therapy, patients’ own polyclonal T cells are engineered to express chimeric antigen receptors to achieve tumor-specific cytotoxicity. Nevertheless, current T cell-based cancer immunotherapy benefits only a fraction of cancer patients [5].
With the advances in early diagnosis and treatment of cancer, about 90% of cancer deaths are caused by metastasis rather than the primary tumors [6]. Tumor metastasis refers to the spread of cancer from its original site to other parts of the body, and can occur years or even decades after primary tumor resection. The immune system often shows the double-edge nature in controlling cancer metastasis. For instance, Natural killer (NK) cells are the major immune cell type that inhibits the metastatic process by killing circulating tumor cells [7]. By contrast, tumor-associated macrophages, tumor-associated neutrophils, myeloid-derived suppressor cells, and Foxp3+ regulatory T cells have been shown to facilitate cancer metastasis [8-10]. T cell-based cancer immunotherapy can be utilized to treat patients with metastatic cancers [11,12], but its role in preventing metastasis at later time points remains poorly investigated.
A key aspect of T cell immunity is the generation of immunological memory. Following exposure to antigen, antigen-specific naïve T cells are clonally selected, expanded, and developed into effector T (Teff) cells. After antigen clearance, the majority of Teff cells die and a small portion of them become memory T (Tmem) cells, which provide long-term protection against re-encountered antigens [13]. Compared with short-lived Teff cells, Tmem cells exhibit “stemness” features and have the potential to self-renew and differentiate into Teff cells [14]. Adoptive transfer of different types of Tmem cells have been shown to inhibit the primary tumor progression in experimental models [15,16]. However, little is known whether adoptive T cell therapy can generate immunological memory and provide continuous protection against cancer metastasis.
In the current study, we first showed that resection of primary B16F10 melanoma inhibited the growth of later re-challenged B16F10 melanoma in the same mouse, in support of anti-tumor immunological memory. Next, WT B6 mice were adoptively transferred with anti-melanoma Pmel-1 CD8+ Teff cells either on the same day of or 3 weeks before intravenous injection of B16F10 cells. Pmel-1 CD8+ Teff cells transferred 3 weeks prior to B16F10 cell inoculation were as potent as Pmel-1 CD8+ Teff cells transferred on the same day in inhibiting B16F10 metastasis. Therefore, our results suggest that adoptive Teff cell therapy generates immunological memory that provides continuous protection against tumor metastasis.
Materials and methods
Mice
C57BL/6 (B6) and Pmel-1 TCR-transgenic mice (B6 background) were purchased from Jackson Laboratory (Bar Harbor, MA). CD8+ T cells in Pmel-1 mice express a transgenic TCR that recognizes the melanoma-associated antigen gp10025-33. All animal experiments in this study were approved by the Houston Methodist Animal Care Committee in accordance with institutional animal care and use guidelines.
Cell lines
The B16F10 melanoma cell line was purchased from ATCC (Manassas, VA). The B16F10-Fluc cell line was purchased from Imanis Life Sciences (Rochester, MN). B16F10-Fluc cells express the luciferase that can be detected by a non-invasive in vivo imaging system. B16F10 and B16F10-Fluc cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin at 37°C and 5% CO2.
In vitro activation of Pmel-1 T cells
Splenocytes from Pmel-1 mice were stimulated with hgp10025-33 peptide (2 µg/ml; GenScript) and murine IL-2 (10 ng/ml; PeproTech) for 24 hours in complete RPMI-1640, which was supplemented with 10% heat-inactivated FBS, 1% penicillin-streptomycin, and 0.1% 2-Mercaptoethanol.
