

Summary
Insulin is an essential growth factor for the survival and self-renewal of human embryonic stem cells (hESCs). Although it is best known as the principal hormone promoting glycolysis in somatic cells, insulin's roles in hESC energy metabolism remain unclear. In this report, we demonstrate that insulin is essential to sustain hESC mitochondrial respiration that is rapidly decreased upon insulin removal. Insulin-dependent mitochondrial respiration is stem cell specific, and mainly relies on pyruvate and glutamine, while glucose suppresses excessive oxidative phosphorylation. Pharmacologic and genetic manipulations reveal that continuous insulin signal sustains mitochondrial respiration through PI3K/AKT activation and downstream GSK3 inhibition. We further show that insulin acts through GSK3 inhibition to suppress caspase activation and rescue cell survival. This study uncovers a critical role of the AKT/GSK3 pathway in the regulation of mitochondrial respiration and cell survival, highlighting insulin as an essential factor for accurate assessment of mitochondrial respiration in hESCs.
Keywords: insulin, AKT, GSK3, mitochondrial respiration, caspase, cell survival, human embryonic stem cells
Graphical Abstract
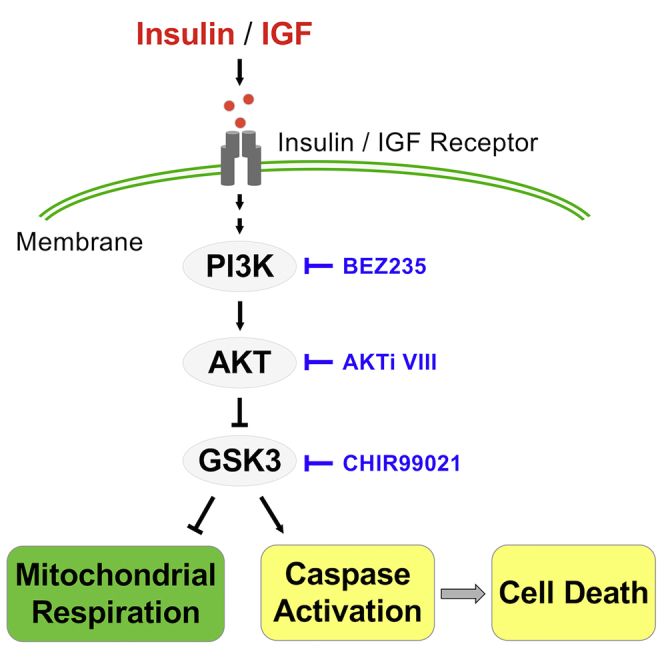
Highlights
-
•
Insulin is continuously required to sustain mitochondrial respiration in hESCs
-
•
Insulin-dependent mitochondrial respiration is substrate specific
-
•
GSK3 is a major regulator of insulin-dependent respiration and cell survival
-
•
Insulin is essential for accurate assessment of mitochondrial respiration in hESCs
Mitogenic regulation of respiration in hESCs is not well understood. Ren et al. find that insulin is continuously required to sustain mitochondrial respiration in hESCs. Both respiration and cell survival are modulated by insulin through AKT/GSK3 pathway. This study reveals an important regulatory mechanism of energetic metabolism and provides a consistent approach to accurately evaluate mitochondrial activities in hESCs.
Introduction
Human embryonic stem cells (hESCs) are capable of unlimited proliferation and have the potential to generate all cell types in our body. hESCs hold great promise for regenerative medicine, and also serve as an indispensable model system to understand cellular physiology in early embryogenesis (Gepstein, 2002; Gerecht-Nir and Itskovitz-Eldor, 2004; Takahashi and Yamanaka, 2013; Thomson et al., 1998). Insulin plays essential roles in hESC maintenance and differentiation (Chen et al., 2011; Godoy-Parejo et al., 2019; Wang et al., 2007; Yang et al., 2019) and is also well known for its regulatory roles in glycolysis in somatic cells. However, the molecular mechanisms of insulin regulation in hESCs remain unclear for many essential cellular processes such as energetic metabolism.
In hESCs, cellular energy is mainly generated through glycolysis and mitochondrial respiration. Although glycolysis produces the majority of ATP in hESCs, mitochondrial respiration is still indispensable for stem cell functions (Folmes and Terzic, 2016; Gu et al., 2016; Teslaa and Teitell, 2015). Mitochondrial respiration not only contributes to substantial energy production but also plays essential roles in fundamental processes such as cell survival and differentiation (Birket et al., 2011; Chung et al., 2007; Leese and Barton, 1984; Mandal et al., 2011; Schieke et al., 2008; Zhang et al., 2018). Excessive mitochondrial respiration has been shown to cause DNA damage and epigenetic changes (Zhang et al., 2017). The manipulation of mitochondrial respiration could affect the pluripotent status and even induce differentiation (Birket et al., 2011; Chung et al., 2007; Mandal et al., 2011; Schieke et al., 2008; Zhang et al., 2018). Due to its important functions, mitochondrial respiration has to be tightly controlled in hESCs; however, it is unknown how signal transduction is involved in mitochondrial respiration regulation in hESCs.
Insulin exerts pleiotropic effects on a variety of cellular processes in the human body. In somatic cells insulin is involved in metabolism, protein synthesis, cell survival, growth control, and apoptosis (Beitner and Kalant, 1971; Haeusler et al., 2018; Kimball et al., 1994; Lawlor and Rotwein, 2000; Morgan et al., 1971; Taha and Klip, 1999). Insulin's functions are often carried out through the PI3K/AKT pathway and its key downstream effectors such as glycogen synthase kinase 3 (GSK3) and FOXO (Brunet et al., 1999; Cross et al., 1995; Manning and Toker, 2017; Romorini et al., 2016; Yu and Cui, 2016; Zhang et al., 2011). Insulin is best known as the principal metabolic regulator promoting glycolysis in somatic cells where insulin stimulates the translocation of the glucose transporter within minutes (Beitner and Kalant, 1971; Dimitriadis et al., 2011; Osawa et al., 1996; Saltiel and Kahn, 2001; Summers et al., 1999; Wu et al., 2005). In hESCs, insulin is essential for maintenance by promoting cell survival and self-renewal (Chen et al., 2011; Godoy-Parejo et al., 2019). Insulin activates PI3K/AKT to suppress caspase cascade and promote cell survival, and AKT overexpression suppresses cell death in the absence of insulin (Godoy-Parejo et al., 2019; Hossini et al., 2016). In embryonic development, insulin has also been shown to stimulate metabolic activities, including glucose consumption and protein synthesis (Baroffio et al., 1986; Pantaleon and Kaye, 1996; Rao et al., 1990). However, it is unclear whether insulin is involved in the regulation of mitochondrial respiration in hESCs.
Despite its essential functions in metabolic regulation in somatic cells, the role of insulin in hESC metabolism remains largely unexplored. We hypothesize that insulin could also play important roles in energy metabolism regulation in stem cells. In this study, we demonstrate that insulin is continuously required to maintain mitochondrial respiration in hESCs while modulations of the insulin pathway lead to rapid energetic responses. Pyruvate and glutamine synergistically contribute to insulin-dependent mitochondrial respiration, which is negatively regulated by glucose. Insulin elevates mitochondrial respiration within minutes through the PI3K/AKT pathway and downstream GSK3 inhibition. We reveal that insulin is required for accurate assessment of energetic metabolism in hESCs. We further show that GSK3 inhibition is required to suppress the caspase cascade and to promote cell survival, but respiration regulation is independent of caspase regulation. This study not only establishes insulin as the chief regulator of stem cell respiration but also highlights GSK3 as a key component in the regulation of mitochondrial respiration and hESC survival.
