

Abstract
Hyperactivation of the NFκB pathway is a distinct feature of inflammatory breast cancer (IBC), a highly proliferative and lethal disease. Gene expression studies in IBC patient tissue have linked EGFR (EGFR/HER2)-mediated MAPK signaling to NFκB hyperactivity, but the mechanism(s) by which this occurs remain unclear. Here, we report that the X-linked inhibitor of apoptosis protein (XIAP) plays a central role in linking these two pathways. XIAP over-expression correlated with poor prognoses in breast cancer patients and was frequently observed in untreated IBC patient primary tumors. XIAP drove constitutive NFκB transcriptional activity, which mediated ALDH positivity (a marker of stem-like cells), in vivo tumor growth, and an IBC expression signature in patient-derived IBC cells. Using pathway inhibitors and mathematical models, we defined a new role for the MAPK interacting (Ser/Thr)-kinase (MNK) in enhancing XIAP expression and downstream NFκB signaling. Furthermore, targeted XIAP knockdown and treatment with a MNK inhibitor decreased tumor cell migration in a dorsal skin fold window chamber murine model that allowed for intravital imaging of local tumor growth and migration. Together, our results indicate a novel role for XIAP in the molecular cross-talk between MAPK and NFκB pathways in aggressive tumor growth, which has the potential to be therapeutically exploited.
Significance:
Signaling by the MNK kinase is essential in inflammatory breast cancer, and it can be targeted to inhibit XIAP–NFκB signaling and the aggressive phenotype of this malignancy.
Introduction
The primary cause of breast cancer morbidity and mortality is metastasis to surrounding tissues and distant organs, a process dependent on hyperproliferation and hypermotility of cells derived from the primary tumor. Among all breast cancer types, inflammatory breast cancer (IBC) is a highly aggressive subtype characterized clinically by extremely motile tumor cell clusters that exhibit localized dermal invasion and frequent lymph node involvement (1–3). Despite an aggressive multimodal treatment regimen, tumor recurrence and metastatic progression are unmet challenges in IBC patients (4).
Comparative gene expression studies from preclinical models and pretreatment patient samples have attempted to define molecular profiles specific to IBC. They reveal highly activated MAPK and NFκB transcriptional profiles associated with increased proliferation in IBC compared with other locally advanced breast cancers (5–8); however, the mechanism for the linkage between these two pathways in IBC tumors has not been described. We sought to determine how EGFR-mediated MAPK activation and NFκB hyperactivity were coordinated to enhance cancer cell survival and proliferation with the goal of elucidating targets for therapeutic intervention. Our previous studies have shown that IBC tumor cells escape from various cell death and oxidative stress stimuli, including EGFR inhibition, through upregulation of the X-linked inhibitor of apoptosis protein (XIAP; refs. 9–13). XIAP, through its multiple domains, not only directly inhibits the initiation and execution phases of the caspase cascade during programmed cell death, but also regulates, in a caspase-independent manner, a range of cellular activities that enhance survival signaling, including NFκB activity (14, 15). Recent studies reveal that translational regulation of select survival proteins is regulated by MAPK signaling, protecting cancer cells during cellular stress (16). However, the understanding of mechanisms linking MAPK signaling and survival signaling remains limited.
In this study, we demonstrate that elevated XIAP expression in patient IBC tumors is associated with aggressive biology and poor clinical outcome. Using IBC cell lines derived from previously untreated primary tumors combined with modulation of XIAP expression, we found that XIAP drives activation of NFκB and its target genes, leading to enhanced tumor growth. Furthermore, we discover that MAPK interacting kinase (MNK) signaling, downstream of EGFR/HER2 activation, promotes XIAP expression and NFκB activity. Collectively, our findings indicate a role for XIAP as a central regulatory node connecting MAPK and NFκB signals, which governs IBC tumor-specific gene signatures, survival, and tumorigenesis.
Materials and Methods
Human breast tumor mRNA expression studies
Gene expression datasets previously published were used to generate a combined total of 1032 breast cancer patients [GEO datasets GSE6532, GSE9195, GSE16391, GSE16446, GSE17907, GSE20685, GSE20711, and GSE21653]. A total of 1018 patients had nonzero event-free survival time and were available for analysis for the expression of two probesets (206536_s_at and 206537_at, Affymetrix), which targeted the XIAP ORF. Patients were grouped into high or low expression of XIAP using the top quartile versus remaining patients by probe set expression values and compared using Kaplan–Meier plots with 95% confidence intervals of event-free survival (earliest event provided within each dataset) by log-rank test. For correlation of XIAP expression with lymph node involvement, two available GEO datasets, GSE6532 and GSE9195, were identified that included lymph node status for a total of 164 patients with IBC.
Breast cancer tissue microarrays
The TMA sections used in this study, with prior patient consent and approval from the Institutional Review Boards from each center, and their clinical characteristics are available in refs. 17–19.
IHC
Four-micron–thick paraffin sections were deparaffinized, rehydrated, and antigen retrieval performed using EDTA buffer at 95°C for 30 minutes. Slides were incubated in a 1:60 dilution of mouse anti-human XIAP (BD Biosciences) overnight at 4°C, washed, and incubated in anti-mouse secondary (Dako anti-mouse Envision kit) for 30 minutes at room temperature. Imaging was performed on a Zeiss Axio Observer A1 microscope and images analyzed with MetaMorph. Scoring of slides was carried out by a board-certified surgical pathologist in a blinded manner. Staining intensity was graded on a qualitative scale [no staining (negative), very focal or very weak staining (borderline), and positive]. For the purpose of statistical analysis, the data were dichotomized as negative or positive (including borderline).
Cell lines
SUM149 and SUM190 cells were obtained from Asterand, Inc. and were cultured as previously described (9). The rSUM149 cell variant is derived from SUM149 and cultured as described previously (10, 20). SUM149 cells stably expressing wtXIAP, shXIAP, and shXIAP+wtXIAP were generated using a lentiviral expression system (kindly provided by Dr. Colin Duckett, University of Michigan, Ann Arbor, MI) and previously reported (13). HeLa cells were grown in DMEM + 10% FBS. For over-expression of MNK1 T344D mutant, cells were transfected with 0.5 μg of pcDNA3.1 (empty vector) or pcDNA3.1 MNK1 T344D (previously described in ref. 21), and harvested 48 hours later for immunoblot analysis. Characterization and authentication of the purchased cell lines were done at Asterand. Additionally, short tandem repeat polymorphism analysis was performed at regular intervals on all cell lines at the Duke Sequencing facility. Cells were cultured at 37°C and 5% CO2.
In vivo tumor xenograft studies
All animal experiments were performed in accordance with protocols approved by the Duke University Institutional Animal Care and Use Committee. IBC cells (5× 106) were suspended in 50-μL PBS and 50-μL Matrigel and injected orthotopically into the fourth mammary fat pad of female SCID mice. Mice were monitored twice weekly and tumor volume measured using the formula V = (L × W2)/2, where L is the longer measurement. Tumor doubling time was found by fitting a nonlinear regression model to the tumor volumes. Mice were euthanized when tumors reached a humane end-point of ~1,500 mm3, at the first sign of morbidity, or at end of study. Tumors were removed and tissue harvested for RNA and Western immunoblot analysis.
In vivo window chamber studies
A dorsal skin-fold window chamber was implanted in Nu/Nu mice as described previously (22). Briefly, the dorsal skin was tented and sutured to a c-frame to hold it in position. Three ~1 mm diameter holes were cut, through which the window frame could be secured, and a 12-mm diameter full-thickness skin punch was removed from the superior skin fold. The titanium window frame (Small Dorsal Kit, APJ Trading) was sutured in place and a total of 1× 105 tumor cells were injected into the fascia in the center of the window in a 20-μL volume, using a 30-gauge needle. Sterile saline was used to fill the window, over which a coverslip was placed and affixed by a retaining ring.