LDH cytotoxicity assay
B16F10 target cells (1 × 104/well) were co-cultured with freshly isolated or in vitro stimulated Pmel-1 splenocytes at various effector (CD8+ Pmel-1 cells) to target cell ratios (5:1, 10:1, 20:1, 40:1) in complete RPMI-1640 in a 96-well plate for 10 hours. The plate was assayed using the Pierce LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific, 88954). Percent cytotoxicity of target cells was determined using the formula: % cytotoxicity = (Experimental value - Effector Cells Spontaneous Control - Target Cells Spontaneous Control)/(Target Cell Maximum Control - Target Cells Spontaneous Control) × 100.
Flow cytometry analysis
Fluorochrome-conjugated antibodies specific for mouse CD45 (clone 30-F11), CD8 (53-6.7), CD44 (IM7), CD69 (H1.2F3), CD25 (PC61), CD62L (MEL-14), PD1 (29F.1A12), Tim3 (B8.2C12), Lag3 (C9B7W), CXCR6 (SA051D1), KLRG1 (MAFA), IFN-γ (XMG1.2), Perforin (eBioOMAK-D), GzmB (GB11), and Ki67 (SolA15) were purchased from BioLegend or Thermo Fisher Scientific.
In brief, freshly isolated or cultured Pmel-1 splenocytes (with or without co-culture with B16F10 cells) were stained with the above antibodies and analyzed on an LSR II or Fortessa flow cytometer (BD Biosciences) by using a previously described method [17]. Dead cells were excluded from the analysis by using the Zombie Aqua Fixable Viability Kit (BioLegend). Intracellular expression of Ki67 was determined by using the Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific). For intracellular staining of cytokines or cytotoxic molecules, cells were re-stimulated for 4 hours with 50 ng/ml phorbol 12-myristate 13-acetate (Sigma-Aldrich) and 500 ng/ml ionomycin (Sigma-Aldrich) in the presence of GolgiStop (BD Biosciences). Intracellular expression of cytokines and cytotoxic molecules were determined by the Cytofix/Cytoperm solution (BD Biosciences) as previously described [17]. The data were processed by using the FlowJo v10 software (Tree Star, Inc.).
B16F10 tumor resection and re-challenge model
Wild type (WT) B6 mice were subcutaneously injected with 5 × 105 B16F10 melanoma cells in the right flank, followed by tumor resection 10 days later when tumors reached to about 8 mm in diameter. Mice in control group received sham surgery (anesthesia and skin incision) in right flank on the same day. Three weeks after tumor resection, mice were subcutaneously injected with 5 × 105 B16F10 cells in the left flank. Tumor growth was monitored daily. Tumor volume was measured by a caliper and calculated using the formula: Volume = (length × width2)/2.
B16F10-Fluc metastasis model and adoptive T cell therapy
WT B6 mice were injected intravenously with 2.5 × 105 B16F10-Fluc melanoma cells and 5 × 106 in vitro activated Pmel-1 CD8+ T cells at different time points, as indicated in the figure legends. In brief, in Teff groups, B16F10-Fluc cells and Pmel-1 CD8+ Teff cells were injected on the same day. In Tmem groups, Pmel-1 CD8+ Teff cells were injected three weeks prior to B16F10-Fluc cell inoculation. Control groups received B16F10-Fluc cell inoculation but without T cell transfer.
In vivo bioluminescence imaging was used to weekly monitor lung metastasis after B16F10-Fluc cell inoculation. In brief, mice were anesthetized with 3% isoflurane, followed by intraperitoneally injection of 150 mg/kg body weight D-luciferin (PerkinElmer). 10 minutes after substrate injection, B16F10-Fluc metastasis in mice were determined by using the IVIS Spectrum in Vivo Imaging System (PerkinElmer). Data analysis were performed using the Living Image 4.7.4 (PerkinElmer).
Statistical analysis
Data were presented by mean ± SD and calculated using the Prism 8 (GraphPad). The P values of animal survival in the B16F10-Fluc metastasis models was determined by The Log-rank test. Other measurements were performed using unpaired Student’s t-test. Differences were considered significant when P < 0.05.