Results
Insulin Is Required to Maintain Mitochondrial Respiration in hESCs
To establish the relationship between mitochondrial respiration and mitogens in hESCs, we compared the metabolic phenotypes of H1 cells under specific mitogens. We first focused on insulin because of its well-known metabolic functions in somatic cells. When H1 cells were cultured in E8 medium without insulin for 24 h, cell proliferation was suppressed in comparison with that in E8 containing insulin (Figure 1A). Oxygen consumption rate (OCR) was measured in those cells by a standard Mito Stress Test on a Seahorse flux analyzer (Figure S1A). The absence of insulin for 24 h led to significant decrease in key parameters of mitochondrial respiration, including basal, maximal respiration, and spare respiratory capacity (Figures 1B and 1C). This suggests that insulin is essential to sustain mitochondrial respiration in hESC maintenance.
Figure 1.

Insulin Is Required to Maintain Mitochondrial Respiration in hESCs
(A) Survival of H1 hESCs after 24 h of treatments with or without insulin (10 μg/mL). The cell numbers were determined using a BD Accuri C6 flow cytometer (n = 3 independent replicates).
(B) Oxygen consumption rate (OCR) of H1 hESCs measured with Mito Stress Test at baseline and in response to oligomycin (Oligo), FCCP, and rotenone plus antimycin A (Rote/AA). Cells were pre-treated for 24 h in E8 medium with or without insulin, followed by 1-h pre-incubation in assay medium (no insulin) before the assay. Data were normalized to the cell confluence. The cell confluence was determined using a Celigo image cytometer (n = 3 independent replicates).
(C) Individual parameters, including basal, maximal respiration, and spare respiratory capacity, calculated from (B).
(D) OCR of H1 hESCs after 1 h of pre-incubation in assay medium with or without insulin (n = 5 independent replicates).
(E) Individual parameters, including basal, maximal respiration, and spare respiratory capacity, calculated from (D).
(F) OCR of H1 hESCs after 1 h of pre-incubation in assay medium with individual essential factors from E8 medium (insulin, 2 ng/mL TGFβ, 100 μg/L FGF2, 10 mg/L holo-transferrin, 64 mg/L ascorbic acid, and 14 μg/L sodium selenite) (n = 3 independent replicates).
(G) OCR measured in H1 hESCs after 1 h of pre-incubation in assay medium with insulin, IGF1 (100 ng/mL), IGF2 (100 ng/mL), or none as indicated.
(H) Glycolysis stress test results of H1 hESCs following pre-incubation with or without insulin for 1 h (n = 4 independent replicates).
(I) Individual parameters, including glycolysis, glycolytic capacity, and glycolytic reverse, calculated from (H).
Data are presented as means ± SD.
We further examined the timescale of respiratory activity changes due to insulin. We noticed that 1-h absence of insulin did not lead to significant changes in cell hESC survival (Figure S1C). A standard Mito Stress Test is usually conducted without insulin (Figure S1A), as it is for all the other Seahorse flux analyses. Under such conditions, cells are kept in insulin-free medium for about 2.5 h, including a pre-incubation stage (1 h) and a measurement stage (1.5 h). Addition of insulin during the pre-incubation stage of Mito Stress Test (Figure S1B) significantly elevated basal, maximal respiration, and spare respiratory capacity (Figures 1D and 1E). This suggests that 1-h absence of insulin is sufficient to suppress mitochondrial respiration. Thus, insulin is continuously required to maintain mitochondrial respiration in hESCs.
We further examined whether other essential stem cell factors are also necessary to maintain mitochondrial respiration. Individual essential culture components, including fibroblast growth factor 2 (FGF2), transforming growth factor β (TGFβ), transferrin, ascorbic acid, and sodium selenite (Chen et al., 2011), were applied to hESCs during the pre-incubation stage and then subjected to respiration analysis with the Mito Stress Test. No insulin-like effect was observed in cells treated with other essential factors from E8 medium, including FGF2, TGFβ, transferrin, ascorbic acid, and sodium selenite (Figures 1F and S1D). In addition, insulin alone was sufficient to promote respiration to the same level as the condition with all E8 components combined (Figure S1E). Meanwhile, insulin-like growth factor 1 (IGF1) and IGF2 proteins were able to sustain mitochondrial respiration at the same level as insulin did when they were added during the pre-incubation stage (Figure 1G). We also showed that respiration was sustained by insulin in both H9 hESCs and NCRM-4 induced pluripotent stem cells (iPSCs) (Figures S1F and S1G). These data suggest that insulin-dependent respiration is maintained through insulin/IGF signaling in human PSC lines.
Given that glycolysis is promoted by insulin in somatic cells, we examined whether insulin could influence glycolysis in hESCs with a Seahorse analyzer (Figure S1H). Insulin was added to hESCs at pre-incubation stage in a glycolysis stress test, but no significant effect was observed with insulin treatment (Figures 1H and 1I). These results indicate that insulin is not necessary to maintain glycolysis in hESCs during short-term treatment.
Insulin Promotes Mitochondrial Respiration through Specific Energy Substrates
Various metabolic substrates could contribute to mitochondrial respiration, but their functions have not been systematically analyzed in hESCs. We inspected their energetic contribution to maintaining respiration with or without insulin. Glucose (Glc), glutamine (Gln), and pyruvate (Pyr) are the main energy substrates in stem cell media and in Mito Stress assay (Figure S1A). We first compared the impact of various substrate combinations on respiration in the standard Mito Stress Test without insulin (Figure S2A). Mitochondrial respiration was greatly influenced by the choice of energetic substrates (Figures 2A and S2B). The consumption of glucose alone (Glc) led to a relatively low level of respiration. The addition of pyruvate (Glc + Pyr) significantly increased both maximal respiration and spare respiratory capacity, but glutamine (Glc + Gln) only had a small positive effect on spare respiratory capacity. Adding both glutamine and pyruvate to glucose (Glc + Pyr + Gln) synergistically increased all the respiration parameters. Surprisingly, when glucose was removed, glutamine and pyruvate (Pyr + Gln) led to the highest levels of respiration (Figures 2A and S2B). These data suggest that glutamine and pyruvate are the main substrates for mitochondrial respiration in hESCs, but glucose suppresses excessive oxidation.
Figure 2.

Insulin Promotes Mitochondrial Respiration through Specific Energy Substrates
(A and B) OCR of H1 hESCs measured after 1 h of pre-incubation with different substrate (glucose-Glc, glutamine-Gln, pyruvate-Pyr) combinations in the absence (A) or presence (B) of insulin (n = 3 independent replicates).
(C) Maximal respiration calculated from (A) and (B).
(D) Basal respiration measurement with injections of UK5099 (2 μM) or BPTES (4 μM).
(E and F) OCR measured following 1 h of pre-incubation with different substrate combinations or under the substrate-free condition (Base) in the presence or absence of insulin as indicated.
(G) OCR measured following 1 h of pre-incubation with substrate-free condition (Base), glucose (Glc), or glucose plus the glycolysis inhibitor 2-DG (Glc + 2-DG) in the presence of insulin.
(H) Basal respiration measurement with injections of different energy substrates or energy-free control (Base).
