

Abstract
Bleeding and altered iron distribution occur in multiple gastrointestinal diseases, but the significance or regulation of these changes remains unclear. Here we report that hepcidin, the master regulator of systemic iron homeostasis, is required for tissue repair in the intestine following experimental damage. This effect was independent of hepatocyte-derived hepcidin or systemic iron levels. Rather, we identified that conventional dendritic cells (cDCs) are a source of hepcidin that is induced by microbial stimulation, prominent in the inflamed intestine of humans, and essential for tissue repair. Mechanistically, cDC-derived hepcidin acted on ferroportin-expressing phagocytes to promote local iron sequestration, which regulated the microbiota and subsequently facilitated intestinal repair. Collectively, these results identify a novel pathway whereby cDC-derived hepcidin promotes mucosal healing in the intestine via nutritional immunity.
One Sentence Summary:
Dendritic cells produce an iron-regulatory factor that modulates the intestinal microbiota to promote mucosal healing.
Inflammatory bowel disease (IBD), colorectal cancer, and gastrointestinal infections cause tissue inflammation that drives bleeding, malabsorption, and diarrhea (1–3). As a result, patients frequently exhibit anemia that is difficult to treat, and bleeding introduces a new source of iron to the intestine (4, 5). Hepcidin is the master regulator of systemic iron homeostasis that is produced as a peptide hormone from the liver and promotes degradation of the cellular iron efflux transporter ferroportin (4, 6–10). Ferroportin is expressed on red pulp macrophages and the basolateral surface of duodenal enterocytes, where it facilitates iron recycling from senescent red blood cells and import of dietary iron, respectively (4, 6–10). Despite these advances, it remains unclear whether hepcidin impacts gastrointestinal health or disease.
To address this, we exposed wild-type (Hamp+/+) and hepcidin-deficient (Hamp−/−) mice to a model of intestinal tissue damage, inflammation, and repair by administering dextran sodium sulfate (DSS) in the drinking water. During DSS administration, both Hamp+/+ and Hamp−/− mice exhibited similar weight loss (Fig. 1A), indicative of comparable inflammation and tissue damage. However, upon removal of DSS, Hamp−/− mice exhibited persistent weight loss, continued disruption of epithelial crypt architecture, and significantly reduced colon lengths relative to controls (Fig. 1A–C). Surprisingly, we observed significant DSS-dependent reductions in liver hepcidin expression, and reduced systemic hepcidin protein levels, relative to naïve controls (Fig. S1A, B). These data are consistent with negative feedback on hepcidin production due to anemia and erythropoiesis (11, 12). To test the role of hepatocyte-derived hepcidin, we bred mice with a floxed gene (HampF/F) to mice expressing Cre recombinase under the control of the albumin promoter, to generate HampΔliver mice that develop systemic iron overload comparable to hepcidin-deficient mice (13). Following exposure to DSS, HampΔliver mice and controls exhibited comparable recovery of body weight, tissue architecture, and colon length (Fig. 1D–F). Thus, hepcidin is essential for mucosal healing, but this occurs independent of hepatocyte expression and systemic iron regulation.
Fig. 1. Extra-hepatic hepcidin promotes mucosal healing.