In vivo CGP57380 treatment
Female nude mice (around 10 weeks old) were randomized into two groups (vehicle and CGP57380 group, n = 2 for each group) after the installment of dorsal window chamber and implantation of GFP-tagged SUM149 IBC cells. Tumor-bearing mice were treated with either CGP57380 (25 mg/kg, i.p.) or vehicle for 4 doses on days 0, 2, 4, 6 after the surgery. A stock solution of CGP57380 in DMSO was made up and further diluted into PBS for administration; vehicle solution contained the same percentage of DMSO in PBS as the CGP57380 solution. The experiment was repeated twice.
In vivo imaging
Mice were anesthetized and mounted to a microscope stage with a custom-made mouse window chamber slide mount. The Zeiss Axio Observer Z1 microscope was used for all imaging with a 5× objective and Apotome (Carl Zeiss AG). GFP fluorescence was excited and acquired with a 488 nm/509 nm excitation/emission filter set, as well as a bright-field transmission image, all recorded by a CMOS camera (C11440, HAMAMATSU photonics K.K). A whole window chamber image was acquired using the tiling function within Zen Pro software. Tumor growth was monitored with fluorescent imaging at designated time points.
RNA isolation
Total RNA isolation from adherent cells was completed using the Ambion mirVana miRNA isolation kit (Invitrogen) following manufacturer’s instructions. Tissue samples were homogenized in the provided lysis buffer (mirVana kit) and total RNA isolated following instructions.
Affymetrix GeneChip analysis
RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies) and total RNA profiled using the U133A 2.0 Human Gene microarrays at the Duke Institute for Genome Sciences & Policy Microarray facility. Expression data were quantile-normalized and summarized using GCRMA express (23). Probe sets with a fluorescence intensity above log2(100) in at least two samples were considered informative. Expression levels were compared using generalized linear models on log2 expression data and probe sets with nominal P values less than 0.05 were considered significant. Differentially expressed genes were translated into pathways using Expression2Kinases (24). Transcription factors and kinases with a combined enrichment score of at least 10 were included for the protein–protein interaction (PPI) network construction. The PPI network was then analyzed for enriched pathways using the Reactome FI plugin from Cytoscape. Pathways with a prior probability of less than 1% were considered relevant. Unsupervised hierarchical cluster analysis of the gene expression data was performed using the Manhattan distance as the dissimilarity metric and the Ward linkage as the dendrogram drawing method. Application of the IBC signature to the expression data of SUM149, wtXIAP, and shXIAP tumors was done as described before (7).
Gene-set enrichment analysis
Expression data of wtXIAP (XIAP high) cells was compared with the combined parental and shXIAP cell lines (XIAP low) using default parameters with gene-set level permutations and signal2noise used to rank genes. Gene-set enrichment visualization was performed using Cytoscape 2.8.3 and a P < 0.001, Q-value cutoff 0.006, similarity cutoff of 0.5, and false discovery rate of 0.1. Gene sets examined were from the current molecular signature (MSigDB) versions 4.0.
Treatment of cells for viability and caspase activity
Cells were seeded and allowed to reach approximately 80% confluence. Cell viability was determined by Trypan blue exclusion assay as described previously (9). Caspase-3/7 activity was determined in cells untreated and treated with 50 ng/mL TRAIL, using the Caspase-Glo Assay (Promega) as per manufacturer’s instructions.
Western immunoblot analysis
Western immunoblots were carried out as described previously (10). Cells were harvested after indicated treatments and times. Tissue samples were homogenized in lysis buffer in a Bullet Blender Storm 24 (Next Advance). Membranes were incubated at 4° C overnight with antibodies against NFκB (P65), p-NFκB (P65), ERK, p-ERK, p-eIF4E, eIF4E, p-p38, p38, survivin, MNK1 (Cell Signaling Technology, all 1:1,000), SOD2 (1:1,000), Bcl-2, (1:1,000), XIAP (1:2,000; BD Biosciences), c-Myc (Sigma-Aldrich), or GAPDH (Santa Cruz Biotechnology, 1:4,000). Densitometric analysis was performed using the NIH ImageJ software (25); for western quantitation measuring effects of MNK modulation on XIAP and NFκB signaling a LI-COR Odyssey FC imager with Image Studio software (LI-COR) was used.
NRAGE peptide treatment
The NRAGE peptide was purchased from NeoBioLab and used as previously described by our lab (13). For all experiments, unpurified NRAGE peptide was added to cells for 24 hours with 6 μmol/L EndoPorter delivery reagent (GeneTools LLC).
Anchorage-independent growth assay
Anchorage-independent growth assay was performed as described previously (26). Indicated treatments were applied for 24 hours, after which cells were harvested and counted. Once visible colonies had formed, they were counted under a micro-scope, and colony counts were normalized to the untreated sample. Images of representative fields were taken with 5× magnification using a Zeiss Axio Observer A1 microscope (Zeiss), Hamamatsu Orca ER digital camera, and MetaMorph software (Molecular Devices).
ALDEFLUOR assay
ALDH enzymatic activity was evaluated using the ALDEFLUOR kit (Stem Cell Technologies) according to the manufacturer’s instructions. Cells were incubated with provided ALDH substrate for 35 minutes at 37° C. The specific ALDH inhibitor diethyl-aminobenzaldehyde (DEAB) was used as a negative control. Sorting gates were established using 7-AAD for viability DEAB-treated, ALDEFLUOR-stained cells as negative controls.
Quantitative PCR analysis
Total RNA was subjected to reverse transcription using the iScript Reverse Transcription SuperMix Kit (Bio-Rad) and oligo d(T) primers as per manufacturer’s instructions. cDNA and SYBR Green were added to a custom PrimePCR plate (Bio-Rad) containing primer pairs for the indicated NFκB target genes and β-actin as a loading control. Further information on primers can be found in Supplementary Table S1. For MNK qRT-PCR studies, qRT-PCR was performed using Applied Biosystems MYC, BIRC5, IL1B, and BCL2 primers and probes using the RNA to CT one step RT-PCR reagent kit (Invitrogen), following the manufacturer’s instructions. The PCR was conducted on an iCycler instrument (Bio-Rad) using the following conditions: [95°C × 2 minutes, (95°C × 5 seconds, 60°C × 30 seconds) × 40 cycles] and fold changes calculated by the 2(−ΔΔCt) method, except for MNK qRT-PCR studies where primer efficiency correction was deemed necessary and the 2(−ΔΔCP) method was used (27).
Mathematical model construction
Simulations were performed in MATLAB (Mathworks Inc.), and bifurcation diagrams were drawn using MATCONT (28). The model formulation for the interactions of MNK, XIAP, and NFκB based on this study and previous work is given by:
where N, M, and X denote NFκB, MNK, and XIAP levels, respectively. gN and gX are the respective production rates for NFκB and XIAP, and kN and kX are their respective degradation rates. Shifted Hill functions, denoting the effect of X on Y, are defined as:
where H− (X) is the inhibitory Hill function, H+ (X) is the excitatory Hill function, and λX,Y denote the equivalent of fold-change in production of Y due to X (29). The parameters used here are: kN = 1.0, kX = 1.2, gN = 120, gX = 150; λN,X = 4, λX,N = 5, and λM,X = 2. For different Hill functions used in the model, , nN,X = nX,N = 4; nM,X = 2, where nX,Y nX,Y denotes the Hill coefficient for the Hill function corresponding to the effect of X on Y, and represents the respective half-maximal concentration. Degradation rates (represented in per unit hour) for XIAP and NFκB are estimated from experimental data on their half-lives (30, 31). The fold-change for effect of MNK on XIAP has been estimated from our results, while the effect of XIAP on NFκB and vice versa were gathered from existing data (15, 32). Production rate (represented in 1,000 molecules per hour) estimation is based on total number of protein molecules per cell reported for signaling molecules (~100,000; ref. 33). For considering the effect of NRAGE mimic, a shifted Hill function Hs (Nr, λNr,N) is included in the first term of equation for NFκB levels, since NRAGE is not supposed to alter the levels of XIAP, but its interaction with NFκB. Parameters used are: λNr,N = 0:5 (estimated from our data), and , nNr,N = 2.