(I and J) Fatty acid oxidation (FAO) assay in H1 hESCs without insulin (I) or with insulin (J) (n = 3 independent replicates).
(K) Endogenous FAO calculated by maximal respiration without or with insulin.
Data are presented as means ± SD.
We then examined the impact of insulin on the substrate-specific respiration. The overall trend is similar to that observed without insulin. When insulin was added during the pre-incubation stage, it did not significantly increase respiration when only glucose was in the assay (Figures 2A and 2B). However, insulin stimulated maximal respiration and spare respiration capacity when glutamine or pyruvate was supplied in the assay (Figures S2C and 2C). Similar to the profiles without insulin, the combination of pyruvate and glutamine (Pyr + Gln) led to the highest OCR, but the level was significantly suppressed by the addition of glucose (Glc + Pyr + Gln). These results indicate that insulin promotes mitochondrial respiration mainly through pyruvate and glutamine oxidation, but glucose suppresses insulin-dependent respiration.
We further evaluated individual substrates' impact on mitochondrial respiration. In the standard Mito Stress assay (Glc + Pyr + Gln), the mitochondrial pyruvate carrier inhibitor UK5099 suppressed basal respiration more than the glutaminase inhibitor bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) (Figure 2D), suggesting that pyruvate and glutamine have a distinctive contribution to respiration in the assay. Similar levels of mitochondrial respiration were observed with either pyruvate (Pyr) or glutamine (Gln) alone (Figure 2E); however, glucose differentially suppressed mitochondrial respiration depending on the availability of other substrates. Pyruvate-based respiration (Pyr + Glc) was less sensitive to glucose suppression than glutamine-based respiration (Gln + Glc) (Figure 2E). The increased glutamine did not relieve glucose suppression (Figure S2D). These results were consistent with the observation in Figure 2B that pyruvate contributed a larger proportion than glutamine to mitochondrial respiration when all three energy substrates were present.
We then inspected how glucose suppresses mitochondrial respiration in hESCs. Besides suppressing pyruvate- and glutamine-based respiration (Figures 2A, 2B and 2E), glucose alone led to lower respiration than base medium without other energy substrates, which could not be rescued by insulin (Figure 2F). This result indicates that glucose represses mitochondrial respiration independent of insulin stimulation. We also showed that the glycolysis inhibitor 2-deoxy-D-glucose (2-DG) rescued maximal respiration (Figure 2G). We further showed that glucose suppressed basal respiration in a few minutes in base medium (green line) (Figure 2H). Although mitochondrial respiration was enhanced by pyruvate and glutamine, it was significantly repressed by glucose. Collectively, these data demonstrate that glucose suppresses mitochondrial respiration in hESCs and has differential impacts on energetic metabolism of pyruvate and glutamine.
In addition to the substrates in standard Mito Stress assays, fatty acids are also utilized in mitochondrial respiration, so we investigated whether insulin influences fatty acid oxidation (FAO). The assay was conducted by combining the Mito Stress Test with XF Palmitate-BSA substrate and etomoxir (ETO), a carnitine palmitoyl-transferase 1A inhibitor. The FAO assay medium contains glucose as a carbon source. We first examined FAO in the absence of insulin (Figure S2E). Basal respiration was not suppressed by ETO, indicating that endogenous FAO (difference between BSA and BSA + ETO) does not contribute to basal respiration. In comparison, ETO significantly inhibited maximal respiration and spare respiratory capacity, suggesting that endogenous FAO is involved in maximal respiration and spare respiratory capacity (Figures 2I and S2F). Exogenous fatty acid palmitate (Palm:BSA) did not further increase the respiration levels in comparison with the control condition (BSA) (Figures 2I and S2F). When insulin was added during the pre-incubation stage, maximal respiration and spare respiratory capacity were significantly increased in both the control condition (BSA) and the one with ETO (BSA + ETO), but basal respiration was not affected by insulin (Figures 2I, 2J, and S2F). The presence of insulin enhanced the endogenous FAO (the difference between BSA and [BSA + ETO]) as indicated by maximal respiration measurement (Figure 2K). Moreover, exogenous palmitate did not enhance OCR in the presence of insulin. These results suggest that insulin upregulates endogenous FAO, which contributes to maximal respiration and spare respiratory capacity in hESCs.
Insulin-Dependent Mitochondrial Respiration Is Cell-type Specific
We next examined whether insulin-dependent mitochondrial respiration is associated with pluripotency. The insulin-dependent mitochondrial respiration diminished after hESCs were induced to differentiate for 2 days, including mesendoderm lineage (Figures 3A and S3B), ectoderm lineage (Figures 3B and S3C), and spontaneous differentiation (Figures S3A and S3D) conditions. The insulin-dependent mitochondrial respiration was also absent in fibroblasts (CCD-1139Sk) (Figure 3C). These data indicate that insulin-dependent respiration is specific to PSCs.
Figure 3.

Insulin-Dependent Mitochondrial Respiration Is Cell-type Specific
(A–C) OCRs were measured by Mito Stress Test in mesendoderm progenitors (A), ectoderm progenitors (B), and fibroblasts (C) after 1 h of pre-incubation with or without insulin.
(D) OCR of fibroblasts measured with different substrate (glucose-Glc, glutamine-Gln, pyruvate-Pyr) combinations.
(E) Spare respiratory capacity calculated from (D).
(F) Spare respiratory capacity calculated from (D) and Figure 2A.
Data are presented as means ± SD.
We then focused on fibroblasts to inspect the utilization of energy substrates in somatic cell respiration. Similar to hESCs, basal respiration of fibroblasts also relied on pyruvate and glutamine and was suppressed by glucose (Figures 3D and S3E). However, unlike hESCs, fibroblasts displayed fewer differences in maximal respiration between different conditions (Figure S3E). The absence of glucose (Pyr + Gln) led to a decrease in spare respiratory capacity, whereas the removal of pyruvate (Glc + Gln) or glutamine (Glc + Pyr) resulted in a small increase (Figure 3E). Even though fibroblasts had much larger spare respiratory capacity than hESCs under standard Mito Stress Test conditions (Glc + Pyr + Gln), the difference diminished under the glucose-free (Pyr + Gln) condition (Figure 3F). These results suggest that the respiratory profiles of hESCs and differentiated cells are both substrate dependent, but the insulin-dependent mitochondrial respiration is only observed in pluripotent stem cells. Unlike in somatic cells, insulin is necessary to accurately measure energetic metabolism in hESCs.
Insulin-Dependent Mitochondrial Respiration Is Rapidly Regulated through the PI3K/AKT Pathway
As mitochondrial respiration is upregulated by insulin and IGFs (Figure 1G), we interrogated how insulin/IGF signal transduction regulates mitochondrial respiration. When hESCs are switched from insulin-containing E8 medium to insulin-free assay medium, the phosphorylation of AKT (Ser473) was significantly decreased within 1 h (Figure 4A). The addition of insulin maintained AKT phosphorylation at the same level as in E8 medium. The insulin-dependent AKT phosphorylation was suppressed by the PI3K inhibitor BEZ235. In the Mito Stress Test, BEZ235 significantly decreased insulin-dependent mitochondrial respiration when it was added during pre-incubation stage (Figures 4B and S4A). To closely observe the immediate effects of insulin on mitochondrial respiration, we injected specific pathway modulators during the measurement of basal respiration (Figure S4B). Within minutes, insulin significantly elevated basal respiration in comparison with the insulin-free condition (Figure 4C). BEZ235 not only suppressed insulin's positive influence but also further decreased basal respiration. These data suggest that insulin maintains mitochondrial respiration through the PI3K/AKT pathway.