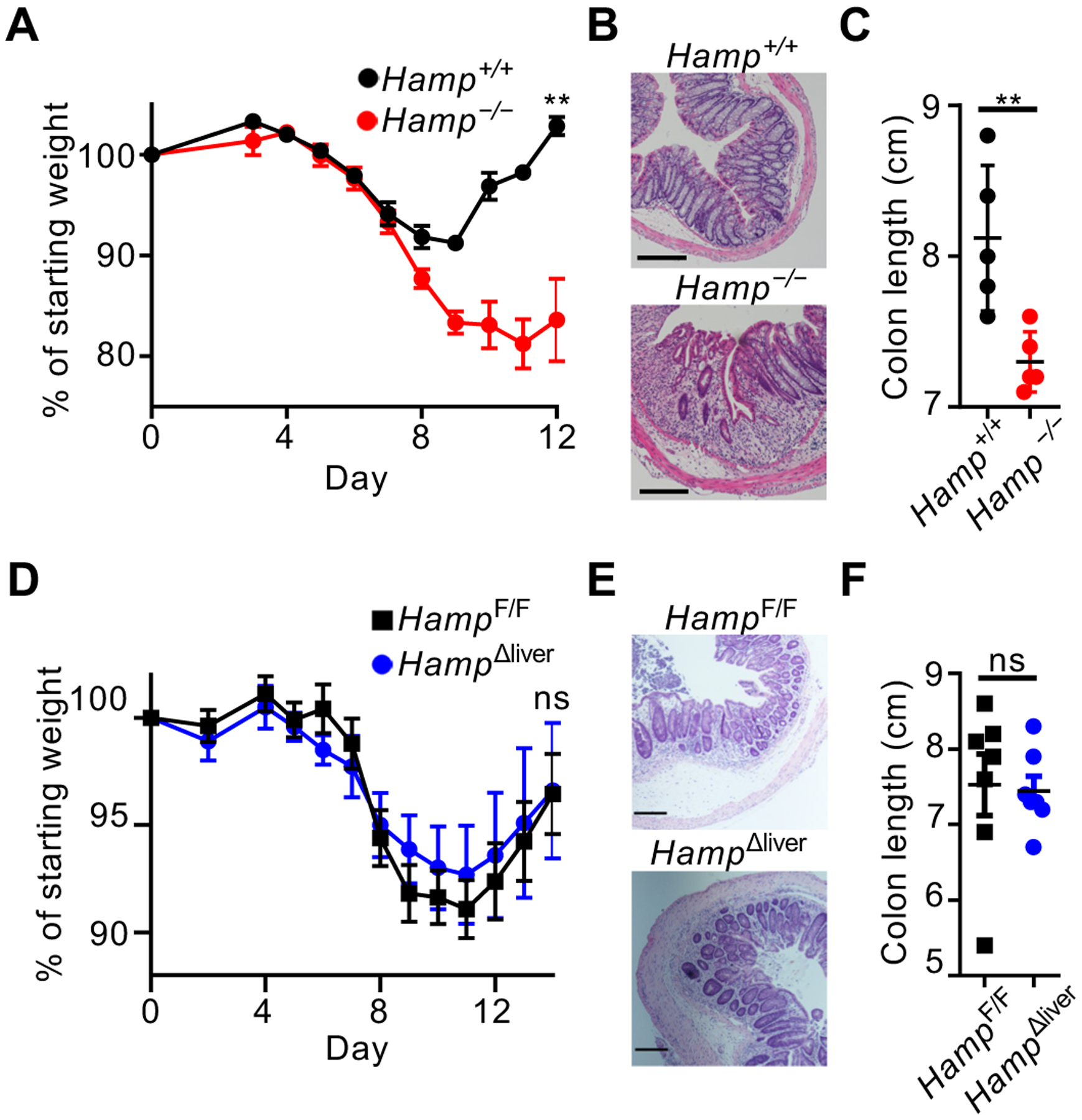
Mice were given DSS for 7 days, and disease and recovery was monitored by weight loss (A), H&E staining of distal colon (B), and colon shortening (C) at day 12. Mice were given DSS for 9 days and recovery was monitored by weight loss (D), H&E staining of the distal colon (E), and colon shortening (F). All scale bars are 200 μm. Data in (A-C, E) are representative of n=3–5 mice per group replicated in two or more independent experiments, and data in D and F are pooled from two independent experiments with n=3–4. Data are shown as the mean ± SEM. Statistics comparing groups used unpaired two-tailed Student’s t-test (**:p<0.01; ***:p<0.001; ****:p<0.0001). In (A) and (D), weights at sacrifice, normalized to starting weight, were analyzed by unpaired two-tailed Student’s t-test.
To interrogate the source(s) of hepcidin that promote mucosal healing, we analyzed tissues of naïve mice. We observed expression within the mesenteric lymph node (mLN) and lamina propria of the colon (cLP), which were maintained upon administration of DSS (Fig. 2A, S1C, D). Previous in vitro studies indicated that macrophages produce hepcidin (14). Surprisingly, we observed that type 2 conventional dendritic cells (cDC2s), and not macrophages or type 1 cDCs (cDC1s), were the dominant myeloid source of hepcidin in the colon following DSS administration (Fig. 2B, C). Bacteria and bacteria-derived molecules were potent inducers of hepcidin expression in both bone marrow-derived DCs and sort-purified cDC2s (Fig. 2D and S2). Intestinal biopsies from Crohn’s disease (CD) and ulcerative colitis (UC) patients revealed a significant increase in hepcidin expression as compared to healthy controls, and significant correlations with DC-associated genes (Fig. 2E, S3). Further, a recently described antibody for hepcidin (15), revealed that cDCs were major producers of hepcidin in the inflamed intestine of IBD patients (Fig. 2F, G). Thus, cDCs are a previously unappreciated source of hepcidin in the intestine of mice and humans that is induced by microbes.
