

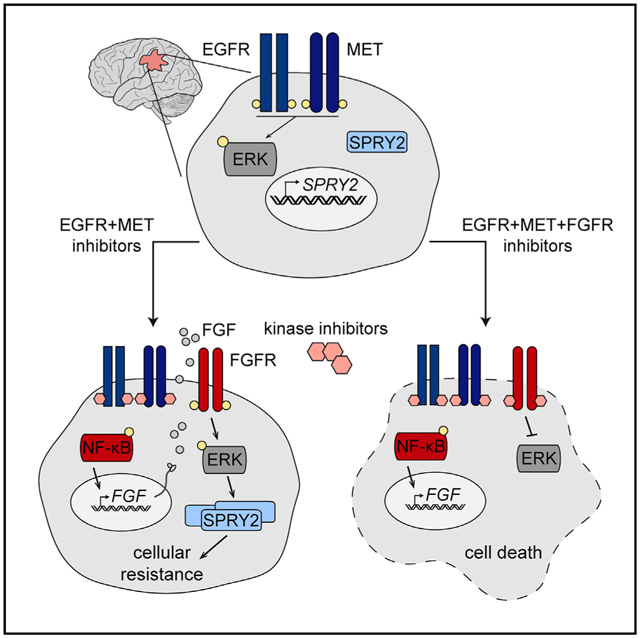
SUMMARY
SPRY2 is a purported tumor suppressor in certain cancers that promotes tumor growth and resistance to receptor tyrosine kinase inhibitors in glioblastoma. Here, we identify a SPRY2-dependent bypass signaling mechanism in glioblastoma that drives resistance to EGFR and MET inhibition. In glioblastoma cells treated with EGFR and MET inhibitors, SPRY2 expression is initially suppressed but eventually rebounds due to NF-κB pathway activation, resultant autocrine FGFR activation, and reactivation of ERK, which controls SPRY2 transcription. In cells where FGFR autocrine signaling does not occur and ERK does not reactivate, or in which ERK reactivates but SPRY2 cannot be expressed, EGFR and MET inhibitors are more effective at promoting death. The same mechanism also drives acquired resistance to EGFR and MET inhibition. Furthermore, tumor xenografts expressing an ERK-dependent bioluminescent reporter engineered for these studies reveal that this bypass resistance mechanism plays out in vivo but can be overcome through simultaneous FGFR inhibition.
Graphical Abstract
In Brief
The poor efficacy of receptor tyrosine kinase inhibitors in glioblastoma may stem from the ability of tumor cells to rewire signaling to maintain expression of SPRY2, a driver of glioblastoma survival. Day et al. identify combination therapies that efficiently and durably suppress SPRY2 expression to potentially treat glioblastoma more effectively.
INTRODUCTION
The standard of care for glioblastoma multiforme (GBM) has not changed substantially in decades and still includes a largely ineffective combination of surgical resection, radiotherapy, and chemotherapy (Stupp et al., 2005). Receptor tyrosine kinase (RTK) inhibitors have yielded disappointing results in GBM clinical trials (Sathornsumetee et al., 2007; De Witt Hamer, 2010), despite the clear involvement of RTKs in glioblastoma pathogenesis (Furnari et al., 2015; Brennan et al., 2013; Stommel et al., 2007). One factor contributing to the failure of RTK inhibitors is the dynamic rewiring of signaling processes that allow GBM cells to evade therapy, in some instances via the compensatory activation of other RTKs (Clark et al., 2012; Ma et al., 2016). For example, in a mouse model of GBM, tumors evaded epidermal growth factor receptor (EGFR) inhibition through the upregulation and phosphorylation of the hepatocyte growth factor receptor (MET) (Jun et al., 2012). Heterogeneity in the response to targeted therapies, either across tumors of different molecular subtypes or within tumors due to variable degrees of oncogene mutation or amplification, has also been a challenging obstacle for durable and complete reduction of tumor burden in GBM (Furnari et al., 2015; Patel et al., 2014). Clearly, new molecular targets are desperately needed that have broad relevance across different GBM cell and tumor contexts.
Several years ago, our lab reported that Sprouty2 (SPRY2), a multifunctional signaling adaptor and purported tumor suppressor in liver, lung, and breast cancers (Masoumi-Moghaddam et al., 2014; Fong et al., 2006; Sutterlüty et al., 2007), surprisingly promotes GBM tumor progression and resistance to therapy (Walsh et al., 2015). Specifically, in differentiated GBM cell lines and glioma stem cells, SPRY2 knockdown antagonized proliferation and anchorage-independent growth and promoted death in response to EGFR and MET inhibitors. In mouse tumor xenografts, SPRY2 knockdown substantially impaired tumor growth, if tumors grew at all. Moreover, analysis of The Cancer Genome Atlas (TCGA) data revealed that the expression of SPRY2 above the population median was a negative prognostic indicator for GBM survival, especially in younger patients. While SPRY2 expression was elevated in tumors expressing the variant III mutant of EGFR (EGFRvIII), SPRY2 protein and transcripts were found in every sample tested, and the effects of SPRY2 knockdown on cell growth and therapeutic resistance were significant and qualitatively identical in many GBM cell backgrounds. Thus, SPRY2 may be the type of broadly relevant target in GBM needed to improve survival. However, SPRY2 lacks intrinsic catalytic domains, and no drugs are yet known that block SPRY2 protein-protein interactions. Thus, for the foreseeable future, targeting SPRY2 may require indirect approaches that exploit the signaling mechanisms responsible for its expression.
In the present study, we identified a SPRY2-dependent bypass signaling mechanism that rescues GBM cells from death in response to EGFR and MET inhibition. In a panel of GBM cell lines, co-inhibition of EGFR and MET led to sustained antagonism of the receptors themselves, but only transient inhibition of extracellular signal-regulated kinase (ERK) phosphorylation and SPRY2 expression, which is strongly regulated by ERK. ERK was reactivated via a nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB)-dependent feedback that led to the autocrine activation of fibroblast growth factor receptors (FGFRs). Activation of this bypass loop explained cell-to-cell variability in survival response to EGFR and MET inhibition and resistance to EGFR and MET inhibition in vivo. An important consequence of ERK reactivation for GBM cell survival of RTK inhibitor treatment was the re-expression of SPRY2. In addition to this intriguing contrast to the more typically reported interaction between SPRY2 and ERK, wherein SPRY2 serves as a negative regulator of ERK activity (Yusoff et al., 2002), this study points to several targets for co-inhibition with EGFR and MET in GBM.
RESULTS
ERK Phosphorylation and SPRY2 Expression Are Only Transiently Suppressed in Response to EGFR and MET Inhibition
U87MG cells expressing EGFRvIII, referred to as U87MG throughout for simplicity, and U118MG cells were treated for up to 48 h with gefitinib and PHA665752, inhibitors of EGFR and MET, respectively (Figures 1A and 1B). This combination was used because it more efficiently reduces GBM cell viability than either inhibitor alone (Huang et al., 2007). By the 1-h time point, phosphorylation of the receptors and the downstream effectors ERK and protein kinase B (AKT) were efficiently inhibited. While receptor phosphorylation remained inhibited throughout the time course, ERK phosphorylation strongly rebounded by 24 h. A similar transient suppression was observed for SPRY2 expression, which is consistent with the reported transcriptional regulation of SPRY2 by ERK (Walsh et al., 2015; Ozaki et al., 2001).
Figure 1. ERK Reactivation Drives SPRY2 Expression and Cellular Survival in Response to EGFR and MET Inhibition.
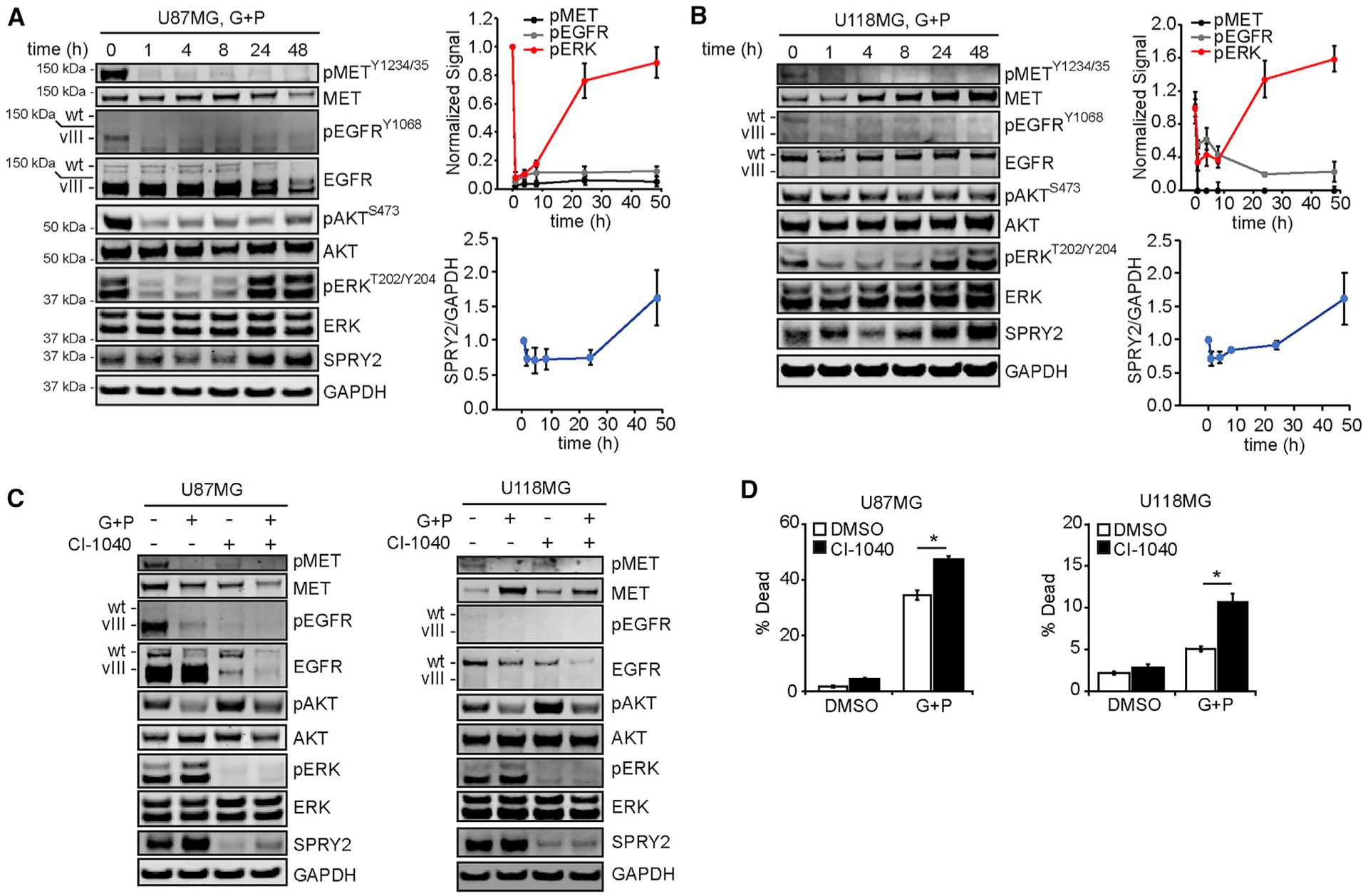
(A and B) U87MG (A) and U118MG cells (B) were treated for ≤48 h with 10 μM gefitinib (G) + 3 μM PHA665752 (P). Cell lysates were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry and normalized to values at t = 0 h.
(C) U87MG and U118MG cells were treated for 48 h with 10 μM G + 3 μM P, 10 μM CI-1040, G + P + CI-1040, or DMSO. Cell lysates were generated and analyzed by western blotting using antibodies against the indicated proteins.
(D) For the same treatments in (C), cell death was measured by flow cytometry.
Throughout the figure panels, representative blot images are shown, and error bars indicate means ± SEMs for three replicates; *p < 0.05 for the indicated comparisons.
Treatment of the same cell lines with gefitinib or PHA665752 alone revealed that both inhibitors were needed to efficiently antagonize ERK and AKT phosphorylation (and produce some degree of cell death in U87MG cells) and that MET inhibition by PHA665752 had the greater effect on ERK (Figure S1A). To test the generality of the observed ERK reactivation effect with other MET inhibitors, we repeated the experiments using JNJ-38877605 or crizotinib (Figure S1B). The same dynamics in ERK phosphorylation and SPRY2 expression were observed.
To test the role of ERK reactivation and SPRY2 re-expression on cell survival, the mitogen-activated protein kinase kinase (MEK) inhibitor CI-1040 was combined with gefitinib and PHA665752 (Figures 1C and S1C). MEK inhibition prevented ERK reactivation and SPRY2 re-expression and augmented cell death (Figures 1D and S1D). MEK inhibition alone was insufficient to induce cell death, potentially due to the lack of inhibition of AKT or some other downstream effector controlled by EGFR and/or MET. In fact, AKT phosphorylation increased in response to MEK inhibition alone, relative to control treatment (Figures 1C and S1C).