Figure 4.

Insulin Regulates Mitochondrial Respiration via PI3K/AKT in hESCs
(A) Western blot analysis of p-AKT (Ser473) and total AKT in H1 hESCs after 1 h of incubation in E8 or Mito Stress Test assay medium supplemented with vehicle ± insulin, or BEZ235 (BEZ, 2.5 μM) ± insulin. GAPDH was used as loading control.
(B) OCR of H1 hESCs after 1 h of pre-incubation with vehicle ± insulin or BEZ235 ± insulin, measured with Mito Stress Test at baseline and in response to Oligo, FCCP, and Rote/AA (n = 3 independent replicates).
(C) Kinetic OCR response to vehicle ± insulin or BEZ235 ± insulin as indicated.
(D) OCR of H1 hESCs following 1 h of pre-incubation with vehicle ± insulin or AKTi VIII (10 μM) ± insulin, measured with Mito Stress Test (n = 3 independent replicates).
(E) Kinetic OCR response to vehicle ± insulin or AKTi VIII (10 μM) ± insulin as indicated.
(F and G) OCR of AKT1-overexpressing (F) and AKT2-overexpressing (G) H1 cell lines following 1 h of pre-incubation with or without insulin, measured with Mito Stress Test. The data were normalized to the cell numbers. The cell numbers were determined using a Celigo image cytometer (n = 3 independent replicates).
(H) Western blot analyses of p-AKT (Ser473) and total AKT in H1 hESCs, and AKT1- and AKT2-overexpressing H1 cell lines after 2 h of incubation in E8 or Mito Stress Test assay medium. GAPDH was used as loading control.
(I) Western blot analyses of p-AKT (Ser473) and AKT in whole-cell lysate and the mitochondrial fraction from H1 hESCs after 2 h of incubation in Mito Stress Test assay medium with or without insulin. COX IV and GAPDH were used as loading controls.
(J) Immunostaining of p-AKT (Ser473) in H1 hESCs (green) after 2 h of incubation in Mito Stress Test assay medium supplemented with insulin. Mitochondria were stained with MitoTracker (red) and nuclei with Hoechst (blue). Vehicle, DMSO. Scale bar, 5 μm.
Data are presented as means ± SD.
Similar to BEZ235, AKT inhibitor VIII (AKTi VIII) efficiently suppressed insulin-dependent basal and maximal respiration (Figures 4D and S4C). AKTi VIII also suppressed insulin-dependent OCR within a few minutes in basal respiration analysis (Figures S4B and S4E). When constitutively active AKT1 or AKT2 was overexpressed in hESCs (Godoy-Parejo et al., 2019), mitochondrial respiration was significantly enhanced in the absence of insulin (Figures 4F, 4G, S4D, and S4E). Western blot analysis confirmed that AKT and p-AKT protein levels of AKT1- and AKT2-overexpressing cell lines were significantly higher than that of wild-type hESCs in insulin-containing E8 condition and insulin-free assay medium (Figure 4H). Because the action of insulin or the pathway inhibitors takes effect in minutes, we speculate that AKT acts directly in mitochondria. We showed that AKT was present in mitochondria and responded to insulin stimulation (Figure 4I). Immunostaining showed that phosphorylated AKTs (Ser473) were observed in mitochondria (Figure 4J). These data indicate that the insulin-dependent mitochondrial respiration is rapidly regulated through the PI3K/AKT pathway, most likely through the action of AKT on mitochondrial targets related to energetic metabolism.
Because various enzymes are involved in energetic metabolism, we inspected whether insulin/AKT acts by altering either protein levels or phosphorylation levels of key metabolic enzymes in the tricarboxylic acid cycle and glycolysis pathway. The phosphorylation of key enzymes, such as pyruvate dehydrogenase E1 component subunit α, pyruvate kinase M2, and acetyl-coenzyme A carboxylase α, were reported to affect mitochondrial respiration in tumor cells (Cerniglia et al., 2015; Jones et al., 2017; Luan et al., 2015). We examined the phosphorylation levels of these enzymes, whereby our data showed that their phosphorylation was not significantly affected by insulin in hESCs (Figure S4F). We further showed that the protein levels of additional metabolic enzymes, including hexokinase 1 (HK1), HK2, and pyruvate dehydrogenase kinase 4, were not significantly changed by insulin either (Figure S4G). These results suggest that the insulin-dependent respiration is not modulated by changing the abundance and phosphorylation of the above metabolic enzymes, but through other targets.
GSK3 Inhibition Promotes Mitochondrial Respiration in hESCs
Besides the metabolic enzymes, we further explored whether AKT-associated signal transducers were involved in mitochondrial respiration in hESCs. GSK3 and FOXO proteins are known AKT substrates involved in signal transduction, and their activities are regulated by AKT-dependent phosphorylation (Brunet et al., 1999; Cross et al., 1995; Manning and Toker, 2017; Romorini et al., 2016; Yu and Cui, 2016; Zhang et al., 2011). We showed that GSK3 and FOXO phosphorylation decreased within 1 h after the removal of insulin (Figures 5A and S5A). In the Mito Stress Test, the inhibition of GSK3 by CHIR99021 significantly elevated OCR, especially maximal respiration, to a level similar to that seen with insulin treatment, and no additive effect was observed for insulin and CHIR99021 (Figure 5B). When CHIR99021 was applied during the measurement of basal respiration it significantly increased the respiration level, although not as potently as insulin (Figure 5C). When CHIR99021 was added during the pre-incubation stage, it rescued both basal and maximal respiration that were suppressed by BEZ235 or AKTi VIII in the Mito Stress Test (Figures 5D and 5E). In contrast, the FOXO inhibitor did not elevate respiration (Figure S5B), although it was able to induce differentiation quickly (Figure S5C). peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) is a known regulator of mitochondrial biogenesis and is downstream of GSK3 regulation (Anderson et al., 2008; Lin et al., 2005; Martin et al., 2018; Souder and Anderson, 2019). We showed that the PGC1α inhibitor ZLN005 suppressed mitochondrial respiration in the presence of insulin (Figure S5D). These results indicate that GSK3 is a key effector in mitochondrial respiration in hESCs.
Figure 5.

GSK3 Inhibition Promotes Mitochondrial Respiration in hESCs
(A) Western blot analysis of p-GSK-3α/β and GSK-3α/β in H1 ESCs following 1 h of incubation in E8 or Mito Stress Test assay medium with or without insulin. β-Actin was used as loading control.
(B) OCR of H1 hESCs following 1 h of pre-incubation with vehicle ± insulin or CHIR99021 (CHIR, 5 μM) ± insulin, measured with Mito Stress Test.
(C) Kinetic OCR response to vehicle, vehicle + insulin, or CHIR99021 as indicated.
(D) OCR of H1 hESCs following 1 h of pre-incubation with vehicle, CHIR99021, AKTi VIII ± CHIR99021, or BEZ235 ± CHIR99021 (n = 3 independent replicates).
(E) Basal and maximal respiration calculated from (D). Vehicle, DMSO.
Data are presented as means ± SD.