Fig. 2. Conventional dendritic cells are a source of hepcidin in the inflamed intestine.

Hepcidin expression was analyzed by qPCR in naïve mouse tissue (A). Mice were provided DSS for 7 days, colon lamina propria myeloid cells were sorted as noted (B), and hepcidin expression was analyzed by qPCR (C). cDC2s sorted from spleen were stimulated and hepcidin expression was analyzed (D). Hepcidin expression was quantified from intestinal biopsies of humans (E). Lamina propria cells from the inflamed ileum of pediatric CD patients were analyzed for hepcidin protein (F-G). For (A, C, and D), representative data with n=3–5 per group are shown, and data were replicated in at least two independent trials. In (E), n=5 for the healthy group and n=21 for the UC and CD groups. In (F), representative histograms are shown. In (G), four independent patients were tested and all data was pooled. All data are shown as mean ± SEM. In (A, D, and E) data were analyzed by unpaired two-tailed Student’s t-test; in (C), data were analyzed by the Mann–Whitney U test; in (G), data were analyzed by one-way ANOVA with Tukey’s multiple comparisons test (*:p<0.05; **:p<0.01; ***:p<0.001).
We deleted hepcidin in cDCs by crossing CD11cCre mice with HampF/F mice. HampΔCD11c mice exhibited a selective loss of hepcidin expression in cDCs (Fig. 3A, S4A). DC development and systemic iron was comparable in HampΔCD11c mice versus controls (Fig. S4B–D). Furthermore, lymphocyte, myeloid, and granulocyte responses were similar in HampΔCD11c mice and controls during naïve conditions and following administration of DSS (Fig. S4E–F, S5). Global transcriptional profiling also revealed minimal changes in cDC subsets from HampΔCD11c mice versus controls (Fig. S6). Thus, cDC-derived hepcidin does not impact immune responses in these contexts. By contrast, HampΔCD11c mice exhibited significantly reduced body weight following removal of DSS, abnormal colon tissue architecture, and shortened colons relative to controls (Fig. 3B–D). Zbtb46Cre-mediated deletion of hepcidin in cDCs resulted in a similar impairment of tissue repair relative to controls (Fig. S7). Thus, cDC-derived hepcidin is essential for mucosal healing.