Resurgent Expression of SPRY2, Driven by ERK Reactivation, Is Critical for Resistance to EGFR and MET Inhibition
To demonstrate the role of SPRY2 in cell survival in response to EGFR and MET inhibition, we repeated the time course experiments from Figure 1 using cells expressing a SPRY2-targeting short hairpin RNA (shRNA) or a control shRNA (Figure 2A). ERK reactivation dynamics were identical with or without SPRY2 knockdown. However, consistent with previous observations (Walsh et al., 2015), SPRY2 knockdown promoted cell death in response to the inhibitors (Figures 2B and S2A). Together with the results of Figure 1, these data demonstrate that in GBM cells, one important role of ERK activity in regulating cell survival is to promote SPRY2 expression, and that the role of SPRY2 as a suppressor of ERK activity observed in other cellular contexts is not as prevalent.
Figure 2. SPRY2 Is Unique among ERK-Regulated Transcripts in Promoting Resistance to EGFR and MET Inhibition.
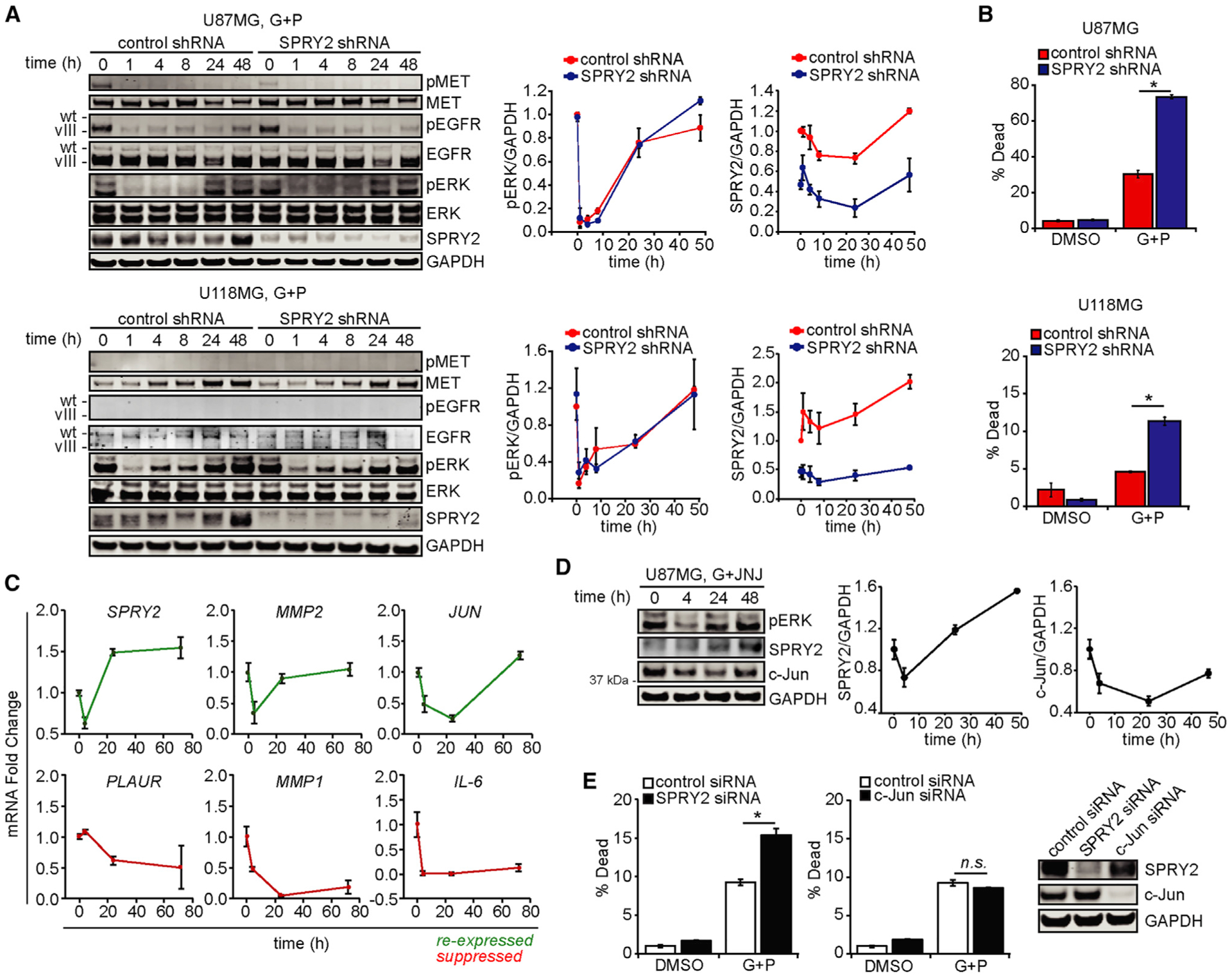
(A) U87MG and U118MG cells expressing control or SPRY2-targeting shRNA were treated for ≤48 h with 10 μM gefitinib (G) + 3 μM PHA665752 (P). Cell lysates were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry and normalized to values at t = 0 h.
(B) For the same cell treatments for 48 h, cell death was measured by flow cytometry.(C) U87MG cells were treated for ≤72 h with 10 μM G + 3 μM P, and RNA was extracted. qRT-PCR was performed using primers against the indicated transcripts.
(D) U87MG cells were treated for ≤48 h with 10 μM G + 5 μM of the MET inhibitor JNJ-38877605 (JNJ). Cell lysates were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry and normalized to values at t = 0 h.
(E) U87MG cells were transfected with SPRY2 or c-Jun siRNAs, or a control siRNA, for 24 h, and then treated with 10 μM G + 3 μM P or DMSO for 24 h. Cell death was quantified by flow cytometry. From a parallel set of cells, lysates were prepared and analyzed by western blotting.
Throughout the figure panels, representative blot images are shown, and error bars indicate means ± SEMs for three replicates; *p < 0.05 for the indicated comparisons.
To demonstrate the particular importance of SPRY2, among other possible ERK-regulated transcripts, for determining cell response to EGFR and MET inhibition, we evaluated the phosphorylation of an ERK-regulated transcription factor, fos-related antigen 1 (FRA1), in response to gefitinib and PHA665752 (Figure S2B). As with ERK, FRA1 phosphorylation was only transiently suppressed and then rebounded by 48 h. Over the same time course, the transcript abundances of five FRA1 target genes (predicted by the NCBI BioSystems Database) that have been implicated in contributing to kinase inhibitor resistance (Geer et al., 2010; Wykosky et al., 2015; Ramsdale et al., 2015; Kim et al., 2012; Kesanakurti et al., 2012; Saito et al., 2018) were measured (Figures 2C and S2C). MMP2 (matrix metalloproteinase 2) and JUN displayed dynamics similar to SPRY2, while PLAUR (plasminogen activator, urokinase receptor), MMP1, and IL-6 (interleukin 6) displayed durable suppression. We chose to characterize the effects of JUN expression on resistance to EGFR and MET co-inhibition because of its cell-intrinsic ability to drive kinase inhibitor resistance, as compared to MMP2, which contributes to kinase inhibitor resistance in the context of the tumor microenvironment. c-Jun, the gene product of JUN, displayed expression dynamics similar to SPRY2 in response to EGFR and MET inhibition (Figure 2D). However, c-Jun knockdown did not alter the cell death response to EGFR and MET inhibition (Figures 2E and S2D), suggesting a unique role for SPRY2 in cell survival. We note that c-Jun knockdown did produce an unanticipated but relatively modest suppression of SPRY2 expression that was apparently insufficient to alter cellular response to EGFR and MET inhibition. Knockdown of SPRY2 or c-Jun did not significantly perturb baseline pMET, pEGFR, or pERK signals, ruling out the possibility that baseline differences in these pathways due to small interfering RNA (siRNA) transfection influenced cell death responses (Figure S2E).
ERK-Mediated SPRY2 Re-expression Is Driven by the Activation of FGFR Family Members
We hypothesized that the observed reactivation of ERK was due to the activation of an alternative RTK. Using an antibody array for 28 different phosphorylated RTKs and 11 downstream effectors, we tested the lysates of U87MG and U118MG cells treated with gefitinib and PHA665752 (Figures 3A and S3A). In both cell lines, the top candidate RTK was FGFR3. The array also detected the anticipated dynamics for ERK phosphorylation. We independently validated the array results for FGFR3 by measuring the phosphorylation of FGFR substrate 2α (FRS2α), a key FGFR adaptor (Kouhara et al., 1997). FRS2α phosphorylation displayed trends in response to EGFR and MET inhibition similar to FGFR3 phosphorylation detected by antibody array (Figure 3B), confirming that functionally relevant amounts of FGFR had been activated in the cells.
Figure 3. FGF Receptors Drive ERK Reactivation and Resistance to EGFR and MET Inhibitors.
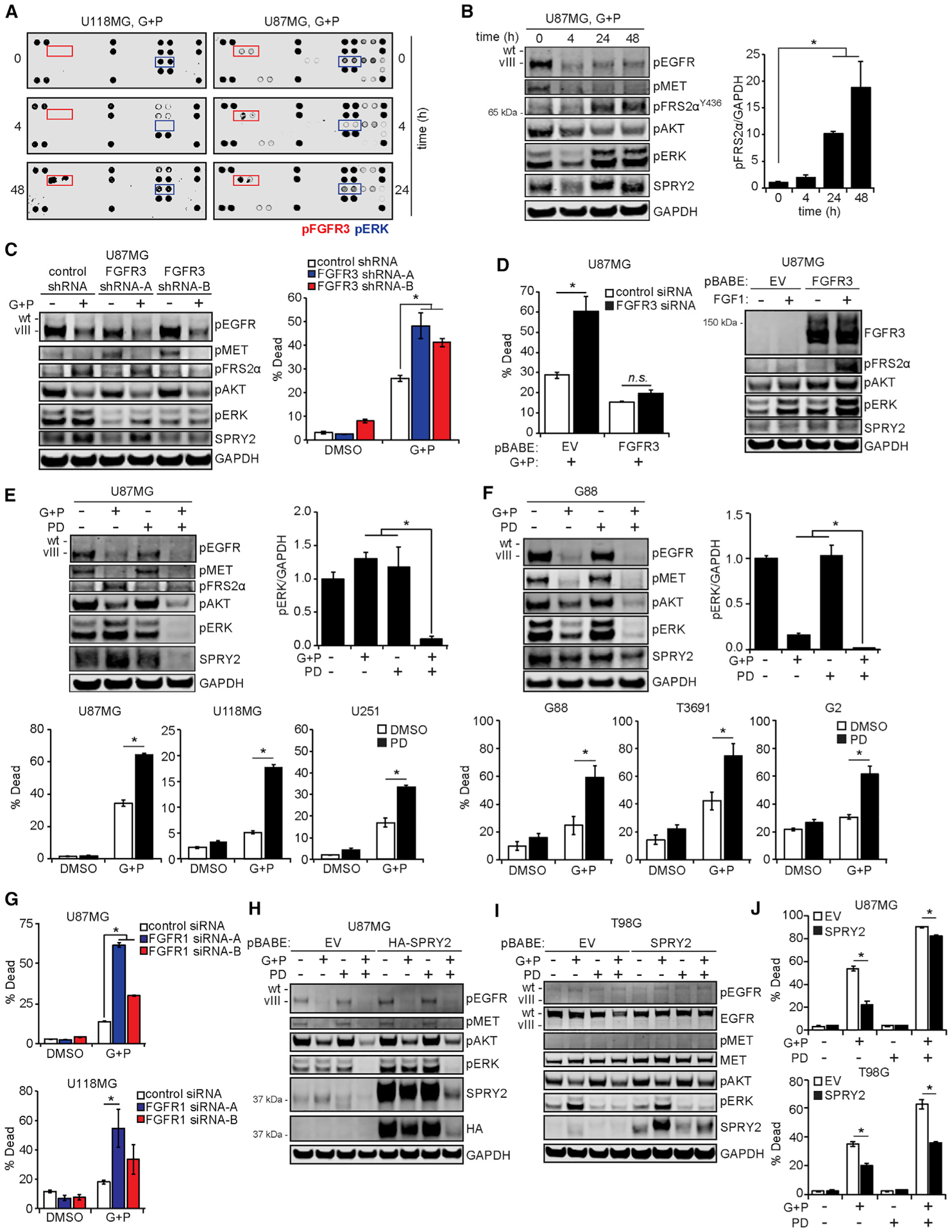
(A) U87MG and U118MG cells were treated with 10 μM gefitinib (G) + 3 μM PHA665752 (P) for the indicated times. Lysates were prepared and incubated with antibody microarrays, with signals for U87MG quantified by densitometry in Figure S3A.
(B) The U87MG lysates from (A) were analyzed by western blotting using antibodies against the indicated proteins, with the pFRS2α signal quantified by densitometry and normalized to values at t = 0 h.
(C) U87MG cells expressing control or FGFR3-targeting shRNA were treated with 10 μM G + 3 μM P or DMSO for 24 h. Cell lysates were analyzed by western blotting using antibodies against the indicated proteins. Cells treated in parallel for 48 h were analyzed by flow cytometry for cell death.
(D) U87MG cells transduced with a vector encoding an RNAi-resistant FGFR3 or an empty vector (EV) were transfected with control or FGFR3-targeting siRNA for 48 h. Cells were then treated for 24 h with 10 μM G + 3 μM P, and cell death was measured by flow cytometry. Cell death data for DMSO-treated cells are shown in Figure S3E. In parallel, untransfected cells were treated with 50 ng/mL FGF1 for 5 min, and cell lysates were analyzed by western blotting using antibodies against the indicated proteins.