Although fibroblasts did not display insulin-dependent mitochondrial respiration (Figure 3C), insulin was required to sustain phosphorylation of AKT and GSK3 proteins in fibroblasts in the same manner as observed in hESCs (Figures S5E, 4A, and 5A). However, AKTi VIII did not suppress respiration in the presence of insulin in fibroblasts (Figure S5F). At the same time, the GSK3 inhibitor CHIR99021 did not increase respiration in insulin-free conditions (Figure S5G). These data indicate that mitochondrial respiration is differentially modulated in hESCs and fibroblasts. We also showed that GSK3 protein levels in hESCs were significantly higher than those in fibroblasts (Figure S5H). It is possible that the low GSK3 level in fibroblasts could bypass the requirement for AKT-dependent phosphorylation to relieve the GSK3 suppression on mitochondrial respiration and other functions. However, more studies are necessary to explore the molecular mechanism.
GSK3 Inhibition Promotes hESC Survival
In addition to sustaining respiration, insulin is necessary for AKT-dependent cell survival during maintenance (Godoy-Parejo et al., 2019). However, we note that insulin is not required for hESC survival when the GSK3 inhibitor CHIR99021 is applied to initiate differentiation (Lin et al., 2017; Yang et al., 2019). We examined whether GSK3 could be involved in the caspase cascade and hESC cell death. The phosphorylation of GSK3 was enhanced without insulin in AKT-overexpressing cell lines, especially in the AKT2-overexpressing cell line (Figure 6A). Caspase-3 cleavage was suppressed in insulin-containing E8 medium. After hESCs were cultured in insulin-free Mito Stress Test assay medium for 2 h, significantly more caspase-3 was cleaved (Figure 6B). The caspase cleavage was suppressed by the addition of either insulin or the GSK3 inhibitor CHIR99021. We then demonstrated that the caspase-3 cleavage was suppressed by AKT overexpression (Figure 6B). However, caspase-3 inhibition with Z-DEVD-FMK did not significantly enhance mitochondrial respiration (Figures 6C and S6A). This result indicates that caspase inhibition by insulin does not contribute to enhanced respiration.
Figure 6.

GSK3 Inhibition Promotes the Survival of hESCs
(A) Western blot analysis of p-GSK-3α/β and GSK-3α/β in H1 ESCs and AKT1- and AKT2-overexpressing H1 cell lines following 2 h of incubation in E8 or Mito Stress Test assay medium. β-Actin was used as loading control.
(B) Western blot analysis of cleaved caspase-3 in H1 ESCs and AKT1- and AKT2-overexpressing H1 cell lines after 2 h of incubation in E8 or Mito Stress Test assay medium with vehicle, CHIR99021 (5 μM), or insulin as indicated. β-Actin was used as loading control.
(C) OCR of H1 hESCs following 1 h of pre-incubation with vehicle or Z-DEVD-FMK (a caspase-3 inhibitor), measured with Mito Stress Test (n = 3 independent replicates).
(D) Images of dissociated H1 hESCs 24 h after plating with vehicle ± insulin or CHIR99021 (1 μM) ± insulin.
(E) Survival of dissociated H1 hESCs 24 h after plating with vehicle ± insulin or CHIR99021 (1 μM) ± insulin. The cell numbers were determined using a BD Accuri C6 flow cytometer. Vehicle, DMSO (n = 4 independent replicates).
Data are presented as means ± SD.
We finally examined whether GSK3 inhibition is associated with hESC survival. Individualized hESCs usually suffered severe cell death in 24 h without insulin after passaging. Insulin and AKT overexpression rescued cell survival (Figures 6D, 6E, S6B, and S6C). Meanwhile, GSK3 inhibition by CHIR99021 also enhanced cell survival significantly (Figures 6D and 6E). Our data suggest that insulin-induced GSK3 inhibition promotes hESC survival and sustains mitochondrial respiration.
Discussion
Insulin signaling plays crucial roles in early embryonic development. Besides regulating growth and differentiation, insulin stimulates metabolic activities, including glucose consumption and protein synthesis (Baroffio et al., 1986; Pantaleon and Kaye, 1996; Rao et al., 1990; Spaventi et al., 1990). As an essential factor for hESC survival, insulin is a common presence in various hESC culture media (Chen et al., 2011; Godoy-Parejo et al., 2019; International Stem Cell Initiative et al., 2010). This study demonstrates that insulin is required to maintain mitochondrial respiration in hESCs and reveals the interaction between energy substrates and insulin signal transduction in respiration. Insulin promotes both mitochondrial respiration and survival of hESCs through the PI3K/AKT/GSK3 pathway.
Unlike somatic cells, hESCs require continuous insulin stimulation to sustain mitochondrial respiration. Insulin promotes respiration through PI3K/AKT activation and subsequent GSK3 inhibition. Suppressed respiration is observed within minutes after the inhibition of the insulin pathway. This fast-mode effect of insulin is distinct from its conventional roles in regulation of transcription, translation, proliferation, and differentiation, which often take hours or days (Colosia et al., 1988; Feldman et al., 1997; Haeusler et al., 2018; Kimball et al., 1997; Lawlor and Rotwein, 2000; Lian et al., 2013). Judging from its immediate impact, insulin probably stimulates kinase cascades to directly activate metabolic enzymes in the mitochondrial respiratory chain. GSK3 may also affect mitochondrial respiration through downstream effectors, such as PGC1α. We showed that the PGC1α inhibitor significantly downregulates mitochondrial respiration, which is consistent with PGC1α′s crucial role in regulating mitochondrial functions (Anderson et al., 2008; Martin et al., 2018; Souder and Anderson, 2019). More studies are necessary to discern the details of respiration regulation by signal transduction.
Besides mitogen stimulation, the combination of metabolic substrates is also critical in maintaining a proper respiration level in hESCs. Pyruvate and glutamine are the main exogenous substrates for respiration, and they synergistically enhance basal and maximal respiration. Meanwhile, endogenous fatty acids also contribute to cellular respiration. Insulin acts as a positive enforcer to sustain the substrate-specific respiration while glucose suppresses oxidation of pyruvate and glutamine. Insulin and glucose are essential for a balanced cellular respiration to support normal stem cell functions. Besides generating cellular energy, glucose could potentially prevent metabolic defects and DNA damage by suppressing excessive respiration (Zhang et al., 2017).
hESCs and somatic cells maintain distinctive energetic balance and also respond differently to mitogen regulation in metabolism. Insulin is best known to promote rapid glycolytic response in adipocytes and hepatocytes (Dimitriadis et al., 2011; Noguchi et al., 2013; Saltiel and Kahn, 2001). Emerging proof also reveals that insulin signaling is involved in the mitochondrial functions (Cheng et al., 2010; Nisr and Affourtit, 2014; Zhao et al., 2014). In myoblasts, insulin elevates the levels of both mitochondrial respiration and glycolysis (Nisr and Affourtit, 2014). We showed that insulin regulates glycolysis and mitochondrial respiration quite differently in hESCs. Insulin does not enhance glycolysis in the fast mode; however, it is required to sustain mitochondrial respiration that requires pyruvate and glutamine. The insulin-dependent mitochondrial respiration diminishes in differentiated cells, such as fibroblasts. It would be interesting to further explore the molecular mechanism of cell-type-specific energy metabolism regulated by insulin and its functional impacts.
