

Abstract
Background: Amyotrophic lateral sclerosis (ALS) can result in the dysfunction of upper and lower motor neurons. A previous study has indicated that TBK1 mutation (hTBK1-c.978T>A) is involved in progression of ALS. However, the mechanism by which TBK1 mutation mediates the progression of ALS remains unclear. Methods: NSC-34 cells with hTBK1-c.978T>A mutation (TBK1 mutation status) was used to mimic ALS in vitro. In addition, cell proliferation was detected by Ki67 staining. Gene and protein expressions in NSC-34 cells were detected by RT-qPCR and western blot, respectively. ROS and PGSK levels in NSC-34 cells were detected by flow cytometry. Results: hTBK1-c.978T>A mutation significantly inhibited the proliferation of NSC-34 cells via inducing cell ferroptosis, while the effect of TBK1 mutation was notably reversed by Ferrostatin-1 or p62 siRNA. Meanwhile, hTBK1-c.978T>A mutation significantly increased the expression of KEAP1 in NSC-34 cells, while this phenomenon was partially reversed by p62 knockdown. Conclusion: hTBK1-c.978T>A mutation promoted promotes the ferroptosis in NSC-34 cells via regulation of KEAP1/NRF2/p62 signaling. Thus, hTBK1-c.978T>A mutation may serve as a possible target for the treatment of ALS.
Keywords: ALS, p62, hTBK1-c.978T>A mutation, KEAP1/NRF2/p62 signaling
Introduction
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease which is characterized by death of motoneurons (MN) and early denervation of the neuromuscular junction (NMJ) [1]. The retraction of MN nerve terminals at striated muscles leads to the phenomenon that patients with ALS suffer muscle weakness and paralysis [2,3], and these symptoms seriously disturb their lives. Nowadays, the main treatment of ALS is neurotrophic drug therapy, while the therapeutic effect is still not ideal [4]. Although some efforts have been made to study ALS [5,6], the outcomes remain limited. Thus, it is urgent to find a new method for the treatment of ALS.
It has been demonstrated gene mutation is known to be participated in ALS. For instance, TAR DNA-binding protein 43 (TDP-43) mutation is correlated with DNA repair in ALS [7]. Watanabe Y et al found that C21ORF2 mutation is involved in progression of ALS [8]. TANK-binding kinase1 (TBK1) has been confirmed as a risk gene of ALS [9]. Meanwhile, a recent research has indicated that TBK1 mutation (TBK1-c.1069C>T, TBK1-c.4C>T et al) could act as a key mediator in ALS [10], and it has been verified that a new TBK1 mutation (hTBK1-c.978T>A mutation) is involved in ALS [11]. However, the mechanism by which hTBK1-c.978T>A mutation mediates the progression of ALS remains unclear.
Ferroptosis is known to be an iron-dependent mechanism of cell death [12]. It is a programmed cell death process which is different from autophagy, necrosis, and apoptosis [13,14]. In addition, it has been indicated that ferroptosis can act as an important mediator in ALS [15-17]. However, the correlation between hTBK1-c.978T>A mutation and ferroptosis in ALS is largely unknown. In the current research, we sought to explore the function of hTBK1-c.978T>A mutation in ALS in vitro. We hope our research would provide a new idea for ALS treatment.
Material and methods
Cell culture
Mouse motor neuron-like hybrid cell line (NSC-34) was purchased from Vecscience Co. Ltd (Wuhan, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F-12K Nutrient Medium (a mixture of 1:1; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 15 mM HEPES (Sigma-Aldrich), 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), 100 U/ml penicillin and 100 µg/ml streptomycin (Sigma-Aldrich). In addition, cells were incubated at 37°C in 5% CO2.
Cell transfection
hTBK1-c.978T>A mutation pcDNA-3.1 or negative control (pcDNA3.1-NC, 10 nM) were transfected into NSC-34 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). hTBK1-c.978T>A mutation plasmids and pcDNA-3.1-NC were purchased from GenePharma (Shanghai, China). For p62 knockdown, siRNAs targeted against p62 (p62 siRNA1, p62 siRNA2 and p62 siRNA3; 10 nM; RiboBio, Guangzhou, China) and a negative control siRNA (siRNA-NC) were transfected into NSC-34 cells using Lipofectamine® 2000 (Invitrogen). Cells were incubated at 37°C for 6 h before subsequent experiments were performed. The sequences of the siRNAs were as follows: Negative control siRNA, 5’-UUCUCCGAACGUGUCACGUTT-3’; p62 siRNA1, 5’-GGAAUGAAGCAACUGAGAUUU-3’; p62 siRNA2, 5’-GGGTTACGATTGCCCAGAT-3’ and p62 siRNA3, 5’-CCGGGAAGCCCGTCCCGUT-3’.