(E) U87MG cells were treated for 24 h with 10 μM G + 3 μM P, 0.5 μM PD173074 (PD), G + P + PD, or DMSO. Lysates were analyzed by western blotting using antibodies against indicated proteins, with the pERK signal quantified by densitometry and normalized to DMSO-treated values. U87MG, U118MG, and U251 cells were treated with the same inhibitors for 48 h, and cell death was measured by flow cytometry.
(F) G88 cells were treated for 48 h with 5 μM G + 1 μM P, 0.5 μM PD, G + P + PD, or DMSO before lysis and western blot analysis. G88, T3691, and G2 cells were treated with the same inhibitors for ≤72 h, and cell death was measured by flow cytometry.
(G) U87MG and U118MG cells were transfected with control or FGFR1-targeting siRNA for 48 h and then treated for 24 h with 10 μM G + 3 μM P, or DMSO, and cell death was measured by flow cytometry.
(H and I) U87MG cells (H) or T98G cells (I) were transduced with vectors encoding SPRY2 or an EV and treated for 24 h with 10 μM G + 3 μM P, 0.5 μM PD, G + P + PD, or DMSO. Lysates were analyzed by western blotting using antibodies against indicated proteins. Hemagglutinin (HA)-tagged SPRY2 was used in U87MG cells to differentiate abundant endogenous SPRY2 from ectopic SPRY2.
(J) SPRY2-overexpressing U87MG or T98G cells were treated with the same inhibitors for 48 h, and cell death was measured by flow cytometry. Throughout the figure panels, representative blot or array images are shown, and error bars indicate means ± SEMs for three replicates; *p < 0.05 for the indicated comparisons.
In U87MG and U118MG cells, expression of one of two independent, non-overlapping shRNAs targeting FGFR3 reduced FRS2α and ERK phosphorylation and augmented cell death in response to EGFR and MET inhibition (Figures 3C, S3B, and S3C). These effects were also observed by transfecting cells with siRNA targeting FGFR3 (Figure S3D). To further demonstrate the importance of FGFR3, we transduced U87MG cells with a vector encoding FGFR3, which lacked the 3′ untranslated region targeted by the FGFR3 siRNA sequences. Expression of this siRNA-resistant FGFR3 rescued cellular resistance to EGFR and MET inhibition, even in the presence of FGFR3 siRNA (Figures 3D and S3E). It was not surprising that cells transduced with the FGFR3 vector displayed higher levels of pERK and pFRS2α in response to FGF1 than control. In U87MG cells, pharmacological inhibition of FGFR3 using PD173074 produced effects on ERK phosphorylation, FRS2α phosphorylation, and cell death similar to those observed with FGFR3 RNA interference (Figures 3E and S3F). In fact, the combined effects of FGFR inhibition with EGFR and MET inhibition were observed in three differentiated GBM lines (U87MG, U118MG, and U251) and three glioma stem cell lines (G88, T3691, and G2), suggesting broad importance of the mechanism identified (Figures 3E and 3F).
The Cancer Cell Line Encyclopedia and qRT-PCR analysis qualitatively suggested that FGFR1 transcripts were more abundant than FGFR3 in at least two of the cell lines studied (U87MG, U118MG; Figures S3G and S3H). Thus, even though FGFR1 was not identified by the array, we hypothesized that the mechanisms activating FGFR3 may also activate FGFR1. In differentiated and glioma stem cell lines, transient FGFR1 knockdown antagonized FRS2α and ERK phosphorylation and promoted cell death in response to EGFR and MET inhibition (Figures 3G and S3I–S3K). Furthermore, simultaneously targeting FGFR1 and FGFR3 augmented cell death in response to EGFR and MET inhibition compared to effects observed with targeting either receptor alone (Figure S3L). Since pharmacological inhibitors of FGFR3 have biochemical activity against multiple family members, results using those drugs could be due to the inhibition of both family members.
To further demonstrate the importance of SPRY2 as a downstream effector of FGFR in regulating cell survival, we transduced U87MG and T98G cells with retroviral vectors encoding SPRY2, followed by treatment with combinations of EGFR, MET, and FGFR inhibitors (Figures 3H and 3I). In both cell backgrounds, the overexpression of SPRY2 promoted resistance to EGFR and MET inhibition and EGFR, MET, and FGFR inhibition (Figure 3J), even with similar suppression of ERK phosphorylation. The effect of ectopic SPRY2 expression was more pronounced in T98G cells, which maintained higher levels of SPRY2 in response to the three-inhibitor combination than U87MG (Figure S3M). In aggregate, these data support a model wherein FGFR-mediated resistance to EGFR and MET inhibition depends at least partially on ERK-driven SPRY2 expression.
FGFR Activation in Response to EGFR and MET Inhibition Results from an NF-κB-Mediated Upregulation of FGF Ligands
We hypothesized that a common ligand could explain the simultaneous activation of FGFR1 and FGFR3. Therefore, we measured transcripts for FGF1 and FGF2, which are known to bind both FGFR family members (Zhang et al., 2006) (Figure S4A). In U87MG and U118MG cells, FGF1, FGF2, or both transcripts were significantly elevated in response to EGFR and MET inhibition for 4–72 h (Figures 4A and S4B). In U87MG cells, changes in transcript abundance corresponded to an increased concentration of FGF1 in conditioned medium (Figure 4A). Analysis of conserved transcription factor binding sequences in the FGF1 gene promoter, in addition to previous studies of transcriptional regulation of FGF2 (Lee and Kay, 2012), suggested that the increased transcriptional activity of NF-κB could be responsible for the upregulated ligand expression observed (Figure 4B). Therefore, we measured the phosphorylation of NF-κB at serine 536, a surrogate measurement for NF-κB transcriptional activity, in response to EGFR and MET inhibition (Figure 4C). In both U87MG and U118MG cells, an early (1–4 h) increase in phosphorylated NF-κB was observed, which is consistent with the dynamics of FGF1 and FGF2 mRNA upregulation. NF-κB phosphorylation trends were identical in cells with or without SPRY2 knockdown, reinforcing the notion that differential SPRY2 expression does not influence the resistance mechanism upstream of ERK activation. In addition, increased phosphorylation of NF-κB was coincident with its increased nuclear localization and FGF1 promoter binding 4 h after cell treatment (Figures 4D, S4C, and S4D).
Figure 4. FGF Ligands Are Expressed Due to NF-κB Activity.
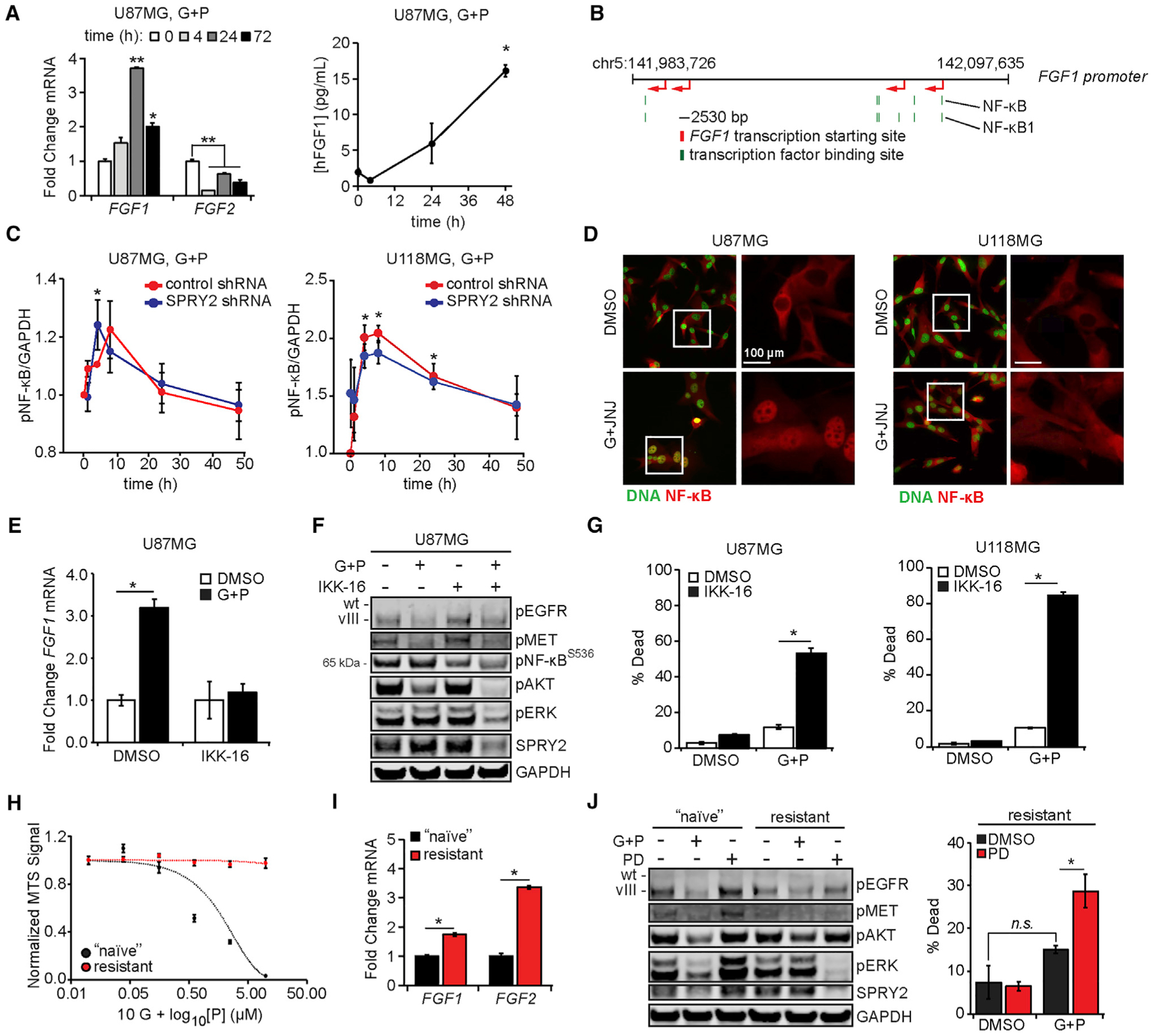
(A) U87MG cells were treated with 10 μM gefitinib (G) + 3 μM PHA665752 (P) for the indicated times, and RNA was extracted. qRT-PCR was performed using primers for FGF1 and FGF2. In parallel, conditioned medium was collected from cells treated with 10 μM G + 3 μM P and analyzed for human FGF1 by ELISA.(B) The schematic shows the positions of NF-κB and NF-κB1 binding sites (green) in the FGF1 promoter relative to predicted FGF1 transcription start sites (red).
(C) U87MG and U118MG cells expressing control or SPRY2-targeting shRNA were treated with 10 μM G + 3 μM P for ≤48 h. Cell lysates were analyzed by western blotting, with pNF-κB signal quantified by densitometry.
(D) U87MG and U118MG cells were treated for 4 h with 10 μM G + 5 μM JNJ-38877605 (JNJ) or DMSO. Cells were stained for NF-κB and DNA. Quantification of NF-κB nuclear intensity is shown in Figure S4C.
(E) U87MG cells were treated with 10 μM G + 3 μM P, 0.5 μM IKK-16, G + P + IKK-16, or DMSO for 6 h, and RNA was extracted. qRT-PCR was performed using primers for FGF1.
(F) U87MG cells were similarly treated for 24 h. Lysates were analyzed by western blotting using antibodies against the indicated proteins. Quantifcation of pERK by densitometry is shown in Figure S4F.
(G) U87MG and U118MG cells were similarly treated (24 h for U87MG, 48 h for U118MG), and cell death was measured by flow cytometry.
(H) G + P-resistant or -naive U87MG cells were treated with 10 μM G + P (10 nM–10 μM) for 24 h. Cell viability was measured by MTS assay.
(I) RNA was extracted from G + P-resistant or -naive U87MG cells, and qRT-PCR was performed using primers for FGF1 and FGF2.
(J) G + P-resistant or -naive U87MG cells were treated with 10 μM G + 3 μM P or 0.5 μM PD173074 (PD) for 4 h. Cell lysates were analyzed by western blotting using antibodies against the indicated proteins. In parallel, G + P-resistant cells were treated with G + P, PD, G + P + PD, or DMSO for 48 h, and cell death was measured by flow cytometry.
Throughout the figure panels, representative blot images are shown, and error bars indicate means ± SEMs for three replicates; *p < 0.05 and **p < 0.01 for comparison to t = 0 h, unless comparison indicated.
To probe the role of NF-κB in FGF ligand synthesis, we measured FGF1 and FGF2 transcripts in cells treated with EGFR and MET inhibitors, with or without the inhibition of IκB kinase (IKK), a kinase upstream of NF-κB (Figures 4E and S4E). In U87MG cells, IKK inhibition using IKK-16 abrogated the increase in FGF1 observed in response to EGFR and MET inhibition, an effect that coincided with decreased ERK activation and SPRY2 expression (Figures 4F and S4F). IKK inhibition also promoted cell death in response to EGFR and MET inhibition (Figure 4G). Similar effects on cell death, ERK activation, and SPRY2 expression in response to EGFR and MET inhibition were observed with transient NF-κB knockdown (Figure S4G). In addition, similar effects on ERK activation, SPRY2 expression, and cell death due to IKK inhibition in combination with EGFR and MET inhibition were also seen in G88 and T3691 glioma stem cell lines (Figures S4H and S4I). Inhibition of IKK alone (Figure 4F) or knockdown of NF-κB alone (Figure S4G) caused increased ERK phosphorylation and SPRY2 expression in U87MG cells. However, inhibition of IKK alone in G88 cells had a qualitatively opposite effect on baseline SPRY2 expression (Figure S4H), suggesting a possible cell-type-specific mechanism whereby NF-κB regulates SPRY2 expression in a way that occurs outside the setting of simultaneous EGFR and MET targeting, as observed previously for other SPRY proteins (Macià et al., 2014).