Many insulin-associated functions are carried out through the PI3K/AKT kinase cascade, and various downstream effectors have been reported (Cantley, 2002; Manning and Toker, 2017; Vivanco and Sawyers, 2002). In hESCs, our data indicate that GSK3 is the main downstream effector of the PI3K/AKT pathway that controls insulin-dependent mitochondrial respiration and cell survival. Complexes I, II, III, and IV of the respiratory chain are phosphorylated and suppressed by GSK3 (Gong et al., 2017; Sieber et al., 2016; Yang et al., 2017), and insulin probably promotes respiration by relieving this GSK3-related suppression. At the same time, GSK3 inhibition suppresses the caspase cascade in the absence of insulin. However, caspase inhibition does not rescue mitochondrial respiration, suggesting that GSK3 regulates parallel pathways for stem cell energy production and cell survival. It is important to note that fibroblasts are insensitive to AKT/GSK3 modulation in both respiration and cell survival. The key downstream effectors of AKT/GSK3 pathway are still to be identified.
Although only the insulin/IGF pathway exerts immediate effect in sustaining respiration, FGF2 and TGFβ may also affect mitochondrial respiration. FGF2 and TGFβ removal for 2 days leads to the loss of insulin-dependent respiration (Figure S3A), coinciding with the exit of pluripotency (Figure S3D). This phenomenon highlights the complexity of metabolic activities in stem cells. When hESCs are cultured without essential growth factors for extended periods of time, changes in transcription and translation could take place to cause metabolic changes, which go beyond signal transduction through kinase cascades. It is important to understand whether the metabolic change is caused by lack of growth factor stimulation or by the loss of pluripotency. Further studies by combining metabolic flux analysis with more sophisticated analyses of pluripotency and metabolic enzymes will likely reveal interesting facts about respiration regulation by those signals.
The understanding of insulin-dependent respiration is crucial for accurate assessment of energetic metabolism in human PSCs. The balance of energetic metabolism is shifted toward mitochondrial respiration upon differentiation, but reprogramming brings the balance toward glycolysis in iPSCs (Zhang et al., 2012a). Metabolic flux analyses on the Seahorse XF platform are the key tools for assessment of energetic metabolism, but most standard assays are conducted in growth-factor-free conditions (Sperber et al., 2015; Xu et al., 2016; Zhang et al., 2012b). In the absence of insulin, mitochondrial respiration in stem cells will be underestimated by more than 20%, while differentiated cells are not affected. When insulin is provided in the assay, mitochondrial respiration is sustained and the difference between hESCs and somatic cells is not as large as previously estimated. At the same time, insulin in the assay suppresses caspase cascade and maintains cellular integrity, which further improves the measurement of respiration. We believe that insulin is an essential factor for accurate energetic analyses of hESCs, and improved metabolic assays based on the findings from this study will help researchers to better understand the relationship between metabolism and pluripotency.
In short, this report shows that insulin sustains hESC mitochondrial respiration and cell survival through the PI3K/AKT/GSK3 axis. Our study highlights the integral role of the insulin pathway in cellular metabolism and reveals that GSK3 is the central factor controlling energetic regulation and cell survival. Understanding of the insulin dependency of hESC respiration will allow us to evaluate the coordination between signal transduction and metabolism in pluripotent and somatic cells and to explore their metabolic dynamics in a more accurate way.
Experimental Procedures
Cell Culture
The use of hESCs and hiPSCs were approved by the Institutional Review Board at the University of Macau. H1 and H9 hESCs (Thomson et al., 1998) and NCRM-4 human iPSCs (hiPSCs) (https://commonfund.nih.gov/stemcells/lines) were used in this study. The majority of experiments were conducted with the H1 line unless otherwise specified. hESCs and hiPSCs were maintained in E8 medium on Matrigel-coated plates (Corning) (Chen et al., 2011). Cells are passaged with Dulbecco’s PBS/EDTA in the presence of Y-27632 (Selleck) as previously described (Chen et al., 2010). Fibroblast (CCD-1139Sk, ATCC) cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco) for 4–5 days and were passaged with TrypLE Select Enzyme (Life Technologies).
Metabolic Flux Assay
Metabolic flux assay was performed using a Seahorse XF96 analyzer (Agilent). hESCs (2 × 104 cells/well) were seeded in the XF96 microplate pre-coated with Matrigel in growth medium. The medium was changed every day. In the Mito Stress Test, on the day of the assay the growth medium was replaced with assay medium (Agilent) or assay medium supplemented with drugs as indicated. Cells were incubated in an incubator without CO2 for 1 h before initiation of the test. The assay medium for the standard Mito Stress Test was Seahorse XF base medium supplemented with glucose (10 mM, Sigma-Aldrich), pyruvate (1 mM, Thermo Fisher) and glutamine (2 mM, Thermo Fisher). The final concentrations of specific chemicals are oligomycin at 1 μM, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) at 1 μM, and rotenone/antimycin A at 0.5 μM. Other drugs or chemicals applied during Mito Stress Test as indicated in the figures include recombinant human TGFβ (R&D), FGF2 (Chen Lab), holo-transferrin (Sigma), ascorbic acid (Sigma), sodium selenite (Sigma-Aldrich), IGF1 (PeproTech), IGF2 (PeproTech), BPTES (Sigma), UK5099 (Sigma), DMSO (Sigma-Aldrich), AKTi VIII (Calbiochem), NVP-BEZ235 (LC Laboratories), CHIR99021 (Selleck), Z-DEVD-FMK (R&D), and ZLN005 (Selleck). The assay medium for the Glycolysis Stress Test was Seahorse XF base medium supplemented with glutamine (2 mM). The final concentrations of specific chemicals were glucose at 10 mM, oligomycin at 1 μM, and 2-DG at 50 mM. In the FAO assay, cells were cultured in E8 medium and then changed to KHB medium (111 mM NaCl [Sigma], 4.7 mM KCl [Sigma], 1.25 mM CaCl2 [Sigma], 2 mM MgSO4 [Sigma], and 1.2 mM NaH2PO4 [Sigma]) supplemented with glucose (2.5 mM), carnitine (0.5 mM, Sigma) and HEPES (5 mM, Sigma) 45 min before the assay. ETO (40 μM) was added 15 min before the assay. BSA or Palm:BSA was added immediately before the assay, followed by sequential injections of oligomycin (1 μM), FCCP (1 μM), and rotenone/antimycin A (0.5 μM). All assay media were adjusted to pH 7.4 at 37°C.
Statistical Analysis
The data are presented as mean ± standard deviation (SD). Student's t test was used for experiments with two groups; ANOVA followed by Tukey’s post hoc test was used for experiments with multiple groups. Significance is indicated in figures as ∗p < 0.05; n.s., not significant.
Author Contributions
Z.R. and G.C. designed the experiments; Z.R., H.Z., C.S., and W.L. conducted Seahorse analysis; Z.R. and C.D. performed western blot and caspase related experiments; Z.R. and H.Z. conducted immunostaining; Z.R. and H.-T.H conducted cell survival experiments; Z.R. and G.C. analyzed the data; Z.R., W.L., and G.C. wrote the paper. All authors read and approved the manuscript. Funding acquisition was by G.C.
Acknowledgments
We thank Drs. William Chao, Edwin Cheung, Qi Zhao, and Garry Wong for the discussion. This project was funded by University of Macau (file nos. MYRG2015-00228-FHS, MYRG2018-00135-FHS, and MYRG2019-00147-FHS), and also by the Science and Technology Development Fund, Macau SAR (file nos. 131/2014/A3, 056/2015/A2, 0059/2019/A1, and 0123/2019/A3).