Quantitative real time polymerase chain reaction (RT-qPCR)
Total RNA was extracted from NSC-34 cells with TRIzol reagent (Takara) and converted into cDNA using PrimeScript RT-PCR Kit (TaKaRa). The level of p62 was tested in triplicate using SYBR Premix Ex Taq (Takara) on a Real-Time PCR System (Applied Biosystems 7500) and normalized to ACTIN. The amplification protocol was set as below: Incubation at 95°C for 5 min, then 40 cycles of 95°C for 10 s and 60°C for 30 s. The primer for p62 was purchased from GenePharma (Shanghai, China). P62 forward, 5’-AGCAGTCAGTAGTTGGTCCTTTG-3’ and reverse 5’-CCATCAGTCCCGTCTTGAAAC-3’. GAPDH: forward, 5’-CATCATCCCTGCCTCTACTGG-3’; reverse 5’-GTGGGTGTCGCTGTTGAAGTC-3’.
Western blot
Cell lysates were prepared using RIPA lysis buffer. Equal amounts (20 µg) of protein from each group were separated by SDS-PAGE electrophoresis. Separated proteins were transferred on to a PVDF (polyvinylidene fluoride) membrane. Then, primary antibodies against NRF2 (1:1000), NOX1 (1:1000), GPX4 (1:1000), FTH1 (1:1000), p62 (1:1000), COX-2 (1:1000), KEAP1 (1:1000) and corresponding secondary antibody (1:5000) were used to incubate with the membrane after blocking. β-actin was utilized as endogenous control. All the antibodies were purchased from Abcam (Cambridge, MA, USA).
CCK-8 assay
The viability of NSC-34 cells in each group was determined by Cell Counting Kit-8 (CCK8) assay using CCK-8 kit of Beyotime. The optical density (OD) values at 450 nm were detected by a microplate reader to indicate cell viability.
Immunofluorescence
Ki67 staining was performed with Ki67 cell proliferation kit (Abcam) to determine the newly proliferated NSC-34 cells in each group. All procedures were followed manufacturer’s protocol. The number and the percentage of Ki67 positive cells were counted and calculated.
ROS detection
Cell suspensions were collected and supplemented with the ROS probe DCFDA (Beyotime, Shanghai, China) [18]. After 20 min of incubation, cells were centrifuged at 300 g, washed with PBS, and resuspended. Finally, the relative ROS level was measured by FACS.
PGSK level detection
Cell suspensions were collected and supplemented with the PGSK probe (Beyotime). After 20 min of incubation, cells were centrifuged at 300 g, washed, and resuspended. Finally, the relative PGSK level was measured by FACS, and the ferroptotic cells were observed under a confocal microscope.
Measurement of mitochondrial membrane potential (∆Ψm)
Cell suspensions were collected and supplemented with the JC-1 buffer (Beyotime). After 20 min of incubation, cells were centrifuged at 600 g, washed with JC-1 buffer, and resuspended. Finally, the mitochondrial membrane potential was measured by FACS.
Statistical analysis
All data were expressed as Mean ± SD. Difference between groups were compared using GraphPad Prism 8 software with one-way ANOVA analysis and Tukey’s tests. Differences were considered significant when P < 0.05.
Results
In vitro model of ALS was successfully established
According to a previous finding [19], TBK1 mutation (hTBK1-c.978T>A) may contribute to the occurrence of ALS. Therefore, NSC-34 cells with hTBK1-c.978T>A mutation was used to mimic ALS in vitro (Supplementary Figure 1). As indicated in Figure 1A, cell viability of NSC-34 was significantly decreased by hTBK1-c.978T>A mutation. Consistently, the proliferation of NSC-34 cells was notably inhibited in the presence of hTBK1-c.978T>A mutation (Figure 1B and 1C). All these data revealed that in vitro model of ALS was successfully established.
Figure 1.
