

Abstract
Mitochondrial dysfunction plays a critical role in the pathophysiology of Parkinson’s disease (PD). The inner mitochondrial membrane (IMM) protein, Mitofilin or Mic60, has been shown to play a key role in controlling and maintaining mitochondrial cristae morphology, and its dysregulation induces cyto-deleterious effects. Here, we investigated the mechanism underlying Mitofilin degradation in dopaminergic neuron death using N27-A cells, and Human Dopamine Neuronal Primary cells treated with PD stressors, Dopamine (DA) or Rotenone (Rot). We found that both PD stressors increased mitochondrial Parkin translocation and interaction with Mitofilin that promotes Mitofilin degradation via ubiquitination, which is responsible for reduced mitochondrial membrane potential and increased ROS production. These effects were concomitant with abnormal mitochondrial structure and increased neuronal death. DA-induced degradation of Mitofilin enhances mitochondrial calpain activity, increases the release of AIF into the cytosol, and promotes apoptosis via an AIF-PARP dependent mechanism. We found that Rot-treated cells exhibit excessive mitophagy, while DA does not trigger mitophagy. In addition, overexpressing USP30, a mitochondrial deubiquitinase, attenuated cell death induced by Rot, but not by DA-treated cells. Together, our study reveals the impact of Parkin-Mitofilin interaction in PD stressor-induced neurotoxicity, which leads to the degradation of Mitofilin, resulting in mitochondrial structural damage and dysfunction that is responsible for neuronal death by apoptosis via an AIF-PARP pathway.
Keywords: Parkinson’s disease (PD), dopamine (DA), rotenone (Rot), Mitofilin/Mic60, Parkin, ubiquitination, mitochondrial dysfunction, mitophagy, cell death
Introduction
Parkinson’s disease (PD) is a neurodegenerative disease characterized by the gradual and preferential loss of dopaminergic neurons in the substantia nigra pars compacta in the midbrain. Dopaminergic neurons release dopamine, which is a neurotransmitter important for movement, cognition, and motivation [1]. The resultant loss of dopaminergic neurons leads in dopamine deficiency in the basal ganglia, which is the target region of these neurons, affecting the neuronal circuitry, and ultimately leading to the classical parkinsonian motor symptoms, and cognitive decline observed in PD patients [1,2]. Several genetic mutations that lead to PD have been identified [3]; however, the majority of PD cases are mostly sporadic or idiopathic with heterogeneous etiology [4]. Environmental parkinsonian neurotoxins or PD stressors, especially pesticides and herbicides, including rotenone (Rot) and 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP), the byproduct of a synthetic analgesic opioid 1-methyl-4-propionoxypiperidine (MPPP) have been shown to induce dopaminergic neuron death [5-7]. Surprisingly, dopamine leakage into the cytosol or impaired dopamine homeostasis may lead to mitochondrial dysfunction and subsequent death of dopaminergic neurons [8,9], pointing to a crucial role of mitochondria in dopaminergic neurons. Dopamine packaged in vesicles prior to release is protected from oxidation; however, reuptake of excess dopamine from terminal buttons through the dopamine transporter (DAT) could result in free cytosolic dopamine that undergoes spontaneous oxidation to generate semiquinonic and quinonic species (DAQ), which can act as an endogenous toxin [9,10].
Mitochondrial dysfunction in dopaminergic neurons is a widely accepted phenomenon in the pathology of both idiopathic and familial Parkinson’s disease. Several studies have demonstrated that exogenous PD stressors, most of which are mitochondrial toxins, induce mitochondrial dysfunction via targeting electron transport chain protein complexes [5,11,12]. Such dysfunctions include abnormal mitochondrial structure, decreased complex I activity, increased reactive oxygen species (ROS) generation, decreased ATP production, and impaired mitochondrial sensitivity to calcium overload [13]. More so, proteomic analysis revealed that proteins involved in various cellular processes, such as mitochondrial function, energetic metabolism, cytoskeleton structure regulation, protein synthesis, and neuronal plasticity are altered in SH-SY5Y cells and rat brains after exogenous dopamine exposure [14-16]. Brain mitochondrial exposure to DAQ leads to covalent modification and rapid loss of the inner mitochondrial membrane protein (Mitofilin), opening of mitochondrial permeability transition pore, and ultimately dopaminergic neuron degeneration [15,16]. However, the exact mechanism by which Mitofilin is degraded and how it initiates dopaminergic neuron death remains unknown.
Mitofilin, or Mic60 is a ubiquitously expressed mitochondrial protein that is critical in the mitochondrial contact site and cristae organizing system (MICOS) assembly, which determines mitochondrial morphology and mitochondrial DNA (mtDNA) organization [17]. Mitofilin reduction has been implicated in many disease conditions, including Parkinson’s disease, diabetes, Down’s syndrome, and ischemic heart damage [18,19]. We have recently shown that Mitofilin knockdown by siRNA in H9c2 myoblasts and HEK293 cells causes cell death by apoptosis via an AIF-PARP mechanism [20]. Others and we have also shown that the downregulation of Mitofilin results in the disruption of MICOS organization, which led to mitochondrial membrane potential dissipation, increased reactive oxygen species (ROS) generation, and subsequently causing cell death [17,20,21]. Here, we will determine the impact of Parkin-Mitofilin interaction in PD stressors-induced cell death.
Parkin is an E3 ubiquitin ligase that covalently attaches ubiquitin to specific substrates [22]. Several mutations in the Parkin gene have been associated with autosomal recessive PD [23,24]. Parkin KO mice have been reported not to exhibit dopaminergic neuron loss, and the administration of MPTP, or 6-hydroxydopamine (6-OHDA) to Parkin KO mice, did not enhance dopaminergic neuron loss [25,26]. Also, Parkin upregulation in stress conditions has been associated with increased cell death [27], while transient overexpression of Parkin in several toxic conditions conferred multivalent protective functions [28]. These findings suggest a complex and misunderstood contribution of Parkin in the mechanism of PD. Therefore, it is important to clarify the exact function of Parkin in the mechanism of pathogenesis of PD. Parkin has been revealed to play an important role in mitophagy, a process that removes damaged or dysfunctional mitochondria [29,30]. Since mitochondrial dysfunction is a major hallmark in the pathogenesis of PD, this has widened the scope of the role of Parkin in PD pathogenesis. Here, we postulate that Parkin physically interacts with Mitofilin in the IMM and causes Mitofilin degradation via ubiquitination. We propose that Mitofilin might act as a novel substrate for Parkin and that Parkin-Mitofilin interaction may play a key role in mitochondrial dysfunction and dopaminergic Neuron death observed in PD.
In this study, using differentiated N27-A dopaminergic cell line and Human Dopamine Neuronal Primary cells, we investigated the mitochondrial-dependent mechanism by which PD stressors (Dopamine and Rotenone) induce dopaminergic neuron death. We report that both stressors increase Parkin levels in mitochondria where it interacts with Mitofilin, and increases its degradation by ubiquitination. Mitofilin loss is believed to cause mitochondrial structural damage that is responsible for increased ROS production and dissipation of mitochondrial membrane potential. We revealed here that DA-induced increased Parkin-Mitofilin interaction promotes dopaminergic neuron death via an AIF-PARP dependent mechanism. In contrast, Rot-induced elevation of Parkin levels in mitochondria causes Mitofilin loss that subsequently promotes neuron death via an increase in mitochondrial clearance by excessive mitophagy.
Materials and methods
Institutional approval
Protocols followed the Guide for the Care, and Use of Laboratory Animals (US Department of Health, NIH), and received UT Health Science Center at San Antonio Institutional Animal Care and Use Committee (IACUC) institutional approval. Animals were housed in the animal specific pathogen free facility at UTHSCSA main campus in cages with standard wood bedding and space for five mice. The animals had free access to food and drinking water and a 12-hour shift between light and darkness. The animals were selected randomly, and the data analysis was performed by a blinded investigator.
Cell culture and differentiation
N27-A Rat Dopaminergic Neural Cell Line was obtained from Dr. Curt Freed (UC Denver). N27-A cells were cultured in RPMI 1640 medium (Gibco™ 11875093) supplemented with 10% fetal bovine serum (FBS; GIBCO-BRL, Grand Island, NY), 2 mM L-glutamine, and 100 U/ml penicillin and streptomycin (dose). Cells were differentiated according to manufacturer’s prescription. Briefly, Cells at 75% confluency were differentiated by replacing culture medium with RPMI medium supplemented with 60 µg/mL Dehydroepiandrosterone (DHEA) (Sigma D063) and 2 mg/mL dibutyryl cyclic AMP (Sigma D0260) for 3 days at 37°C. Human Dopamine Neuronal Primary Cells (HDNP) were purchased from Celprogen (Cat# 36064-01). Cells were cultured on pre-coated flasks with Human Dopamine Neuronal Primary Cell Culture Extra-cellular Matrix plates (Cat# E36064-01-T25) using Dopamine Neuronal Primary Cell Culture Media (Cat# M36064-01). Cells were grown in an atmosphere of 5% CO2-95% humidified air at 37°C and were used between passage 4 and 7, at 80% confluence. Cells were regularly tested for mycoplasma using Lookout Mycoplasma PCR detection kit (MP0035, Sigma).
Treatment of cells with PD stressors (DA and Rot)
Cultured N27-A and HDNP cells were incubated for 24 hours in the presence of DA (250 μM) (Dopamine HCl, Sigma; H8502-5 g), or Rot (1 μM) (Rotenone, Sigma 45656-250 mg), or Vehicle (Veh) (0.01% DMSO in media). 24 hours post treatment; cells were trypsinized and collected for further applications.
Flow cytometry analysis of cell death
Cell death was determined using Annexin V-PE/7-AAD Apoptosis Detection Kit (BD Bioscience, BD PharMingen, catalog no. 556547) according to the manufacturer’s instructions with Annexin V PE replaced with Annexin V APC dye. Cells treated with Veh, DA or Rot were washed twice in PBS and resuspended in 400 μl of 1× binding buffer. The cells were stained with 5 μl APC-conjugated Annexin V (BD PharMingen, catalog no. 550475) and 7-aminoactinomycin-D (7-AAD; BD PharMingen, catalog no. 559763) and analyzed using a BD LSR II flow cytometer (BD Biosciences, San Jose, CA, UTHSCSA flow cytometry core).
Cell viability
Cell viability was determined using the tetrazolium dye 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay by following standard protocols. Briefly, cells were cultured in a 96 well plate and treated with either Veh, DA, Rot as described above. At the end of the treatment, cells were placed in 50 µL of serum-free media supplemented with 50 µL of MTT solution into each well for 3 h incubation. After that, 150 µL of DMSO was added into each well before absorbance was read at 590 nm.
Mitochondria isolation from mice brain
Methods described in [31] were adapted to isolate mitochondria from mice brains. Briefly, mice brains were excised and immediately placed in cold Krebs-Henseleit buffer. The brain tissue was transferred to isolation buffer A (mM), 210 mannitol, 70 sucrose, 1 EDTA and 50 Tris-HCl, pH 7.4. Brains were finely minced and homogenized in the same buffer A (1 ml buffer/0.15 g of tissue) using a Kontes and Potter-Elvehjem tissue grinder. The homogenate was centrifuged at 1,300×g for 3 min, and the supernatant was filtered through a cheesecloth and centrifuged at 10,000×g for 10 min. The pellet was gently washed 3 times with 500 µl of buffer B (in mM): 150 sucrose, 50 KCl, 2 KH2PO4, 5 succinic acid, and 20 Tris/HCl, pH 7.4, and resuspended with 50 µl of the same buffer. Mitochondrial protein concentration was assayed using the Bradford method.
