

Abstract
Here we reported a new strategy to construct synthetic metabolons using dCas9-guided assembly. Three orthogonal dCas9 proteins were exploited to guide the independent and site-specific assembly of their fusion partners onto a single DNA scaffold. This new platform was applied towards the construction of a two-component cellulosome. Because of the superior binding affinity, the resulting structures exhibited both improved assembly and reducing sugar production. Conditional enzyme assembly was made possible by utilizing toehold-gated sgRNA (thgRNA), which blocks cellulosome formation until the spacer region is unblocked by a RNA trigger. This platform is highly modular owing to the ease of target synthesis, combinations of possible Cas9-fusion arrangements, and expansion to other metabolic pathways.
For the past few decades, enzymes have been exploited for the production of a variety of products, ranging from both bulk and fine chemicals to active pharmaceutical agents and next generation biofuels1,2 In nature, enzymes are frequently clustered together to coordinate and optimize their collective action via a process called substrate channeling3,4 Rather than random clustering, a protein scaffold is typically used to organize multiple enzyme partners in close proximity to facilitate their concerted actions. 5
One way to mimic this native enzyme assembly is through the design of synthetic metabolons,6which are artificial multi-enzyme complexes that spatially organize enzymatic pathways in close proximity 7-9 These synthetic metabolons offer many advantages such as limiting the diffusion of transient intermediates, facilitating the fast turnover of toxic intermediates, preventing crosstalk between competitive metabolic pathways, as well as having the ability to concentrate reactants which can drive unfavorable reactions.10-13
Most popular synthetic scaffolds for enhanced pathway efficiency are based on the modular metazoan signalling proteins, SH3, PDZ, and GBD, which specifically recruit pathway enzymes tagged with their cognate interaction peptide ligands14. However, one major drawback is the relative low binding affinity (Kd in the range of 0.1 to 8 μM). As a result, a very large protein scaffold containing 1 GBD, 6 SH3, and 2 PDZ domains was required to provide the maximum product titer for glucaric acid synthesis15. However, it is often difficult to vary the required enzyme density since a new protein scaffold must be redesigned.14,15 Moreover, more complex proteins scaffolds tend to be either truncated or mis-folded.10
Nucleic acids represent another attractive biological scaffold for enzyme assembly because of our ability to manipulate the base-pairing property to create defined Structures16 More importantly, DNA templates can be easily modulated for simple and quick optimization of enzyme stoichiometry, order, and spacing. Although enzymes can be chemically conjugated with DNA oligos for site-specific hybridization,12 this often leads to activity loss and limits the strategy to in vitro applications.17 One alternative is based on enzyme fusions with zinc finger proteins (ZFPs) for site-specific binding onto a double-stranded DNA template.9,13 Artificial cellulosomes based on ZFPs have been reported with levels of enhancement in cellulose hydrolysis similar to those achieved using protein scaffolds.13 While ZFP fusions offer the ease of assembly, the relatively modest binding affinity (Kd ~ 100 nM) makes it difficult to control the actual enzyme binding ratio onto each individual scaffold.
Inspired by the ease of ZFPs to guide the formation of enzyme cascades, we present here a new and potent approach that enables specific and high-affinity binding to DNA scaffolds using the CRISPR/Cas9 system. The Cas9 protein is ideally suited for modular enzyme assembly as it naturally offers high-affinity, sequence-specific DNA binding (Kd values ~ 0.05 to 0.5 nM).18-20 While the native function of Cas9 is for RNA-guided DNA cleavage, nuclease-null Cas9 (dCas9) proteins have been generated while preserving the same high-affinity binding capability.21 Three orthogonal dCas9 protein pairs, each capable of using its own unique sgRNA for independent and specific targeting, were exploited as a new way to create synthetic enzyme cascades. Because of our past success on using DNA scaffolds to create artificial cellulosome structures.12 we used this platform as an example to demonstrate the effectiveness of our new dCas9-guided assembly strategy (Fig. 1).
Figure 1.