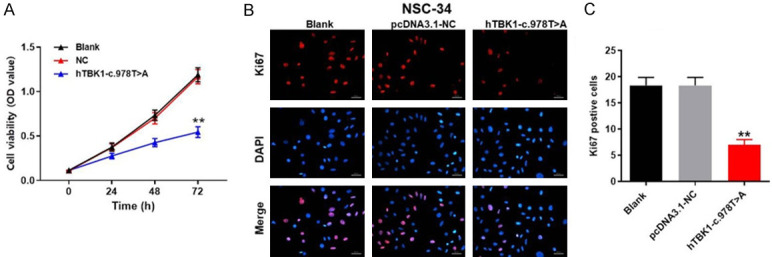
In vitro model of ALS was successfully established. NSC-34 cells were treated with hTBK1-c.978T>A mutation or NC for 0, 24, 48 or 72 h. A. Cell viability was tested by CCK-8 assay. B. The proliferation of NSC-34 cells was measured by Ki67 staining. Red indicates Ki67; Blue indicates DAPI. C. The positive cell rate of Ki67 staining was calculated. **P < 0.01 compared to control, n = 3.
hTBK1-c.978T>A mutation inhibited the proliferation of NSC-34 cells via inducing ferroptosis
In order to test the cell viability, CCK-8 assay was performed. As expected, the viability of NSC-34 cells was significantly decreased by hTBK1-c.978T>A mutation, while this phenomenon was partially reversed by ferroptosis inhibitor (Ferrostatin-1) (Figure 2A). Additionally, the relative ROS level of NSC-34 cells was obviously increased by hTBK1-c.978T>A mutation (Figure 2B). However, hTBK1-c.978T>A mutation-induced increase of ROS in NSC-34 cells was significantly reversed by Ferrostatin-1 as well (Figure 2B). Consistently, Ferrostatin-1 significantly inhibited hTBK1-c.978T>A mutation-induced decrease of mitochondrial membrane potential in NSC-34 cells (Figure 2C). Moreover, hTBK1-c.978T>A mutation-induced ferroptosis in NSC-34 cells was notably inhibited in the presence of Ferrostatin-1 (Figure 2D). Furthermore, hTBK1-c.978T>A mutation greatly decreased the PGSK level in NSC-34 cells, while the inhibitory effect of hTBK1-c.978T>A mutation on PGSK level was significantly reversed by Ferrostatin-1 (Figure 2E). Taken together, hTBK1-c.978T>A mutation inhibited the proliferation of NSC-34 cells via inducing ferroptosis.
Figure 2.

hTBK1-c.978T>A mutation inhibited the proliferation of NSC-34 cells via inducing ferroptosis. NSC-34 cells were treated with hTBK1-c.978T>A mutation, pDNA3.1-NC (NC) or hTBK1-c.978T>A mutation + Ferrostatin-1 for 48 h. Then, (A) cell viability was tested by CCK-8 assay. (B) ROS level in NSC-34 cells was measured by flow cytometry. The relative ROS level was calculated. (C) Mitochondrial membrane potential in NSC-34 cells was tested by JC-1 assay. (D, E) Iron level of NSC-34 cells was measured by PGSK detection. Green fluorescence indicates PGSK. The relative PGSK level was calculated. **P < 0.01 compared to control, ##P < 0.01 compared to hTBK1-c.978T>A mutation, n = 3.
hTBK1-c.978T>A mutation regulated ferroptosis-related protein expressions in NSC-34 cells
Next, the ferroptosis-related protein expressions in NSC-34 cells with or without hTBK1-c.978T>A mutation were detected with western blot. As showed in Figure 3A-D, the protein expressions of p62, COX-2 and NOX1 in NSC-34 cells were significantly increased by hTBK1-c.978T>A mutation, while the effect of hTBK1-c.978T>A mutation on these three proteins was partially revered by Ferrostatin-1. In addition, hTBK1-c.978T>A mutation-induced inactivation on GPX4 and FTH1 in NSC-34 cells was reversed by Ferrostatin-1 as well (Figure 3A, 3E and 3F). Since COX-2, NOX1, GPX4 and FTH1 are known to be the ferroptosis-related proteins [20-23], these data confirmed that hTBK1-c.978T>A mutation inhibited the proliferation of NSC-34 cells via inducing ferroptosis.
Figure 3.