To further demonstrate the role of FGFR signaling in GBM cell resistance to EGFR and MET inhibition, we grew cells in medium containing EGFR and MET inhibitors for >2 months, which produced a population of cells that were no longer responsive to gefitinib and PHA665752 over a range of concentrations (Figure 4H). FGF1 and FGF2 transcripts were significantly upregulated in the drug-resistant cells (Figure 4I). In addition, cells with acquired resistance remained sensitive to combined EGFR-MET-FGFR inhibition with respect to cell death and displayed both lowered ERK phosphorylation and SPRY2 expression in response to FGFR inhibition alone (Figures 4J and S4J).
Activation of the FGFR-ERK-SPRY2 Signaling Axis Explains Heterogeneous Cellular Responses to RTK Inhibition
To understand whether cell-to-cell heterogeneity in FGFR-driven ERK activation explained variation among cells in their ability to resist EGFR and MET inhibition, we engineered U87MG cells to express a previously described fluorescent reporter of ERK activity (Albeck et al., 2013). The reporter consists of mVenus fused to a nuclear localization sequence (NLS) and the PEST domain of FRA1, which is stabilized upon phosphorylation by ERK (Figures 5A and S5A). A second control reporter consists of a fusion of Cerulean with an NLS and a PEST domain that is not regulated by ERK (called d2). These reporters are referred to as the FIRE (FRA1-based integrative reporter of ERK) system, with the ratio of mVenus to Cerulean intensity typically reported.
Figure 5. Single-Cell Measurements Reveal That Cell-to-Cell Variability in FGF-Driven Autocrine Signaling Explains Heterogeneous Cell Responses to the Inhibition of EGFR and MET.
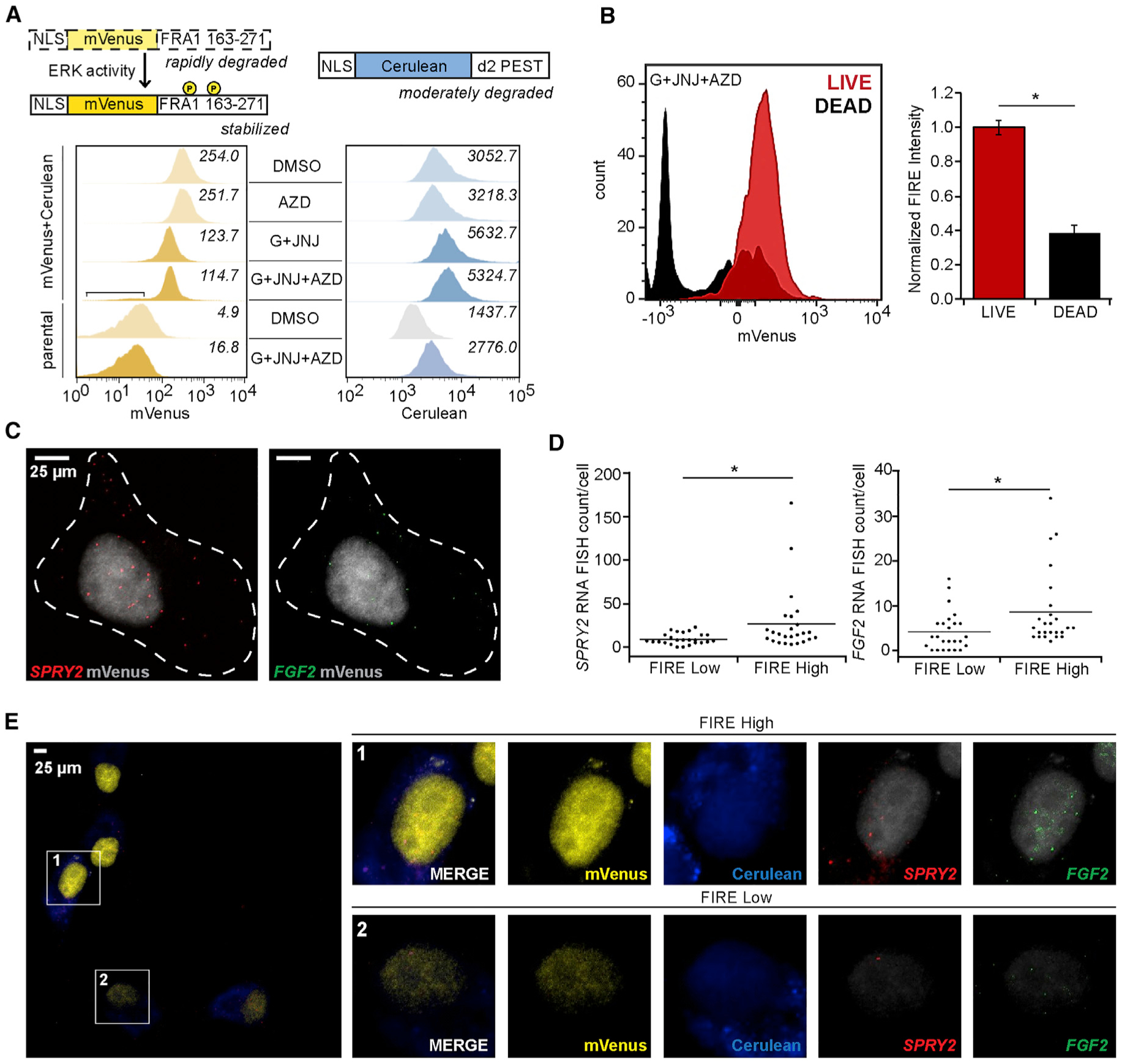
(A) The schematic shows the domain structures of the FIRE reporters designed by Albeck et al. (2013) (mVenus, ERK regulated; cerulean, control). U87MG cells expressing the reporters were treated with 10 μM gefitinib (G) + 5 μM JNJ-38877605 (JNJ), 0.5 μM AZD4547 (AZD), G + JNJ + AZD, or DMSO for 24 h. The square bracket indicates a population of cells for the G + JNJ + AZD condition displaying mVenus fluorescence consistent with untransduced parental cells. Italicized values indicate the average geometric mean fluorescence intensities for three separate histograms per sample or condition.
(B) U87MG cells expressing FIRE reporters were treated for 24 h with 10 μM G + 5 μM JNJ + 0.5 μM AZD, and cell death was measured by flow cytometry. FIRE intensity, normalized to live cells, are shown for TO-PRO-3− (live) or TO-PRO-3+ (dead) cells. Representative histograms for mVenus signal distributions are shown. Median signals were used to compute mVenus-cerulean intensity ratios.
(C) Representative images of a single U87MG cell with single-molecule RNA FISH probes recognizing SPRY2 or FGF2 mRNA are shown. Dotted lines indicate representative cell boundaries used for quantifying single-molecule RNA FISH spots per cells.
(D) U87MG cells expressing the FIRE reporter were treated for 24 h with 10 μM G + 5 μM JNJ before single-molecule RNA FISH probe hybridization. Cerulean and mVenus intensities, in addition to single-molecule RNA FISH spot counts, were calculated for n > 100 cells across 3 biological replicates. SPRY2 and FGF2 single-molecule RNA FISH spot counts for lowest- or highest-quartile FIRE populations are plotted.
(E) Representative images of cells treated for 24 h with G + JNJ, including cells with both high (1) and low (2) FIRE intensities, show differences in single-molecule RNA FISH signals with respect to FIRE intensity. Quantification of matched DMSO-treated cells are found in Figure S5D.
Throughout the figure panels, representative images are shown, and error bars indicate means ± SEMs of three replicates, unless otherwise noted; *p < 0.05 for the indicated comparisons.
U87MG cells expressing FIRE were treated for 24 h with combinations of EGFR, MET, and FGFR inhibitors and subjected to flow cytometry (Figures 5A and S5B). The MET inhibitor JNJ-38877605 and the FGFR inhibitor AZD4547 were used for these studies based on their low autofluorescence. JNJ-38877605 and AZD4547 produced effects on cell death similar to those observed with other MET and FGFR inhibitors (Figure S5C). Cells treated with the combination of gefitinib, JNJ-38877605, and AZD4547 displayed the lowest mVenus intensities (Figure 5A). These cells displayed approximately equal expression of Cerulean as compared to cells treated with all other combinations, suggesting that the loss of mVenus signal did not result solely from the antagonism of protein synthesis with three inhibitors. To assess how differences in mVenus intensity (ERK activity) correlated with cell death, we included a live-cell impermeable dye in flow cytometry. Dead cells displayed lower mVenus intensities and a lower normalized FIRE intensity (Figure 5B). Thus, individual cells with lower ERK activity were more likely to die than cells with high ERK activity.
To explore the basis for cellular heterogeneity in ERK activation, U87MG cells expressing the FIRE reporter system were treated with gefitinib and JNJ-38877605 for 24 h, fixed, and hybridized overnight with single-molecule RNA fluorescence in situ hybridization (FISH) probes recognizing FGF2 and SPRY2 mRNA transcripts (Figure 5C). Cells with higher FIRE intensity expressed more SPRY2 than those with lower FIRE intensity (Figures 5D and 5E). In addition, cells with higher FIRE intensity displayed increased FGF2 (Figures 5D and 5E). Median mRNA counts for SPRY2 and FGF2 and median FIRE intensity for treated versus untreated conditions were qualitatively consistent with population-level measurements made by qRT-PCR or flow cytometry (Figure S5D versus Figures 2C, 4A, and 5A).
A Bioluminescent Reporter of ERK Activity Demonstrates Relevance of FGFR-ERK-SPRY2-Mediated Bypass Resistance In Vivo
To assess the relevance of FGFR-mediated bypass signaling through ERK in vivo, we modified the previously described FIRE reporter to create a bioluminescence-based reporter of ERK activity (Figure 6A). mVenus was replaced by click beetle green (CBGreen) luciferase in the ERK-dependent construct, and Cerulean was replaced by click beetle red (CBRed) luciferase in the ERK-independent construct, to create the CB-FIRE reporter system. d-Luciferin is a substrate for both CBGreen and CBRed, but CBRed produces longer wavelengths than CBGreen, enabling the detection of two independent signals (with spectral unmixing) using a single substrate injection. Before the in vivo experiments, U87MG and G88 cells expressing the CB-FIRE reporter system were used for in vitro validation of the ability of CB-FIRE to reflect ERK dynamics accurately in response to the inhibitors (Figures 6B and S6A–S6C). CB-FIRE signals mirrored trends in ERK activity measured by phospho-specific western blotting (Figure S6D).
Figure 6. FGFR-ERK-SPRY2 Bypass Signaling Drives Resistance to EGFR and MET Inhibition In Vivo.
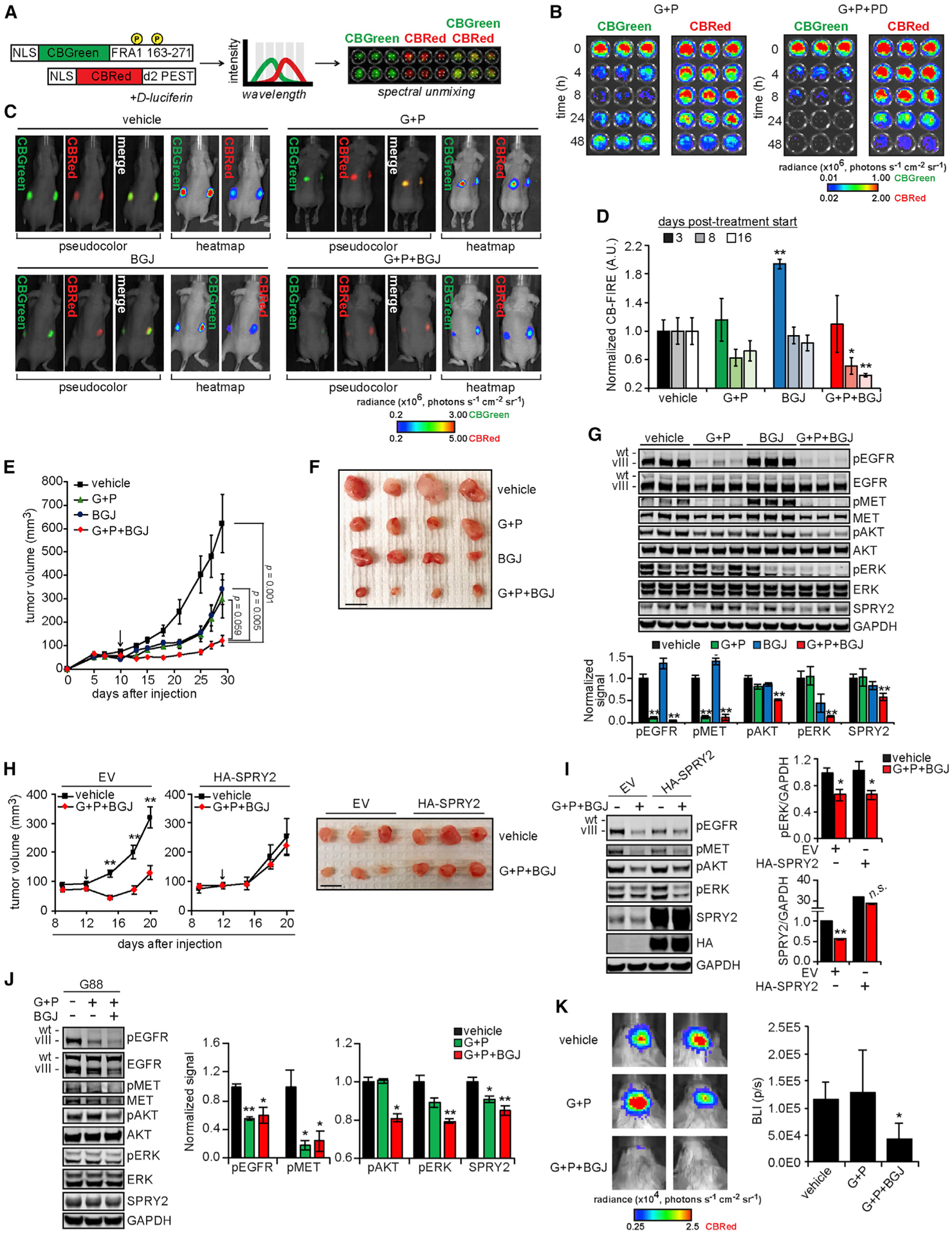
(A) The design of click beetle (CB) luciferase-based reporters of ERK activity (CB-FIRE system) was based on the design of the FIRE reporter system. Bioluminescent imaging of cells expressing the CB-FIRE reporters (CBGreen, ERK dependent; CBRed, control) with spectral unmixing allowed for the simultaneous detection of signals from both luciferases.