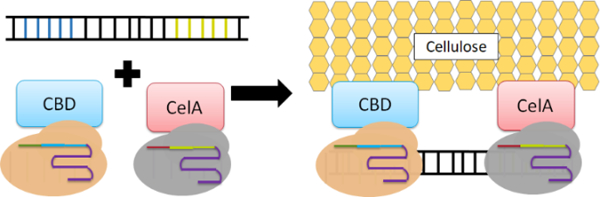
Formation of artificial cellulosome structures by dCas9-mediated assembly onto a double-stranded DNA template. The cellulose-binding domain (CBD) is used to recruit the endogluconase.
To demonstrate the feasibility of our new dCas9-guided assembly strategy, three dCas9 orthologs from Streptococcus pyogenes (SpdCas9), Streptococcus thermophiles (St1dCas9), and Streptococcus aureus (SadCas9) were used. These dCas9 proteins require not only different PAM sequences for DNA targeting but also different scaffold domains in their sgRNAs for dCas9 binding.22 The first step toward achieving dCas9-guided assembly is to demonstrate that these dCas9 orthologs can bind specifically onto DNA targets in a single, dual, and triplicate manner. A Cas beacon assay, developed previously to determine SpdCas9 binding to their corresponding DNA targets, was adapted for this purpose (Fig. 2A) 23 Binding of the dCas9:sgRNA complex unwinds the DNA strands, causing displacement of the quencher oligo 2, which can be monitored by an increase in fluorescence (Fig. 2A).
Figure 2.
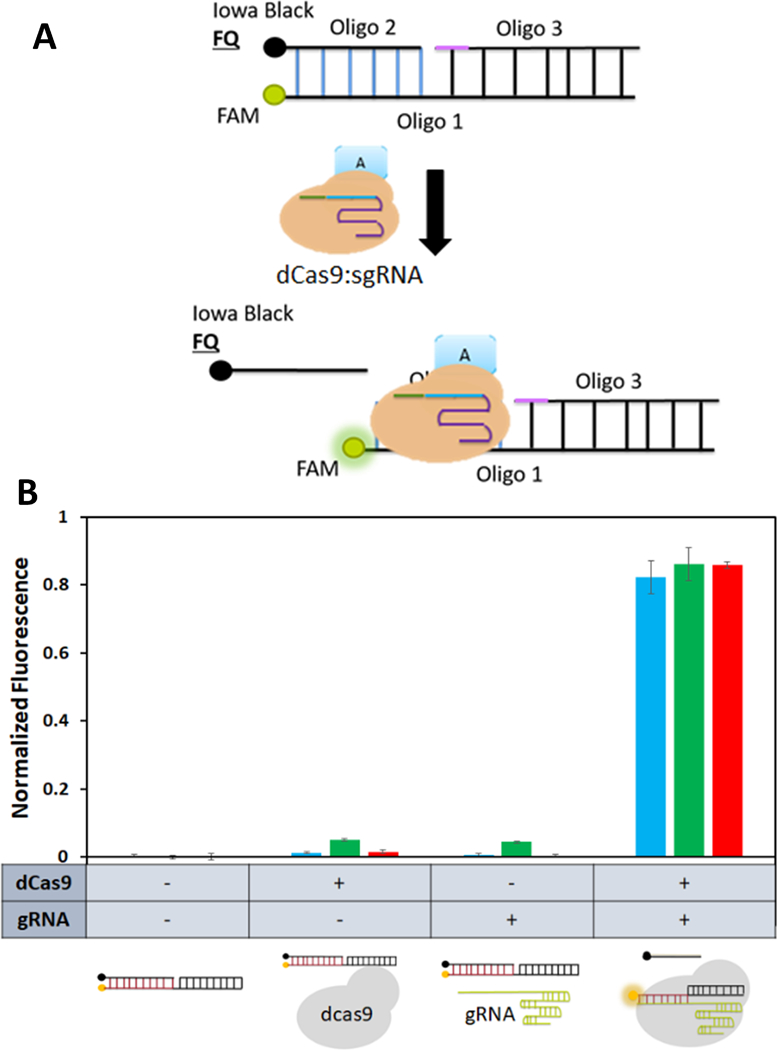
Functionality of individual dCas9 proteins using the Cas beacon assay. (A) Binding of a dCas9 fusion protein to a Cas beacon target and the subsequent displacement of the quencher strand. (B) Binding efficiencies of three different dCas9 fusion proteins to their corresponding Cas beacon targets. In all cases, dCas9 binding resulted in a significant increase in fluorescence. Blue: SPdCa s9, Green: ST1dCas9, and Red: SadCas9.