Statistical analysis
The statistical analyses were conducted using GraphPad Prism (GraphPad Software, Inc.) Student two-tailed t test, Fisher exact test (IHC), and Mantel–Cox log-rank test (survival). Differences were considered significant at P < 0.05. When comparing multiple groups, ANOVA protected Tukey HSD test was performed. Comparison of XIAP mRNA expression using the two probesets were analyzed with respect to their expression distribution using the rank-based Mann–Whitney test.
Results
High XIAP levels are associated with aggressive breast cancer characteristics
To explore whether XIAP expression correlates with survival outcomes in breast cancer, we queried expression data from a collection of 1,032 breast cancers, using microarray probe sets targeting the open reading frame (ORF) of XIAP (Supplementary Fig. S1). Breast cancer patients with elevated expression (top quartile) of XIAP mRNA had decreased event-free survival compared with patients with lower XIAP mRNA levels (Fig. 1A). We further examined whether expression of XIAP was independently associated with disease-free survival adjusting for available covariates, which included PAM50-based molecular subtype and lymph node status. High XIAP expression was associated with increased HR of recurrence (HR 1.68, P < 0.001) after adjusting for PAM50 subtype and lymph node status, while expected trends were observed with increased recurrence noted for nonluminal molecular subtypes and decreased recurrence risk noted for lymph node–negative disease. Given the role of IAP proteins in metastasis (34), we hypothesized that XIAP expression would be increased in breast cancer patients with lymph node–positive disease (LN+). We observed increased expression of XIAP among patient tumors with lymph node involvement versus no lymph node involvement at diagnosis (Fig. 1B and C).
Figure 1.

High XIAP mRNA expression correlates with poor event-free survival and lymph node involvement in advanced breast cancer patients. A, Event-free survival for patients with IBC (n = 1018), separated as high (top quartile) versus low XIAP expression, as determined by probe set 1 (left) 206536_s_at and probe set 2 (right) 206537_at from the Affymetrix GeneChip U133A 2.0 Array [top quartile vs. remaining patients, log-rank test for probe set 1 - 206536_s_at: P < 0.0001, HR 1.80 (95% CI, 1.53–2.62) and probe set 2 - 206537_at: P = 0.0006, HR 1.40 (95% CI, 1.11–1.87), both P < 2e – 6]. B and C, Increased XIAP expression among breast patient tumors (n = 164) with lymph node (LN) involvement versus no lymph node involvement at diagnosis. Box and whisker plots (B) and histograms (probeset 1, P = 0.005; probeset 2, P = 0.02; C) with expression distribution analyzed using the rank-based Mann-Whitney test.
Lymph node involvement and the presence of tumor cell clusters (tumor emboli) in the dermal lymphatic vessels is a classic feature of IBC presentation at diagnosis. Therefore, we conducted IHC analysis for XIAP protein expression in breast tissue microarrays, which included benign and malignant samples (n = 198) of different stages and grades from non-IBC and IBC patients (Table 1). Overall, positive cytoplasmic staining of XIAP was only observed in invasive breast tumors and triple-negative samples. IBC samples characterized by tumor emboli showed strong staining for XIAP in >90% cells along with positive staining in the identified tumor emboli. Representative images are shown in Fig. 2A–F. We performed a multivariate analysis to determine whether XIAP over-expression in IBC is related to other clinicopathologic features [e.g., histologic grade, hormone receptor status, HER2 status, and triple-negative breast cancer (TNBC) status - n = 158]. In univariate analysis, XIAP expression was significantly associated with high histologic grade (grade 3 vs. grade 1; HR = 1.305; P < 0.001), ER status (ER+ vs. ER−; HR = 0.887; P = 0.036), PR status (PR+ vs. PR−; HR = 0.887; P = 0.010), TNBC status (TNBC+ vs. TNBC−; HR = 1.178; P = 0.012) and tumor phenotype status (IBC vs. non-IBC; HR = 1.299; P = 0.010). In multivariate analysis, including all parameters associated with XIAP expression in univariate analysis, only histologic grade (grade 3 vs. grade 1; HR = 1.232; P = 0.016) and tumor phenotype (IBC vs. non-IBC; HR = 1.230; P = 0.049) remained significant. These data suggest that XIAP expression is correlated with breast cancer of higher histologic grade and that XIAP over-expression is specifically associated with IBC, independent of the classical clinicopathologic determinants of IBC.
Table 1.
Correlation of XIAP expression with clinicopathologic parameters in invasive breast carcinomas (IHC analysis of tissue microarrays)
| Grade | Positive | Negative |
|---|---|---|
| 1,2 | 42 | 71 |
| 3 | 43 | 27 |
| P | 0.0022 | |
| Stage | ||
| 1, 2 | 54 | 68 |
| 3, 4 | 13 | 2 |
| P | 0.0021 | |
| Molecular subtype | ||
| TNBC | 20 | 7 |
| Other types | 60 | 73 |
| P | 0.0103 |
Figure 2.
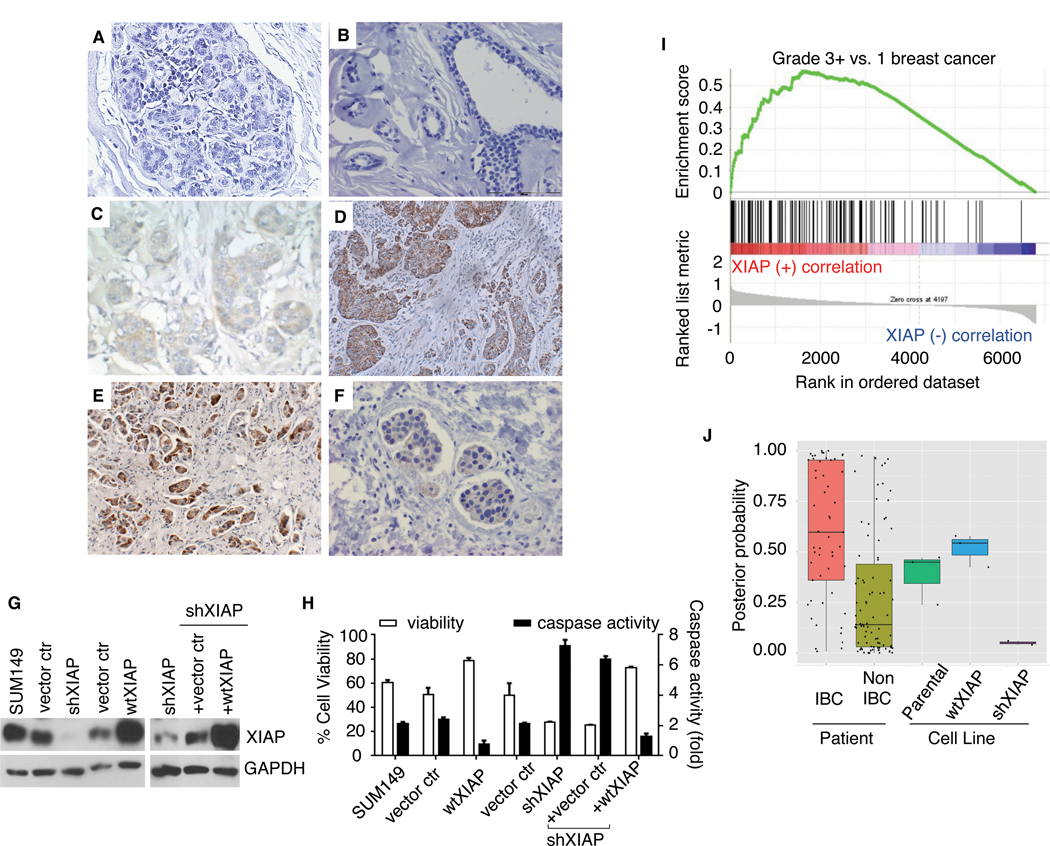
XIAP protein expression in high-grade breast cancers and higher cellular levels associated with proliferative signature. A-F, XIAP levels were analyzed by IHC analysis in TMAs of breast cancers from human patients. A, No expression in normal breast lobule. B, No expression in benign duct (right) and normal acini (left). C, Borderline staining in >50% of tumor cells. D, Positive staining in >90% of invasive tumor cells. E, Infiltrating IBC with tumor cell clusters showing strong positive staining in >80%–90% of cells. F, Staining in a representative intralymphatic tumor emboli identified in IBC specimens. Magnification, × 400. See Table 1 for full histopathology results. G, Immunoblot analysis for expression of XIAP in indicated parental and XIAP-modulated cell lines. H, Functional effects of XIAP expression or depletion evaluated by cell viability (left axis, white bars) and caspase activity (right axis, black bars) of indicated cell lines after administration of 50 ng/mL TRAIL; viability bars represent mean ± SEM (n = 3–4), caspase-3/7 activity bars represent mean ± SEM fold change normalized to untreated (n = 2–3). I, Enrichment plot showing correlation of XIAP over-expression with published features of high-grade breast cancer (37) from GSEA analysis. J, Application of the IBC-specific patient gene signature to the expression data from SUM149, wtXIAP, and shXIAP cells. The figure shows, in boxplot format, the posterior IBC probability on the y-axis for all samples including patients with IBC (red, positive control), patients with non-IBC (moss green, negative control), SUM149 cells (green), wtXIAP (blue), and shXIAP (purple).