(B) U87MG cells expressing CB-FIRE reporters were treated with 10 μM gefitinib (G) + 3 μM PHA665752 (P) ± 0.5 μM PD173074 (PD) for the indicated times before the addition of d-luciferin and bioluminescent imaging.
(C) Subcutaneous xenograft tumors were formed in mice using U87MG cells expressing the CB-FIRE reporters, and mice were imaged 8 days after starting indicated treatments (G, 100 mg/kg; P, 30 mg/kg; BGJ398 [BGJ], 20 mg/kg). Representative images of unmixed luciferase signals, with radiance pseudo-colored for qualitative comparison or as a heatmap for quantitative comparison, overlaid with photographs of animals are shown.
(D) CBGreen-CBRed photon flux values, normalized to values for vehicle treatments are shown for 3, 8, or 16 days after treatment began.
(E) Tumor volumes were measured every 2 to 3 days for ≤29 days after cell injection (n = 10 tumors for each treatment group). Indicated statistical comparisons are for final tumor volumes. Arrow indicates treatment start.
(F) Representative excised tumors from each group are shown. Scale bar, 10 mm.
(G) Tumor lysates (n = 3 per group) were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry.
(H) Subcutaneous xenograft tumors were formed in mice using U87MG cells expressing HA-tagged SPRY2 (HA-SPRY2) or transduced with empty vector (EV), and mice were treated with the indicated treatments (G, 100 mg/kg; P, 30 mg/kg; BGJ, 20 mg/kg). Tumor volumes were measured every 3 days for ≤20 days after cell injection (n = 8 tumors for each treatment group). The indicated statistical comparisons are between vehicle and G + P + BGJ group tumor volumes. The arrow indicates treatment start. Representative excised tumors from each group are shown. Scale bar, 10 mm.
(I) Tumor lysates (n = 3 per group) were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry.
(J) Orthotopic xenograft tumors were generated in mice using G88 cells expressing the CB-FIRE reporters. Mice were treated daily (5 days on, 2 days off, 4 days on) with the indicated treatments (G, 100 mg/kg; P, 30 mg/kg; BGJ, 30 mg/kg) before euthanasia and harvest of mouse brain hemispheres containing tumors. Lysates generated from mouse brains (n = 4 for vehicle and G + P + BGJ; n = 3 for G + P) were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry.
(K) Representative images of unmixed, CBRed luciferase signals from mice treated as described. The range of luminescence values for heatmap images was chosen to demonstrate the differences between vehicle and G + P + BGJ-treated animals most clearly. Quantified bioluminescence intensities (BLI) plotted as photons per second (p/s) are shown.
Throughout the figure panels, representative images are shown, and error bars represent means ± SEMs. *p < 0.05 or **p < 0.01 for comparisons against vehicle control.
With the reporter validated, U87MG cells expressing the CB-FIRE reporter system were subcutaneously injected in both flanks of NU/NU mice. Xenografts were engineered subcutaneously to avoid any issues of differential blood-brain barrier penetrance among the drugs being used. After 10 days of tumor growth, animals were randomly assigned for treatment with gefitinib + PHA665752, FGFR inhibitor BGJ398, gefitinib + PHA665752 + BGJ398, or vehicle. BGJ398 was used for in vivo studies due to its recent evaluation in a GBM clinical trial (NCT01975701). In vitro, BGJ398 produced effects similar to other FGFR inhibitors (Figure S6E). In vivo, bioluminescent imaging revealed a time-dependent decrease in CB-FIRE signal only when all three inhibitors were combined (Figures 6C, 6D, and S6F). In addition, the combination of all three inhibitors resulted in tumors that were smaller than those for control treatments, gefitinib + PHA665752, or BGJ398 alone (Figures 6E, 6F, and S6G). Ex vivo analyses of harvested tumors confirmed that tumors treated with three inhibitors also displayed the lowest levels of phosphorylated ERK and AKT and the lowest expression of SPRY2 (Figure 6G). No significant weight loss was observed in animals across treatment groups, including the triple combination of EGFR, MET, and FGFR inhibitors (Figure S6H).
To further demonstrate the importance of SPRY2 expression in mediating resistance to inhibitors in vivo, we performed similar subcutaneous xenograft experiments using U87MG cells with the ectopic expression of SPRY2. SPRY2-overexpressing tumors displayed almost no response to the combination of EGFR, MET, and FGFR inhibitors, in clear contrast to tumors formed from empty vector-transduced cells (Figure 6H). Ex vivo analyses of harvested tumors confirmed that tumors expressing ectopic SPRY2 maintained higher expression levels in response to the three-inhibitor combination, even with similar decreases in ERK phosphorylation, further highlighting the importance of SPRY2 expression in driving resistance to inhibitors in vivo (Figure 6I). In contrast to observations in vitro, the combination of EGFR, MET, and FGFR inhibitors did not have a large effect on ectopic SPRY2 expression levels.
We next sought to demonstrate the feasibility of the proposed combination therapy in an orthotopic model of glioblastoma. G88 cells were injected intracranially in BALB/c SCID mice and allowed to form tumors for 4 days, after which mice began daily treatment with gefitinib + PHA665752, gefitinib + PHA665752 + BGJ398, or vehicle control. Analyses of tumor lysates demonstrated that the three-drug combination of EGFR, MET, and FGFR inhibitors suppressed AKT and ERK phosphorylation, as well as SPRY2 expression, more than the two-drug combination of EGFR and MET inhibitors (Figure 6J). Bioluminescence imaging 5 days after starting treatment revealed that tumors treated with all three inhibitors were smaller than those treated with EGFR and MET inhibitors or vehicle (Figure 6K). Overall, these data suggest that the combination of EGFR, MET, and FGFR inhibitors reduced GBM orthotopic tumor growth via the suppression of an ERK-SPRY2 resistance mechanism.
DISCUSSION
The bypass signaling mechanism elucidated here (summarized in Figure 7) provides new understanding of the sequence of events that are required for SPRY2 to drive GBM resistance to EGFR and MET inhibitors. This mechanism was at play in differentiated GBM cells expressing EGFRvIII or wild-type EGFR alone and in glioma stem cells, pointing to the broad relevance of the bypass mechanism. Our prior study identifying the role of SPRY2 in GBM began to elucidate the specific SPRY2-regulated processes (e.g., p38 signaling) that explained the observed effects of SPRY2 knockdown (Walsh et al., 2015). While work remains to further understand those processes, the present study highlights the importance of identifying the druggable signaling pathways that enable SPRY2 to be expressed at all.
Figure 7. ERK and SPRY2 Are at the Center of a Bypass Resistance Mechanism That Enables GBM Cells to Evade Treatment with EGFR and MET Inhibitors.
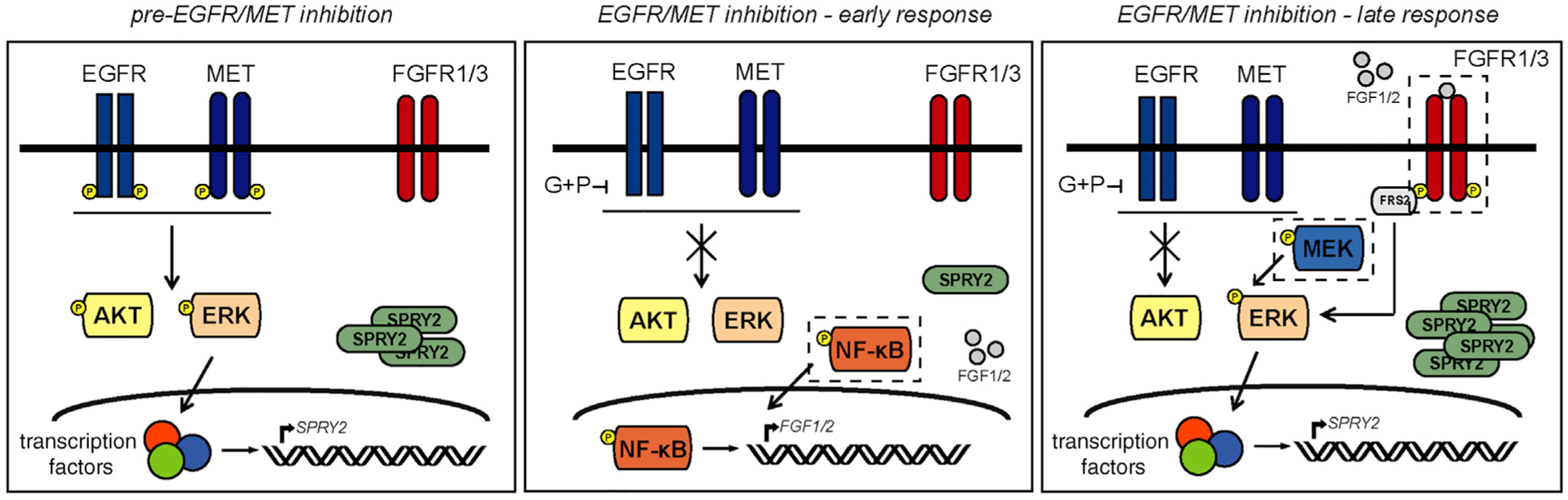
In the model of signaling regulation proposed here, combined inhibition of EGFR and MET abrogates ERK and AKT activities, but the effect on ERK is short-lived. Activation of NF-κB promotes FGF1 and FGF2 production and autocrine activation of FGFR. The resultant reactivation of ERK drives SPRY2 expression and rescues GBM cells from death. The dashed boxes indicate proteins that, when targeted in combination with EGFR and MET, could be useful targets to pursue in future combinatorial strategies.
The cascade that ultimately leads to SPRY2 re-expression is initiated by the activation of the NF-κB pathway as a response to EGFR and MET inhibition, which may be viewed as a surprising effect, given that EGFR is a known driver of NF-κB signaling. However, NF-κB pathway activation is also a fairly common cellular response to stress (Mercurio and Manning, 1999), including chemotherapeutics (Godwin et al., 2013) and even EGFR inhibition (Blakely et al., 2015). In fact, IL-6-mediated activation of NF-κB signaling in response to EGFR kinase inhibitors is a reported mechanism of resistance in GBM (Zanca et al., 2017). We do not believe that the specific IL-6-dependent mechanism is at play in our studies, however, because IL-6 transcript abundance fell precipitously and did not rebound in response to EGFR and MET inhibition (Figure 2C). Whatever the driver of NF-κB, the production of FGF ligands that follows activates FGFR, a receptor that drives resistance to kinase inhibitors in melanoma, gastrointestinal stromal tumors, and non-small-cell lung cancer (Yadav et al., 2012; Li et al., 2015; Ware et al., 2010). In GBM, FGFR has more typically been viewed as a target in the small subset of tumors that harbor FGFR1 and FGFR3 oncogenic fusions (FGFR-transforming acidic coiled-coil fusions), and not as a primary driver of kinase inhibitor resistance (Singh et al., 2012). Of course, reactivation of ERK, driven by any number of upstream processes, has been previously implicated as driving resistance to kinase inhibitors in cancer (e.g., Yadav et al., 2012; Ercan et al., 2012; Tricker et al., 2015). However, the importance of ERK signaling specifically as a driver of SPRY2 expression is unique to the GBM setting and is surprising given that ERK has numerous substrates (Wortzel and Seger, 2011).