Results
Resection of primary B16 melanoma establishes protective immunological memory against re-challenged B16 melanoma
To determine whether immunological memory can protect against tumor recurrence/rechallenge, we assessed the growth of re-challenged B16F10 melanoma following the resection of primary B16F10 melanoma. In brief, 5 × 105 B16F10 cells were subcutaneously injected in the right flank of B6 mice on day -31. On day -21, when tumors reached to about 8 mm in diameter, these primary tumors were surgically removed. On day 0, the memory group of mice with the resection of primary tumors were re-challenged with 5 × 105 B16F10 cells in the left flank. The control group of B6 mice were also subcutaneously injected in the left flank with 5 × 105 B16F10 cells on day 0, but without primary tumor inoculation and resection (Figure 1A). We found that the tumor growth in the memory group was significantly slower than that in the control group (Figure 1B). As shown in Figure 1C, the tumor sizes in the memory group were still exceedingly small on day 16 after re-challenge of B16 cells, whereas the mice in the control group need to be euthanized as tumors reached maximum allowable size (Figure 1C). Therefore, although B16F10 melanoma is a tumor type with relatively low immunogenicity, resection of primary B16 melanoma establishes protective immunological memory against re-challenged B16 melanoma.
Figure 1.
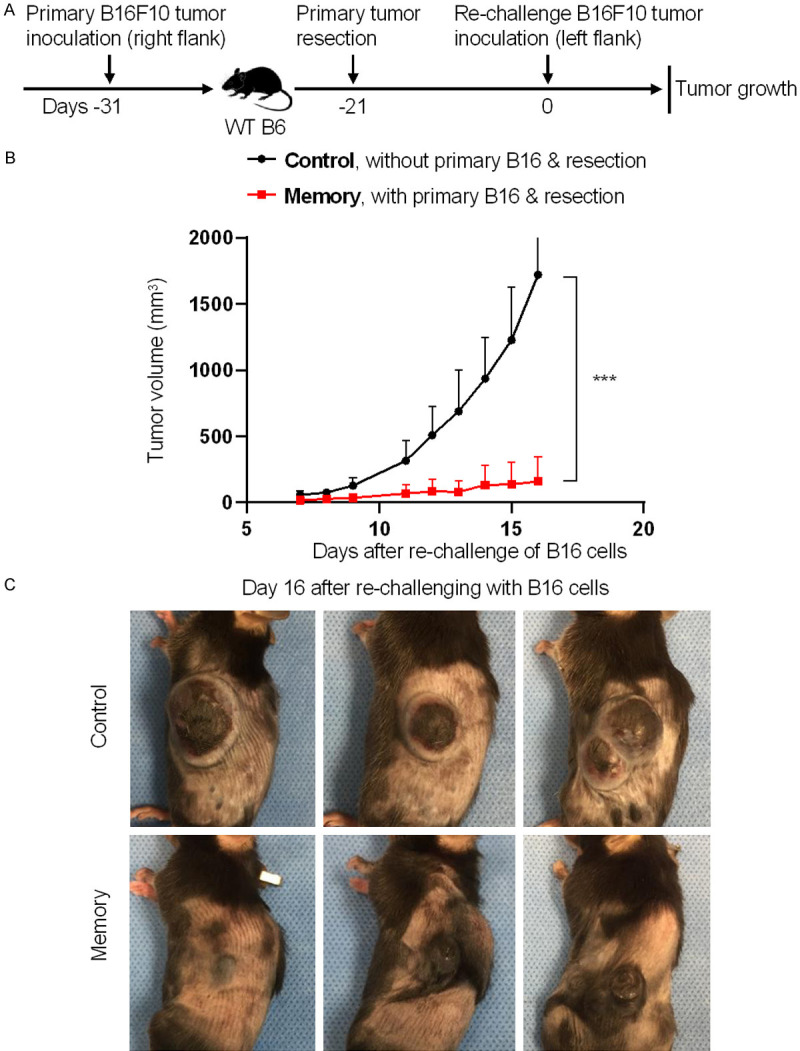
Resection of primary B16 melanoma establishes immunological memory against re-challenged B16 melanoma. 5 × 105 B16F10 cells were subcutaneously injected in the right flank of B6 mice on day -31. The primary tumors were surgically removed on day -21. On day 0, the memory group of mice with the resection of primary tumors were re-challenged with 5 × 105 B16F10 cells in the left flank. Mice in the control group were also subcutaneously injected in the left flank with 5 × 105 B16F10 cells on day 0, but without primary tumor inoculation and resection. The growth of re-challenged tumors was monitored daily. A. Schematic of the experimental design. B. Tumor volumes of the re-challenged B16 melanoma. C. Representative images of mice with tumor outgrowth on day 16 post B16 cell re-challenge. Data are presentative of two independent experiments (mean ± SD, n=5). ***P < 0.001; unpaired student’s t-test.