We next investigated whether XIAP upregulation contributes to the hyperproliferative phenotype in IBC. To explore this, we used SUM149 (basal-like, constitutively activated EGFR) and SUM190 (luminal-like, HER2-over-expressing) tumor cells, which are considered true IBC-like models derived from primary tumors of IBC patients before treatment (35, 36). To assess the global effects of modulating XIAP expression in IBC cells we conducted transcriptome profiling of genetically modified derivatives with XIAP over-expression (wtXIAP), depletion (shXIAP), and reconstitution (shXIAP+XIAP), along with appropriate vector controls. XIAP expression and function in these variants was validated by immunoblot (Fig. 2G) and measurement of caspase activation and cell viability after treatment with TRAIL (Fig. 2H). GSEA analysis and GNF expression atlas ontologies revealed a network of related gene sets enriched in XIAP-over-expressing cells (Supplementary Fig. S2) reported to be associated with cell-cycle regulation and proliferation, response to cell stress and stem cell maintenance, and resistance to hypoxic and oxidative stress. The list of the top 100 genes that are differentially expressed in XIAP high versus low samples are provided in Supplementary Table S2A and S2B. Of interest was a strong and positive correlation between XIAP over-expression and gene sets (37) enriched for high-grade breast cancer (Fig. 2I), corroborating the observed increased XIAP staining in IBC specimens, in particular IBC, which are already at stage III or higher at diagnosis.
XIAP knockdown abrogates IBC-specific gene signature
To further investigate the clinical relevance of XIAP in IBC, we evaluated how the gene expression profile of the XIAP-over-expressing versus knockdown cells compared with a published IBC-patient derived gene signature from a comparative analysis of untreated primary tumors from stage- and subtype-matched IBC and non-IBC patients, which includes the largest collection of IBC tumors from the World IBC Consortium (7). As expected, the patient-derived SUM149 cell line is IBC-like with an average posterior probability (similarity) of 44.7%; likewise, the XIAP-over-expressing (wtXIAP) cells also show a significant posterior IBC probability (i.e. 51.5%). In contrast, we observed that knockdown of XIAP (shXIAP) abolished the IBC-specific patient gene expression profile (i.e., posterior IBC probability of 0.05%; Fig. 2J). Taken together, these results indicate that XIAP maintains an IBC-like phenotype and associated gene expression signatures, which are dominated by proliferative and prosurvival gene network.
XIAP over-expression enhances in vivo IBC tumor growth
Despite the correlation of XIAP over-expression with proliferative genes, modulating XIAP expression alone in vitro does not have a significant effect on cellular proliferation (10). Therefore, to determine a possible in vivo role for XIAP over-expression, we characterized the tumor growth kinetics of the XIAP modulated (wtXIAP, shXIAP, and vector control) cells. Tumor cells were implanted in the mammary fat pad of nude mice and tumor growth measured over time (Fig. 3A). Initially all mice formed tumors with similar kinetics; however, the growth in mice bearing wtXIAP-over-expressing tumors was significantly increased compared with vector control tumors (doubling time of 6.9 days, wtXIAP; 10.3 days, vector controls). In contrast, although shXIAP tumors grew to palpability, most regressed (10/12) or plateaued in size compared with vector control tumors in the study period. Vector controls are shown combined in Fig. 3A as they had similar growth kinetics (separate in Supplementary Fig. S3A). Demonstrating specificity of these phenotypes to XIAP, robust tumor growth similar to wtXIAP was observed in tumors expressing an shRNA-resistant XIAP construct (shXIAP+wtXIAP; Fig. 3A).
Figure 3.
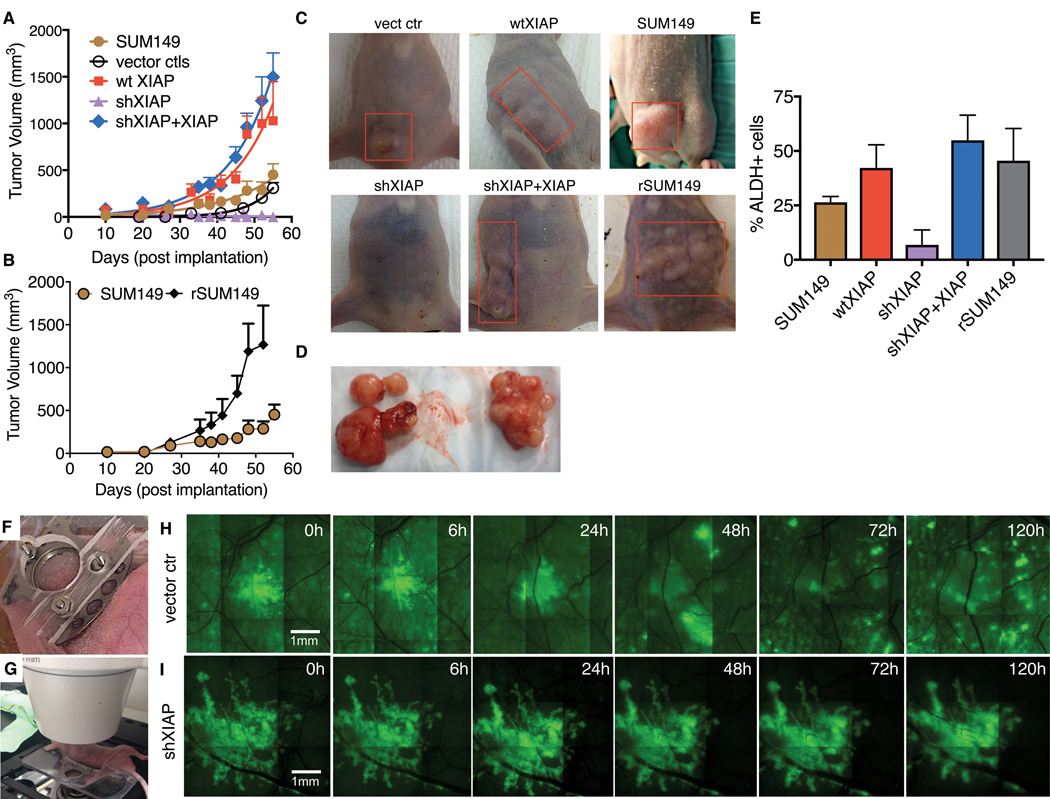
XIAP depletion suppresses in vivo tumor growth and motility. A, Tumor growth curves of indicated XIAP-modulated tumor xenografts implanted orthotopically. B, Tumor growth curves of SUM149 (gold) and rSUM149 (black) xenografts. C and D, Representative images of mice implanted with tumors (C) and extracted tumor clusters (D). E, Flow cytometric analysis of ALDH activity in population of cells indicated. Bars represent mean ± SEM of ALDEFLUOR-positive cells as a percentage of the total number of cells analyzed (n = 2–3; *, P < 0.05). F and G, Representative image showing position of the window chamber implanted in the dorsal skin of the nude mice and live imaging. H and I, Time-course imaging of the local tumor growth and migration in the window chamber of GFP labeled SUM149-vector control (H) and shXIAP (I) cells.