Co-IP assays
Cells treated with Veh, DA, or Rot were incubated in the lysis buffer containing 150 mM NaCl, 10 mM HEPES, 50 mM Tris, 5 mM EDTA, 0.25% sodium deoxycholate, 0.1% octylphenyl-polyethylene glycol (IGEPAL CA-630), pH 7.4, plus Complete Protease Inhibitor Cocktail Tablets). Lysates were centrifuged (10 min, 13,000×g, 4°C), and the supernatants were precleared with 10 μl protein A/G resin/mg protein (1 h, 4°C, shaking) (Pierce Biotechnology, Inc.) and centrifuged 2 min at 2,000×g. For the co-IP of Flag-Parkin and Myc-Mitofilin agarose beads (80 µl) were conjugated with either Anti-Flag antibody (Cell Signaling Technology Cat# 14793, RRID:AB_2572291) or Anti-Myc antibody (Cell Signaling Technology Cat# 2272, RRID:AB_10692100). 1 mg protein of the precleared lysates were incubated overnight at 4°C with conjugated beads to pull down Flag-Parkin and Myc-Mitofilin, respectively in a final volume of 500 μl lysis buffer. For the endogenous Parkin and Mitofilin, 5 µg of the anti-Parkin antibody (Cell Signaling Technology Cat# 4211, RRID:AB_2159920) and anti-Mitofilin antibody (Proteintech Cat# 10179-1-AP, RRID:AB_2127193) were used, respectively. The mouse purified IgG was used as a negative control. The immunoprecipitated proteins were eluted from the beads with 30 μl 3× Laemmli sample buffer (37°C, 1 h). After centrifugation (3 min, 13,000×g, 4°C), immunoprecipitated proteins, as well as lysates (input), were analyzed by SDS/PAGE and immunoblotting (Western blot).
Western blot assay
Equal amounts of protein (40 μg) were loaded in each well of 4-20% Tris-glycine gels (Bio-Rad) as recently described [32]. The gel was electrophoresed for 90 min at 125 V of constant voltage, and blotted onto a nitrocellulose membrane using electrophoretic transfer at 90 V of constant voltage for 1.5 h. The membrane was washed, blocked with 5% blocking buffer, and probed with various primary antibodies at 4°C overnight. The following antibodies: anti-Mitofilin (Proteintech Cat# 10179-1-AP, RRID:AB_2127193), anti-VDAC1 (Millipore Cat# MABN504, RRID:AB_2716304), anti-Parkin (Cell Signaling Technology Cat# 4211, RRID:AB_2159920), Total OXPHOS Rodent WB Antibody Cocktail (Abcam Cat# ab110413, RRID:AB_2629281), anti-p62 (Cell Signaling Technology Cat# 5114, RRID:AB_10624872), anti-Beclin-1 (Cell Signaling Technology Cat# 3495, RRID:AB_1903911), anti-Ubiquitin (Proteintech Cat# 10201-2-AP, RRID:AB_671515), anti-LC3B (Novus Cat# NB100-2220, RRID:AB_10003146) (Santa Cruz Biotechnology Cat# sc-365467, RRID:AB_10847086), anti-AIF (Cell Signaling Technology Cat# 5318, RRID:AB_10634755), anti-PARP (Cell Signaling Technology Cat# 9532, RRID:AB_659884), and anti-GAPDH (Cell Signaling Technology Cat# 2118, RRID:AB_561053), were used. After washing, membranes were incubated at room temperature for 1 h with the corresponding fluorophore-conjugated secondary antibodies (goat anti-mouse IR Dye 800CW, 10 ng/ml and goat anti-rabbit Alexa 680, 20 ng/ml). The total protein loading was measured by immersing the membrane in the ponceau S solution (CAS No.: 6226-79-5). The membranes were washed thrice and bands were visualized using an infrared fluorescence system (Odyssey Imaging System, Li-COR Biosciences).
Mitochondrial membrane potential (ΔΨm) measurement
ΔΨm was assessed using MitoTracker Red CMXROS assay kit (ThermoFisher Scientific, catalog no. M7512) by flow cytometry and fludorescent microscopy according to manufacturer’s protocol. N27-A cells were plated in six-well plates for MitoTracker Red assay or on coverslips for labeling and allowed to reach 70-80% confluence, after which cells were treated with vehicle, DA or Rot for 24 h. Live cells were harvested and incubated with MitoTracker Red (150 nM) for 1 h at 37°C. CCCP was used as a control.
Immunofluorescence staining
Cells were cultured on coverslips and treated as described above. After treatment, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.25% Triton X-100. After blocking in 3% BSA for 30 min. Slides were incubated with first antibody diluted in 1% BSA for overnight. After washing with PBS, coverslips were incubated with primary antibodies for Parkin, Mitofilin and Ubiquitin antibodies (Abcam Cat# ab53042, RRID:AB_2042572), (Proteintech Cat# 10179-1-AP, RRID:AB_2127193), (Proteintech Cat# 10201-2-AP, RRID:AB_671515) followed by secondary antibodies Alexa Fluor 488 Goat anti-rabbit (Abcam Cat# ab150077, RRID:AB_2630356) and Goat anti-mouse (Abcam Cat# ab150113, RRID:AB_2576208). Images was taken on a Zeiss Axiovert 200M inverted motorized fluorescence microscope (Carl Zeiss Microscope, Jena, Germany).
Mitochondrial ROS production
Mitochondrial ROS production was assessed as previously described [33]. Mitochondrial ROS generation was measured spectrofluorometrically (560-nm excitation and 590-nm emission). Around 500,000 treated cells were treated with Veh, DA or Rot for 24 h, and incubated in a solution containing: 20 mM Tris, 250 mM sucrose, 1 mM EGTA, 1 mM EDTA, and 0.15% bovine serum albumin adjusted to pH 7.4 at 30°C with continuous mixing. ROS was measured with the H2O2-sensitive dye Amplex red (10 μM) according to the manufacturer’s instructions (ThermoFisher, catalog no. A12222). H2O2 levels were measured from a calibration curve obtained from the fluorescence emission intensity as a function of H2O2 concentration. The sodium salt of glutamate/malate (3 mM) was used to activate complex I of the mitochondrial electron transport chain.
Mitochondrial structural integrity
N27-A cells treated with PD stressors were imaged by electron microscopy to observe mitochondrial quality and morphology. Cells were fixed in 2.5% (wt/vol) glutaraldehyde (Fluka), stored in the same solution at 4°C overnight. Cells were washed with PBS, fixed in 2% (wt/vol) osmium tetroxide for 2 h at room temperature. Cells were dehydrated in a graded alcohol series and embedded in Eponate 12 medium and the blocks were cured at 60°C for 48 h. Sections (70 nm) were cut with an RMC ultramicrotome and mounted on Formvar-coated grids. The sections were double-stained with uranyl acetate and lead citrate, and finally examined and imaged with a 100CX JEOL transmission electron microscope.
ATP assay
Intracellular ATP levels in cells treated with PD stressors were quantified using an ATP Bioluminescence Assay Kit (Roche Applied Science, catalog no. A22066) according to the manufacturer’s protocol. The luminescence of the cells was measured using a plate reader. The concentration of ATP in each group was obtained using an ATP standard curve and normalized to the protein concentrations of the samples, which were determined using the BCA assay.
Mitophagy flux
Mitophagy in mitochondria was detected by using a Mitophagy Detection Kit according to the manufacturer (Dojindo Molecular Technology, Rockville, MD, USA). This kit is composed of Mitophagy Dye, reagent for detection of mitophagy, and Lyso Dye, reagent for staining of lysosomes allowing accurate quantification of the damaged mitochondria fusing to the lysosomes. The signal was detected in N27-A cells using flow cytometry analysis at the following wavelengths: Mitophagy Dye: 561 nm (Ex) and 650 nm (Em); Lyso Dye: 488 nm (Ex) and 550 nm (Em) as previously described in [34].
siRNA and plasmid transfection
The siRNAs against rat Park2 (Parkin), rat IMMT (Mitofilin), and scrambled siRNA were purchased from Life Technologies (Parkin siRNA, catalog no. 4390771, siRNA ID: s132779; Mitofilin siRNA, catalog no. AM16708, siRNA ID: 281926; and scrambled siRNA, catalog no. AM16708, siRNA ID: 4390843). N27-A cells passage 4-7, at 70-80% confluence were transfected with either Parkin or Mitofilin siRNAs (200 nM; Parkin siRNA, sense sequence 200 nM; TTCCAAACCGGATGAGTGG-3 and antisense sequence CCACTCATCCGGTTTGGAA Mitofilin siRNA, sense sequence GGUGGUGUCUCAAUAUCAUtt and antisense AUGAUAUUGAGACACCACCtt) using Lipofectamine 3000 (ThermoFisher Scientific, catalog no. L3000015) according to the manufacturer’s instructions. Myc-USP30 plasmid (USP30 (NM_001033202) Mouse Tagged ORF Clone, Myc-Mitofilin plasmid (Immt (NM_029673)) Mouse Tagged ORF Clone, entry plasmid Myc-PCMV6 (NM_010849) Mouse Tagged ORF Clone purchased from Origene (Catalog no: MR226441, MR216091 & MR227353), Flag-Parkin plasmid (PARK2 cDNA ORF Clone), Human, N-DYKDDDDK (Flag®) tag purchased from Sino Biological (catalog no: HG12092-NF). Cells were transfected using Lipofectamine 3000 (ThermoFisher Scientific, catalog no. L3000015) according to manufacturer protocols.
Calpain activity assay
To measure calpain activity, we used the Calpain Activity Fluorometric Assay Kit (Biovision Catalog #: K240) following the supplier’s instructions. Cells treated cells with vehicle, DA or Rot collected, and suspended in Extraction buffer, lysed, and the concentration was determined. Equal concentrations of lysates were incubated with the reaction buffer, calpain substrate and/or inhibitor for 1 h at 37°C. Readings were then taken on a spectrofluorometer at the excitation/emission wavelengths of 400/505 nm.
Statistical analysis
Data presented in bar graphs are expressed as means, and error bars are the standard errors of the mean (± SEM) for a minimum of three independent trials (n ≥ 3). Comparisons were conducted using the Student’s t-test and one-way ANOVA with post-hoc Dunnett’s or Tukey’s corrections for multiple comparisons, where appropriate, using Prism 8 (Graphpad Software). A difference of P < 0.05 was considered to be statistically significant.
Results
Parkinsonian neurotoxins (PD stressors) induce cell death in N27-A cells
We examined whether exogenous dopamine (DA) and Rot cause an increase in differentiated N27-A cell death. N27-A cells were treated with DA (250 µM), Rot (1 µM), or vehicle (Veh) (0.01% DMSO) as a negative control. We found that PD stressors treatment for 24 hours increased N27-A cell death when compared with Veh. In fact, we observed that the level of cells in the early apoptotic stage (Annexin V+/7AAD-) was increased in the DA and Rot treated cells compared to Veh group (21.84±2.52% in DA and 29.29±4.42% in Rot, respectively, Figure 1A and 1B). Further, DA and Rot increased cell death (Annexin V+/7AAD+) by 43.15±4.8% and 39.8±2.7%, respectively, when compared to Veh at 12.4±1.5% (n = 12/group, P < 0.05; Figure 1A and 1C). Conversely, we performed MTT assay, which determines the level of metabolic activity in eukaryotic cells and can be used as an estimate of live cells, on cells treated with these PD stressors. We found that DA and Rot reduced cell viability compared to Veh (25%±5.2% in DA and 31.26%±4.1% in Rot versus 100% in Veh, n = 9/group, P < 0.001; Figure 1E). Taken together, these results indicate that PD stressors (DA and Rot) treatment induce N27-A cell death.