Activated anti-tumor Pmel-1 CD8+ T cells exhibit an effector T-cell phenotype and mediate potent cytotoxicity against B16 melanoma cells
To explore the potential of immunological memory in treating cancers, we employed the adoptive T-cell therapy approach. TCR transgenic Pmel-1 CD8+ T cells from Pmel-1 mice recognize the melanoma-associated antigen gp10025-33, and are commonly used to study the efficacy and mechanisms of adoptive T-cell therapy. Herein, we first characterize the in vitro activation and cytotoxic function of Pmel-1 cells. Splenocytes from Pmel-1 mice were stimulated in vitro with hgp10025-33 peptide for 24 hours, followed by flow cytometry and cytotoxic assay analyses (Figure 2A). Almost all CD8+ T cells were sufficiently activated as indicated by the high expression of early activation marker CD69 and CD25 (Figure 2B). Freshly isolated CD8+ T cells expressed significantly lower level of CD69 than did activated CD8+ T cells (Figure 2C).
Figure 2.

In vitro activated Pmel-1 CD8+ T cells mediate potent cytotoxic activity against B16F10 cells. Pmel-1 splenocytes were stimulated with hgp10025-33 peptide for 24 hours, followed by flow cytometry and cytotoxic assay analyses. Freshly isolated Pmel-1 splenocytes were used as control cells. A. Schematic of the experimental design. B. Gating strategies for analyzing CD8+ T cells. C. % CD69 expression of CD8+ T cells. D. The freshly isolated or in vitro stimulated Pmel-1 splenocytes were co-cultured with B16F10 target cells at different effector (CD8+ cells) to target cell ratio for 10 hours, followed by LDH cytotoxicity analysis. The graph shows % cytotoxicity of target cells. Data are presentative of two independent experiments (mean ± SD, n=3). ***P < 0.001; unpaired student’s t-test.
To investigate the cytotoxic activity of Pmel-1 CD8+ T cells against B16 melanoma, we used the LDH cytotoxic assay which measures the amount of LDH released from target cells. The freshly isolated or the above activated Pmel-1 CD8+ T cells were co-cultured with B16F10 target cells at different ratio for 10 hours, followed by calculating the % cytotoxicity of target cells. Activated Pmel-1 CD8+ T cells exhibited potent cytotoxic activity even at low effector/target ratios, whereas the freshly isolated Pmel-1 CD8+ T cells displayed significantly lower cytotoxic activity (Figure 2D).