We first investigated the binding of individual dCas9 proteins to their unique DNA targets. All assays were performed using purified dCas9 proteins and in vitro transcribed sgRNAs (Fig. 2B). For each dCas9:sgRNA pair, no detectable increase in fluorescence was observed when either dCas9 or sgRNA was added. In contrast, a significant increase in fluorescence was observed when both were added, indicative of dCas9 binding (Fig. 2B). Binding of each dCas9 protein onto the corresponding beacon was further validated using the electro-mobility shift assay (Fig. S1), and a slower mobility band was detected in all cases.
Having confirmed the binding ability of all three dCas9 proteins, we next explored their orthogonality to their respective targets. To do so, we expanded upon the single beacon format and developed a new dual beacon approach. Two different fluorophores (FAM and Cy5) were used to detect simultaneous binding of two dCas9 fusions onto the same dual beacon (Fig. 3A). Independent of the dCas9 combinations tested, a significant increase in the respective fluorescent signal was observed only when the corresponding dCas9A:sgRNA complex was added (Fig. 3B). These results support correct displacement of the quencher strand, indicative of the orthogonality of the three dCas9:sgRNA pairs. More importantly, addition of both dCas9:sgRNA complexes did not hinder binding of either dCas9 protein as a similar increase in fluorescent signal was detected (Fig. 3B). These findings were further supported using the EMSA assay, in which single and dual binding were clearly demonstrated (Fig. S2). It should be noted that a 25-bp spacing between the two binding sites is necessary to minimize the steric effect of dual dCas9 binding as a much smaller increase in fluorescence was observed using a shorter 15-bp spacer (Fig. S3).
Figure 3.
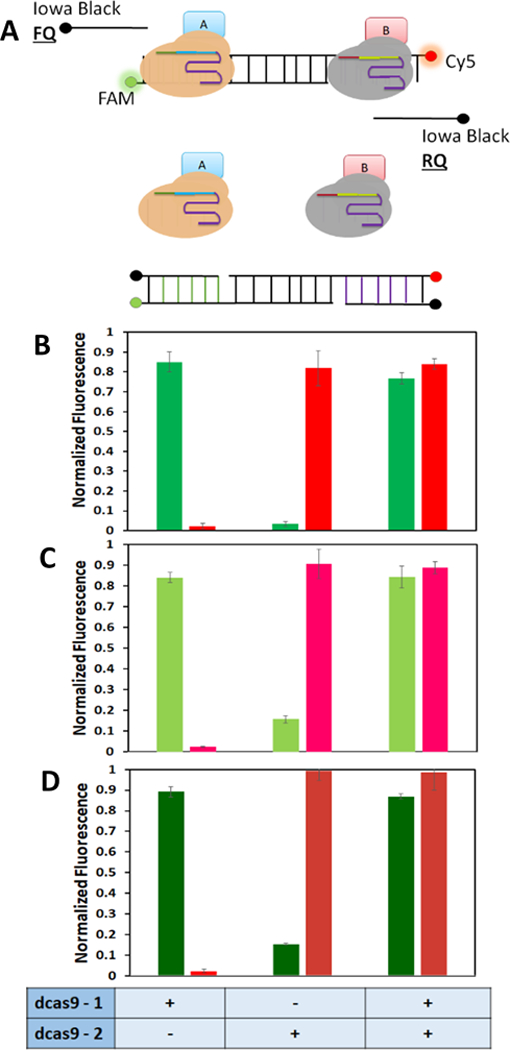
(A) Schematic of the dual beacon assay. (A) Fully hybridized dual beacon has only background fluorescence. Addition of dCas9A/sgRNA complex results in displacement of the quencher Oligo 2 and an increase in FAM fluorescence. While incubation with dCas9B:/sgRNA complex displaces only Oligo 4 resulting in Cy5 fluorescence. When both dCas9 fusion proteins and the cognate sgRNAs are present, both quencher strands are displaced causing an increase in both FAM and Cy5 fluorescence. (B) Binding efficiencies of three dCas9 fusion protein combinations to their corresponding dual beacon targets. (B) SpdCas9/StidCas9, (C) SpdCas9/SadCa9Green, (D) SadCas9/St1dCas9.