Figure 1.
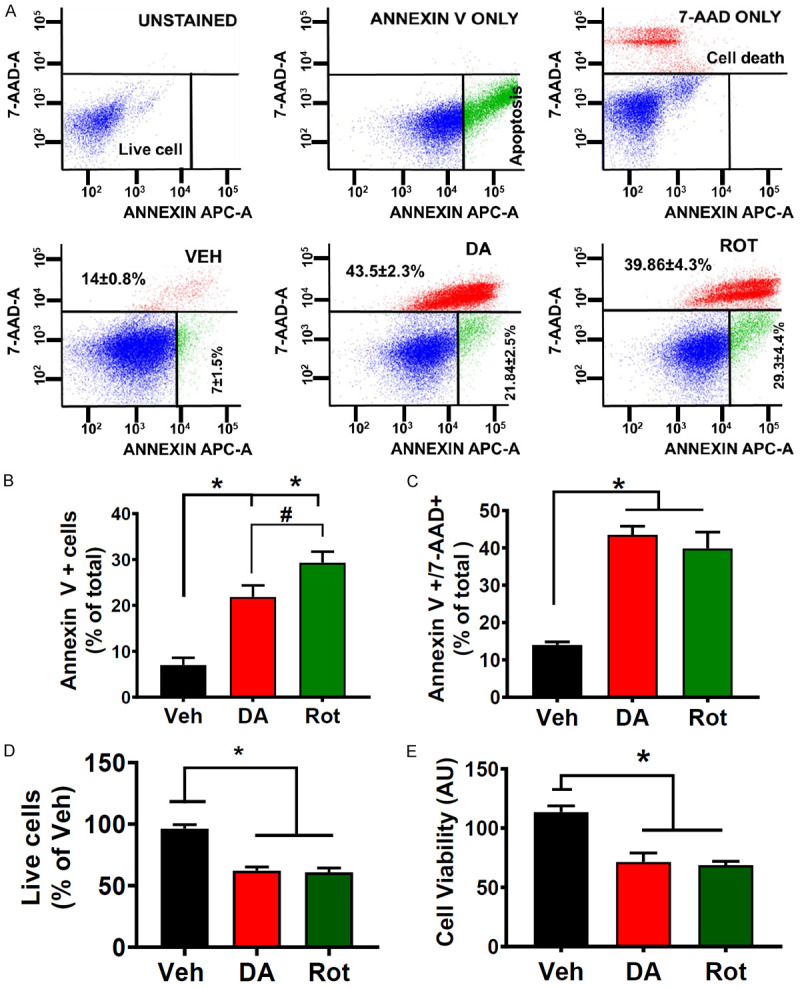
PD Stressors treatment in differentiated N27-A cells increases cell death. A. Top: Flow cytometry with unstained (blank, blue cells), Annexin V only (early apoptosis, green cells), and 7-aminoactinomycin D (7-AAD) only (dead cells and/or late apoptosis, red cells) used for calibration. Bottom: flow cytometry analysis with Annexin V/7-AAD was used to assess cell death after dopamine (250 μM) treatment, rotenone (1 μM), versus vehicle (0.01% DMSO). Values are mean ± SEM; *P < 0.001, n = 9/group. B. Bar graph showing an increase in early apoptosis (Annexin V+) in DA and Rot treated cells compared with Vehicle. Values are expressed as means ± SEM, *P < 0.001, n = 9/group. C. Bar graph showing an increase in cell death (7-AAD+) in DA and Rot treated cells compared with Vehicle (Veh). Values are expressed as means ± SEM; *P < 0.001, n = 9/group. D. Bar graph showing a reduction in live in DA and Rot treated cells compared to Vehicle Values are expressed as means ± SEM, *P < 0.001, n = 9/group. E. Bar graph showing a reduction in cell viability (MTT assay) in DA and Rot treated cells compared with sham. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group.
PD stressors increase mitochondrial structural damage, ROS production, and decrease mitochondrial membrane potential in N27-A cells
Mitochondrial dysfunction is a major hallmark in Parkinson’s disease [35]. To determine the effect of PD stressors on mitochondrial morphology and function, we measured mitochondrial membrane potential (ΔΨm), and ROS production as well as unveiled the mitochondrial structural integrity. N27-A cells were treated with PD stressors, and CCCP (10 µM), a mitochondrial oxidative phosphorylation uncoupler, known to dissipate mitochondrial membrane potential. ΔΨm was measured by fluorescence microscopy using MitoTracker Red (CMXRos) dye (150 nM), a cell-permeant dye that accumulates in mitochondria with intact membrane potentials and with flow cytometry analysis. Retention of the MitoTracker Red dye indicates high ΔΨm and serves as an indication of good mitochondrial integrity. We measured relative fluorescence in live cells that retain the MitoTracker dye. Figure 2A shows flow cytometry analysis of mitochondria from DA, Rot, and Veh groups indicating that cells treated with DA and Rot were more uncoupled compared to Veh. In fact, we found that the level of MitoTracker Red dye in mitochondria of cells treated with DA and Rot was reduced compared to Veh (37% and 46.6% in DA and Rot, respectively, versus 75% in Veh, n = 9/group *P < 0.05), (Figure 2A). We further assessed mitochondrial ΔΨm with fluorescence microscopy. In fact, we found that cells treated with DA. Rot or CCCP displayed similar reduction in fluorescence intensity of MitoTracker Red versus Veh (Figure 2B). (n = 3/group, Figure 2A and 2B).
Figure 2.
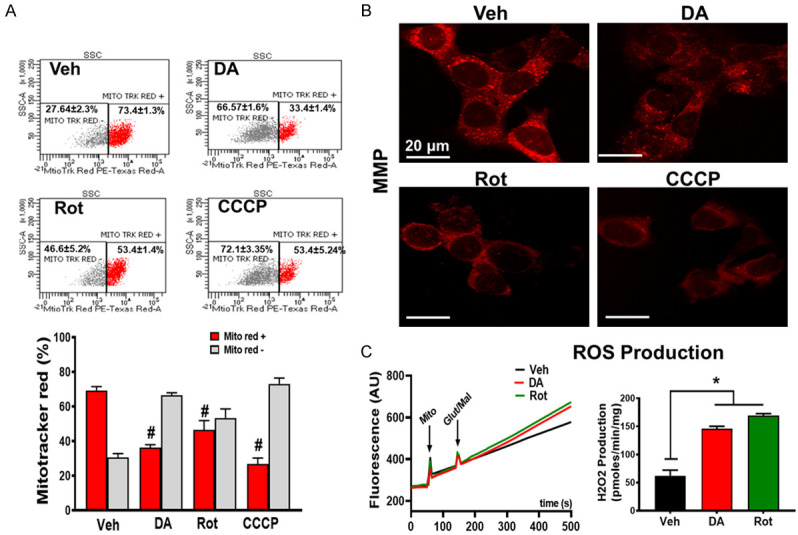
PD stressors treatment causes mitochondrial dysfunction. A. Top: Mitochondrial membrane potential (ΔΨm) was measured by flow cytometry using MitoTracker Red. MitoTracker Red positive (red) and increase in MitoTracker Red negative (grey) cells treated with DA and Rot compared with vehicle. Bottom: corresponding graph showing the reduction in ΔΨm (Red bar: MitoTracker positive cells, grey bar: MitoTracker negative cells) in PD stressor treated cells. B. Fluorescent microscope images measured by MitoTracker Red. Representative images collected from N27-A cells treated with Vehicle, DA, Rot, or CCCP (positive control). Red color denotes MitoTracker Red staining. C. Left: Representative recording of mitochondria reactive oxygen species (ROS) production in PD stressor treated cells using Amplex red in the presence of horseradish peroxidase after stimulation of complex I with glutamate/malate. Right: Graph showing increase in ROS production in N27-A cells treated with DA and Rot compared with vehicle (Veh). Values are expressed as means ± SEM of three independent experiments, *P < 0.05 (n = 5/group).
Since the reduction in ΔΨm has been implicated in increased reactive oxygen species (ROS) production [36], we queried whether PD stressor-treatment on N27-A cells increases ROS production. We found that PD stressors treatment significantly increased ROS production. In fact, ROS production in Veh treated cells was 62.33±9.82 pmol·min-1·mg protein-1 versus 145.98±4.23 pmol·min-1·mg protein-1 in DA and 168.94±3.71 in Rot treated cells (n = 6/group P < 0.05, Figure 2C). To define the effects of PD stressors on the structure and morphology of mitochondria, treated cells were collected and processed for electron microscopy imaging. We observed that mitochondria from PD stressor cells were swollen and characterized with ruptured and fragmented cristae morphology versus Veh in which cristae were mostly normal and continuous (Figure 3A and 3B). This observation indicates that PD stressors increase mitochondrial cristae damage, which is associated with reduced mitochondrial membrane potential, and increased ROS production observed in Figure 2A and 2B.
Figure 3.
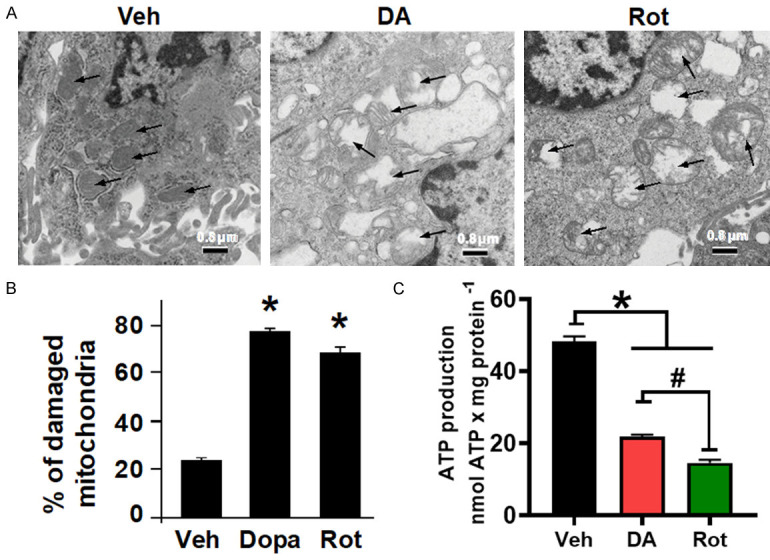
PD stressors treatment increases mitochondrial structural damage. A. Electron microscopy images of N27-A cells showing damaged mitochondria cristae after DA and Rot treatment, compared with Vehicle (Veh). B. Bar graph showing percentage of damaged mitochondria in each group. Fragmented or disrupted cristae with empty spaces (in the matrix) were considered damaged mitochondria, while mitochondria with dense continuous cristae were considered as good or undamaged. A minimum of 100 mitochondria were counted in each group (132, 107, and 116 in Veh, DA, and Rot groups, respectively). C. Bar graph showing a reduction in ATP production in cells treated with DA and Rot compared with control. Values are expressed as means ± SEM of five independent experiments, *P < 0.05, n = 5/group.
The cristae organization and structure have been shown to modulate the function of the OXPHOS system [37]. We, therefore, sought to determine whether the disruption of mitochondrial cristae morphology after PD stressor treatment perturbed ATP production. We observed that PD stressors decreased the levels of ATP production when compared with Veh-treated cells (21.83±1.61 nmol/mg-1 in DA-treated, and 14.65 nmol/mg-1 in Rot-treated cells compared to 48.32±2.135 nmol/mg-1 in Veh treated cells) (Figure 3C). Together, these results indicate that exogenous DA and Rot treatment in N27-A cells induces mitochondrial structural damage and dysfunction that is responsible for the increase in ROS production and reduction in membrane potential and ATP production.