To investigate why activated Pmel-1 CD8+ T cell mediate potent cytotoxic activity, the freshly isolated or the activated Pmel-1 CD8+ T cells were co-cultured with B16F10 target cells at an effector-to-target ration of 40:1 for 10 hours, followed by flow cytometry analysis. The activated CD8+ T cells highly expressed the activation marker CD44 and displayed a shift toward CD44+CD62L-, whereas the majority of CD8+ T cells in the freshly-isolated group were CD44-CD62L+ (Figure 3A and 3B). Compared with the freshly-isolated group, CD8+ T cells from activated group expressed significantly higher levels of effector T (Teff) cell markers CXCR6 and KLRG1, proliferation marker Ki67, inflammatory cytokine IFN-γ, and cytotoxicity molecules perforin and granzyme B (Figure 3C-F). Activated CD8+ T cells transiently express inhibitory receptors. Indeed, CD8+ T cells from the activated group but not freshly-isolated group expressed the inhibitor receptors PD-1, Tim-3, and Lag-3 (Figures 3C and 3D, S1). Taken together, activated anti-tumor Pmel-1 CD8+ T cells exhibit a Teff cell phenotype and mediate potent cytotoxicity against B16 melanoma cells.
Figure 3.

Pmel-1 CD8+ displays an effector T cell phenotype during killing of B16F10 cells. The freshly isolated or the activated Pmel-1 splenocytes were co-cultured with B16F10 target cells at a CD8+ effector to target ration of 40:1 for 10 hours, followed by flow cytometry analysis. A. Representative plots of CD44 and CD62L expressions on CD8+ T cells. B. Quantitation of % CD44+ cells among Pmel-1 CD8+ cells. C. Histograms show CXCR6, KLRG1, PD-1 and Ki67 expressions on Pmel-1 CD8+ T cells. D. Bar graphs display mean fluorescence intensity (MFI) of CXCR6, KLRG1, PD-1 and Ki67 of Pmel-1 CD8+ T cells; E. Representative contour plots show % IFN-γ+ and % perforin+GzmB+ cells among Pmel-1 CD8+ T cells; F. Quantitation of % IFN-γ+ and % perforin+GzmB+ cells among CD8+ T cells. Data are presented by mean ± SD (n=5). ***P < 0.001; unpaired student’s t-test.
Adoptive transfer of Pmel-1 CD8+ Teff cells inhibits B16F10 metastasis
Metastasis is the leading cause of death in cancer. Therefore, we next determined the anti-metastasis efficacy of in vitro generated Pmel-1 CD8+ Teff cells. WT B6 mice were intravenously injected with 0.25 × 106 B16F10-Fluc cells, and adoptively transferred with (Teff group) or without (control group) 5 × 106 in vitro generated Pmel-1 CD8+ Teff cells on the same day. Animal survival was monitored daily. Tumor metastasis was assessed on day 24 in euthanized mice or monitored weekly by using the IVIS Spectrum in vivo imaging system (Figure 4A). Mice in the Teff group survived significantly longer than control mice (Figure 4B). At 24 days post B16F10-Fluc cell injection, there were numerous metastatic nodules on the lung surfaces of control mice, whereas metastatic nodules were not detected in the Teff group (Figure 4C). Consistently, the IVIS Spectrum images showed that mice in the control group but not in the Teff group developed obvious lung metastasis on day 21. One mouse in the Teff group did not develop obvious metastasis even on day 49 (Figure 4D). Therefore, adoptive transfer of Pmel-1 CD8+ Teff cells inhibits B16F10 metastasis.
Figure 4.

Adoptive transfer of Pmel-1 CD8+ Teff cells inhibits melanoma metastasis. WT B6 mice were intravenously injected with 0.25 × 106 B16F10-Fluc cells, and adoptively transferred with (Teff group) or without (control group) 5 × 106 in vitro generated Pmel-1 CD8+ Teff cells on the same day. Melanoma metastasis and animal survival was monitored. A. Schematic of the experimental design. B. Animal survival after B16F10-Fluc cell injection. C. Images show metastasis nodules on lungs on day 24 post B16F10-Fluc cell inoculation. D. Luminescence images of mice on days 14, 21, 35 and 49 post B16F10-Fluc cell inoculation. Data are presentative of two independent experiments (mean ± SD, n=5). **P < 0.01; log-rank test.