The bypass signaling mechanism also identifies several potential targets whose inhibition along with EGFR and MET may lead to more meaningful responses in GBM patients: FGFR, MEK/ERK, and the NF-κB pathway. While GBM clinical trials involving FGFR inhibitors have typically only enrolled patients with FGFR fusions or activating mutations, our findings motivate the evaluation of FGFR inhibitors, in combination with EGFR and MET inhibitors, even for patients lacking FGFR genetic alterations. Such approaches may be enabled by the recent US Food and Drug Administration (FDA) approval of the FGFR inhibitor erdafitinib for the treatment of metastatic bladder cancer. Partial responses to erdafitinib have been reported in some GBM patients enrolled in a phase I clinical trial (Tabernero et al., 2015). Along the same lines, the FDA-approved MEK inhibitors trametinib and binimetinib should also be evaluated in combination with EGFR and MET inhibitors. While none are yet FDA approved, investigational inhibitors of the NF-κB pathway have been deployed in clinical trials, including the IKK inhibitor SAR113945 and curcumin (NCT01113333 and NCT01269203, respectively). Proteasome inhibitors, such as the approved myeloma and mantle cell lymphoma drug bortezomib, also efficiently block the NF-κB pathway. Marizomib, another proteasome inhibitor, displays good blood-brain barrier penetration (Di et al., 2016) and is in clinical trials for GBM (NCT03345095).
A major obstacle to studies involving three independent kinase inhibitors is the potential for intolerable drug toxicities. While two-inhibitor strategies have been evaluated, such as the combination of erlotinib and sorafenib for the treatment of brain tumors (Peereboom et al., 2013), three-inhibitor clinical trials have not yet been initiated. However, an ongoing phase I trial is evaluating the safety of combining three (mebendazole, bevacizumab, irinotecan) or even four (mebendazole, vincristine, carboplatin, temozolomide) drugs for treating brain tumors (NCT01837862). Thus, there is a clear precedent for combining three or more drugs in medical oncology, even if those drugs have not been kinase inhibitors. It is encouraging that animals in the current study, which were treated simultaneously with EGFR, MET, and FGFR inhibitors, did not display significant evidence of toxicity, as evaluated by weight loss during the study (Figure S6H). Furthermore, the availability of bi-specific antibodies that target EGFR and MET simultaneously (Emdal et al., 2017), in addition to an inhibitor (S49076) that has activity against both MET and FGFR (Rodon et al., 2017), may increase options for safely targeting three kinases simultaneously. As new inhibitors are developed with improved blood-brain barrier penetrance (e.g., EGFR inhibitors osimertinib and AZD3759), it may also eventually be possible to effectively target kinases in GBM with lower dosages (Heffron, 2016).
The tendency for cell signaling pathways to become rewired in response to therapy in ways that drive resistance makes it useful to follow tumors longitudinally as treatment continues. The most common method is to euthanize different populations of animals at each time point of interest (Germann et al., 2017). Here, a bioluminescent reporter of ERK activity was developed. By expressing the reporters in GBM cells used to engineer xenografts, repeated evaluation of ERK activity was possible within a single population of mice over time, greatly reducing the number of animals needed and eliminating a source of variability in the measurements. The reporter system also requires far less technical expertise than intravital microscopy and only requires injection of a single substrate, versus other dual-luciferase combinations. Furthermore, while most previously developed bioluminescence-based reporters of signaling pathways are designed to detect transcriptional changes downstream of one or many kinases (Kocher and Piwnica-Worms, 2013), our reporter measures the activation of ERK post-translationally through a kinase-substrate interaction. Eventually, the basic principle used here to develop an ERK reporter could be extended to other kinases with known substrate interactions regulating protein stability.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew Lazzara (mlazzara@virginia.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were conducted according to protocols approved by the University of Virginia Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines. For subcutaneous tumor models, NU/NU Nude mice (strain code:088) were obtained from Charles River Laboratories. For intracranial brain tumor models, BALB/c scid mice were obtained from The Jackson Laboratory (Stock No. 001803). All mice used for both tumor models were female, aged 4–8 weeks and weighed 20–25 g at the start of experiments. Mice were randomly assigned to experimental groups. All mice were kept at 3–5 mice per cage and maintained under standard husbandry procedures in a vivarium at the University of Virginia.
Cell lines and Primary Cultures
Differentiated GBM cell lines, U87MG stably expressing EGFRvIII (referred to as U87MG herein, source: Dr. Frank Furnari; male), U118MG (source: ATCC catalog# HTB-15; male), U251 (source: Dr. Gary Kuo; male), and T98G (source: ATCC catalog# CRL-1690; male), in addition to amphotropic Phoenix cells (source: Dr. Gary Nolan; female) and 293FT (source: ATCC catalog# PTA-5077; female) used for virus production, were maintained as adherent cultures in DMEM supplemented with 10% fetal bovine serum (VWR), 1 mM L-Glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (GIBCO). Glioma stem cells G88 (Mineo et al., 2016), G2 (Mineo et al., 2016), and T3691 (Mathew et al., 2014) were maintained as suspension cultures in Neurobasal Medium with B27 and N-2 supplements (GIBCO) with 50 ng/mL human recombinant EGF (Peprotech) and human recombinant basic FGF (Peprotech). U87MG, U118MG, and U251 parental or transduced cell lines were tested for mycoplasma using the MycoAlert system (Cambrex) and used within a maximum of 15 passages from testing. G2 and G88 cells were authenticated by short tandem repeat profiling within six months of their use in live culture. All cells were kept in a humidified 5% CO2 incubator at 37°C.
METHOD DETAILS
shRNA and ectopic expression vectors
Oligonucleotides for SPRY2-targeting short hairpin RNA (shRNA) and corresponding non-targeting control shRNA were previously cloned into the lentiviral pSicoR-puro plasmid (Tyler Jacks, MIT) and used to generate U87MG and U118MG cells stably expressing shRNA (Walsh et al., 2015). Oligonucleotides for FGFR3 shRNA and corresponding non-targeting control shRNA were cloned into the lentiviral pLKO.1-puro plasmid (Broad Institute, The RNAi Consortium). shRNA sequences are listed in Table S1. pBABE-puro-FGFR3c-WT (Liao et al., 2013) was a gift from Dr. Matthew Meyerson (Dana Farber Cancer Institute). Versions of the pBABE-SPRY2-hygro vector, either with or without an N-terminal hemagglutinin (HA) tag, were subcloned from pHM6-HA-SPRY2 kindly provided by Dr. Tarun Patel (Yigzaw et al., 2001).
FIRE and CB-FIRE reporters cloning
pMSCV-NLS-mVenus-FIRE-puro and pMSCV-NLS-mCerulean-d2-puro (Albeck et al., 2013) were generously provided by Dr. John Albeck (University of California Davis). The plasmid encoding Cerulean was modified by replacing the puromycin resistance gene with a hygromycin B resistance gene, to allow for selection of cells expressing both vectors. These vectors were further modified for bioluminescence applications. Specifically, the gene encoding click beetle green (CBGreen) luciferase was amplified using PCR primers compatible for insertion into pMSCV-NLS-mVenus-FIRE-puro. The gene encoding for click beetle red (CBRed) luciferase was amplified using PCR primers compatible for insertion into pMSCV-NLS-mCerulean-d2-hygro. After PCR amplification, both the PCR amplification products and the fluorescent protein containing vectors were digested, purified via agarose gel electrophoresis, and ligated prior to transformation into competent E. coli. Restriction enzymes, T4 DNA ligase, and Phusion® polymerase were all purchased from New England Biolabs. Subcloning Efficiency DH5α competent cells were purchased from Invitrogen. All plasmids were submitted for Sanger sequencing prior to retrovirus generation, to confirm successful recombination of DNA sequences. Plasmids pNB777 and pNB778 containing the genes for CBGreen and CBRed (Mazo-Vargas et al., 2014) were provided by Nicolas Buchler (Duke University).
Viral production and cell line engineering
Lentivirus was produced by calcium phosphate-mediated co-transfection of 293FT cells with lentiviral plasmids (pSicoR-puro or pLKO.1-puro) and viral packing vectors pCMV-VSVg, pMDL-gp-RRE, and pRSV-Rev. Virus was harvested 48 h post-transfection and used to infect target cells. Retrovirus was similarly produced by calcium phosphate-mediated transfection of Amphotropic Phoenix cells (Gary Nolan, Stanford University) with retroviral plasmids (pBABE or pMSCV). Virus was harvested 48 h post-transfection and used to infect target cells. U87MG, U118MG, T98G, and U251 cells transduced with virus were selected in 2 μg/mL puromycin or 150 μg/mL hygromycin B (Gemini Biosciences). G88 cells expressing retroviral luminescent reporter constructs were generated similarly, but in the absence of FBS, and selected with 50 μg/mL puromycin or 300 μg/mL hygromycin B.
siRNA transfection
Commercially available siRNA for knockdown of FGFR1, FGFR3, c-Jun, or SPRY2, in addition to control siRNA, was transfected using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer-recommended protocol. In brief, siRNA was added to cells at a final concentration of 10 nM for 1–3 days prior to treatment with inhibitors or lysis. Target sequences, vendors, and catalog numbers for siRNAs can be found in Table S2.
Phospho-RTK antibody array
PathScan® RTK Signaling Antibody Array Kits were purchased from Cell Signaling Technology and used according to manufacturer’s recommendation. In brief, arrays were incubated with lysates overnight at 4°C, washed, and incubated with a biotinylated detection-antibody cocktail at room temperature. Infrared dye-conjugated streptavidin was added, and slides were scanned using a LI-COR Odyssey CLx system.
Inhibitors and growth factors
Geftinib (LC Laboratories), CI-1040 (LC Laboratories), PHA665752 (Santa Cruz Biotechnology), PD173074 (ApexBio), JNJ-38877605 (Selleck Chemicals), IKK-16 (Selleck Chemicals), crizotinib (Cell Signaling Technoloy), trametinib (ApexBio), AZD4547 (ApexBio), and BGJ398 (ApexBio) were reconstituted in DMSO for use in vitro. Recombinant human FGF1 and FGF2 (PeproTech) were used at 50 ng/mL. FGF incubation time was 5 min unless otherwise noted.
Western blotting
Whole-cell lysates were prepared in a standard cell extraction buffer (Life Technologies) supplemented with protease and phosphatase inhibitors (Sigma). Lysates were cleared by centrifugation at 20,000 × g for 10 min at 4°C, and total protein concentrations were determined by micro-bicinchoninic assay (Thermo Fisher Scientific). Approximately 20 μg of total protein was loaded per lane on 4%–12% gradient polyacrylamide gels (Thermo Fisher Scientific) under denaturing and reducing conditions and transferred to 0.2 μm nitrocellulose membranes (Bio-Rad). After probing with antibodies, membranes were imaged on a LI-COR Odyssey CLx system. Densitometry measurements were calculated using Image Studio software version 5.2.5 (LI-COR). Antibodies used for western blotting included pEGFR (Tyr 1068, #3777), EGFR (#2232), pMET (Tyr 1234/35, #3126), MET (#8198), pERK (Thr 202/Tyr 204, #4377), ERK (#4695), SPRY2 (#14954), HA (#2367), pFRS2α (Tyr 436, #3861), FGFR1 (#9740), FGFR3 (#4574), pNF-κB (Ser 536, #3033), NF-κB (#8242), pFRA1 (Ser 265, #5841), pAKT (Ser 473, #4060), AKT (#9272), and c-Jun (#9165) from Cell Signaling Technology in addition to GAPDH (sc-32233) from Santa Cruz Biotechnology. Infrared dye-conjugated secondary antibodies for western blotting were purchased from Rockland Immunochemicals.
Quantitative reverse transcription PCR (qRT-PCR)
RNA was extracted from cells using the RNeasy kit (QIAGEN) with on-column DNase I digestion. Equal amounts of RNA were reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) on an Applied Biosystems 7300 or StepOnePlus Real-Time PCR System. Relative amounts of mRNA were determined using the comparative CT method using GAPDH as an endogenous reference. qRT-PCR primer sequences are provided in Table S3.
Immunofluorescence microscopy and image analysis
Cells were plated on glass coverslips and maintained in 6-well dishes during inhibitor treatments. After treatment, cells were fixed in 4% paraformaldehyde in PBS for 20 min, followed by permeabilization with 0.25% Triton X-100 in PBS for 5 min, and stained overnight at 4°C with an anti-NF-κB primary antibody (CST#8242) at the manufacturer’s recommended dilution. The next day, samples were washed and stained with an anti-rabbit IgG conjugated to Alexa Fluor 594 (Thermo Fisher Scientific) and the DNA dye Hoechst (Thermo Fisher Scientific). Samples were imaged at 20 × magnification using a Zeiss AxioObserver Z1 widefield microscope and Axiocam 506 digital camera. To quantify NF-κB subcellular localization, nuclei were selected using the Hoechst signal as a mask in the ImageJ software. The integrated density of the NF-κB signal within this region was calculated and reported on a per cell basis. The mean signal was reported for n > 100 cells, across three biological replicates, for each condition.