hTBK1-c.978T>A mutation regulated ferroptosis-related protein expression in NSC-34 cells. A. The protein expressions of p62, COX2, NOX1, GPX4 and FTH1 in NSC-34 cells were detected by western blot. B-F. The relative expressions of p62, COX-2, NOX1, GPX4 and FTH1 were quantified by normalizing to β-actin. **P < 0.01 compared to control, ##P < 0.01 compared to hTBK1-c.978T>A mutation, n = 3.
hTBK1-c.978T>A mutation-induced ferroptosis in NSC-34 cells was inhibited by p62 knockdown
To further confirm the interaction of hTBK1-c.978T>A mutation and ferroptosis in NSC-34 cells, p62 knockdown rescue experiment was performed. As indicated in Figure 4A-C, the expression of p62 in NSC-34 cells was significantly decreased when transfected with p62 siRNAs. These data revealed that p62 siRNAs were stably transfected into NSC-34 cells. Since NSC-34 cells were more sensitive to p62 siRNA2, p62 siRNA2 was selected of use in following experiments. Next, the CCK-8 experiment indicated hTBK1-c.978T>A mutation-induced reduction of cell viability was reversed by p62 siRNA2 (Figure 4D). Meanwhile, silencing of p62 notably reversed hTBK1-c.978T>A mutation-induced increase of ROS in NSC-34 cells (Figure 4E). Consistently, hTBK1-c.978T>A mutation-induced decreases of PSGK level in NSC-34 cells was significantly rescued by p62 knockdown (Figure 4F). Altogether, knockdown of p62 reversed hTBK1-c.978T>A mutation-induced ferroptosis in NSC-34 cells.
Figure 4.

Knockdown of p62 reversed hTBK1-c.978T>A mutation-induced ferroptosis in NSC-34 cells. NSC-34 cells were transfected with p62 siRNA1, p62 siRNA2 or p62 siRNA3. Then, (A) gene expression of p62 was investigated by RT-qPCR. (B) Protein expression of p62 in NSC-34 cells was tested by western blot. (C) The relative protein level of p62 was quantified by normalizing to β-actin. (D) CCK-8 assay was performed to test the cell viability. (E) ROS and (F) PGSK level in NSC-34 cells were detected by flow cytometry. **P < 0.01 compared to control, ##P < 0.01 compared to hTBK1-c.978T>A mutation, n = 3.
hTBK1-c.978T>A mutation promoted the ferroptosis in NSC-34 cells via mediation of KEAP1/NRF2/p62 signaling
To further explore the mechanism by which hTBK1-c.978T>A mutation promoted the ferroptosis in NSC-34 cells, western blot was used. As we expected, the expressions of p62 and KEAP1 in NSC-34 cells were notably increased by hTBK1-c.978T>A mutation, while this phenomenon was significantly reversed by p62 siRNA2 (Figure 5A-C). To sum up, hTBK1-c.978T>A mutation induced the ferroptosis in NSC-34 cells via mediation of KEAP1/NRF2/p62 axis.
Figure 5.
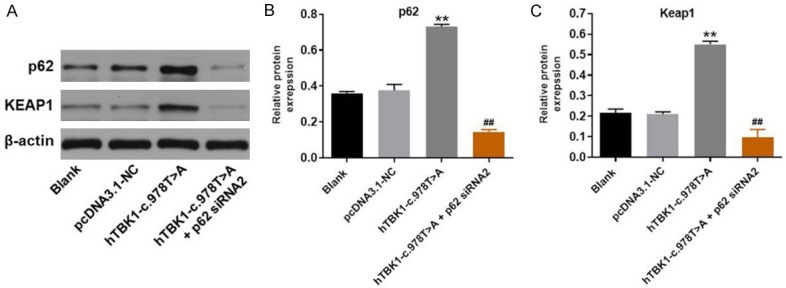
hTBK1-c.978T>A mutation promoted the ferroptosis in NSC-34 cells via mediation of KEAP1/NRF2/p62 signaling. A. The protein expressions of p62 and KEAP1 in NSC-34 cells were tested by western blot. B, C. The relative protein levels of p62 and KEAP1 were quantified by normalizing to β-actin. **P < 0.01 compared to control, ##P < 0.01 compared to hTBK1-c.978T>A mutation, n = 3.
Discussion
It has been previously reported that TBK1 mutation is involved in ALS [24]. In our study, we found hTBK1-c.978T>A mutation contributed to the occurrence of ALS via inducing ferroptosis and mediating KEAP1/NRF2/p62 signaling. This finding further supplemented the mechanism by which TBK1 mutation modulated the progression of ALS.