PD stressors increase Parkin-mitochondrial co-localization
PINK1 and Parkin have been reported to be recruited in depolarized mitochondria and initiate the elimination of damaged mitochondria through mitophagy [29,30]. We performed Western blot analysis in the whole cell lysate of treated N27-A cells and found that the levels of Parkin are increased in DA and Rot treated cells compared to Veh group in both differentiated N27-A cells and HDNP cells (Figures 4A and 10D). In fact, DA and Rot treatment increased Parkin levels by 48.4±6%, and 35.8±4%, respectively, compared to Veh (100%). We also observed via fluorescence microscopy that Parkin coats the mitochondria (Figure 4B). We further determined whether the upregulation of Parkin and its mitochondrial localization is associated with an increase in mitophagy. To this end, we determined the level of mitophagy flux in cells treated with PD stressors. We observed that median fluorescence intensity for Lyso dye, which stains all the lysosomes in cells, was increased in Rot treated cells, but unchanged in DA group compared to Veh (Figure 4C). We thereafter revealed the mechanism of this mitophagy by examining changes in LC3, Beclin-1, and p62 (SQSTM1) expression. We surprisingly observed that the levels of p62, LC3BII/LC3BI ratio and Beclin-1, were only slightly (but not significantly) changed in DA treated cells (Figure 4D), indicating no change in autophagic/mitophagic flux. Although in Rot treated cells, p62 protein levels and the ratio LC3BII/LC3BI were increased, while Beclin-1 protein levels were reduced compared to Veh. These results suggest that the mitophagy process is more aggressive in Rot treated cells, but less active in DA treated cells. Consistently, the median fluorescence intensity of Mitophagy dye in Rot treated cells, while it was reduced in DA group compared to Veh (Figure 4D). These results suggest that Rot induces mitochondrial clearance via mitophagy in N27-A cells, while damaged mitochondria in DA treated cells are not removed by mitophagy.
Figure 4.
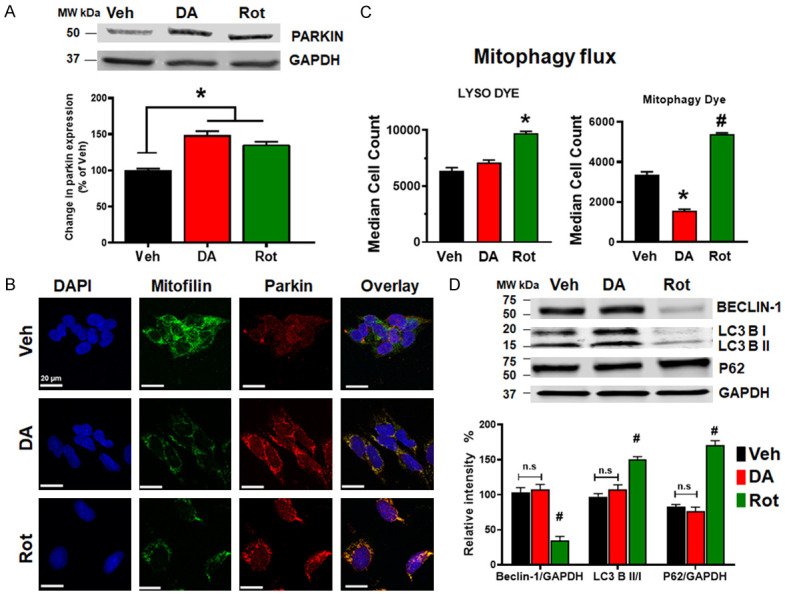
PD stressors increase Parkin levels and impact mitophagy. A. Immunoblots and bar graph (as a percent of vehicle (Veh)) showing an increase in Parkin protein level after DA and Rot treatment as compared to vehicle. B. Confocal microscopy images showing N27-A cells treated with Vehicle, DA, or Rot and labeled with DAPI (blue), Mitofilin (green) & Parkin (red) as well as the overlay. Note the increase in Parkin-Mitofilin co-localization (yellow) in DA and Rot groups. C. Bar graphs showing the median cell count of cells fluorescing with the lysosome dye, and mitophagy dye. D. Immunoblots and corresponding Bar graph showing the levels of mitophagy related proteins Beclin-1, p62/SQSTM1, and LC3B I & II. Note that both Beclin-1 and LC3B I & II levels are unchanged in Veh and DA treated cells, while Beclin-1 and LC3B I & II are reduced and p62 is increased in Rot-treated cells.
Figure 10.
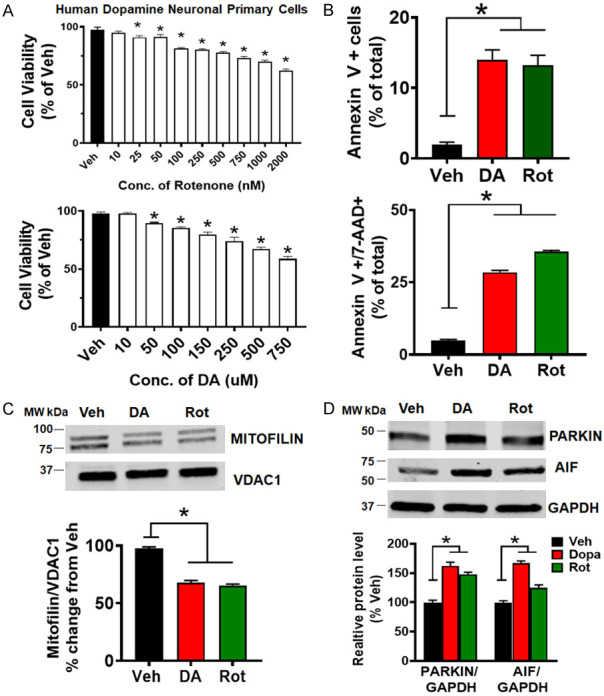
PD stressors treatment in HDNP cells produces similar results as observed in N27-A cells. A. Bar graph showing dose-response of HDNP cells to different concentrations of Rot (top) & DA (bottom) compared with Vehicle. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group. B. Top: bar graph showing an increase in early apoptosis (Annexin V+) in PD stressor treated cells versus Veh. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group. Bottom: bar graph showing an increase in cell death (Annexin V+ & 7-AAD +) in PD stressors-treated cells versus Vehicle. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group. C. Immunoblots and bar graph (as a percent of vehicle) showing a decrease in Mitofilin protein level after DA, and Rot treatment compared with Vehicle. Values are expressed as means ± SEM of three independent experiments, *P < 0.05, n = 3/group. D. Immunoblots and bar graph (as a percent of vehicle) showing an increase in Parkin and AIF protein level after DA and Rot treatment compared with Vehicle. Values are expressed as means ± SEM of three independent experiments, *P < 0.05, n = 3/group.
PD stressors increase Parkin-Mitofilin interaction
Mitofilin has been found as one of the mitochondrial proteins significantly reduced after exposure of isolated brain mitochondria, PC12 cells or SH-5YSY cells to dopamine quinone [15,16]. Consistently, Western blot (Figure 5A) and immunostaining (Figure 5C) performed in N27-A cells revealed that Mitofilin levels were reduced by 35% upon DA exposure compared to Veh. To assess whether Mitofilin reduction does not result from a general decrease in the number of mitochondria, we determined the level of other inner mitochondrial proteins such as Electron Transport Chain (ETC) proteins, Cyclophilin D (CyPD), and TOMM40. Using equal amount of mitochondrial protein, Western blot analysis showed no significant changes in the levels of these proteins after DA or Rot treatment versus Veh (Figure 5B). Given that the increase in Parkin is associated with decreased Mitofilin levels, we investigated the impact of the regulation of the link between these two proteins. To this aim, we transiently expressed Flag-tagged Parkin, or Parkin siRNA in N27-A cells and found that Parkin overexpression strongly reduced the basal levels of Mitofilin, while Parkin knockdown with siRNA increased basal Mitofilin levels, suggesting that Parkin might act upstream of Mitofilin (Figure 5D; top). We also co-expressed Myc-tagged Mitofilin and Flag-tagged Parkin in N27-A cells and performed immunoprecipitation to confirm Parkin and Mitofilin interaction. Interestingly, we found that Myc-Mitofilin co-precipitated with Flag-Parkin, which was revealed in the pulled-down by Western blot analysis (Figure 5D; bottom). We next determined whether DA and Rot increase Parkin interaction with Mitofilin in the IMM. Using immunostaining approach, we observed that Parkin co-localization with Mitofilin was increased in response to PD stressors compared to control (Figure 4B). The Pearson’s correlation coefficients were higher in PD stressors treated groups (DA = 0.36, and Rot = 0.48, n = 3/group *P < 0.05) when compared with Veh-treated cells (Veh = 0.03). To confirm this observation, we performed co-immunoprecipitation (co-IP) with Mitofilin antibody, followed by Western blot analysis with Parkin antibody in mitochondrial fractions to define whether the interaction of the Parkin with Mitofilin increases upon PD stressor treatment. We observed an increase in Parkin pulled down after DA and Rot treatment when IP was performed with Mitofilin antibody (Figure 5E). Reverse co-IP with anti-Parkin antibody showed an increase in Mitofilin in the pull-down assessed by Western blot analysis (Figure 5E). To confirm these results, we treated another cell line (Human Dopamine Neuronal Primary Cell, HDNPC) with PD stressors as described above. Consistent with our results in N27-A cells, Western blot analysis showed that Parkin protein levels were increased, and Mitofilin levels were reduced in HDNP cells treated with DA and Rot compared to cells treated with Veh (Figure 10C and 10D). Together, these results indicate that DA and Rot upregulate Parkin in mitochondria to favor its interaction with Mitofilin.
Figure 5.
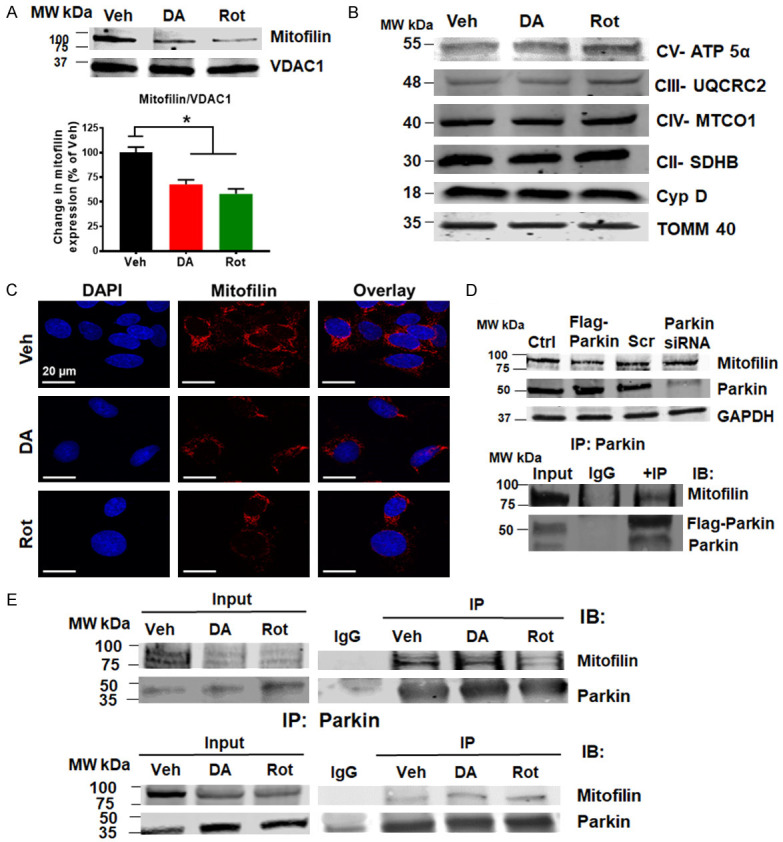
PD stressors increases Parkin interaction with Mitofilin. A. Immunoblots and bar graph (as a percent of vehicle) showing a decrease in Mitofilin protein level after DA and Rot treatment compared with vehicle. B. Immunoblots showing no changes in various mitochondrial protein levels after DA, and Rot treatment compared with Veh. C. Confocal images showing immunolabeling of N27-A cells treated with Veh, DA, or Rot, and labeled with DAPI (blue), Mitofilin (red), and overlay. Note the reduction in Mitofilin intensity in DA and Rot treated cells. D. Top: Immunoblots showing Mitofilin reduction after Parkin overexpression and Mitofilin increase after Parkin knockdown. Cells were transfected with either PCMV6 entry plamid (control) or Flag-Parkin or scrambled (Scr) siRNA or Parkin siRNA. Bottom: Immunoblots of Co-IP showing Myc-Mitofilin interacting with Flag-Parkin in N27-A cells. Cells were co-transfected with Flag-Parkin and Myc-Mitofilin expression vectors for co-IP assays using the anti-Flag antibody. E. Immunoblots of Co-IP analysis using anti-Mitofilin antibody (top) and anti-Parkin antibody (bottom) showing increase in endogenous Parkin interaction with endogenous Mitofilin in N27A-cells in response DA and Rot treatments.