Fig. 3. Dendritic cell-derived hepcidin acts on ferroportin-expressing phagocytes to facilitate mucosal healing.
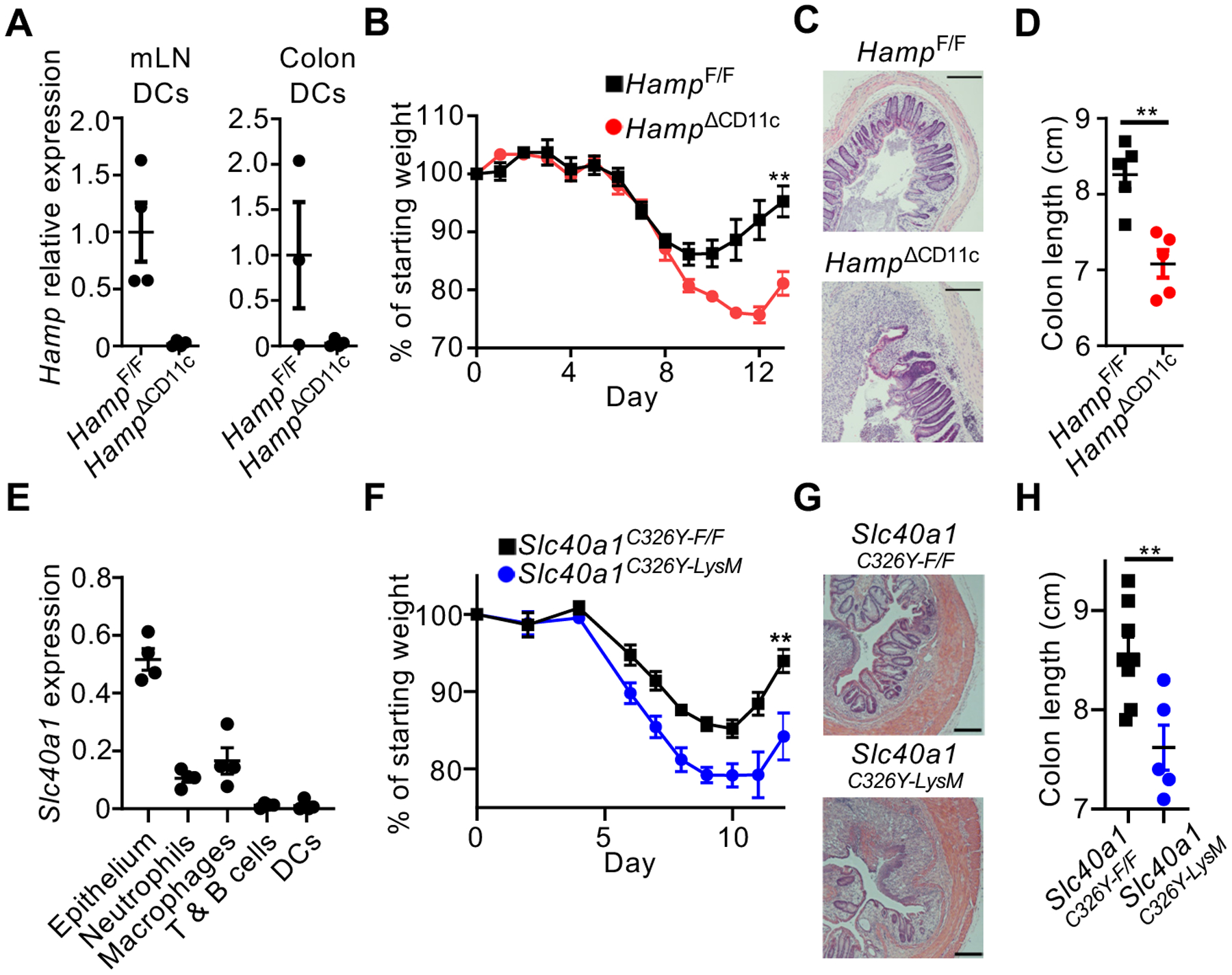
Hepcidin expression was determined by qPCR in mice exposed to DSS for 7 days (A). Mice were given DSS for 8 days, and recovery was monitored by weight change (B), H&E staining of distal colon (C), and colon shortening (D). Sort-purified cells from the naïve mouse colon were analyzed for Slc40a1 expression by qPCR (E). Mice were given DSS for 7 days and recovery was monitored by weight change (F), H&E staining of distal colon (G), and colon shortening (H). All scale bars are 200 μm. All data are shown as mean ± SEM. Data in (D) and (H) were analyzed by unpaired two-tailed Student’s t-test. In (B) and (F), weights at sacrifice, normalized to starting weight, were analyzed by unpaired two-tailed Student’s t-test. For all statistical tests, *:p<0.05; **:p<0.01; ***:p<0.001; ****:p<0.0001. Data in (A-D) are representative of at least two independent experiments with n=3–5 per group. Data in (F) and (H) are pooled from, and data in (J) is representative of, three independent experiments with n=1–3 per group.
We next profiled the colonic expression of ferroportin (Slc40a1) and observed high expression in epithelium, neutrophils, and macrophages (Fig. 3D). To determine whether these are the targets of hepcidin that facilitate mucosal healing, we utilized mice in which a hepcidin-resistant ferroportin variant, Slc40a1C326Y, is expressed from the endogenous locus after Cre-mediated recombination (Fig. S8A) and (16). The expression of Slc40a1C326Y in DCs or intestinal epithelial cells had no impact on mucosal healing (Fig. S8B–E). By contrast, Slc40a1C326Y expression in macrophages and neutrophils via LysMCre resulted in significantly reduced body weight, abnormal colonic tissue architecture, and shortened colons relative to controls and following removal of DSS (Fig. 3F–H). Consistent with post-translational regulation of ferroportin, DC-derived hepcidin did not impact Slc40a1 or Hmox1 mRNA levels in macrophages (Fig. S8F). Thus, ferroportin-expressing macrophages and/or neutrophils are a critical target for hepcidin-mediated mucosal healing.