In addition to our exogenously XIAP-modulated cell lines, we also compared parental SUM149 cells to rSUM149, a highly apoptotic-resistant cell line with endogenous XIAP over-expression dependent on XIAP IRES-mediated translation (10). High XIAP levels in rSUM149 correlate with multidrug resistance to chemo-, immuno-, and targeted therapy–mediated apoptosis (20); however, the in vivo growth characteristics of this cell line has not been studied. Similar to wtXIAP tumors, mice bearing rSUM149 tumors showed an aggressive growth pattern with formation of multiple tumor cell clusters (Fig. 3B). Representative pictures from tumor-bearing mice and related statistical analysis for SUM149 and rSUM149 are shown in Fig. 3C and D, respectively; additional images are shown in Supplementary Fig. S3.
Collectively, these results reveal that XIAP over-expression (endogenous or exogenous) enhances tumor growth, possibly explaining why XIAP expression is associated with aggressive features in patient tumors.
XIAP depletion reduces expression of ALDH+, a cancer stem-like marker, and decreases IBC tumor cell motility
A pathologic hallmark of IBC tumors is the formation of tumor emboli enriched with ALDH+ cells [enzymatic marker of cancer stem-like cells reported to be high in IBC cells and patient tumors (38) and postulated to reflect collective tumor cell migration of IBC cells (39)]. On the basis of our evidence of a proliferative signature corresponding with enhanced tumorigenicity in vivo, we investigated whether XIAP expression modulates cancer stem-like characteristics. Indeed, both wtXIAP and rSUM149 cells exhibited increased proportions of ALDH+ cells compared with significantly reduced ALDH positivity in XIAP-silenced (shXIAP) cells (Fig. 3E). This effect was specific to XIAP, as reconstitution of XIAP in shXIAP cells enabled reemergence of ALDH+ cells.
To test whether XIAP inhibition affects IBC tumor cell motility, we employed a dorsal skin fold window chamber model in nude mice that allows for intravital imaging of local tumor growth and migration (Fig. 3F and G). GFP-SUM149 (vector ctr) IBC tumors grew as multiple tumor cell clusters inside the window chamber (Fig. 3H), similar to that observed in IBC patients (39, 40), as opposed to single solid masses observed with other breast cancer lines (22). Using this approach, we compared the short-term (0–120 hours) growth and migration pattern of the SUM149-derived vector control and shXIAP-implanted cells, which revealed significant inhibition of motility in the tumors arising from XIAP-depleted cells (Fig. 3I).
XIAP-over-expressing tumor cells exhibit high NFκB target gene expression
Gene expression profiles identified 933 differentially expressed genes between control and wtXIAP tumors (n = 3 each genotype, Supplementary Table S3). Those genes were enriched for biological processes of transcription, RNA biosynthesis, and protein metabolism among others. As IBC patient tumor profiles are dominated by NFκB target genes, we investigated whether IBC tumors generated with XIAP-over-expressing cells have increased NFκB activity. Expression2Kinases (X2K) analysis revealed that this gene list was enriched for target genes of two transcription factors in the NFκB family (RELA and NFκB1). In addition to target gene enrichment analysis, X2K also builds a protein–protein interaction (PPI) network that provides signal transduction pathways capable of explaining observed gene expression differences. Analysis of this PPI network identified a subnetwork regulated by NFκB activity (Fig. 4A). These data were further confirmed with qRT-PCR analysis: increased expression of several known NFκB target genes (NFΚB1, MYC, and TNFAIP3) was observed in wtXIAP and shXIAP+XIAP tumors, while their expression was reduced in shXIAP tumors (Fig. 4B). Immunoblot analysis of both wtXIAP and shXIAP+XIAP tumors confirmed enhanced activation of the nuclear transcription factor NFκB (phospho-p65; Supplementary Fig. S3).
Figure 4.

Functional partnership of XIAP and NFκB signaling in IBC tumor cell survival. A, Subnetwork of the PPI network identified by E2K to regulate the gene expression profile identified by comparing SUM149 and wtXIAP primary tumors (n = 3). The subnetwork shows potential signaling mechanisms controlling NFκB transcriptional activity. In the network, transcription factors are color-coded red, activating kinases are color-coded green, and cytoplasmic signal transduces are color-coded yellow. The size of the nodes relates to the number of interactions each node has within the identified PPI network. B, Bar graphs showing quantitative PCR analysis of indicated NFκB target mRNAs in tumor samples from indicated xenografts. Bars represent mean SEM in fold compared with FG9 (n = 2; *, P < 0.05). C, NRAGE-treated cells subjected to quantitative PCR analysis for indicated NFκB target mRNAs. Bars represent average fold expression (compared with FG9 control) SEM. *, P < 0.05; **, P < 0.005 compared with EndoPorter (EP) alone. D, Anchorage-independent growth assay of cells treated with EndoPorter alone or NRAGE peptide+EP. Bars represent mean ± SEM colonies formed in soft agar as a percentage of untreated (n = 3; **, P < 0.005 compared with EndoPorter alone). E, Representative images of the indicated treatments in C and D.
Targeting the XIAP–NFκB interaction inhibits anchorage-independent growth
To further investigate the role of XIAP-mediated NFκB activity in IBC cells, we employed a small peptide mimetic modeled after the NRAGE repeat domain (41), which blocks the XIAP–BIR1 domain interaction with TAB1, interrupting XIAP-NFκB signaling (15). Treatment of cells with NRAGE peptide, delivered using Endoporter, led to decreased transcriptional activity of NFκB as measured by target gene expression (Fig. 4C). wtXIAP cells exhibited a modest increase in anchorage-independent growth relative to control cell lines (Fig. 4D and E). Treatment with NRAGE peptide reduced anchorage-independent growth in control and wtXIAP cells in a dose-dependent manner (Fig. 4D and E). Collectively, these results reveal that XIAP drives activation of NFκB and its target genes, demonstrating a functional interplay in advanced breast cancers like IBC, which are characterized by an increased proliferative state. Furthermore, use of the NRAGE peptide highlights the potential for developing BIR1 domain antagonists that can target the XIAP–NFκB interaction and/or signaling to potentiate therapeutic apoptosis in IBC cells.
XIAP is regulated by the MAPK-interacting kinase, MNK
Our results demonstrate a role for XIAP in the proliferative phenotype of IBC through a functional partnership with NFκB, which we have shown to be caspase-independent (13). Although previous reports indicate pervasive NFκB activity in IBC and suggest that NFκB activity may be downstream of the EGFR/HER2–MAPK signaling (6), the link between the two remains ill-defined. MAPK signaling is a critical regulator of stress response, including control of protein synthesis machinery driving cancer cell survival (42). To evaluate the effects of ERK1/2 signaling on XIAP expression, we treated SUM149 and SUM190 IBC cells with the MEK1/2 inhibitor UO126, which effectively reduced ERK1/2 MAPK phosphorylation, but led to only modest decreases in XIAP levels (Fig. 5A). As expected, p38 MAPK phosphorylation, which is downstream of MKK3/4/6, was relatively unchanged by UO126. Both p38 and ERK1/2 MAPK signaling intersect translation machinery through the MNK, which phosphorylates the cap-binding protein, eIF4E (43). Likely due to the maintenance of p38 MAPK signaling during UO126 treatment, MNK signaling to eIF4E was also only modestly reduced (Fig. 5A). Given MNK’s known roles in oncogenesis and survival signaling (44), we next investigated whether MNK functions downstream of MAPK signaling to promote XIAP–NFκB signaling. Treatment with the prototypical MNK inhibitor CGP57380, abolished eIF4E (S209) phosphorylation, indicating MNK signaling interruption. Strikingly, MNK inhibition led to a significant decrease in XIAP protein levels in both cell lines, an effect more robust than that of UO126 (Fig. 5B). The intensity of XIAP reduction by MNK inhibition, relative to ERK1/2 inhibition, suggests that MNK more directly controls XIAP than ERK1/2 MAPK. Confirming this effect on XIAP expression, MNK depletion by RNAi also led to a decline in XIAP protein levels (Fig. 5C), and over-expression of a constitutively active MNK1 mutant (T344D) in HeLa cells induced XIAP expression concomitant with increased eIF4E (S209) phosphorylation (Fig. 5D). Quantitation of the effects of MNK modulation on XIAP protein levels (Fig. 5A–D) is shown in Fig. 5E. Taken together, these results suggest that MNK activity controls XIAP expression.