According to Dobson-Stone C et al [25], CYLD mutation (c.2155A4G, p.M719V) could promote the progression of ALS via mediation of p62. Our research was consistent to this data. CYLD is known to interact with TBK1 [26]. However, the correlation between TBK1 hTBK1-c.978T>A mutation and CYLD in ALS remains unclear and more investigation are needed.
It has been confirmed that ferroptosis is involved in the progression of ALS [16,27]. In addition, our findings revealed that TBK1 mutation inhibited the expressions of COX-2 and NOX1 and upregulated the protein levels of GPX4 and FTH1 in NSC-34 cells. COX-2, NOX1, GPX4 and FTH1 were key mediators in ferroptosis [12,21,28,29]. Taken together with the backgrounds, hTBK1-c.978T>A mutation induced the ferroptosis via mediation of COX-2, NOX1, GPX4 and FTH1. In addition, TBK1 mutation was confirmed to promote the ferroptosis in NSC-34 cells via mediation of KEAP1/NRF2/p62 signaling in this study. P62 is known to be participated in oxidative stress, autophagy and cell signaling [30]. Additionally, KEAP1 could serve as a NRF2 activity repressor [31]. Besides, p62 can remove ubiquitinated proteins and modulate KEAP1/NRF2 axis [32]. It has been indicated that p62 can regulate KEAP1/NRF2 pathway [33]. Our study was consistent to this recent finding. On the other hand, D’Amico AG et al revealed that pituitary adenylate cyclase activating polypeptide (PACAP) modulates ALS process via mediation of MAPK/ERK signaling [34], and PACAP dysregulation may be related to the altered hippocampal function [35]. However, the relationship between TBK1 mutation and PACAP dysregulation needed to be explored in the future.
Frankly speaking, there are some limitations in this research. For instance, the correlation between p62 and ferroptosis remains unclear. In addition, this research focused only on KEAP1/NRF2/p62 so far, and other more pathways should be investigated in the future. In conclusion, hTBK1-c.978T>A mutation promotes the ferroptosis in NSC-34 cells via mediation of KEAP1/NRF2/p62 signaling. Thus, hTBK1-c.978T>A mutation could serve as a novel target ALS treatment.
Acknowledgements
This research was supported by Natural Science Foundation of China (30871359).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Starikov L, Kottmann AH. Diminished ventral oligodendrocyte precursor generation results in the subsequent over-production of dorsal oligodendrocyte precursors of aberrant morphology and function. Neuroscience. 2020 doi: 10.1016/j.neuroscience.2020.05.027. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 2.Arbour D, Vande Velde C, Robitaille R. New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J Physiol. 2017;595:647–661. doi: 10.1113/JP270213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hossain MI, Marcus JM, Lee JH, Garcia PL, Singh V, Shacka JJ, Zhang J, Gropen TI, Falany CN, Andrabi SA. Restoration of CTSD (cathepsin D) and lysosomal function in stroke is neuroprotective. Autophagy. 2020:1–19. doi: 10.1080/15548627.2020.1761219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciesler J, Sari Y. Neurotrophic peptides: potential drugs for treatment of amyotrophic lateral sclerosis and Alzheimer’s disease. Open J Neurosci. 2013;3:21–29. doi: 10.13055/ojns_3_1_2.130408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morren JA, Galvez-Jimenez N. Current and prospective disease-modifying therapies for amyotrophic lateral sclerosis. Expert Opin Investig Drugs. 2012;21:297–320. doi: 10.1517/13543784.2012.657303. [DOI] [PubMed] [Google Scholar]
- 6.Silani V, Braga M, Cardin V, Scarlato G. The pathogenesis of ALS: implications for treatment strategies. Neurol Neurochir Pol. 2001;35:25–39. [PubMed] [Google Scholar]