NF-κB Chromatin Immunoprecipitation
Crosslinked chromatin was extracted from cells using the SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology) according to manufacturer recommendations. In short, cells treated with inhibitors were fixed with 1% formalin for 10 min and lysed. Resulting crosslinked chromatin was fragmented by partial digestion with Micrococcal Nuclease to a size of 1 to 5 nucleosomes. 8 μg of fragmented, crosslinked chromatin was incubated overnight at 4°C with 1 μg of antibody against NF-κB (CST#8242) or IgG control (CST#2729). Antibody-chromatin complexes were harvested by addition of ChIP-Grade Protein G magnetic beads. Resulting immunoprecipitated chromatin was analyzed via qPCR using primers designed to amplify regions proximal to NF-κB binding sites in the FGF1 promoter. Percent input values were calculated using CT values for immunoprecipitated chromatin reactions and matched 2% input samples using the Equation . Primer binding regions and sequences are listed in Table S4.
Flow cytometry
For cell death and reporter assays, cells were plated in 6-well dishes and treated with inhibitors. At the end of an experiment, floating and adherent cells were collected, resuspended in PBS containing TO-PRO-3 (Thermo Fisher Scientific), and analyzed within 1 h of collection on a BD Biosciences FACSCalibur or LSR II cytometer. Data analysis was performed using FlowJo, FCS Express, or Flowing Software. An example gating strategy for experiments combining live/dead staining with a fluorescent reporter is provided in Figure S7A. In brief, cellular debris was excluded from analysis by gating on the forward and side scatter dot plot. Within the whole cell population, live and dead cells were gated based on TO-PRO-3 permeability and analyzed separately for mVenus and Cerulean fluorescence. Untreated cells expressing only mVenus or Cerulean were used as software compensation controls to correct for spectral overlap.
FGF1 ELISA
Conditioned medium collected from cells was centrifuged at 500 × g to remove cellular debris and then used with the Human FGF-acidic Quantikine ELISA kit (R&D Systems) according to manufacturer recommendations. FGF1 concentrations were calculated using a standard curve of known FGF1 concentrations. Untreated (t = 0 h) conditioned medium samples were collected from cells grown in the absence of inhibitor for the maximum treatment time to account for possible differences in cell number and secreted factor accumulation over time.
Generation of resistant cells and MTS assay
To generate U87MG cells resistant to gefitinib and PHA665752, cells were grown in medium containing 10 μM gefitinib + 3 μM PHA665752 for > 2 months, with inhibitor replenishment every 3–5 days. Naive cells and cells resistant to gefitinib and PHA665752 were then seeded in 96-well plates. After 24 h, medium from each well was replaced with medium including the MTS reagent (Promega) at the manufacturer-recommended concentration. Conversion of the MTS reagent into a soluble formazan product was allowed to proceed for 1–4 h, after which the absorbance at 490 nm was measured. To ensure signal linearity, the absorbance was measured at three different times. Values from the median time point were reported.
Single-molecule RNA fluorescence in situ hybridization (FISH)
Single-molecule RNA FISH probes (Stellaris) against FGF2 and SPRY2 were conjugated to Atto647N and Alexa Fluor 594, respectively, and purified via HPLC. Cells were plated on #1 LabTek chambered coverglass, and, after various treatments, were fixed in 4% paraformaldehyde for 10 min, washed with 1X PBS, and permeabilized in 70% ethanol. RNA FISH probes were hybridized and added to samples as described previously (Shaffer et al., 2017). Samples were imaged at 63 × magnification using a Zeiss AxioObserver Z1 widefield microscope and Axiocam 506 camera. Multiple z-stack images were acquired across three biological replicates per treatment condition to ensure single, representative z-slice images were consistent with the entire cell. RNA FISH spots were counted using StarSearch (https://rajlab.seas.upenn.edu/StarSearch/launch.html), and mVenus/Cerulean intensity was quantified using ImageJ. Individual nucleotide sequences for single-molecule RNA FISH probe sets are listed in Table S5.
IVIS Spectrum imaging and analysis
For in vitro bioluminescence assays, cells were plated in 96-well plates and treated with inhibitors as indicated. After treatment, cells were incubated with 150 μg/mL d-luciferin (Gold Biotechnology) for 10–15 min prior to imaging. An IVIS Spectrum imager (Perkin Elmer) was used to acquire images at six wavelengths (540, 560, 580, 620, 640, and 660 nm), selected based on software-specified settings for CBRed and CBGreen luciferases. Samples with expression of only one luciferase were used as controls for the spectral unmixing performed by the Living Image software version 4.5.5 (Perkin Elmer). For in vivo assessment of tumor luminescence, mice were injected with d-luciferin at 150 mg/kg, anesthetized, and imaged 10–15 min post injection. To confirm the maximum luciferase emission plateau was reached, successive images were acquired from 5–20 min after luciferin injection.
For in vivo experiments, one mouse with two independent tumors containing cells expressing only one luciferase each was used as a control for the spectral unmixing. Since the detected signal at each wavelength is a combination of the signals produced by the two different types of luciferase, the imaging control mouse is necessary to determine the emission spectra of CBGreen and CBRed independently. The images in Figure S7B of an imaging control mouse demonstrate how most of the detected luminescence shifted from the left to right tumor as wavelength increased, consistent with the expected emission wavelengths of CBGreen and CBRed. This information was used to generate a normalized emission spectrum, which was then used to unmix signals from tumors that lacked spatially resolved green and red signal. Representative unmixed images of the imaging control mouse, and another mouse with dual-luciferase expressing tumors, are shown in Figure S7C and demonstrate the ability of the software to distinguish the green and red luciferase signals from one another. While the magnitude of photon flux increased as the tumors grew (demonstrated by the differences in y axis scaling in the spectra in Figure S7C), the normalized spectra generated from images unmixed on separate days were practically identical. Therefore, the software-based spectral unmixing algorithm reliably differentiated CBRed from CBGreen luciferase throughout the study. For the data presented in Figure 6D, tumors with a CBGreen intensity value lower than the negative control tumor, consisting entirely of cells expressing only CBRed, were excluded from the analysis (excluded tumors per group: vehicle, n = 0/8; G+P, n = 1/10; BGJ, n = 0/10; G+P+BGJ, n = 3/10).
Subcutaneous mouse tumor xenografts
1.0 × 106 engineered U87MG cells were injected subcutaneously into each flank of 4–8-week-old, female, Nu/Nu mice (Charles River). When tumors reached an average volume of 50–100 mm3, animals were randomly assigned to treatment groups of gefitinib + PHA665752, BGJ398, gefitinib + PHA665752 + BGJ398, or vehicle. Gefitinib and BGJ398 were resuspended in corn oil for oral administration at a concentration of 100 mg/kg and 20 mg/kg, respectively. PHA665752 was resuspended in an aqueous solution containing 1% dimethyl acetamide, 10% propylene glycol, and 1.05 moles L-lactic acid per mole of PHA665752 and was delivered at 30 mg/kg by intraperitoneal injection. Animals were treated three days per week. Tumors were measured with calipers before and during treatment, and tumor volumes were calculated as π/6 × A × B2, where A and B are the larger and smaller tumor diameters, respectively. In addition, bioluminescence measurements using an IVIS Spectrum imager were performed one or two times per week to assess changes in luciferase expression in response to the different drug treatments. Excised tumors were homogenized in lysis buffer before western blotting. All experiments were approved by the University of Virginia Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines.
Intracranial mouse brain tumor xenografts
1,000 G88 cells were stereotactically injected into the right striatum of 4–8-week-old, female, BALB/c scid mice (The Jackson Laboratory). After recovery from the surgical procedure, animals were randomly assigned to treatment groups of gefitinib + PHA665752, gefitinib + PHA665752 + BGJ398, or vehicle control. Drugs were prepared as listed above for subcutaneous experiments, with the exception of the concentration of BGJ398 (30 mg/kg). Animals were treated daily for five days, followed by two days of rest, and an additional four days of treatment prior to euthanasia. After the first five days of treatment, animals were imaged with an IVIS imager as described above for subcutaneous experiments. For the data presented in Figure 6L, one vehicle-treated animal was excluded from the analysis due to noted leakage of cells during injection surgery (excluded tumors per group: vehicle, n = 1/4; G+P, n = 0/3; G+P+BGJ, n = 0/4). Harvested mouse brains were dissected into right (tumor-containing) and left (normal tissue) hemispheres and homogenized separately in lysis buffer before western blotting. All experiments were approved by the University of Virginia Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with a two-tailed Student’s t test calculated in Microsoft Excel where significance was determined by a p-value < 0.05. The exact details of these statistical comparisons, including values for n, are found in the figure legends. Unless stated otherwise in the figure legend, all data are representative of results obtained from 3 independent replicates. The figure legends also list definitions for dispersion and precision measurements, stating error bar definitions throughout. Rationale for instances of excluding individual tumors from statistical comparisons for in vivo mouse data can be found within the Method Details section above.
DATA AND CODE AVAILABILITY
This study did not generate or analyze datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-EGF Receptor (Tyr1068) (D7A5) XP Rabbit mAb | Cell Signaling Technology | RRID:AB_2096270 |
| EGF Receptor | Cell Signaling Technology | RRID:AB_331707 |
| Phospho-Met (Tyr1234/1235) | Cell Signaling Technology | RRID:AB_331713 |
| Met (D1C2) XP Rabbit mAb | Cell Signaling Technology | RRID:AB_10858224 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (197G2) Rabbit mAb | Cell Signaling Technology | RRID:AB_331775 |
| p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb | Cell Signaling Technology | RRID:AB_390779 |
| Spry2 (D3G1A) Rabbit mAb | Cell Signaling Technology | RRID:AB_2798658 |
| Phospho-FRS2-alpha (Tyr436) | Cell Signaling Technology | RRID:AB_2231950 |
| FGF Receptor 1 (D8E4) XP Rabbit mAb | Cell Signaling Technology | RRID:AB_11178519 |
| FGF Receptor 3 (C51F2) | Cell Signaling Technology | RRID:AB_2246903 |
| Phospho-NF-KappaB p65 (Ser536) (93H1) Rabbit mAb | Cell Signaling Technology | RRID:AB_331284 |
| NF-kappaB p65 (D14E12) XP Rabbit mAb | Cell Signaling Technology | RRID:AB_10859369 |
| Phospho-FRA1 (Ser265) (D22B1) Rabbit mAb | Cell Signaling Technology | RRID:AB_10835210 |
| Phospho-Akt (Ser473) | Cell Signaling Technology | RRID:AB_2315049 |
| Akt antibody | Cell Signaling Technology | RRID:AB_329827 |
| c-Jun (60A8) Rabbit mAb | Cell Signaling Technology | RRID:AB_2130165 |
| HA-Tag (6E2) Mouse mAb | Cell Signaling Technology | RRID:AB_10691311 |
| GAPDH (6C5) antibody | Santa Cruz Biotechnology | RRID:AB_627679 |
| Goat Anti-Mouse IgG Antibody, IRDYE700DX Conjugated | Rockland Immunochemicals | RRID:AB_220121 |
| Goat Anti-RABBIT IgG Antibody DyLight 800 Conjugated | Rockland Immunochemicals | RRID:AB_1660964 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | RRID:AB_2534079 |
| Normal Rabbit IgG antibody | Cell Signaling Technology | RRID:AB_1031062 |
| Bacterial and Virus Strains | ||
| Subcloning Efficiency DH5α | Thermo Fisher Scientific | Cat # 18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gefitinib | LC Laboratories | Cat # G-4408 |
| PHA665752 | Santa Cruz Biotechnology | Cat # sc-203186 |
| CI-1040 | LC Laboratories | Cat # P-8499 |
| PD173074 | ApexBio | Cat # A8253 |
| JNJ-38877605 | Selleck Chemicals | Cat # S1114 |
| IKK-16 | Selleck Chemicals | Cat # S2882 |
| crizotinib | Cell Signaling Technology | Cat # 4401 |
| trametinib | ApexBio | Cat # A3018 |
| AZD4547 | ApexBio | Cat # A8350 |
| BGJ398 | ApexBio | Cat # A3014 |
| Recombinant human FGF1 | Peprotech | Cat # 100–17A |
| Recombinant human FGF2 | Peprotech | Cat # AF-100–18B |
| Recombinant human EGF | Peprotech | Cat # AF 100–15 |
| D-luciferin | Gold Biotechnology | Cat # LUCNA-1G |
| TO-PRO-3 | Thermo Fisher Scientific | Cat # T3605 |
| Lipofectamine™ RNAiMAX | Thermo Fisher Scientific | Cat # 13778500 |
| Puromycin | Gemini Biosciences | Cat # 400–128P |
| Hygromycin B | Gemini Biosciences | Cat #400–123 |
| Critical Commercial Assays | ||
| SimpleChIP® Enzymatic Chromatin IP Kit | Cell Signaling Technology | Cat # 9003 |
| PathScan RTK Signaling Antibody Array Kit | Cell Signaling Technology | Cat # 7949 |
| Human FGF-acidic Quantikine ELISA kit | R&D Systems | Cat # DFA00B |
| CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) | Promega | Cat # G5421 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat # 23225 |
| Experimental Models: Cell Lines | ||
| U87MG stably expressing EGFRvIII (referred to as U87MG herein) | Dr. Frank Furnari | N/A |
| U251 | Dr. Gary Kuo | N/A |
| U118MG | ATCC | RRID:CVCL_0633 |
| T98G | ATCC | RRID:CVCL_0556 |
| G88 | (Mineo et al., 2016) | N/A |
| G2 | (Mineo et al., 2016) | N/A |
| T3691 | (Mathew et al., 2014) | N/A |
| Amphotropic Phoenix cells | Dr. Gary Nolan | N/A |
| 293FT | ATCC | RRID:CVCL_6911 |
| Experimental Models: Organisms/Strains | ||
| NU/NU Nude mouse | Charles River | Strain code: 088 |
| BALB/c scid mouse | The Jackson Laboratory | Stock No: 001803 |
| Oligonucleotides | ||
| pLKO.1-control shRNA; targeting sequence: ATCACAGAATCGTCGTATGCA | This paper; See Table S1 | N/A |
| pLKO.1-FGFR3 shRNA-A; targeting sequence: TGCGTCGTGGAGAACAAGTTT | This paper; See Table S1 | N/A |
| pLKO.1-FGFR3 shRNA-B; targeting sequence: GACAAGGAGCTAGAGGTTCTC | This paper; See Table S1 | N/A |
| pSicoR-control shRNA; targeting sequence: GTCATATAGACCATAGTTA | (Walsh et al., 2015); See Table S1 | N/A |
| pSicoR-SPRY2 shRNA; targeting sequence: GATGCATATGTCCAATATA | (Walsh et al., 2015); See Table S1 | N/A |
| siRNA targeting FGFR1 | Thermo Fisher Scientific; See Table S2 | Cat # AM51331 |
| siRNA targeting FGFR3, c-Jun, NF-κB | Santa Cruz Biotechnology; See Table S2 | Cat # sc-29314, sc-29223, sc-29410 |
| siRNA targeting SPRY2 | Dharmacon (GE); See Table S2 | Cat # M-005206–01 |
| qRT-PCR primers | This paper; See Table S3 | N/A |
| NF-kB ChIP primers for FGF1 promoter | This paper; See Table S4 | N/A |
| Single-molecule RNA FISH probes for SPRY2 and FGF2 | Dr. Arjun Raj; See Table S5 for complete sequences | Stellaris |
| Recombinant DNA | ||
| pSicoR-puro | Dr. Tyler Jacks | N/A |
| pLKO.1-puro | Broad Institute, The RNAi Consortium | N/A |
| pMSCV-NLS-mVenus-FIRE-puro | (Albeck et al., 2013) | N/A |
| pMSCV-NLS-mCerulean-d2-hygro | This paper | N/A |
| pMSCV-NLS-CBGreen-FIRE-puro | This paper | N/A |
| pMSCV-NLS-CBRed-d2-hygro | This paper | N/A |
| pBABE-puro-FGFR3c-WT | (Liao et al., 2013) | Addgene #45711 |
| pBABE-HA-SPRY2-hygro | This paper | N/A |
| pBABE-SPRY2-hygro | This paper | N/A |
| pHM6-HA-SPRY2 | (Yigzaw et al., 2001) | N/A |
| pCMV-VSVg | Dr. Mehul Shah | N/A |
| pMDL-gp-RRE | Dr. Mehul Shah | N/A |
| pRSV-Rev | Dr. Mehul Shah | N/A |
| pNB777 | (Mazo-Vargas et al., 2014) | Addgene #60153 |
| pNB778 | (Mazo-Vargas et al., 2014) | Addgene #60154 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Living Image version 4.4.5 | Perkin Elmer | RRID:SCR_014247 |
| StarSearch RNA FISH software | Dr. Arjun Raj | https://rajlab.seas.upenn.edu/StarSearch/launch.html |
| Image Studio software version 5.2.5 | LI-COR | RRID:SCR_015795 |
Highlights.