Adoptive transfer of Pmel-1 CD8+ Teff cells generates immunological memory against B16F10 metastasis
We next used two models to determine whether adoptive Pmel-1 Teff cell transfer generates immunological memory against B16F10 metastasis. In the first model, WT B6 mice were adoptively transferred with 5 × 106 in vitro generated Pmel-1 CD8+ Teff cells, and intravenously injected with 0.25 × 106 B16F10-Fluc cells either on the same day (Teff group) or 21 days later (Tmem group). Control WT B6 mice did not receive T-cell transfer, and were injected with B16F10-Fluc cells on the same day as either the Teff group (Control-A group) or the Tmem group (Control-B group). Post B16F10-Fluc cell injection, animal survival was monitored daily and tumor metastasis was assessed weekly by using the IVIS Spectrum imaging system (Figure 5A). Almost all mice in both control groups died within 30 days after injection of B16F10-Fluc cells. Mice in Teff and Tmem groups had comparable survival curves and survived significantly longer than mice in control groups (Figure 5B). Obvious lung metastasis was detected in control groups but not in Teff and Tmem groups on day 21 post B16F10-Fluc cell injection. Even on day 35, some mice in Teff and Tmem groups did not develop obvious lung metastasis (Figure 5C).
Figure 5.

Early adoptive Pmel-1 CD8+ Teff cell transfer inhibits later melanoma metastasis (model 1). WT B6 mice were adoptively transferred with 5 × 106 in vitro generated Pmel-1 CD8+ Teff cells, and intravenously injected with 0.25 × 106 B16F10-Fluc cells either on the same day (Teff group) or 21 days later (Tmem group). Control WT B6 mice did not receive T-cell transfer, and were injected with B16F10-Fluc cells on the same day as either the Teff group (Control-A group) or the Tmem group (Control-B group). Post B16F10-Fluc cell injection, animal survival and tumor metastasis were assessed. A. Schematic of the experimental design. B. Animal survival post B16F10-Fluc cell injection. C. Luminescence images of mice on days 14, 21, 35 and 49 post B16F10-Fluc cell inoculation. Data are presentative of two independent experiments (mean ± SD, n=5). **P < 0.01; log-rank test.
To avoid variance in B16F10 cell conditions, we developed a second model in which all groups of WT B6 mice were intravenously injected with 0.25 × 106 B16F10-Fluc cells on day 0. The Teff group or Tmem group of mice were adoptively transferred with 5 × 106 Pmel-1 CD8+ Teff cells either on day 0 or day -21, receptively. The control group did not receive T-cell transfer and were only injected with B16F10-Fluc cells (Figure 6A). Compared to the survival in control group, the survival in both Teff and Tmem groups was significantly prolonged. However, there was no significant difference in survival between Teff and Tmem groups (Figure 6B). Severe lung metastasis was observed by in vivo imaging in the control group but not in Teff and Tmem groups on day 21 post B16F10-Fluc cell injection. Indeed, in both Teff and Tmem groups, lung metastasis was dramatically delayed (Figure 6C). Taken together, adoptive transfer of Pmel-1 CD8+ Teff cells generates effective immunological memory that provides continuous protection against B16F10 metastasis.
Figure 6.

Early adoptive Pmel-1 CD8+ Teff cell transfer inhibits later melanoma metastasis (model 2). WT B6 mice were intravenously injected with 0.25 × 106 B16F10-Fluc cells on day 0. The Teff group or Tmem group of mice were adoptively transferred with 5 × 106 Pmel-1 CD8+ Teff cells either on day 0 or day -21, receptively. The control group did not receive T-cell transfer and were only injected with B16F10-Fluc cells. A. Schematic of the experimental design. B. Animal survival post B16F10-Fluc cell injection. C. Luminescence images of mice on days 21, 28, 35 and 49 post B16F10-Fluc cell inoculation. Data are presentative of two independent experiments (mean ± SD, n=5). **P < 0.01; log-rank test.