To test whether this intestinal hepcidin-ferroportin axis regulates local iron distribution in the gut, we employed quantitative mass spectrometry imaging. Strikingly, iron levels within the cecal tissue of DSS-treated HampΔCD11c mice were decreased versus controls (Fig. 4A, S9A, B). Consistent with this, non-heme iron levels were increased in the luminal content of HampΔCD11c mice versus controls after DSS-induced damage, but not in naïve mice (Fig. 4B, S9C, D). This likely involves conversion of heme-bound iron from erythrocytes into non-heme-bound iron via heme oxygenase 1 in phagocytes (17), which would then efflux to extracellular space through ferroportin unless regulated by hepcidin. Iron sequestration is a key component of nutritional immunity (4, 18, 19), so we examined whether cDC-derived hepcidin alters the microbiota. HampΔCD11c mice exhibited a significant shift in microbiota composition relative to littermate controls (Fig. 4C). Fecal microbiota transplantation (FMT) from HampΔCD11c mice to wild-type germ free recipients was sufficient to transfer impaired mucosal healing relative to controls (Fig. S10). Catenibacteria and Bifidobacteria were significantly different genera in HampΔCD11c mice relative to controls (Fig. S11A). Notably, Bifidobacteria supports epithelial barrier function, and dietary iron supplementation can suppress Bifidobacteria and exacerbate inflammation (20). We found Bifidobacteria expanded with restricted dietary iron, and oral administration of Bifidobacteria increased expression of intestinal tight junctions in wild-type mice and enhanced mucosal healing in HampΔCD11c mice (Fig. S11B–E). Bifidobacteria only partially restored normal mucosal healing, and the pathways by which DC-derived hepcidin promotes colonization with this microbe remains unclear. In addition, HampΔCD11c mice also exhibited significantly increased levels of tissue-infiltrating bacteria relative to controls following DSS exposure, and antibiotic treatment eliminated DSS-induced phenotypes (Fig. 4D, S12). To determine whether excess extracellular iron impairs healing in HampΔCD11c mice, we administered deferoxamine (DFO), which sequesters extracellular iron from bacteria by chelation (21). DFO treatment in DSS-exposed HampΔCD11c mice was sufficient to completely restore mucosal healing (Fig. 4E, F).
Fig. 4. Dendritic cell-derived hepcidin sequesters iron to shape the intestinal microbiota.

Mice were exposed DSS for 7 days. Whole ceca tissues were analyzed for iron levels by quantitative mass spectrometry imaging (A), and iron levels were quantified in colon lumen contents (B). Fecal microbiota were analyzed by 16S rRNA gene sequencing and principal coordinate analysis (C). Mice were exposed to DSS for 7 days and bacterial CFU were quantified from colon tissue homogenates (D). Mice were given DSS in drinking water for 7 days and treated daily with either PBS vehicle or DFO from day 0 through day 11. DSS-induced disease and recovery was monitored by weight loss (E) and H&E staining of distal colon (F). In (A), two independent experiments with n=1–5 per group were performed and representative data is shown. Data in (B) and (E) are pooled data from two independent experiments, each with n=3–5 per group. Data in (C) and (D) are representative of two independent experiments with n=5 per group. In (B) and (D), groups were compared using unpaired two-tailed Student’s t-test. In (C), p-value was determined using a PERMANOVA test. In (E) weights at sacrifice, normalized to starting weight, were analyzed by one-way ANOVA using Tukey’s multiple comparisons. In (F), representative data are shown from two independent experiments with n=3–5 per group, and scale bars are 200 μm. For all statistical tests, (*:p<0.05; **:p<0.01).
In summary, our results outline a model in which cDCs produce hepcidin in response to microbiota-derived signals, and subsequently limit iron release from intestinal phagocytes to limit tissue infiltration by the microbiota and thus promote mucosal healing (Fig. S13). This contrasts with liver-derived hepcidin, which acts as an endocrine hormone, is induced by inflammatory cytokines, and has the potential to protect against systemic infection (7, 22, 23). Although not observed in our models, it will be important to interrogate whether DC-derived hepcidin has the potential to directly impact the immune response or systemic iron homeostasis in other contexts. Furthermore, our results indicate that hepcidin mimetics could be a beneficial therapeutic strategy in the context of FMT or gastrointestinal diseases where mucosal healing is an emerging therapeutic goal.
Supplementary Material
Acknowledgments:
We thank members of the Sonnenberg Laboratory for discussions and critical reading of the manuscript, the Epigenomics Core of Weill Cornell Medicine, and Samah Mozumder and Kihwan Kim for technical assistance.