- 7.Konopka A, Whelan DR, Jamali MS, Perri E, Shahheydari H, Toth RP, Parakh S, Robinson T, Cheong A, Mehta P, Vidal M, Ragagnin AMG, Khizhnyak I, Jagaraj CJ, Galper J, Grima N, Deva A, Shadfar S, Nicholson GA, Yang S, Cutts SM, Horejsi Z, Bell TDM, Walker AK, Blair IP, Atkin JD. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol Neurodegener. 2020;15:51. doi: 10.1186/s13024-020-00386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe Y, Nakagawa T, Akiyama T, Nakagawa M, Suzuki N, Warita H, Aoki M, Nakayama K. An amyotrophic lateral sclerosis-associated mutant of C21ORF2 is stabilized by NEK1-mediated hyperphosphorylation and the inability to bind FBXO3. iScience. 2020;23:101491. doi: 10.1016/j.isci.2020.101491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10:5. doi: 10.1186/s13041-017-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Majo M, Topp SD, Smith BN, Nishimura AL, Chen HJ, Gkazi AS, Miller J, Wong CH, Vance C, Baas F, Ten Asbroek ALMA, Kenna KP, Ticozzi N, Redondo AG, Esteban-Pérez J, Tiloca C, Verde F, Duga S, Morrison KE, Shaw PJ, Kirby J, Turner MR, Talbot K, Hardiman O, Glass JD, de Belleroche J, Gellera C, Ratti A, Al-Chalabi A, Brown RH, Silani V, Landers JE, Shaw CE. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol Aging. 2018;71:266.e1–266.e10. doi: 10.1016/j.neurobiolaging.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P, Blanc F, Lacomblez L, Sellal F, Fleury MC, Meininger V, Cazeneuve C, Clot F, Flabeau O, LeGuern E, Brice A French Clinical and Genetic Research Network on FTLD/FTLD-ALS. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging. 2015;36:3116.e5–3116.e8. doi: 10.1016/j.neurobiolaging.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Liu P, Wu D, Duan J, Xiao H, Zhou Y, Zhao L, Feng Y. NRF2 regulates the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via FOCAD-FAK signaling pathway. Redox Biol. 2020;37:101702. doi: 10.1016/j.redox.2020.101702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Huang Z, Xie Z, Chen Y, Zheng Z, Wei X, Huang B, Shan Z, Liu J, Fan S, Chen J, Zhao F. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol Med. 2020;160:552–565. doi: 10.1016/j.freeradbiomed.2020.08.029. [DOI] [PubMed] [Google Scholar]
- 14.Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, Jiang SD. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J Cell Physiol. 2020 doi: 10.1002/jcp.30039. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petillon C, Hergesheimer R, Puy H, Corcia P, Vourc’h P, Andres C, Karim Z, Blasco H. The relevancy of data regarding the metabolism of iron to our understanding of deregulated mechanisms in ALS; hypotheses and pitfalls. Front Neurosci. 2018;12:1031. doi: 10.3389/fnins.2018.01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devos D, Moreau C, Kyheng M, Garçon G, Rolland AS, Blasco H, Gelé P, Timothée Lenglet T, Veyrat-Durebex C, Corcia P, Dutheil M, Bede P, Jeromin A, Oeckl P, Otto M, Meininger V, Danel-Brunaud V, Devedjian JC, Duce JA, Pradat PF. A ferroptosis-based panel of prognostic biomarkers for Amyotrophic lateral sclerosis. Sci Rep. 2019;9:2918. doi: 10.1038/s41598-019-39739-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devos D, Cabantchik ZI, Moreau C, Danel V, Mahoney-Sanchez L, Bouchaoui H, Gouel F, Rolland AS, Duce JA, Devedjian JC FAIRPARK-II and FAIRALS-II studygroups. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J Neural Transm (Vienna) 2020;127:189–203. doi: 10.1007/s00702-019-02138-1. [DOI] [PubMed] [Google Scholar]
- 18.Kauffman ME, Kauffman MK, Traore K, Zhu H, Trush MA, Jia Z, Li YR. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React Oxyg Species (Apex) 2016;2:361–370. doi: 10.20455/ros.2016.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corcia P, Lumbroso S, Cazeneuve C, Mouzat K, Camu W, Vourc’h P on Behalf the FILSLAN network. Pre-symptomatic diagnosis in ALS. Rev Neurol (Paris) 2020;176:166–169. doi: 10.1016/j.neurol.2019.07.027. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao J, Ding H, Tan S, Chen A, Zhang F, Zheng S. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16:1482–1505. doi: 10.1080/15548627.2019.1687985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dachert J, Ehrenfeld V, Habermann K, Dolgikh N, Fulda S. Targeting ferroptosis in rhabdomyosarcoma cells. Int J Cancer. 2020;146:510–520. doi: 10.1002/ijc.32496. [DOI] [PubMed] [Google Scholar]