Figure 5.

MNK signaling regulates XIAP and NFκB and facilitates SUM149 cell motility in vivo. A and B, Immunoblot analysis of indicated proteins in SUM149 and SUM190 cells treated with U0126 (10 μmol/L; A) or CGP57380 (10 μmol/L; B) for the designated intervals. C, Analysis of SUM149 cells transfected with control or MNK1 targeting siRNAs harvested 72 hours posttransfection. D, Immunoblots of HeLa cells transfected with vector control or constitutively active MNK1 (T344D). E, Quantitation of Western blot images in A–D correcting each respective control to 1; CGP57380 values were determined for 24-hour time point. F, qRT-PCR quantification of RNA in SUM149 cells that were transfected with siRNA as in C for select NFκB target genes. G and H, SUM149-GFP cells were implanted into a dorsal window chamber and imaged at the designated intervals after treatment with vehicle (G) or CGP57380 (H). Arrows, treatment schedule. Error bars, SEM; *, P < 0.05;*, P < 0.001.
Targeting MNK inhibits NFκB activation
We next investigated whether MNK inhibition could target NFκB (p65) activity. Indeed, using either CGP57380 (Fig. 5B) or direct MNK depletion (Fig. 5C) reduced the levels of both p-p65 and p65 in IBC cells, coincident with a decline in XIAP. Analysis of RNA expression by qRT-PCR following MNK1 depletion revealed a decrease in RNA levels of several select NFκB target genes including MYC and survivin (Fig. 5F). Together, these results indicate that modulating MNK signaling regulates XIAP protein levels and downstream NFκB target genes. Thus, MNK controls XIAP–NFκB signaling and can be targeted to restrain the oncogenic effects of XIAP–NFκB activity.
MNK inhibition decreases IBC cell dissemination in vivo
MNK inhibition has been shown to reduce in vivo tumor growth in several cancer types (45, 46). However, the role of MNK signaling in mediating tumor cell invasion, a characteristic of IBC we show is reliant upon XIAP expression (Fig. 3), has not been tested. Using the window chamber model with GFP-tagged SUM149, we tested infiltration of SUM149 cells within the window chamber in vehicle or CGP57380-treated mice. MNK inhibition with CGP57380 led to a marked reduction in cell dissemination within the window chamber, suggesting that MNK signaling enables IBC migration (Fig. 5G and H). Thus, MNK inhibition reduces XIAP levels and NFκB activity and mirrors the effects of XIAP depletion in IBC cells in vivo (Figs. 3 and 5).
Mathematical modeling suggests how cells might maintain high levels of XIAP and NFκB
Integrating our quantitative immunoblot and qRT-PCR data indicating XIAP regulation of NFκB in a MNK-responsive manner combined with earlier reports that also propose XIAP as a transcriptional target of NFκB, we constructed a quantitative mathematical model to decode the dynamics of the MNK/XIAP/NFκB axis. Mutual activation between NFκB and XIAP leads to bistability in the system: cells display either low XIAP/low NFκB, or high XIAP/high NFκB protein levels (shown by two solid green circles in Fig. 6A). To switch between these two states/phenotypes, cells must cross a “tipping point” or threshold (as shown by hollow green circle in Fig. 6A). Once cells have attained such a threshold (high XIAP, high NFκB), this mutual activation would maintain that state (Fig. 6B and C). Only when a “brake” is significantly applied on either NFκB or XIAP levels (e.g., using an inhibitor like NRAGE peptide), can cells be postulated to exit that state and eventually attain a low XIAP, low NFκB state (Fig. 6D). These findings indicate a homeostatic relationship between XIAP and NFκB and suggest that mutual activation of XIAP/NFκB stabilizes a hyperproliferative phenotype in cancer.
Figure 6.

Simulation of a mathematical model for MNK/XIAP/NFκB axis. A, Nullcline simulations for the mathematical model, where red curve represents the change in NFκB levels on changing XIAP, and black curve represents the change in XIAP levels as a function of NFκB. The solid green circles indicate two possible stable states (phenotypes) of the MNK/XIAP/NFκB network, high XIAP, high NFκB and low XIAP, low NFκB, whereas hollow circle indicates an unstable state. B and C, Bifurcation diagram showing how cells in low XIAP, low NFκB state switch to a high XIAP, high NFκB upon increasing MNK levels. D, Bifurcation diagram showing how cells with high NFκB levels may switch to a low NFκB state upon treatment with NRAGE mimics. In B–D, solid blue lines reflect stable states, dotted red lines denote unstable state, and different colored regions highlight the existence of different phenotype(s) at different values of MNK or NRAGE. E, Schema summarizing the cross-talk between EGFR-mediated MAPK activation, XIAP, and NFκB activity. Activation of the EGFRs, EGFR, and HER2 by exogenous ligand ultimately leads to activation of the ERK1/2 MAPK. ERK may also be activated by other receptor tyrosine kinases or microenvironment stresses, along with the p38 MAPK. The MNK is an eIF4G-associated, eIF4E kinase activated by both p38 and ERK1/2 MAPKs. MNK activation leads to increased XIAP levels. The BIR1 domain of XIAP facilitates a physical interaction with the TGFβ-associated binding protein, TAB1, and its cognate kinase, TAK1. This binding event leads to the phosphorylation of the NFκB-activating kinase, IKKβ, allowing NFκB to translocate to the nucleus and to increase expression of genes that can promote tumor cell proliferation, growth, migration, and disease progression.
Collectively, our findings define XIAP as a signaling intermediate linking the MAPK mitogenic cascade to NFκB prosurvival signaling (schema in Fig. 6E). In doing so, XIAP confers a proliferative signature and phenotype to IBC cells, enabling aggressive tumor growth in one of the most lethal subtypes of breast cancer.
Discussion
The current study uncovers XIAP as an oncogenic signaling intermediate, linking the MAPK and NFκB signaling pathways, with significant implications for locally advanced breast cancer tumor growth. XIAP mRNA levels correlated with lymph node involvement and decreased event-free survival among patients with IBC, and XIAP over-expression was observed in high-grade breast cancers and IBC patient tumors, substantiating previous reports of XIAP over-expression in breast cancer tissue (47) and correlation of XIAP expression with tumor recurrence in basal-like breast cancer patients (48). We show that XIAP is necessary for the constitutive activation of the NFκB pathway in IBC, and demonstrate that the XIAP–NFκB axis directly correlates with the tumor growth rate in vivo. These findings reveal a functional necessity for XIAP expression in the progression of aggressive, locally advanced breast cancers like IBC. Finally, we defined a critical role of XIAP in transducing MAPK signals to NFκB downstream of MNK, possibly explaining the survival and oncogenic phenotypes associated with MNK signaling (44).