Glioblastoma cells evade EGFR and MET inhibition via FGFR-SPRY2 bypass signaling
Autocrine FGFR signaling and SPRY2 synthesis explains heterogeneous drug response
FGFR blockade augments glioblastoma response to EGFR and MET inhibition
A bioluminescent reporter tracks longitudinal ERK response to therapy in vivo
ACKNOWLEDGMENTS
Single-molecule RNA FISH probes were a generous gift from Dr. Arjun Raj (University of Pennsylvania). This work used an IVIS bioluminescence/fluorescence scanner in the University of Virginia (UVA) Molecular Imaging Core, which was purchased with support from NIH grant 1S10RR025694-01 and is supported by the UVA School of Medicine. The Flow Cytometry Facility is supported through the UVA Cancer Center National Cancer Institute grant no. P30-CA044579-23. This work was supported by American Cancer Society Research Scholar grant no. RSG-15-010-01-CDD (to M.J.L.), National Science Foundation Chemical, Bioengineering, Environmental, and Transport Systems (NSF CBET) grant no. 1511853 (to M.J.L.), NSF Graduate Research Fellowship Program (GRFP) grant no. DGE-1321851 (to E.K.D.), and the UVA Cancer Center support grant from the National Cancer Institute (P30CA044579).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.014.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albeck JG, Mills GB, and Brugge JS (2013). Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 49, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, Hrustanovic G, Chan E, Lin L, Neel DS, et al. (2015). NF-κ-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 11, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. ; TCGA Research Network (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PA, Iida M, Treisman DM, Kalluri H, Ezhilan S, Zorniak M, Wheeler DL, and Kuo JS (2012). Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia 14, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witt Hamer PC (2010). Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol. 12, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di K, Lloyd GK, Abraham V, MacLaren A, Burrows FJ, Desjardins A, Trikha M, and Bota DA (2016). Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro Oncol. 18, 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdal KB, Dittmann A, Reddy RJ, Lescarbeau RS, Moores SL, Laquerre S, and White FM (2017). Characterization of In Vivo Resistance to Osimertinib and JNJ-61186372, an EGFR/Met Bispecific Antibody, Reveals Unique and Consensus Mechanisms of Resistance. Mol. Cancer Ther 16, 2572–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, et al. (2012). Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CW, Chua MS, McKie AB, Ling SH, Mason V, Li R, Yusoff P, Lo TL, Leung HY, So SK, and Guy GR (2006). Sprouty 2, an inhibitor of mitogen-activated protein kinase signaling, is down-regulated in hepatocellular carcinoma. Cancer Res. 66, 2048–2058. [DOI] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, and Mischel PS (2015). Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 15, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, and Bryant SH (2010). The NCBI BioSystems database. Nucleic Acids Res. 38, D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ, Hale M, Boucher DM, Sorrell DA, Martinez-Botella G, et al. (2017). Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther 16, 2351–2363. [DOI] [PubMed] [Google Scholar]
- Godwin P, Baird AM, Heavey S, Barr MP, O’Byrne KJ, and Gately K (2013). Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol 3, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron TP (2016). Small Molecule Kinase Inhibitors for the Treatment of Brain Cancer. J. Med. Chem 59, 10030–10066. [DOI] [PubMed] [Google Scholar]
- Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, Furnari FB, and White FM (2007). Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc. Natl. Acad. Sci. USA 104, 12867–12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HJ, Acquaviva J, Chi D, Lessard J, Zhu H, Woolfenden S, Bronson RT, Pfannl R, White F, Housman DE, et al. (2012). Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 31, 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, and Rao JS (2012). Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 3, e445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, Soo RA, Christensen JG, Lee JH, and Cho BC (2012). Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther 11, 2254–2264. [DOI] [PubMed] [Google Scholar]
- Kocher B, and Piwnica-Worms D (2013). Illuminating cancer systems with genetically engineered mouse models and coupled luciferase reporters in vivo. Cancer Discov. 3, 616–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, and Schlessinger J (1997). A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89, 693–702. [DOI] [PubMed] [Google Scholar]
- Lee JG, and Kay EP (2012). NF-κB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest. Ophthal-mol. Vis. Sci 53, 1530–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Huynh H, Li X, Ruddy DA, Wang Y, Ong R, Chow P, Qiu S, Tam A, Rakiec DP, et al. (2015). FGFR-Mediated Reactivation of MAPK Signaling Attenuates Antitumor Effects of Imatinib in Gastrointestinal Stromal Tumors. Cancer Discov. 5, 438–451. [DOI] [PubMed] [Google Scholar]
- Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, Liu Q, Pugh TJ, Pedamallu CS, Hayes DN, et al. (2013). Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 73, 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Tang N, Thompson RC, Mobley BC, Clark SW, Sarkaria JN, and Wang J (2016). InsR/IGF1R Pathway Mediates Resistance to EGFR Inhibitors in Glioblastoma. Clin. Cancer Res 22, 1767–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macià A, Vaquero M, Gou-Fàbregas M, Castelblanco E, Valdivielso JM, Anerillas C, Mauricio D, Matias-Guiu X, Ribera J, and Encinas M (2014). Sprouty1 induces a senescence-associated secretory phenotype by regulating NFκB activity: implications for tumorigenesis. Cell Death Differ. 21, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoumi-Moghaddam S, Amini A, and Morris DL (2014). The developing story of Sprouty and cancer. Cancer Metastasis Rev. 33, 695–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew LK, Skuli N, Mucaj V, Lee SS, Zinn PO, Sathyan P, Imtiyaz HZ, Zhang Z, Davuluri RV, Rao S, et al. (2014). miR-218 opposes a critical RTK-HIF pathway in mesenchymal glioblastoma. Proc. Natl. Acad. Sci. USA 111, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazo-Vargas A, Park H, Aydin M, and Buchler NE (2014). Measuring fast gene dynamics in single cells with time-lapse luminescence microscopy. Mol. Biol. Cell 25, 3699–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F, and Manning AM (1999). NF-kappaB as a primary regulator of the stress response. Oncogene 18, 6163–6171. [DOI] [PubMed] [Google Scholar]
- Mineo M, Ricklefs F, Rooj AK, Lyons SM, Ivanov P, Ansari KI, Nakano I, Chiocca EA, Godlewski J, and Bronisz A (2016). The Long Noncoding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep. 15, 2500–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki K, Kadomoto R, Asato K, Tanimura S, Itoh N, and Kohno M (2001). ERK pathway positively regulates the expression of Sprouty genes. Biochem. Biophys. Res. Commun 285, 1084–1088. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, Nabors LB, Rosenfeld MR, Mikkelsen T, and Grossman SA; New Approaches to Brain Tumor Therapy Consortium (2013). NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 15, 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, Bukczynska PE, Young RJ, Boyle SE, Shackleton M, et al. (2015). The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signal 8, ra82. [DOI] [PubMed] [Google Scholar]
- Rodon J, Postel-Vinay S, Hollebecque A, Nuciforo P, Azaro A, Cattan V, Marfai L, Sudey I, Brendel K, Delmas A, et al. (2017). First-in-human phase I study of oral S49076, a unique MET/AXL/FGFR inhibitor, in advanced solid tumours. Eur. J. Cancer 81, 142–150. [DOI] [PubMed] [Google Scholar]
- Saito R, Miki Y, Ishida N, Inoue C, Kobayashi M, Hata S, Yamada Okabe H, Okada Y, and Sasano H (2018). The Significance of MMP-1 in EGFR-TKI-Resistant Lung Adenocarcinoma: Potential for Therapeutic Targeting. Int. J. Mol. Sci 19, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, and Rich JN (2007). Molecularly targeted therapy for malignant glioma. Cancer 110, 13–24. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, et al. (2012). Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. (2007). Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318, 287–290. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Sutterlüty H, Mayer CE, Setinek U, Attems J, Ovtcharov S, Mikula M, Mikulits W, Micksche M, and Berger W (2007). Down-regulation of Sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol. Cancer Res 5, 509–520. [DOI] [PubMed] [Google Scholar]
- Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, Calvo E, Moreno V, Adamo B, Gazzah A, et al. (2015). Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol 33, 3401–3408. [DOI] [PubMed] [Google Scholar]
- Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, Pratilas CA, Rosen N, Gray NS, Wong KK, and Jänne PA (2015). Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR-Mutant Lung Cancer. Cancer Discov. 5, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh AM, Kapoor GS, Buonato JM, Mathew LK, Bi Y, Davuluri RV, Martinez-Lage M, Simon MC, O’Rourke DM, and Lazzara MJ (2015). Sprouty2 Drives Drug Resistance and Proliferation in Glioblastoma. Mol. Cancer Res 13, 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, Helfrich BA, Doebele RC, and Heasley LE (2010). Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through derepression of FGFR2 and FGFR3 expression. PLoS One 5, e14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortzel I, and Seger R (2011). The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J, Hu J, Gomez GG, Taylor T, Villa GR, Pizzo D, Vanden-Berg SR, Thorne AH, Chen CC, Mischel PS, et al. (2015). A urokinase receptor-Bim signaling axis emerges during EGFR inhibitor resistance in mutant EGFR glioblastoma. Cancer Res. 75, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, Buchanan S, Henry JR, Starling JJ, and Peng SB (2012). Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J. Biol. Chem 287, 28087–28098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigzaw Y, Cartin L, Pierre S, Scholich K, and Patel TB (2001). The C terminus of sprouty is important for modulation of cellular migration and proliferation. J. Biol. Chem 276, 22742–22747. [DOI] [PubMed] [Google Scholar]
- Yusoff P, Lao DH, Ong SH, Wong ES, Lim J, Lo TL, Leong HF, Fong CW, and Guy GR (2002). Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J. Biol. Chem 277, 3195–3201. [DOI] [PubMed] [Google Scholar]
- Zanca C, Villa GR, Benitez JA, Thorne AH, Koga T, D’Antonio M, Ikegami S, Ma J, Boyer AD, Banisadr A, et al. (2017). Glioblastoma cellular cross-talk converges on NF-κB to attenuate EGFR inhibitor sensitivity. Genes Dev. 31, 1212–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, and Ornitz DM (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem 281, 15694–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or code.