Published: November 12, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.10.008.
Supplemental Information
References
- Anderson R.M., Barger J.L., Edwards M.G., Braun K.H., O'Connor C.E., Prolla T.A., Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroffio A., Raddatz E., Markert M., Kucera P. Transient stimulation of glucose metabolism by insulin in the 1-day chick embryo. J. Cell Physiol. 1986;127:288–292. doi: 10.1002/jcp.1041270215. [DOI] [PubMed] [Google Scholar]
- Beitner R., Kalant N. Stimulation of glycolysis by insulin. J. Biol. Chem. 1971;246:500–503. [PubMed] [Google Scholar]
- Birket M.J., Orr A.L., Gerencser A.A., Madden D.T., Vitelli C., Swistowski A., Brand M.D., Zeng X. A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells. J. Cell Sci. 2011;124:348–358. doi: 10.1242/jcs.072272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Cerniglia G.J., Dey S., Gallagher-Colombo S.M., Daurio N.A., Tuttle S., Busch T.M., Lin A., Sun R., Esipova T.V., Vinogradov S.A. The PI3K/Akt pathway regulates oxygen metabolism via pyruvate dehydrogenase (PDH)-E1alpha phosphorylation. Mol. Cancer Ther. 2015;14:1928–1938. doi: 10.1158/1535-7163.MCT-14-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Gulbranson D.R., Hou Z., Bolin J.M., Ruotti V., Probasco M.D., Smuga-Otto K., Howden S.E., Diol N.R., Propson N.E. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Hou Z., Gulbranson D.R., Thomson J.A. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7:240–248. doi: 10.1016/j.stem.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z., Tseng Y., White M.F. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 2010;21:589–598. doi: 10.1016/j.tem.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S., Dzeja P.P., Faustino R.S., Perez-Terzic C., Behfar A., Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007;4(Suppl 1):S60–S67. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosia A.D., Marker A.J., Lange A.J., el-Maghrabi M.R., Granner D.K., Tauler A., Pilkis J., Pilkis S.J. Induction of rat liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase mRNA by refeeding and insulin. J. Biol. Chem. 1988;263:18669–18677. [PubMed] [Google Scholar]
- Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Dimitriadis G., Mitrou P., Lambadiari V., Maratou E., Raptis S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011;93(Suppl 1):S52–S59. doi: 10.1016/S0168-8227(11)70014-6. [DOI] [PubMed] [Google Scholar]
- Feldman E.L., Sullivan K.A., Kim B., Russell J.W. Insulin-like growth factors regulate neuronal differentiation and survival. Neurobiol. Dis. 1997;4:201–214. doi: 10.1006/nbdi.1997.0156. [DOI] [PubMed] [Google Scholar]
- Folmes C.D., Terzic A. Energy metabolism in the acquisition and maintenance of stemness. Semin. Cell Dev. Biol. 2016;52:68–75. doi: 10.1016/j.semcdb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gepstein L. Derivation and potential applications of human embryonic stem cells. Circ. Res. 2002;91:866–876. doi: 10.1161/01.res.0000041435.95082.84. [DOI] [PubMed] [Google Scholar]
- Gerecht-Nir S., Itskovitz-Eldor J. Cell therapy using human embryonic stem cells. Transpl. Immunol. 2004;12:203–209. doi: 10.1016/j.trim.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Godoy-Parejo C., Deng C., Liu W., Chen G. Insulin stimulates PI3K/AKT and cell adhesion to promote the survival of individualized human embryonic stem cells. Stem Cells. 2019;37:1030–1041. doi: 10.1002/stem.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y., Guo H., Zhang Z., Zhou H., Zhao R., He B. Heat stress reduces sperm motility via activation of glycogen synthase kinase-3alpha and inhibition of mitochondrial protein import. Front. Physiol. 2017;8:718. doi: 10.3389/fphys.2017.00718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Gaeta X., Sahakyan A., Chan A.B., Hong C.S., Kim R., Braas D., Plath K., Lowry W.E., Christofk H.R. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell. 2016;19:476–490. doi: 10.1016/j.stem.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler R.A., McGraw T.E., Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018;19:31–44. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossini A.M., Quast A.S., Plotz M., Grauel K., Exner T., Kuchler J., Stachelscheid H., Eberle J., Rabien A., Makrantonaki E. PI3K/AKT signaling pathway is essential for survival of induced pluripotent stem cells. PLoS One. 2016;11:e0154770. doi: 10.1371/journal.pone.0154770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Stem Cell Initiative C., Akopian V., Andrews P.W., Beil S., Benvenisty N., Brehm J., Christie M., Ford A., Fox V., Gokhale P.J. Comparison of defined culture systems for feeder cell free propagation of human embryonic stem cells. In Vitro Cell. Dev. Biol. Anim. 2010;46:247–258. doi: 10.1007/s11626-010-9297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J.E., Esler W.P., Patel R., Lanba A., Vera N.B., Pfefferkorn J.A., Vernochet C. Inhibition of acetyl-CoA carboxylase 1 (ACC1) and 2 (ACC2) reduces proliferation and de novo lipogenesis of EGFRvIII human glioblastoma cells. PLoS One. 2017;12:e0169566. doi: 10.1371/journal.pone.0169566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball S.R., Jurasinski C.V., Lawrence J.C., Jr., Jefferson L.S. Insulin stimulates protein synthesis in skeletal muscle by enhancing the association of eIF-4E and eIF-4G. Am. J. Physiol. 1997;272:C754–C759. doi: 10.1152/ajpcell.1997.272.2.C754. [DOI] [PubMed] [Google Scholar]
- Kimball S.R., Vary T.C., Jefferson L.S. Regulation of protein synthesis by insulin. Annu. Rev. Physiol. 1994;56:321–348. doi: 10.1146/annurev.ph.56.030194.001541. [DOI] [PubMed] [Google Scholar]
- Lawlor M.A., Rotwein P. Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol. Cell. Biol. 2000;20:8983–8995. doi: 10.1128/mcb.20.23.8983-8995.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leese H.J., Barton A.M. Pyruvate and glucose uptake by mouse ova and preimplantation embryos. J. Reprod. Fertil. 1984;72:9–13. doi: 10.1530/jrf.0.0720009. [DOI] [PubMed] [Google Scholar]
- Lian X., Zhang J., Zhu K., Kamp T.J., Palecek S.P. Insulin inhibits cardiac mesoderm, not mesendoderm, formation during cardiac differentiation of human pluripotent stem cells and modulation of canonical Wnt signaling can rescue this inhibition. Stem Cells. 2013;31:447–457. doi: 10.1002/stem.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Handschin C., Spiegelman B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lin Y., Linask K.L., Mallon B., Johnson K., Klein M., Beers J., Xie W., Du Y., Liu C., Lai Y. Heparin promotes cardiac differentiation of human pluripotent stem cells in chemically defined albumin-free medium, enabling consistent manufacture of cardiomyocytes. Stem Cells Transl. Med. 2017;6:527–538. doi: 10.5966/sctm.2015-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan W., Wang Y., Chen X., Shi Y., Wang J., Zhang J., Qian J., Li R., Tao T., Wei W. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget. 2015;6:13006–13018. doi: 10.18632/oncotarget.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S., Lindgren A.G., Srivastava A.S., Clark A.T., Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells. 2011;29:486–495. doi: 10.1002/stem.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning B.D., Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.A., Souder D.C., Miller K.N., Clark J.P., Sagar A.K., Eliceiri K.W., Puglielli L., Beasley T.M., Anderson R.M. GSK3beta regulates brain energy metabolism. Cell Rep. 2018;23:1922–1931.e4. doi: 10.1016/j.celrep.2018.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan H.E., Jefferson L.S., Wolpert E.B., Rannels D.E. Regulation of protein synthesis in heart muscle. II. Effect of amino acid levels and insulin on ribosomal aggregation. J. Biol. Chem. 1971;246:2163–2170. [PubMed] [Google Scholar]