Parkin induces ubiquitination of Mitofilin
Parkin is an E3 ubiquitin ligase implicated in familial Parkinson’s disease [22]. To determine whether Parkin-Mitofilin interaction promotes the ubiquitination and degradation of Mitofilin, we performed immunolabeling with anti-Mitofilin and anti-Ubiquitin antibodies on cells treated with PD stressors. Using confocal microscopy, we observed an increase in the co-localization between Ubiquitin with Mitofilin signals after treatment (Figure 6A). In fact, the degrees of co-localization (Pearson’s correlation coefficients) between Ubiquitin and Mitofilin was higher in DA (0.82±0.06), and Rot (0.87±0.04) treated cells when compared with Veh-treated cells (0.23±0.02) (n = 3/group *P < 0.05). We further confirmed these results by performing (co-IP) with anti-Ubiquitin followed by Western blot analysis and observed that an increase in Mitofilin in the pulled-down after DA treatment when compared with Veh (Figure 6B). However, we only found a little amount of Mitofilin in the pull-down using anti-Ubiquitin antibody after Rot treatment. This result could be the consequence of an increase in mitochondria elimination by mitophagy that we observed after Rot treatment.
Figure 6.
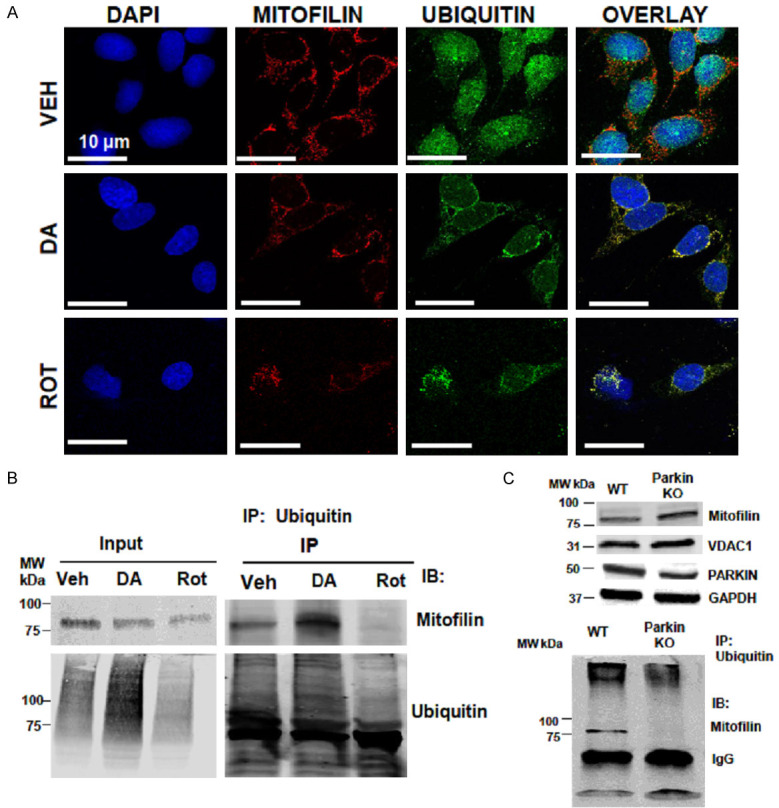
A. Confocal microscopy images showing N27-A cells treated with Veh, DA, or Rot, and labeled with DAPI (blue), Mitofilin (red) & Ubiquitin (green) as well as the overlay (yellow). Note the increase in Mitofilin-Ubiquitin co-localization (yellow) in DA and Rot treated groups compared to Veh. B. Immunoblots of Co-IP assays using anti-Ubiquitin antibody showing an increase in Mitofilin-Ubiquitin interaction in N27-A cells treated DA compared with vehicle and Rot. C. Top: Western blot images showing the increase in Mitofilin level in brain mitochondria from Parkin KO animals compared to WT, and the corresponding VDAC1, Parkin and GAPDH. Bottom: Immunoblots of Co-IP assays using anti-Ubiquitin antibody. Note the increase in Mitofilin pull-down in WT animals compared to Parkin KO.
Parkin-Mitofilin interaction in vivo
To determine whether the Parkin-Mitofilin relationship observed in vitro is physiologically relevant in vivo, we isolated mitochondria from the brains of WT and Parkin KO animals, and we determined the levels of Mitofilin using Western blot analysis. We found that Mitofilin levels were increased in Parkin KO animals compared to wild type (WT) (Figure 6C; top). Further, we performed immunoprecipitation (IP) with anti-Ubiquitin antibody on brain lysates to determine the level of Mitofilin in Parkin KO animals versus WT. Western blot analysis after IP revealed ubiquitinated the absence of Mitofilin in the pull-down with ubiquitin antibody in Parkin KO mice, whereas, in WT, we could observe Mitofilin in the pull-down (Figure 6C; bottom). These results support the hypothesis of mitochondrial Parkin-Mitofilin interaction, which leads to the ubiquitination and degradation of Mitofilin.
Effect of Mitofilin knockdown on PD stressor-induced cell death
To determine the impact of Mitofilin loss in DA-induced cell death, Mitofilin siRNA (200 nM) was transfected to N27-A cells. The level of cell death was assessed by flow cytometry with Annexin V/7-AAD, as previously described. Consistent with our previous report [20], there was about 40% Mitofilin knockdown after 24 hr of treatment with Mitofilin siRNA. We found that this reduction in Mitofilin expression is associated with a 42.4±5.07% increase in cell death by when compared to Scr siRNA (n = 5/group *P < 0.05) (Figure 7A and 7B). We also observed that DA treatment for 24 hr following Mitofilin knockdown, resulted in a significant reduction in DA-induced cell death in cells treated with Mitofilin siRNA compared to cells treated with Scrambled (Scr) siRNA. In fact, while Scr siRNA + DA treatment resulted in 5 fold increase in cell death (compared to Scr SiRNA only treated cells), the level of DA-induced cell death in cells treated with Mitofilin siRNA was only increased by 3 fold when compared to Scr SiRNA only treated cells (n = 9/group *P < 0.05, #P < 0.001 Figure 7C). However, Rot treatment after Mitofilin knockdown did not have a significant effect on cell death (Figure 7C). Taken together, these results confirm a crucial role of Mitofilin regulation in the mechanism of DA-induced cell death.
Figure 7.
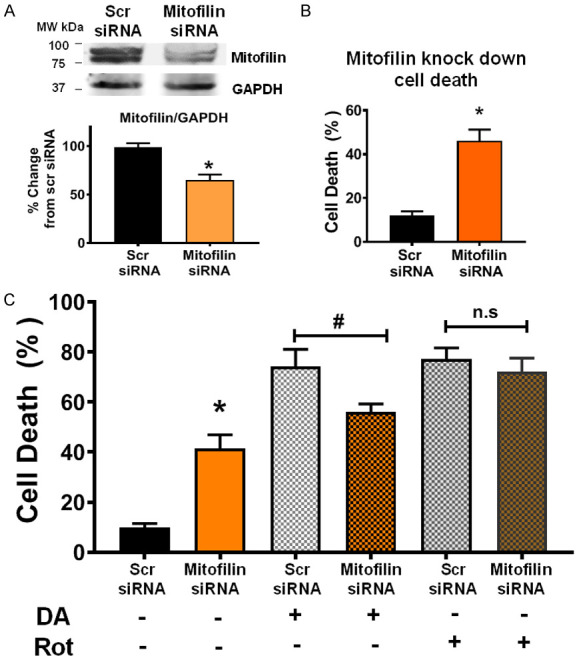
Mitofilin knockdown reduces DA-induced neuron death. A. Top: Flow cytometry analysis with unstained (purple cells), Annexin V only (early apoptosis, green cells), and 7-aminoactinomycin D (7-AAD) only (dead cells and/or late apoptosis, red cells) used for calibration, flow cytometry analysis with Annexin V/7-AAD was used to assess cell death after scrambled siRNA versus Mitofilin siRNA treatment. Bottom left: bar graph showing an increase in early apoptosis (Annexin V+) in Mitofilin siRNA treated cells versus with scramble siRNA. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group. Bottom right: bar graph showing an increase in cell death (7-AAD +) in Mitofilin siRNA treated cells versus with scramble siRNA. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group. B. Top: Western blot analysis showing the reduction in expression of Mitofilin in N27-A cells after transfection with scrambled siRNA versus Mitofilin siRNA. Bottom: Graph showing a decrease in Mitofilin expression. Values are expressed as means ± SEM of three independent experiments, *P < 0.05 Mitofilin siRNA-treated cells versus cells treated with scrambled siRNA (n = 9/group). C. Bar graph showing % change in cell death (7-AAD +) in scrambled siRNA versus Mitofilin siRNA after treatment with DA. Note the difference in cell death between Scrambled siRNA + DA and Mitofilin siRNA + DA groups, normalized to scrambled siRNA. Values are expressed as means ± SEM of five independent experiments, *P < 0.001, n = 9/group.
Parkin knockdown affects Rot-, but not DA-induced cell death
To investigate the impact of Parkin in DA-induced cell death, we transfected N27-A cells with Scr or Parkin siRNA (200 nM) followed by PD stressors treatment for 24 hours. Cells were stained with Annexin V/7-AAD to analyze cell death by flow cytometry. Treatment with Parkin siRNA yielded to about 45% Parkin knockdown efficiency (Figure 8A). We found that Parkin knockdown by siRNA increased cell death by 2 fold compared to Scr siRNA (22.93±1.1% in Parkin siRNA versus Scr siRNA 5.6±0.6%) (Figure 8B). Also, treatment with DA did not affect the levels of cell death in both groups (57.6±3.13% of cell death in Scr siRNA + DA versus 59.1%±2.34% in Parkin siRNA + DA (n = 9/group P < 0.05, Figure 8B). However, treatment with Rot yielded to a significant reduction in cell death in Parkin siRNA group compared to Scr siRNA. In fact, Parkin siRNA + Rot resulted in a 3-fold reduction compared to Scr siRNA + Rot treatment that resulted in a 6-fold increase versus Scr siRNA (n = 9/group P < 0.05, Figure 8B). These results indicate that Parkin knockdown decreases Rot-, but not in DA- induced cell death.