Funding: Research in the Sonnenberg Laboratory is supported by the National Institutes of Health (NIH) fellowship to N.J.B. (F32AI124517), a Crohn’s and Colitis Foundation fellowship to L.Z. (608975) and the following to GFS: NIH (R01AI143842, R01AI123368, R01AI145989, R21CA249274 and U01AI095608), the NIAID Mucosal Immunology Studies Team (MIST), the Searle Scholars Program, the American Asthma Foundation Scholar Award, an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, a Wade F.B. Thompson/Cancer Research Institute (CRI) CLIP Investigator grant, the Meyer Cancer Center Collaborative Research Initiative, and Linda and Glenn Greenberg, and the Roberts Institute for Research in IBD (JRI). G.F.S. is a CRI Lloyd J. Old STAR. Funding support also included the European Research Council (FP7/2011-2015 #261296), the “Fondation pour la Recherche Médicale” (DEQ20160334903), the Laboratory of Excellence GR-Ex (ANR-11-LABX-0051), a labex GR-Ex fellowship (to J.R.R.M. and S.L.), the French National Research Agency (ANR-11-IDEX-0005-02), the “Fondation ARC pour la recherche sur le cancer” to S.Z., the NIH (PP30ES023515 and 1U2CES030859 to C.A. and M.A; and R00HL125899 to S.M.C.), and the Science Foundation Ireland (FRL4862 to S.M.C.). The JRI IBD Live Cell Bank is supported by the JRI, Jill Roberts Center for IBD, Cure for IBD, the Rosanne H. Silbermann Foundation and Weill Cornell Medicine Division of Pediatric Gastroenterology and Nutrition.
Footnotes
Competing interests: G.F.S holds stock and is a member of an advisory board for Celsius Therapeutics Inc. T.A. is an employee of Amgen, Inc.. The other authors declare no competing interests.
Data and materials availability: All data necessary to understand and evaluate the conclusions of this paper are provided in the manuscript and supplementary materials. Microarray and 16S rRNA sequencing data are available from the GEO database with accession numbers GSE143869 and GSE139371. Floxed mice are available with a material transfer agreement.
References and Notes:
- 1.Karin M, Lawrence T, Nizet V, Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124, 823–835 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS, Inflammatory Bowel Disease. Annual Review of Immunology 28, 573–621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khor B, Gardet A, Xavier RJ, Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muckenthaler MU, Rivella S, Hentze MW, Galy B, A Red Carpet for Iron Metabolism. Cell 168, 344–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss G, Goodnough LT, Anemia of Chronic Disease. New England Journal of Medicine 352, 1011–1023 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Donovan A et al. , The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell metabolism 1, 191–200 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Drakesmith H, Nemeth E, Ganz T, Ironing out Ferroportin. Cell metabolism 22, 777–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung E et al. , High-throughput screening of small molecules identifies hepcidin antagonists. Molecular pharmacology 83, 681–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganz T, Nemeth E, Hepcidin and Disorders of Iron Metabolism. Annual Review of Medicine 62, 347–360 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Nemeth E et al. , Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (New York, N.Y.) 306, 2090–2093 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Pasricha SR, McHugh K, Drakesmith H, Regulation of Hepcidin by Erythropoiesis: The Story So Far. Annual review of nutrition 36, 417–434 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Shanmugam NK, Trebicka E, Fu LL, Shi HN, Cherayil BJ, Intestinal inflammation modulates expression of the iron-regulating hormone hepcidin depending on erythropoietic activity and the commensal microbiota. J Immunol 193, 1398–1407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zumerle S et al. , Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 123, 3646–3650 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Peyssonnaux C et al. , TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107, 3727–3732 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasu BJ et al. , Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 115, 3616–3624 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Lakhal-Littleton S et al. , An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayama H et al. , Heme ameliorates dextran sodium sulfate-induced colitis through providing intestinal macrophages with noninflammatory profiles. Proceedings of the National Academy of Sciences, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez CA, Skaar EP, The Impact of Dietary Transition Metals on Host-Bacterial Interactions. Cell host & microbe 23, 737–748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drakesmith H, Prentice AM, Hepcidin and the iron-infection axis. Science (New York, N.Y.) 338, 768–772 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Sarkar A, Mandal S, Bifidobacteria—Insight into clinical outcomes and mechanisms of its probiotic action. Microbiological Research 192, 159–171 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Hider RC, Hoffbrand AV, The Role of Deferiprone in Iron Chelation. New England Journal of Medicine 379, 2140–2150 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Casu C, Nemeth E, Rivella S, Hepcidin agonists as therapeutic tools. Blood 131, 1790–1794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michels K, Nemeth E, Ganz T, Mehrad B, Hepcidin and Host Defense against Infectious Diseases. PLOS Pathogens 11, e1004998 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.