- Nisr R.B., Affourtit C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta. 2014;1837:270–276. doi: 10.1016/j.bbabio.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi R., Kubota H., Yugi K., Toyoshima Y., Komori Y., Soga T., Kuroda S. The selective control of glycolysis, gluconeogenesis and glycogenesis by temporal insulin patterns. Mol. Syst. Biol. 2013;9:664. doi: 10.1038/msb.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa H., Sutherland C., Robey R.B., Printz R.L., Granner D.K. Analysis of the signaling pathway involved in the regulation of hexokinase II gene transcription by insulin. J. Biol. Chem. 1996;271:16690–16694. doi: 10.1074/jbc.271.28.16690. [DOI] [PubMed] [Google Scholar]
- Pantaleon M., Kaye P.L. IGF-I and insulin regulate glucose transport in mouse blastocysts via IGF-I receptor. Mol. Reprod. Dev. 1996;44:71–76. doi: 10.1002/(SICI)1098-2795(199605)44:1<71::AID-MRD8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Rao L.V., Wikarczuk M.L., Heyner S. Functional roles of insulin and insulinlike growth factors in preimplantation mouse embryo development. In Vitro Cell. Dev. Biol. 1990;26:1043–1048. doi: 10.1007/BF02624438. [DOI] [PubMed] [Google Scholar]
- Romorini L., Garate X., Neiman G., Luzzani C., Furmento V.A., Guberman A.S., Sevlever G.E., Scassa M.E., Miriuka S.G. AKT/GSK3beta signaling pathway is critically involved in human pluripotent stem cell survival. Sci. Rep. 2016;6:35660. doi: 10.1038/srep35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel A.R., Kahn C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Schieke S.M., Ma M., Cao L., McCoy J.P., Jr., Liu C., Hensel N.F., Barrett A.J., Boehm M., Finkel T. Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J. Biol. Chem. 2008;283:28506–28512. doi: 10.1074/jbc.M802763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber M.H., Thomsen M.B., Spradling A.C. Electron transport chain remodeling by GSK3 during oogenesis connects nutrient state to reproduction. Cell. 2016;164:420–432. doi: 10.1016/j.cell.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souder D.C., Anderson R.M. An expanding GSK3 network: implications for aging research. Geroscience. 2019;41:369–382. doi: 10.1007/s11357-019-00085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaventi R., Antica M., Pavelic K. Insulin and insulin-like growth factor I (IGF I) in early mouse embryogenesis. Development. 1990;108:491–495. doi: 10.1242/dev.108.3.491. [DOI] [PubMed] [Google Scholar]
- Sperber H., Mathieu J., Wang Y., Ferreccio A., Hesson J., Xu Z., Fischer K.A., Devi A., Detraux D., Gu H. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015;17:1523–1535. doi: 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers S.A., Yin V.P., Whiteman E.L., Garza L.A., Cho H., Tuttle R.L., Birnbaum M.J. Signaling pathways mediating insulin-stimulated glucose transport. Ann. N. Y. Acad. Sci. 1999;892:169–186. doi: 10.1111/j.1749-6632.1999.tb07795.x. [DOI] [PubMed] [Google Scholar]
- Taha C., Klip A. The insulin signaling pathway. J. Membr. Biol. 1999;169:1–12. doi: 10.1007/pl00005896. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development. 2013;140:2457–2461. doi: 10.1242/dev.092551. [DOI] [PubMed] [Google Scholar]
- Teslaa T., Teitell M.A. Pluripotent stem cell energy metabolism: an update. EMBO J. 2015;34:138–153. doi: 10.15252/embj.201490446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Vivanco I., Sawyers C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wang L., Schulz T.C., Sherrer E.S., Dauphin D.S., Shin S., Nelson A.M., Ware C.B., Zhan M., Song C.Z., Chen X. Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood. 2007;110:4111–4119. doi: 10.1182/blood-2007-03-082586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Khan S.A., Lange A.J. Regulation of glycolysis-role of insulin. Exp. Gerontol. 2005;40:894–899. doi: 10.1016/j.exger.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Xu Z., Robitaille A.M., Berndt J.D., Davidson K.C., Fischer K.A., Mathieu J., Potter J.C., Ruohola-Baker H., Moon R.T. Wnt/beta-catenin signaling promotes self-renewal and inhibits the primed state transition in naive human embryonic stem cells. Proc. Natl. Acad. Sci. U S A. 2016;113:E6382–E6390. doi: 10.1073/pnas.1613849113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Chen Z., Gao J., Shi W., Li L., Jiang S., Hu H., Liu Z., Xu D., Wu L. The key roles of GSK-3beta in regulating mitochondrial activity. Cell Physiol. Biochem. 2017;44:1445–1459. doi: 10.1159/000485580. [DOI] [PubMed] [Google Scholar]
- Yang Y., Ren Z., Xu F., Meng Y., Zhang Y., Ai N., Long Y., Fok H.I., Deng C., Zhao X. Endogenous IGF signaling directs heterogeneous mesoderm differentiation in human embryonic stem cells. Cell Rep. 2019;29:3374–3384.e5. doi: 10.1016/j.celrep.2019.11.047. [DOI] [PubMed] [Google Scholar]
- Yu J.S., Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050–3060. doi: 10.1242/dev.137075. [DOI] [PubMed] [Google Scholar]
- Zhang C., Skamagki M., Liu Z., Ananthanarayanan A., Zhao R., Li H., Kim K. Biological significance of the suppression of oxidative phosphorylation in induced pluripotent stem cells. Cell Rep. 2017;21:2058–2065. doi: 10.1016/j.celrep.2017.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Nuebel E., Daley G.Q., Koehler C.M., Teitell M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11:589–595. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Nuebel E., Wisidagama D.R., Setoguchi K., Hong J.S., Van Horn C.M., Imam S.S., Vergnes L., Malone C.S., Koehler C.M. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012;7:1068–1085. doi: 10.1038/nprot.2012.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yalcin S., Lee D.F., Yeh T.Y., Lee S.M., Su J., Mungamuri S.K., Rimmele P., Kennedy M., Sellers R. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol. 2011;13:1092–1099. doi: 10.1038/ncb2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Cui P., Li Y., Feng G., Tong M., Guo L., Li T., Liu L., Li W., Zhou Q. Mitochondrially produced ATP affects stem cell pluripotency via Actl6a-mediated histone acetylation. FASEB J. 2018;32:1891–1902. doi: 10.1096/fj.201700626RR. [DOI] [PubMed] [Google Scholar]
- Zhao X., Bak S., Pedersen A.J., Jensen O.N., Hojlund K. Insulin increases phosphorylation of mitochondrial proteins in human skeletal muscle in vivo. J. Proteome Res. 2014;13:2359–2369. doi: 10.1021/pr401163t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.