Figure 8.
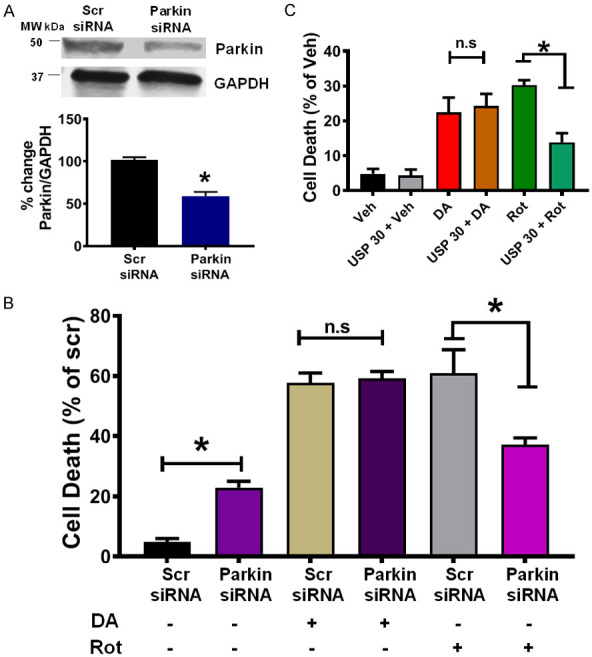
Parkin knockdown attenuates Rot-, but not DA-induced neuron death. A. Top: Immunoblot showing the reduction in expression of Parkin in N27-A cells after transfection with Parkin siRNA versus scrambled siRNA. Bottom: Graph showing a decrease in Parkin expression. Values are expressed as means ± SEM of three independent experiments, *P < 0.05 Parkin siRNA-treated cells versus cells treated with scrambled siRNA (n = 9/group). B. Graph showing the level of cell death after transfection with scrambled siRNA or Parkin siRNA, and treatment with DA, or Rot. Values are expressed as means ± SEM of five independent experiments, *P < 0.001 Parkin siRNA-treated cells versus cells treated with scrambled siRNA (n = 9/group). C. Bar graph showing the level of cell death after overexpression of Myc-USP30, and treatment with DA, or Rot. Values are expressed as means ± SEM of five independent experiments, *P < 0.001. Note that the cell death in Rot treatment resulted in less cell death in USP30 expressing cells.
Counteracting Parkin-mediated mitophagy attenuates Rot- but not DA-induced cell death
To investigate whether lessening Parkin-mediated ubiquitination of Mitofilin mediates DA-induced cell death, N27-A cells were transfected with myc-USP30 for 24 hours, followed by DA, Rot, or Veh treatment. USP30 is a mitochondrial deubiquitinase, which opposes Parkin-mediated mitochondrial protein ubiquitination [38]. We found that while overexpression of exogenous USP30 alone did not significantly affect cell survival, it reduced Rot-induced cell death (Figure 8C). However, USP30 overexpression did not affect DA-induced cell death rate. In fact, the rate of cell death was reduced by 18% when Rot was treated in Myc-USP30 expressing cells compared with control plasmid transfected cells. Instead, cell death remained unchanged in both control plasmids and Myc-USP30 expressing cells when treated with DA (22.42±24.26 versus 24.26±19.48%) (n = 9/group P < 0.05, Figure 8C). Together, these findings indicate that overexpressing USP30 attenuates Rot-, but not DA-induced cell death.
PD stressor-induced Mitofilin loss activates AIF-PARP cell death mechanism
We have recently reported that Mitofilin knockdown induces cell death in an AIF-PARP cleavage dependent mechanism [20]. We determined whether Mitofilin loss in response to PD stressors treatment results in cell death via an AIF-PARP dependent mechanism. We observed that DA treatment upregulated the levels of AIF and PARP cleavage in N27-A cells (Figure 9A). We found a reduction in both AIF and PARP protein levels in Rot treated groups as compared to Veh. This reduction could be attributed to the increased mitophagy observed after Rot treatment. In fact, we found that compared to control (normalized to 100%) DA increased AIF protein levels by (115.42%±7.35%) while AIF levels in Rot treated cells were (98.67±3.55%). Further, PARP cleavage in DA-treated cells was increased to (213.34±6.38%), and to 114.85±5.23% in Rot-treated cells, compared to Veh normalized to 100%. We also determined whether calpain activity was involved in the mechanism of PD stressor toxicity. We found that calpain activity was increased in DA-treated cells compared to Veh (Figure 9B). In fact, there was a 2-fold increase in calpain activity in DA treated cells compared to Veh (Figure 9C). Together, these results suggest that DA induces cell death by increasing calpain activity and activating AIF-PARP pathway.
Figure 9.
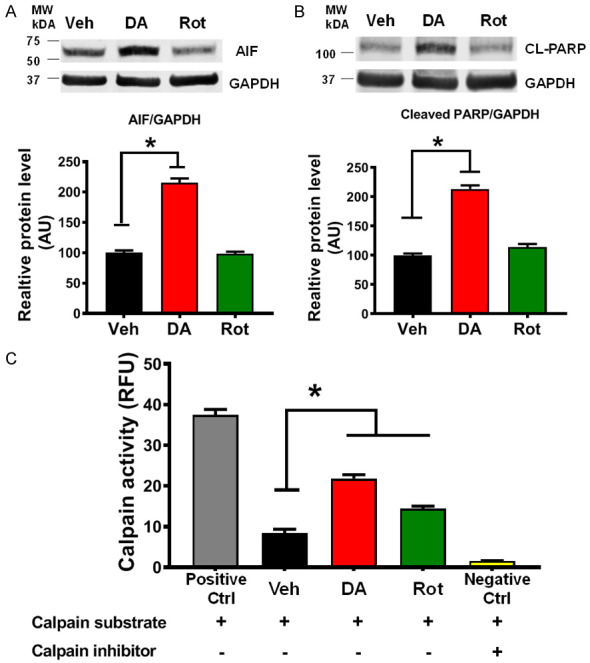
DA, but not Rot, treatment induces activation of AIF-PARP pathway, and increase in calpain activity. A. Western blot analysis and graph showing an increase in apoptosis-inducing factor (AIF) expression in N27-A cells treated with DA compared with Vehicle-, and Rot-treated cells. Values are expressed as means ± SEM of five independent experiments, *P < 0.001 (n = 5/group). B. Immunoblots and graph showing an increase in poly(ADP-ribose),polymerase (PARP) expression in N27-A cells treated with DA compared with Vehicle-, and Rot-treated cells. Values are expressed as means ± SEM of three independent experiments, *P < 0.001 (n = 5/group). C. Graph showing the increase in calpain activity in N27-A cells treated with DA compared with Vehicle-, and Rot-treated cells. Note: Calpain activity was measured using a kit in the presence or absence of calpain substrate and inhibitor. Values are expressed as means ± SEM of five independent experiments, *P < 0.001 (n = 5/group).
PD stressor treatment in human dopamine neuronal primary cell induces similar responses as observed in N27-A cells
We determined whether Parkin-Mitofilin interaction induced by DA and Rot treatments observed in differentiated N27-A cells were reproducible in a dopaminergic neuronal primary cell line. We treated Human Dopamine Neuronal Primary Cells (HDNP cells) with DA and Rot for 24 hours and determined the cytotoxicity effects of these PD stressors on this HDNP cell line using MTT assay. We found that treatment 500 µM DA or 1 µM Rot reduced cell viability by almost 30% (Figure 10A). After 24-hour treatment, cells were stained with Annexin V/7-AAD and analyzed by flow cytometry. We found that DA and Rot treatment increase cell death in HDNP cells (Figure 10B). Furthermore, Western blot analysis on lysates from HDNP cells revealed that Mitofilin levels were reduced in DA, and Rot treated cells compared to Veh (Figure 10C). This reduction was associated with increased Parkin levels, as observed in N27-A cells (Figure 10C). Consistently, we found that DA treatment in HDNP cells also increased AIF protein levels and activated cell death via AIF-PARP dependent mechanism (Figure 10D). Together, these results indicate that DA treatment triggers Mitofilin degradation via Parkin dependent mechanism and induces cell death via AIF-PARP pathway.
Discussion
In this study, we report that PD stressors treatment to in differentiated N27-A and HDNP cells induce dopaminergic neurons death in different mechanisms that include translocation of Parkin into mitochondria and regulation of Mitofilin levels. We found that Parkin mediates a cyto-deleterious effects of PD stressors by increasing its interaction with Mitofilin, which promotes Mitofilin ubiquitination and degradation. Loss of Mitofilin in the inner mitochondrial membrane (IMM), is associated with increased calpain activity, which results in increased mitochondrial structural damage and dysfunction that is postulated to be responsible for the dissipation of mitochondrial membrane potential and increased ROS production. Together, these effects result in the release of mitochondrial AIF into the cytosol, from where its translocation into the nucleus that is facilitated by PARP, yields to DNA condensation and fragmentation that subsequently leads to cell death, as shown in Figure 11.
Figure 11.
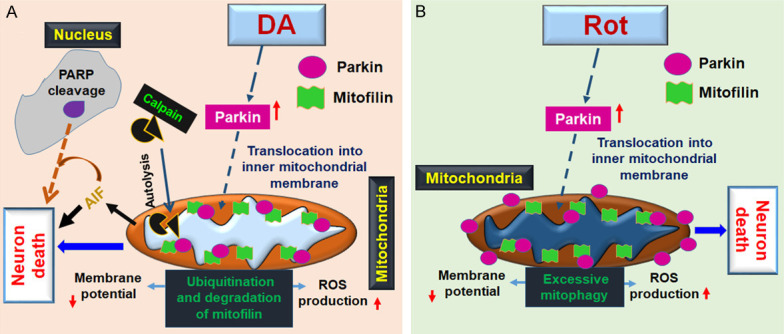
Scheme depicting the mechanism underlying DA-, and Rot-induced N27-A dopaminergic neuron death. A. DA treatment to cultured N27-A dopaminergic neurons induces cell death by increasing Parkin translocation into mitochondria, where it promotes ubiquitination of Mitofilin and its degradation. Loss of Mitofilin in the IMM causes mitochondrial structural damage and dysfunction that is responsible for the reduction of membrane potential as well as an increase in ROS production and mitochondrial calpain activity. Together, the resulted deleterious mitochondrial effects provoke cleavage and release of AIF into the cytosol, where its nuclear translocation, facilitated by PARP, ultimately initiates DNA condensation and fragmentation that subsequently leads to cell death. B. Rot increases cell death by upregulating cytosolic Parkin, which in part translocates into mitochondria where it interacts with Mitofilin to increase mitochondrial structural damage and dysfunction. Damage in mitochondria, associated with ubiquitination of outer membrane proteins by Parkin promote an excessive eliminated by mitophagy that reduces the pool of mitochondria causing cell death.
Parkinson’s disease (PD) is characterized by the neurodegeneration of dopaminergic neurons in the substantia nigra pars compacta. PD is a multifactorial disease that can result from complex combination of genetic factors and/or environmental factors, associated with mitochondria homeostasis [5,11,12,39]. Mitochondria are organelles responsible for energy production, calcium homeostasis, and various metabolic reactions necessary for cell survival and death [40-42]. Disruption of the IMM, as a result of damage to the mitochondrial contact site and cristae junction organizing system (MICOS) complex, results in dysfunction of the mitochondrial ETC and oxidative phosphorylation, increases ROS production, as well as decreases mitochondrial membrane potential and ATP production, which could lead to cell death [42]. Hence, mitochondrial dysfunction plays an important role in the pathogenesis of many disease conditions, including neurodegenerative diseases, ischemia/reperfusion injury, diabetes, hepato-encephalopathy and many others [18].