It has been postulated that in aggressive tumors like IBC, a delicate balance exists between unabridged cellular proliferation, the requirement for cancer stem cell self-renewal, and the ability of cancer stem cell progeny to “self-metastasize” and migrate away, freeing up space for continued tumor expansion (39, 49). Our results showing that XIAP expression directly correlates with the number of ALDH+ cells and cell motility in IBC cells warrants further investigation of the role of XIAP in IBC metastatic progression. Studies overwhelmingly show that XIAP antagonism in established tumors or in cell lines can sensitize tumor cells to therapy-mediated cell death, thereby implicating XIAP as a chemoresistance factor (50). However, other reports suggest that XIAP expression correlates with favorable clinical outcome (51). Perhaps, contributing to these contradictions are the upstream signals regulating XIAP expression and broader cellular context of the signaling landscape.
MNK is well known for its role in regulating IRES-mediated translation (21) and in our study, interruption of MNK signaling led to a significant reduction in XIAP protein expression. Intriguingly, XIAP mRNA contains an IRES (52) and MNK regulation of eIF4G and eIF4E may function to facilitate XIAP translation in IBC, or MNK may act on XIAP through one of its more recently described eIF4E-independent contexts (53). Interestingly, MNK1/2–null (44) like XIAP-null (54) animals are viable and do not exhibit any major defects in growth and ability to undergo apoptosis. MNK1/2–null mice were reported to exhibit delays in tumor development, suggesting a role for MNK1/2 and downstream effectors in tumorigenesis (44). Our current study showing a strong correlation between XIAP expression and tumor growth in vivo in two MAPK-hyperactivated IBC models provides support for XIAP as a possible downstream effector in potentiating the mitogenic effects of MAPK/MNK signaling. Indeed, we found that MNK inhibition restricted IBC tumor invasion/migration, suggesting therapeutic potential in targeting MNK in IBC as a means to target the XIAP–NFκB axis in cancer.
IBC cells over-expressing XIAP were shown to be resistant to immunotherapy-mediated cell death (13). This work revealed a caspase-independent ability of XIAP to activate NFκB and demonstrated that direct targeting of caspase-binding domains may not reverse resistance (unpublished data). The efficacy of the NRAGE peptide, which prevents the XIAP–BIR1 domain from activating NFκB, in inhibiting anchorage-independent growth in IBC cells underscores the recent mounting evidence for a non-apoptotic function of XIAP as a signaling intermediate in tumor growth (14, 55). However, the practical hurdles of delivering a peptide to tumor cells renders this approach clinically difficult, particularly as NRAGE peptide has a short half-life. Our findings demonstrate that inhibition of MNK signaling represents another mechanism to target XIAP–NFκB signaling in IBC. The finding that MNK inhibition disrupts tumor dissemination is especially relevant for IBC and other subtypes of cancer that are highly metastatic (Fig. 5). Importantly, MNK inhibitors are being developed and pursued clinically, making MNK a more practical target for this pathway. This work presents a new druggable pathway consisting of MNK, XIAP, and NFκB (Fig. 6) that can be used to enhance the efficacy of therapeutic agents by pushing the cells below the tipping point (Fig. 6A) and consequently constraining the proliferative advantage. Thus, XIAP serves as a link between MAPK and NFκB signaling to control IBC proliferation and tumor aggression.
Supplementary Material
Acknowledgments
This work was supported by American Cancer Society Research Scholar Grant (to G.R. Devi), P30 Cancer Center Support Grant NIH CA014236 developmental funds (to G.R. Devi), Department of Defense W81XWH-13-1-0047 and -0046 (to G.R. Devi and M.A. Morse); Department of Defense W81XWH-17-1-0297 (to G.R. Devi); Duke University Diversity Enhancement Fellowship (to M.K. Evans), and the Duke School of Medicine Interdisciplinary Inflammatory Breast Cancer Colloquium Funds (to G.R. Devi); Computational Cancer Biology fellowship from Gulf Coast Consortia - CPRIT RP170593 (to M.K. Jolly); and NSF PHY-1427654 (to H. Levine). The authors would like to thank Amy Aldrich, Courtney Edwards, Adrian Ramirez, Arianna Price, Ronnie Shammas Jr, Larissa M. Gearhart-Serna for technical assistance; Drs. Hengtao Zhang, Yulin Zhao, and Scott Sauer for technical support in the window chamber studies. We thank Tao Wang for flow cytometry support, members of the Devi Lab, Drs. Mark Dewhirst, Ashley Chi, Jeffrey Marks, and Sally Kornbluth, for helpful discussions during manuscript preparation, Dr. Donna Crabtree for editorial assistance, and Duke University Core facilities (Light Microscopy, Cancer Center Isolation Facility, Center for Genomic and Computational Biology, Flow Cytometry, Optical Molecular Imaging and Analysis, Preclinical Translational Research Unit).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Nguyen DM, Sam K, Tsimelzon A, Li X, Wong H, Mohsin S, et al. Molecular heterogeneity of inflammatory breast cancer: a hyperproliferative phenotype. Clin Cancer Res 2006;12:5047–54. [DOI] [PubMed] [Google Scholar]
- 2.Woodward WA. Inflammatory breast cancer: unique biological and therapeutic considerations. Lancet Oncol 2015;16:e568–76. [DOI] [PubMed] [Google Scholar]
- 3.Arora J, Sauer SJ, Tarpley M, Vermeulen P, Rypens C, Van Laere S, et al. Inflammatory breast cancer tumor emboli express high levels of antiapoptotic proteins: use of a quantitative high content and high-throughput 3D IBC spheroid assay to identify targeting strategies. Oncotarget 2017;8: 25848–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costa R, Santa-Maria CA, Rossi G, Carneiro BA, Chae YK, Gradishar WJ, et al. Developmental therapeutics for inflammatory breast cancer: biology and translational directions. Oncotarget 2017;8:12417–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Golen KL, Bao LW, Pan Q, Miller FR, Wu ZF, Merajver SD. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin Exp Metastasis 2002;19:301–11. [DOI] [PubMed] [Google Scholar]
- 6.Van Laere SJ, Van der Auwera I, Van den Eynden GG, van Dam P, Van Marck EA, Vermeulen PB, et al. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 over-expression and MAPK hyperactivation. Br J Cancer 2007;97:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Laere SJ, Ueno NT, Finetti P, Vermeulen P, Lucci A, Robertson FM, et al. Uncovering the molecular secrets of inflammatory breast cancer biology: an integrated analysis of three distinct affymetrix gene expression datasets. Clin Cancer Res 2013;19:4685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allensworth JL, Evans MK, Bertucci F, Aldrich AJ, Festa RA, Finetti P, et al. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol Oncol 2015;9:1155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aird KM, Ding X, Baras A, Wei J, Morse MA, Clay T, et al. Trastuzumab signaling in ErbB2-over-expressing inflammatory breast cancer correlates with X-linked inhibitor of apoptosis protein expression. Mol Cancer Ther 2008;7:38–47. [DOI] [PubMed] [Google Scholar]
- 10.Aird KM, Ghanayem RB, Peplinski S, Lyerly HK, Devi GR. X-linked inhibitor of apoptosis protein inhibits apoptosis in inflammatory breast cancer cells with acquired resistance to an ErbB1/2 tyrosine kinase inhibitor. Mol Cancer Ther 2010;9:1432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aird KM, Allensworth JL, Batinic-Haberle I, Lyerly HK, Dewhirst MW, Devi GR. ErbB1/2 tyrosine kinase inhibitor mediates oxidative stress-induced apoptosis in inflammatory breast cancer cells. Breast Cancer Res Treat 2012;132:109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allensworth JL, Aird KM, Aldrich AJ, Batinic-Haberle I, Devi GR. XIAP inhibition and generation of reactive oxygen species enhances TRAIL sensitivity in inflammatory breast cancer cells. Mol Cancer Ther 2012;11: 1518–27. [DOI] [PubMed] [Google Scholar]