Discussion
Adoptive T cell therapy offers a novel treatment option for cancer patients, which leads to a new era for tumor immunology research. In this study, we adoptively transferred Pmel-1 CD8+ Teff cells into mice either three weeks prior to or on the same day of melanoma cell inoculation. At both experimental time points, adoptive Pmel-1 Teff cell therapy significantly inhibited melanoma metastasis and prolonged animal survival. These results highlight the potential of using adoptive T cell therapy to provide continuous protection against cancer metastasis.
Cancer metastasis accounts for the majority of cancer-associated deaths [18]. Many efforts have been made to define how different immune cell types inhibit or facilitate cancer metastasis [10,19]. For instance, NK cells are the major immune cell type that inhibit cancer metastasis through directly killing circulating cancer cells [20]. CD8+ T cells also inhibit cancer metastasis, evident by the fact that depletion of CD8+ T cells results in an increase in metastasis [21]. Moreover, tumor associated macrophages and neutrophils, myeloid-derived suppressor cells, and Foxp3+ regulatory T cells have been shown to facilitate cancer metastasis possibly through inhibiting the function of NK and CD8+ T cells [10]. Nevertheless, it was previously unknown whether anti-tumor CD8+ cells can be used therapeutically to prevent metastasis. A novel finding of the current study was that adoptive CD8+ T cell therapy potently inhibited melanoma metastasis.
Pmel-1 CD8+ T cells are commonly used to investigate the new strategies and mechanisms for adoptive T cell therapy [22,23]. To effectively inhibit the growth of subcutaneous B16 melanoma, multiple approaches need to be combined to maximally increase the activity of adoptively transferred Pmel-1 cells. Such approaches include in vitro activation of Pmel-1 cells prior to cell transfer, using lymphopenic or irradiated hosts, and administration of the hosts with vaccination and IL-2 [24,25]. Different from subcutaneous melanoma, here we found that adoptive transfer of Pmel-1 Teff cells alone into WT mice was sufficient to inhibit the metastasis of intravenously injected melanoma cells. Hence, it is possible that circulating cancer cells are more susceptible to adoptive CD8+ Teff cell therapy than that of local solid tumors. Indeed, current adoptive CAR-T cell therapies also more effectively eliminate hematological cancer cells than they do against solid tumors [4,26]. More investigations are urgently required to define why adoptive T cell therapies exhibit different efficacies against hematological versus solid tumors.
An important finding of the current study was that adoptive Pmel-1 Teff cell transfer three weeks prior to melanoma cell inoculation still significantly inhibited melanoma metastasis. This finding represents a proof of concept that adoptive T cell therapy generates immunological memory to continuously protect against cancer metastasis. Of note, use of cell engineering techniques (e.g., c-Jun overexpression) effectively increases the function of adoptively transferred anti-tumor T cells [27]. Similar techniques can be applied in future to extend the function of therapeutic T cells in preventing cancer metastasis.
In summary, the current study found that adoptive Teff cell therapy provided continuous protection against melanoma metastasis. Cancer metastasis leads to most cancer-associated deaths. In light of the rapid developments of adoptive T cell therapy for cancer (e.g., continuous inventions of CAR-T and engineered TCR therapies), it is important to recognize and emphasize the potential of adoptive T cell therapy for preventing cancer metastasis.