Disruption of MICOS has been shown to play a key role in the neuropathophysiology of PD [11]. Mitofilin (or Mic60) is a key component of MICOS that forms an oligomeric structure with itself and various other proteins to give the IMM its distinct cristae structure and provides a site for respiration [43,44]. Mitofilin is sensitive to oxidative stress and subject to covalent modification and reduction after mitochondria or cell exposure to PD stressors such as dopamine and MPTP [12,15,35,45]. Using rat cardiomyoblasts, we recently showed that knockdown of Mitofilin increases mitochondrial dysfunction and cell death [20]. Also, Wang’s group revealed that Mitofilin loss in drosophila resulted in an arrest of neuronal mitochondrial movement that is associated with functional and structural disruption of neuromuscular junction synapses [44]. However, the mechanisms responsible for the reduction of Mitofilin levels in response to PD stressors still needs to be elucidated. In this study, we used differentiated N27-A dopaminergic neurons and Human Dopamine Neuronal Primary Cell to determine the impact of Mitofilin regulation in PD stressor-induced cell death. We found that exogenous dopamine (DA) and rotenone (Rot) exposure to differentiated N27-A cells and HDNP cells increase cell death (Figures 1A and 10B). Concomitantly, in N27-A cells and HDNP cells treated with PD stressors, we observed a significant reduction in Mitofilin levels that were associated with increased damage of mitochondrial structural integrity (Figures 3, 4A and 4B). These results are in agreement with those reported by [9,46], in which dopamine oxidation increases mitochondrial dysfunction and cell death in SH-SY5Y cells. Therefore, we investigated the molecular mechanisms that may result in decreased Mitofilin protein levels in response to exogenous dopamine and rotenone treatment. Our results showed that PD stressors reduce mitochondrial membrane potential, increase oxidative stress, and mitochondrial structural damage (Figures 2 and 3) as well as decrease mitochondrial ATP production (Figure 3C). These results being consistent with PD stressors impairment of mitochondrial function. Based on these findings, we postulated that reduction of Mitofilin levels might occur via clearance of damaged mitochondria in cells, a process well known as mitophagy by [29]. Consistently, we observed an increase in Parkin levels and its relocalization in mitochondria (Figure 4A and 4B). We found that Rot treatment induces mitophagy in N27-A cells (Figure 4C and 4D). In fact, Rot treatment increased overall mitophagy by reducing the levels of p62, the ratio of LC3BII/I, and increased mitophagy flux (Figure 4C and 4D). The reduction in the overall protein levels of LC3 and Beclin-1 could be explained by their degradation by autophagy, as they are incorporated during autophagosome generation and degraded during mitophagy [47]. Counteracting mitophagy with USP30 overexpression, or knockdown of Parkin was able to attenuate Rot- but not DA-induced cell death (Figure 8B and 8C). The reduction in the overall protein levels of LC3 and Beclin-1 could be explained by their degradation by autophagy, as they are incorporated during autophagosome generation and degraded during mitophagy [47]. Surprisingly, autophagy markers such as p62, protein levels, and LC3BII/I ratio remained unchanged after DA treatment compared to vehicle. In fact, we found that in DA-treated cells, the increase in Parkin, although associated with a decrease in Mitofilin, was not linked with an increase in overall mitophagy (Figure 4B and 4D). This is in agreement with previous reports that DA does not induce mitophagy [9], and hence, we excluded mitophagy as responsible for Mitofilin loss.
Previous studies have demonstrated that ubiquitination occurs in different mitochondrial compartments, including the IMM [48,49]. Some IMM proteins such as succinate dehydrogenase subunit A (SDHA) [50], and MCU complex [51] can be ubiquitinated in response to harmful stimuli. Therefore, we investigated whether Mitofilin is ubiquitinated in the IMM in response to DA treatment. We performed immunoprecipitation with anti-Mitofilin antibody on whole cell lysates from cells treated with DA, Rot, and Veh. We found a strong and increased interaction between Mitofilin with Ubiquitin after DA treatment (Figure 6A). This result was confirmed by reverse Co-IP analysis using anti-Ubiquitin antibody (Figure 6B) that showed a strong co-localization of Ubiquitin, and Mitofilin after DA treatment. We found that the ubiquitination of Mitofilin is associated with an increase in Parkin translocation to mitochondria. Using a biochemical analysis approach in N27-A cells ectopically expressing Myc-Mitofilin and Flag-Parkin, we found that Parkin is a novel interacting partner of Mitofilin. In fact, Myc-Mitofilin was co-precipitated by the anti-flag antibody, indicating that flag-Parkin interacted with Myc-Mitofilin in the IMM (Figure 5D). In fact, we observed that the levels of Mitofilin are significantly higher in Parkin KO mice compared to WT (Figure 6C). Further, Western blot analysis after IP with anti-Ubiquitin antibody in mice brain lysates revealed the absence of Mitofilin in the pull-down from Parkin KO mice compared to WT. This suggests that Parkin-Mitofilin interaction is physiologically relevant in Mitofilin homeostasis and important for the maintenance of mitochondria structure and function. However, this Parkin-Mitofilin interaction is greatly increased in stress conditions resulting in excessive ubiquitination and degradation of Mitofilin (Figures 5E, 6A-C). Although we cannot ignore the existence of other mitochondrial ubiquitin ligases, to the best of our knowledge, there is no report of any other ubiquitin ligase targeting Mitofilin. Nevertheless, further experiments are required to determine whether Mitofilin is a substrate for any of these mitochondrial ubiquitin ligases during stress conditions.
Next, we investigated whether knockdown of Mitofilin using siRNA will exacerbate N27-A cell death following DA and Rot exposure. Van Laar and colleagues have previously reported that Mitofilin knockdown exacerbated cellular vulnerability to DA and Rot, and Mitofilin overexpression protects against DA-induced cell death [52]. Our results revealed that about 40% Mitofilin knockdown alone by siRNA increased cell death by 46%, Mitofilin siRNA treatment for 24 hours, followed by treatment 250 µM DA increased neuronal death to 55-65% as opposed to Scr siRNA co-treatment with 250 µM DA which resulted in 75-85% cell death (Figure 7C). These results were not completely in line with those reported by [52]. In fact, Hastings group reported that Mitofilin knockdown (20-30%) with shRNA did not affect the basal viability of differentiated SH-SY5Y cells. We observed a significant reduction in the number of live and viable N27-A cells after 40% knockdown of Mitofilin with siRNA (Figure 7A and 7B). Our current results are in line with our previous observations in H9c2 cardiomyoblasts and HEK293T cells, indicating that knockdown of Mitofilin promotes cell apoptosis [20]. We show here that, while Scrambled siRNA + DA co-treatment resulted in about 5-fold increase in cell death (compared to Scrambled siRNA treatment alone), Mitofilin siRNA + DA co-treatment resulted in less cell death (3-fold increase compared to scrambled siRNA alone). This apparent discrepancy might be only the result of the transfection efficiency and the difference in the cell line used.
Exogenous DA has been found to induce cell death in neuronal cell lines and primary rat neurons [52,53]. However, the mitochondrial-dependent mechanism underlying its toxicity still needs to be elucidated. DA has been suggested to trigger cell death in SH-SY5Y neuroblastoma cells by activating stress kinases (SAPK/JNK and p38), increasing α-synuclein aggregation and autophagic cell death, without affecting apoptosis inducing factor (AIF) and cytochrome c distribution [53]. The same study suggested that DA does not induce nuclear fragmentation characteristic of apoptosis and that inhibiting the stress kinases partially reverses DA-induced cell death. Although our result corroborates some of these findings, we have observed that the levels of AIF were significantly increased in DA-treated N27-A cells versus Veh. In fact, we report a 2-fold increase in AIF protein levels after N27-A cells exposure to dopamine (Figure 9A). The apparent discrepancy might be explained by the use of SH-SY5Y neuroblastoma cell line, which is not a pure dopaminergic cell line [54]. The SH-SY5Y cell line possesses cancerous properties that may influence its differentiation, metabolic properties, genomic stability, and hence, this cell line displays physiological characteristics that differ greatly from dopaminergic neuronal features [54]. Furthermore, the use of high dose DA (500 μM) in cells could trigger rapid stress response and autophagy, which may also shed light on this discrepancy. In contrast, in this current study, we utilized Human Dopamine Primary cells and purified differentiated N27-A dopaminergic neurons, originally derived from embryonic rat dopamine neurons, that is an improved in vitro model [46].
We recently reported that Mitofilin knockdown with siRNA induces caspase 3-independent cell death via activation of calpains [20]. Calpains are proapoptotic proteases mediating the cleavage and release of AIF [55]. Because our results suggest that DA induces Mitofilin degradation and a 2-fold increase in both AIF protein levels and PARP cleavage (Figure 9A and 9B), we determined whether calpain activity is involved in DA-induced cell death mechanism. We found that PD stressor (DA) treatment increases calpain activity (Figure 9C). We, therefore, postulate that increase in calpain activity, in response to DA actions, promotes Mitofilin-induced mitochondrial structural damage and dysfunction that is responsible for AIF release into the cytosol. In the cytosol, AIF interacts with PARP, which facilitates its translocation into the nucleus, where AIF activation, concomitant with ROS effects, results in DNA condensation and fragmentation that is responsible for cell death by apoptosis [20]. However, initial trials with three calpain inhibitors (ALLN, Calpeptin, and E64) did not lessen DA- or Rot-induced cell death. Therefore, further studies are needed to sort out the exact calpain isoform involved in the DA-induced cell death mechanism.
In this study, we investigated the impact of Parkin-Mitofilin interaction in the cell death-induced by PD stressors (DA and Rot) using two different cell lines. These studies have some limitations. These PD stressors are known to act as an inhibitor of the complex I of the electron transfer chain, which results in increases ROS production. In addition, cytochrome c reduction, observed during dopamine auto-oxidation, attributed to direct interaction with dopamine semiquinone might also induce oxidative stress. This suggests possible alternative deleterious mechanisms in PD stressors actions. Therefore, it does not exclude that these alternative negative impacts in cells could potentiate the effects observed in our studies. Although our studies were performed in two cell lines, additional investigations in intact animals are encouraged due to a much different cell environment in vivo.
Conclusion
We report here that two distinct pathways underly the mechanism of PD stressors (DA and Rot)-induced N27-A dopaminergic neuron and HDPN death via regulation of Parkin-Mitofilin interaction. We found that both PD stressors increase Parkin interaction with Mitofilin, which promotes Mitofilin ubiquitination and further degradation resulting in mitochondrial structural damage and dysfunction, ultimately leading to increased neuron death. DA treatment increases calpain activity, which aggravates mitochondrial structural damage and dysfunction without activating mitophagy, thereby promoting neuronal death by apoptosis via an AIF-PARP dependent mechanism. In contrast, Rot treatment induces mitochondrial elimination and reduction of Mitofilin levels mainly by promoting excessive mitophagy. Further, Parkin knockdown by siRNA, or over-expression of the mitochondrial deubiquitinase (USP30) attenuated Rot-induced cell death. Together, this study reveals the key role of the regulation of Parkin-Mitofilin interaction in the deleterious effects of PD stressor-induced neurotoxicity.