- 13.Evans MK, Sauer SJ, Nath S, Robinson TJ, Morse MA, Devi GR. X-linked inhibitor of apoptosis protein mediates tumor cell resistance to antibody-dependent cellular cytotoxicity. Cell Death Dis 2016;7:e2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis J, Burstein E, Reffey SB, Bratton SB, Roberts AB, Duckett CS. Uncoupling of the signaling and caspase-inhibitory properties of X-linked inhibitor of apoptosis. J Biol Chem 2004;279:9023–9. [DOI] [PubMed] [Google Scholar]
- 15.Lu M, Lin SC, Huang Y, Kang YJ, Rich R, Lo YC, et al. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell 2007;26:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ 2009;16:368–77. [DOI] [PubMed] [Google Scholar]
- 17.Bertucci F, Ueno NT, Finetti P, Vermeulen P, Lucci A, Robertson FM, et al. Gene expression profiles of inflammatory breast cancer: correlation with response to neoadjuvant chemotherapy and metastasis-free survival. Ann Oncol 2014;25:358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heller G, Geradts J, Ziegler B, Newsham I, Filipits M, Markis-Ritzinger EM, et al. Downregulation of TSLC1 and DAL-1 expression occurs frequently in breast cancer. Breast Cancer Res Treat 2007;103:283–91. [DOI] [PubMed] [Google Scholar]
- 19.Sood AK, Saxena R, Groth J, Desouki MM, Cheewakriangkrai C, Rodabaugh KJ, et al. Expression characteristics of prostate-derived Ets factor support a role in breast and prostate cancer progression. Hum Pathol 2007;38: 1628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams KP, Allensworth JL, Ingram SM, Smith GR, Aldrich AJ, Sexton JZ, et al. Quantitative high-throughput efficacy profiling of approved oncology drugs in inflammatory breast cancer models of acquired drug resistance and re-sensitization. Cancer Lett 2013;337:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown MC, Bryant JD, Dobrikova EY, Shveygert M, Bradrick SS, Chandramohan V, et al. Induction of viral, 7-methyl-guanosine cap-independent translation and oncolysis by mitogen-activated protein kinase-interacting kinase-mediated effects on the serine/arginine-rich protein kinase. J Virol 2014;88:13135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer GM, Fontanella AN, Shan S, Hanna G, Zhang G, Fraser CL, et al. In vivo optical molecular imaging and analysis in mice using dorsal window chamber models applied to hypoxia, vasculature and fluorescent reporters. Nat Protoc 2011;6:1355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4: 249–64. [DOI] [PubMed] [Google Scholar]
- 24.Chen EY, Xu H, Gordonov S, Lim MP, Perkins MH, Ma’ayan A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2012;28:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with imageJ. Biophotonics Int 2004;11:36–42. [Google Scholar]
- 26.Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat 2013;137:359–71. [DOI] [PubMed] [Google Scholar]
- 27.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhooge A, Govaerts W, Kuznetsov YA. MATCONT: A MATLAB package for numerical bifurcation analysis of ODEs. ACM Transact Math Soft 2003; 29:141–64. [Google Scholar]
- 29.Lu M, Jolly MK, Gomoto R, Huang B, Onuchic J, Ben-Jacob E. Tristability in cancer-associated microRNA-TF chimera toggle switch. J Phys Chem B 2013;117:13164–74. [DOI] [PubMed] [Google Scholar]
- 30.Gu L, Zhu N, Zhang H, Durden DL, Feng Y, Zhou M. Regulation of XIAP translation and induction by MDM2 following irradiation. Cancer Cell 2009;15:363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipniacki T, Paszek P, Brasier AR, Luxon B, Kimmel M. Mathematical model of NF-kappaB regulatory module. J Theor Biol 2004;228:195–215. [DOI] [PubMed] [Google Scholar]
- 32.Lin MT, Chang CC, Chen ST, Chang HL, Su JL, Chau YP, et al. Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-kappaB-dependent XIAP up-regulation. J Biol Chem 2004;279:24015–23. [DOI] [PubMed] [Google Scholar]
- 33.Milo R, Jorgensen P, Moran U, Weber G, Springer M. BioNumbers–the database of key numbers in molecular and cell biology. Nucleic Acids Res 2010;38:D750–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oberoi-Khanuja TK, Murali A, Rajalingam K. IAPs on the move: role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis 2013;4: e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson FM, Bondy M, Yang W, Yamauchi H, Wiggins S, Kamrudin S, et al. Inflammatory breast cancer: the disease, the biology, the treatment. CA Cancer J Clin 2010;60:351–75. [DOI] [PubMed] [Google Scholar]
- 36.Robertson FM, Chu K, Fernandez SV, Mu Z, Zhang X, Liu H, et al. Genomic profiling of pre-clinical models of inflammatory breast cancer identifies a signature of epithelial plasticity and suppression of TGFb signaling. J Clin Exp Pathol 2012;2:2161. [Google Scholar]
- 37.Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst 2006;98:262–72. [DOI] [PubMed] [Google Scholar]
- 38.Charafe-Jauffret E, Ginestier C, Iovino F, Tarpin C, Diebel M, Esterni B, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res 2010;16:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vermeulen PB, Van Laere SJ, Dirix LY. Inflammatory breast carcinoma as a model of accelerated self-metastatic expansion by intravascular growth. Br J Cancer 2009;101:1028–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arora JSS, Tarpley M, Vermeulen P, Rypens C, Van Laere S, Williams KP, et al. Inflammatory breast cancer tumor emboli express high levels of antiapoptotic proteins: use of a quantitative high content and high-throughput 3D IBC spheroid assay to identify targeting strategies. Oncotarget 2017;8: 25848–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rochira JA, Matluk NN, Adams TL, Karaczyn AA, Oxburgh L, Hess ST, et al. A small peptide modeled after the NRAGE repeat domain inhibits XIAP-TAB1-TAK1 signaling for NF-kappaB activation and apoptosis in P19 cells. PLoS One 2011;6:e20659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol 2012;4. doi: 10.1101/cshperspect.a012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 1997;16:1909–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ueda T, Sasaki M, Elia AJ, Chio II, Hamada K, Fukunaga R, et al. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci U S A 2010;107:13984–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grzmil M, Huber RM, Hess D, Frank S, Hynx D, Moncayo G, et al. MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J Clin Invest 2014;124:742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A 2013;110:E2298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaffer S, Orta L, Sunkara S, Sabo E, Burstein DE. Immunohistochemical detection of antiapoptotic protein X-linked inhibitor of apoptosis in mammary carcinoma. Hum Pathol 2007;38:864–70. [DOI] [PubMed] [Google Scholar]
- 48.Xu YC, Liu Q, Dai JQ, Yin ZQ, Tang L, Ma Y, et al. Tissue microarray analysis of X-linked inhibitor of apoptosis (XIAP) expression in breast cancer patients. Med Oncol 2014;31:764. [DOI] [PubMed] [Google Scholar]
- 49.Enderling H, Hlatky L, Hahnfeldt P. Migration rules: tumours are conglomerates of self-metastases. Br J Cancer 2009;100:1917–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kashkar H. X-linked inhibitor of apoptosis: a chemoresistance factor or a hollow promise. Clin Cancer Res 2010;16:4496–502. [DOI] [PubMed] [Google Scholar]
- 51.Seligson DB, Hongo F, Huerta-Yepez S, Mizutani Y, Miki T, Yu H, et al. Expression of X-linked inhibitor of apoptosis protein is a strong predictor of human prostate cancer recurrence. Clin Cancer Res 2007;13:6056–63. [DOI] [PubMed] [Google Scholar]
- 52.Holcik M, Lefebvre C, Yeh C, Chow T, Korneluk RG. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat Cell Biol 1999;1:190–2. [DOI] [PubMed] [Google Scholar]
- 53.Brown MC, Gromeier M. MNK Controls mTORC1:Substrate association through regulation of TELO2 binding with mTORC1. Cell Rep 2017;18: 1444–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB. Characterization of XIAP-deficient mice. Mol Cell Biol 2001;21:3604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang X, Wu Z, Mei Y, Wu M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J 2013;32:2204–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.