- 22.Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, Ghosh MC, Cox JE, Maschek JA, Tyurin VA, Tyurina YY, Bayir H, Aron AT, Chang CJ, Kagan VE, Philpott CC. Iron chaperone PCBP1 protects murine liver from lipid peroxidation and steatosis. Hepatology. 2020 doi: 10.1002/hep.31328. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werth EG, Rajbhandari P, Stockwell BR, Brown LM. Time course of changes in sorafenib-treated hepatocellular carcinoma cells suggests involvement of phospho-regulated signaling in ferroptosis induction. Proteomics. 2020;20:e2000006. doi: 10.1002/pmic.202000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerbino V, Kaunga E, Ye J, Canzio D, O’Keeffe S, Rudnick ND, Guarnieri P, Lutz CM, Maniatis T. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron. 2020;106:789–805. doi: 10.1016/j.neuron.2020.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Dobson-Stone C, Hallupp M, Shahheydari H, Ragagnin AMG, Chatterton Z, Carew-Jones F, Shepherd CE, Stefen H, Paric E, Fath T, Thompson EM, Blumbergs P, Short CL, Field CD, Panegyres PK, Hecker J, Nicholson G, Shaw AD, Fullerton JM, Luty AA, Schofield PR, Brooks WS, Rajan N, Bennett MF, Bahlo M, Landers JE, Piguet O, Hodges JR, Halliday GM, Topp SD, Smith BN, Shaw CE, McCann E, Fifita JA, Williams KL, Atkin JD, Blair IP, Kwok JB. CYLD is a causative gene for frontotemporal dementia - amyotrophic lateral sclerosis. Brain. 2020;143:783–799. doi: 10.1093/brain/awaa039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X, Kalac M, Markson M, Chan M, Brody JD, Bhagat G, Ang RL, Legarda D, Justus SJ, Liu F, Li Q, Xiong H, Ting AT. Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL) Cell Death Dis. 2020;11:94. doi: 10.1038/s41419-020-2294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, Barnham KJ, Crouch PJ, Ayton S, Donnelly PS, Bush AI. Cu(II) (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177:656–667. doi: 10.1111/bph.14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu MM, Dong R, Hua Z, Lv NN, Ma Y, Huang GC, Cheng J, Xu HY. Therapeutic potential of Liuwei Dihuang Pill against KDM7a and Wnt/beta-catenin signaling pathway in diabetic nephropathy-related osteoporosis. Biosci Rep. 2020;40:178–185. doi: 10.1042/BSR20201778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin PL, Tang HH, Wu SY, Shaw NS, Su CL. Saponin formosanin C-induced ferritinophagy and ferroptosis in human hepatocellular carcinoma cells. Antioxidants (Basel) 2020;9:682. doi: 10.3390/antiox9080682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei R, Enaka M, Muragaki Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci Rep. 2019;9:10366. doi: 10.1038/s41598-019-46824-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Wang W, Chen Q, Zhou Y, Wang L, Huang H. Antinociceptive effects of IL-6R vs. glucocorticoid receptors during rat hind paw inflammatory pain. Neurosci Lett. 2020;738:135356. doi: 10.1016/j.neulet.2020.135356. [DOI] [PubMed] [Google Scholar]
- 32.Chen MJ, Lin PL, Wang L, Cheng YM, Chen CY, Lee H. Cytoplasmic, but not nuclear Nrf2 expression, is associated with inferior survival and relapse rate and response to platinum-based chemotherapy in non-small cell lung cancer. Thorac Cancer. 2020;11:1904–1910. doi: 10.1111/1759-7714.13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Wu YY, Wang X, Shen P, Jia Q, Yu S, Wang Y, Li X, Chen W, Wang A, Lu Y. Lycopene prevents carcinogen-induced cutaneous tumor by enhancing activation of the Nrf2 pathway through p62-triggered autophagic Keap1 degradation. Aging (Albany NY) 2020;12:8167–8190. doi: 10.18632/aging.103132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Amico AG, Maugeri G, Saccone S, Federico C, Cavallaro S, Reglodi D, D’Agata V. PACAP modulates the autophagy process in an in vitro model of amyotrophic lateral sclerosis. Int J Mol Sci. 2020;21:2943. doi: 10.3390/ijms21082943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson GC, Parsons R, May V, Hammack SE. The role of pituitary adenylate cyclase-activating polypeptide (PACAP) signaling in the hippocampal dentate gyrus. Front Cell Neurosci. 2020;14:111. doi: 10.3389/fncel.2020.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.