Acknowledgements
This study was supported by the Career Cornerstone Award from Houston Methodist Research Institute (to W.C.) and by National Institute of Health (NIH) National Cancer Institute R01CA211861 and Houston Methodist Research Institute start-up funding (to B.H.). The authors would like to thank the Houston Methodist Flow Cytometry Core and PreClinical Imaging (Small Animal) Core Facility for excellent services.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, Gao L, Wen Q, Zhong JF, Zhang C, Zhang X. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13:86. doi: 10.1186/s13045-020-00910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, Rao A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567:530–534. doi: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16:151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 6.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32:135–154. doi: 10.1016/j.ccell.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 8.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirström K, West BL, Coussens LM. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, Deng J, Xu M, Briest S, Biragyn A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69:5996–6004. doi: 10.1158/0008-5472.CAN-08-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, Tian T, Wei Z, Madan S, Sullivan RJ, Boland G, Flaherty K, Herlyn M, Ruppin E. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24:1545–1549. doi: 10.1038/s41591-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, Bracarda S, Stadler WM, Donskov F, Lee JL, Hawkins R, Ravaud A, Alekseev B, Staehler M, Uemura M, De Giorgi U, Mellado B, Porta C, Melichar B, Gurney H, Bedke J, Choueiri TK, Parnis F, Khaznadar T, Thobhani A, Li S, Piault-Louis E, Frantz G, Huseni M, Schiff C, Green MC, Motzer RJ IMmotion151 Study Group. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019;393:2404–2415. doi: 10.1016/S0140-6736(19)30723-8. [DOI] [PubMed] [Google Scholar]
- 13.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 14.Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, Drexler I, Hofer T, Riddell SR, Busch DH. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. 2014;41:116–126. doi: 10.1016/j.immuni.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, Mejias-Perez E, Esteban M, Melero I, Hidalgo A, Sancho D. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. 2017;8:16073. doi: 10.1038/ncomms16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang JY, Zhao YL, Lv YP, Cheng P, Chen W, Duan M, Teng YS, Wang TT, Peng LS, Mao FY, Liu YG, Fu XL, Yu PW, Luo P, Zhang WJ, Zou QM, Zhuang Y. Modulation of CD8(+) memory stem T cell activity and glycogen synthase kinase 3beta inhibition enhances anti-tumoral immunity in gastric cancer. Oncoimmunology. 2018;7:e1412900. doi: 10.1080/2162402X.2017.1412900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Zhang H, Shi X, Xiao X, Fan Y, Minze LJ, Wang J, Ghobrial RM, Xia J, Sciammas R, Li XC, Chen W. Ablation of transcription factor IRF4 promotes transplant acceptance by driving allogenic CD4(+) T cell dysfunction. Immunity. 2017;47:1114–1128. e1116. doi: 10.1016/j.immuni.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 19.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lengagne R, Graff-Dubois S, Garcette M, Renia L, Kato M, Guillet JG, Engelhard VH, Avril MF, Abastado JP, Prevost-Blondel A. Distinct role for CD8 T cells toward cutaneous tumors and visceral metastases. J Immunol. 2008;180:130–137. doi: 10.4049/jimmunol.180.1.130. [DOI] [PubMed] [Google Scholar]
- 22.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, Hu J, Hocker JD, Hawk NV, Kapoor V, Telford WG, Gurusamy D, Yu Z, Bhandoola A, Xue HH, Roychoudhuri R, Higgs BW, Restifo NP, Bender TP, Ji Y, Gattinoni L. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nat Immunol. 2019;20:337–349. doi: 10.1038/s41590-018-0311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer DC, Balasubramaniam S, Hanada K, Wrzesinski C, Yu Z, Farid S, Theoret MR, Hwang LN, Klebanoff CA, Gattinoni L, Goldstein AL, Yang JC, Restifo NP. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanada KI, Yu Z, Chappell GR, Park AS, Restifo NP. An effective mouse model for adoptive cancer immunotherapy targeting neoantigens. JCI Insight. 2019;4:e124405. doi: 10.1172/jci.insight.124405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuca G, Reppel L, Landoni E, Savoldo B, Dotti G. Enhancing chimeric antigen receptor T-cell efficacy in solid tumors. Clin Cancer Res. 2020;26:2444–2451. doi: 10.1158/1078-0432.CCR-19-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, Nagaraja S, Granja J, de Bourcy CFA, Majzner R, Satpathy AT, Quake SR, Monje M, Chang HY, Mackall CL. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.