Acknowledgements
We would like to thank Flow Cytometry Shared Resource Facility, which is supported by UT Health San Antonio, NIH-NCI P30 CA054174-20 (CTRC at UTHSCSA) and UL1 TR001120 (CTSA grant) for all the Flow cytometry data generated in the different studies. National Institutes of Health [grant HL138093 (JCB)]; Perry and Ruby Stevens Parkinson’s Disease Center of Excellence pilot grant (JCB); and the American Heart Association (AHA) Scientific Development Grant 17SDG33100000 (JCB).
Disclosure of conflict of interest
None.
Abbreviations
- AIF
Apoptosis-inducing factor
- DAQ
Dopamine quinones
- DAT
Dopamine transporter
- DA
Dopamine
- IMM
inner mitochondrial membrane
- MICOS
Mitochondrial contact site and cristae organizing system
- MPTP
1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine
- PARP
Poly (ADP-ribose) polymerase
- PD
Parkinson’s disease
- ROS
reactive oxygen species
- Rot
Rotenone
- ΔΨm
Mitochondrial Membrane Potential
References
- 1.Rice ME, Patel JC, Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011;198:112–137. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rumpel R, Alam M, Schwarz LM, Ratzka A, Jin X, Krauss JK, Grothe C, Schwabe K. Neuronal firing activity in the basal ganglia after striatal transplantation of dopamine neurons in hemiparkinsonian rats. Neuroscience. 2017;360:197–209. doi: 10.1016/j.neuroscience.2017.07.069. [DOI] [PubMed] [Google Scholar]
- 3.Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rocca WA. The burden of Parkinson’s disease: a worldwide perspective. Lancet Neurol. 2018;17:928–929. doi: 10.1016/S1474-4422(18)30355-7. [DOI] [PubMed] [Google Scholar]
- 5.Nandipati S, Litvan I. Environmental exposures and parkinson’s disease. Int J Environ Res Public Health. 2016;13:881. doi: 10.3390/ijerph13090881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL. The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience. 2007;146:741–755. doi: 10.1016/j.neuroscience.2007.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamel F, Tanner C, Umbach D, Hoppin J, Alavanja M, Blair A, Comyns K, Goldman S, Korell M, Langston J, Ross G, Sandler D. Pesticide exposure and self-reported Parkinson’s disease in the agricultural health study. Am J Epidemiol. 2007;165:364–374. doi: 10.1093/aje/kwk024. [DOI] [PubMed] [Google Scholar]
- 8.Alberio T, Bondi H, Colombo F, Alloggio I, Pieroni L, Urbani A, Fasano M. Mitochondrial proteomics investigation of a cellular model of impaired dopamine homeostasis, an early step in Parkinson’s disease pathogenesis. Mol Biosyst. 2014;10:1332–1344. doi: 10.1039/c3mb70611g. [DOI] [PubMed] [Google Scholar]
- 9.Bondi H, Zilocchi M, Mare MG, D’Agostino G, Giovannardi S, Ambrosio S, Fasano M, Alberio T. Dopamine induces mitochondrial depolarization without activating PINK1-mediated mitophagy. J Neurochem. 2016;136:1219–1231. doi: 10.1111/jnc.13506. [DOI] [PubMed] [Google Scholar]
- 10.Alberio T, Lopiano L, Fasano M. Cellular models to investigate biochemical pathways in Parkinson’s disease. FEBS J. 2012;279:1146–1155. doi: 10.1111/j.1742-4658.2012.08516.x. [DOI] [PubMed] [Google Scholar]
- 11.Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038–3062. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Laar VS, Berman SB. The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson’s disease. Neurobiol Dis. 2013;51:43–55. doi: 10.1016/j.nbd.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol Dis. 2013;51:35–42. doi: 10.1016/j.nbd.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberio T, Bossi AM, Milli A, Parma E, Gariboldi MB, Tosi G, Lopiano L, Fasano M. Proteomic analysis of dopamine and alpha-synuclein interplay in a cellular model of Parkinson’s disease pathogenesis. FEBS J. 2010;277:4909–4919. doi: 10.1111/j.1742-4658.2010.07896.x. [DOI] [PubMed] [Google Scholar]
- 15.Van Laar VS, Dukes AA, Cascio M, Hastings TG. Proteomic analysis of rat brain mitochondria following exposure to dopamine quinone: implications for Parkinson disease. Neurobiol Dis. 2008;29:477–489. doi: 10.1016/j.nbd.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Laar VS, Mishizen AJ, Cascio M, Hastings TG. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol Dis. 2009;34:487–500. doi: 10.1016/j.nbd.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Ruan Y, Zhang K, Jian F, Hu C, Miao L, Gong L, Sun L, Zhang X, Chen S, Chen H, Liu D, Song Z. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016;23:380–392. doi: 10.1038/cdd.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, Madungwe NB, Bopassa JC. Mitochondrial inner membrane protein, Mic60/mitofilin in mammalian organ protection. J Cell Physiol. 2019;234:3383–3393. doi: 10.1002/jcp.27314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Laar VS, Otero PA, Hastings TG, Berman SB. Potential role of Mic60/mitofilin in Parkinson’s disease. Front Neurosci. 2018;12:898. doi: 10.3389/fnins.2018.00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madungwe NB, Feng Y, Lie M, Tombo N, Liu L, Kaya F, Bopassa JC. Mitochondrial inner membrane protein (mitofilin) knockdown induces cell death by apoptosis via an AIF-PARP-dependent mechanism and cell cycle arrest. Am J Physiol Cell Physiol. 2018;315:C28–C43. doi: 10.1152/ajpcell.00230.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tombo N, Imam Aliagan AD, Feng Y, Singh H, Bopassa JC. Cardiac ischemia/reperfusion stress reduces inner mitochondrial membrane protein (mitofilin) levels during early reperfusion. Free Radic Biol Med. 2020;158:181–194. doi: 10.1016/j.freeradbiomed.2020.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, Parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 23.Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s disease genetics study group and the european consortium on genetic susceptibility in Parkinson’s disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- 24.Prestel J, Sharma M, Leitner P, Zimprich A, Vaughan JR, Dürr A, Bonifati V, De Michele G, Hanagasi HA, Farrer M, Hofer A, Asmus F, Volpe G, Meco G, Brice A, Wood NW, Müller-Myhsok B, Gasser T European Consortium on Genetic Susceptibility in Parkinson’s Disease (GSPD) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 25.Perez FA, Curtis WR, Palmiter RD. Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 2005;6:71. doi: 10.1186/1471-2202-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguiar AS Jr, Tristao FS, Amar M, Chevarin C, Lanfumey L, Mongeau R, Corti O, Prediger RD, Raisman-Vozari R. Parkin-knockout mice did not display increased vulnerability to intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Neurotox Res. 2013;24:280–287. doi: 10.1007/s12640-013-9389-0. [DOI] [PubMed] [Google Scholar]
- 27.Park H, Chung KM, An HK, Gim JE, Hong J, Woo H, Cho B, Moon C, Yu SW. Parkin promotes mitophagic cell death in adult hippocampal neural stem cells following insulin withdrawal. Front Mol Neurosci. 2019;12:46. doi: 10.3389/fnmol.2019.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dawson TM, Dawson VL. The role of Parkin in familial and sporadic Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S32–39. doi: 10.1002/mds.22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akabane S, Uno M, Tani N, Shimazaki S, Ebara N, Kato H, Kosako H, Oka T. PKA regulates PINK1 stability and parkin recruitment to damaged mitochondria through phosphorylation of MIC60. Mol Cell. 2016;62:371–384. doi: 10.1016/j.molcel.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 31.Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520:606–611. doi: 10.1016/j.bbrc.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madungwe NB, Feng Y, Imam Aliagan A, Tombo N, Kaya F, Bopassa JC. Inner mitochondrial membrane protein MPV17 mutant mice display increased myocardial injury after ischemia/reperfusion. Am J Transl Res. 2020;12:3412–3428. [PMC free article] [PubMed] [Google Scholar]
- 33.Madungwe NB, Zilberstein NF, Feng Y, Bopassa JC. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am J Cardiovasc Dis. 2016;6:93–108. [PMC free article] [PubMed] [Google Scholar]
- 34.Feng Y, Madungwe NB, da Cruz Junho CV, Bopassa JC. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol. 2017;174:4329–4344. doi: 10.1111/bph.14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139(Suppl 1):216–231. doi: 10.1111/jnc.13731. [DOI] [PubMed] [Google Scholar]
- 36.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review) Int J Mol Med. 2019;44:3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. 2016;41:261–273. doi: 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 38.Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 39.Ryan BJ, Hoek S, Fon EA, Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci. 2015;40:200–210. doi: 10.1016/j.tibs.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 40.El-Hattab AW, Suleiman J, Almannai M, Scaglia F. Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab. 2018;125:315–321. doi: 10.1016/j.ymgme.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 42.Moon HE, Paek SH. Mitochondrial dysfunction in Parkinson’s disease. Exp Neurobiol. 2015;24:103–116. doi: 10.5607/en.2015.24.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfanner N, van der Laan M, Amati P, Capaldi RA, Caudy AA, Chacinska A, Darshi M, Deckers M, Hoppins S, Icho T, Jakobs S, Ji J, Kozjak-Pavlovic V, Meisinger C, Odgren PR, Park SK, Rehling P, Reichert AS, Sheikh MS, Taylor SS, Tsuchida N, van der Bliek AM, van der Klei IJ, Weissman JS, Westermann B, Zha J, Neupert W, Nunnari J. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J Cell Biol. 2014;204:1083–1086. doi: 10.1083/jcb.201401006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai PI, Papakyrikos AM, Hsieh CH, Wang X. Drosophila MIC60/mitofilin conducts dual roles in mitochondrial motility and crista structure. Mol Biol Cell. 2017;28:3471–3479. doi: 10.1091/mbc.E17-03-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burte F, De Girolamo LA, Hargreaves AJ, Billett EE. Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. J Proteome Res. 2011;10:1974–1986. doi: 10.1021/pr101211k. [DOI] [PubMed] [Google Scholar]
- 46.Gao L, Zhou W, Symmes B, Freed CR. Re-cloning the N27 dopamine cell line to improve a cell culture model of Parkinson’s disease. PLoS One. 2016;11:e0160847. doi: 10.1371/journal.pone.0160847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:1865. doi: 10.3390/ijms18091865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, Carr SA. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12:825–831. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner SA, Beli P, Weinert BT, Scholz C, Kelstrup CD, Young C, Nielsen ML, Olsen JV, Brakebusch C, Choudhary C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics. 2012;11:1578–1585. doi: 10.1074/mcp.M112.017905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lavie J, De Belvalet H, Sonon S, Ion AM, Dumon E, Melser S, Lacombe D, Dupuy JW, Lalou C, Benard G. Ubiquitin-dependent degradation of mitochondrial proteins regulates energy metabolism. Cell Rep. 2018;23:2852–2863. doi: 10.1016/j.celrep.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Matteucci A, Patron M, Vecellio Reane D, Gastaldello S, Amoroso S, Rizzuto R, Brini M, Raffaello A, Cali T. Publisher Correction: parkin-dependent regulation of the MCU complex component MICU1. Sci Rep. 2019;9:4665. doi: 10.1038/s41598-018-37929-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Laar VS, Berman SB, Hastings TG. Mic60/mitofilin overexpression alters mitochondrial dynamics and attenuates vulnerability of dopaminergic cells to dopamine and rotenone. Neurobiol Dis. 2016;91:247–261. doi: 10.1016/j.nbd.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 54.Xicoy H, Wieringa B, Martens GJ. The SH-SY5Y cell line in Parkinson’s disease research: a systematic review. Mol Neurodegener. 2017;12:10. doi: 10.1186/s13024-017-0149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ. Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem Biophys Res Commun. 2011;415:533–538. doi: 10.1016/j.bbrc.2011.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]