To demonstrate that all three dCas9 proteins can co-localize simultaneously onto a single DNA scaffold, a new template harboring three binding sites was designed. One of the two oligos is fused to a FAM dye for tracking by EMSA (Fig. S4). Consistent with the beacon results, a gradual decrease in the complex mobility was observed when either one, two, or three dCas9 proteins was bound. No unbound DNA was detected indicating 100% binding in all three cases. This is significantly better than the roughly 50% assembly using the ZFP-guided approach.13 These binding results confirm our ability to recruit multiple dCas9 proteins simply by modulating the number of orthogonal binding sites on a single DNA scaffold.
After confirming our ability to co-localize multiple dCas9 proteins onto a single DNA scaffold, we next explored this strategy to construct artificial cellulosomes. A two-component mini-cellulosome structure was created by fusing CBD to the C-terminus of SpdCas9 and the endoglucanase CelA to the C-terminus of St1dCas9. The ability to co-localize both dCas9 fusions was first tested using the Cas beacon assay. Additon of CBD or CelA had no impact on dCas9 binding as a similar increase in fluorescence was detected (Fig. S5). The two dCas9 fusions were then incubated with a DNA scaffold to generate a CBD-CelA cellulosome structure. As depicted in Fig. 4, a 2.6-fold enhancement in cellulose hydrolysis was observed as compared with unassembled dCas9 fusions. This level of improvement is even better than that obtained using assembly by direct DNA hybridization,12 again highlighting the benefit of the sub- nanomolar binding affinity of dCas9 proteins in optimizing co-localization of the two cellulsomal components.
Figure 4.
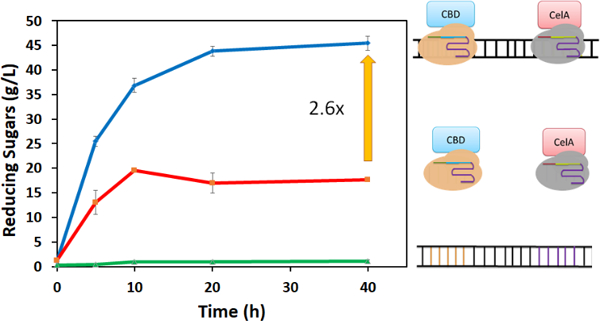
Assembly of a two-component celluosome using SpdCas9-CBD and ST1dCas9-CelA. The production of reducing sugars from DNA alone, free enzyme fusions, and the assembled celluosome were compared.
Since the efficiency of many enzyme cascades can be optimized by tuning enzyme stoichiometry and order, another benefit of our dCas9-guided assembly is the ease of reconfiguring the assembled proteins simply by changing the DNA scaffold. A new DNA scaffold was first designed to change the order from CBD-CelA to CelA-CBD (Fig. S6). A similar level of enhancement in cellulose hydrolysis was obtained since both celluosomes are equally efficient in bringing CelA in proximity to the substrate. A more complex cellulosome structure containing three CBDs and CelAs was also created by changing the number of binding sites on the DNA scaffold (Fig. S6). Although the amount of proteins used was the same as before, complete assembly was achieved even with 1/3 the amount of DNA due to the tight binding of dCas9s. This feature is particularly attractive for assembling in vivo enzyme cascades in E. coli as increasing numbers of binding sites can be inserted into plasmid DNA to enhance co-localization.9,12,13, The feasibility for both in vitro and in vivo enzyme assemblies while providing sub-nanomolar affinity is perhaps the most distinguishing feature of our new approach.
In plant, many native metabolons are dynamic in nature to provide adaptive changes in cellular responses24 While protein scaffolds have succeeded in improving enzymatic functions, they are restricted for dynamic modulation.25 Using a technology newly developed for conditional binding of dCas9,26 we next demonstrated the ability to provide dynamic modulation of cellulosome assembly. A toehold-gated sgRNA (thgRNA), 25 with the spacer region initially blocked by a hairpin, is used. Addition of a RNA trigger unblocks the thgRNA by toehold-mediated strand displacement and reactivates the spacer for dCas9 binding to the DNA target (Fig. 5A).
Figure 5.
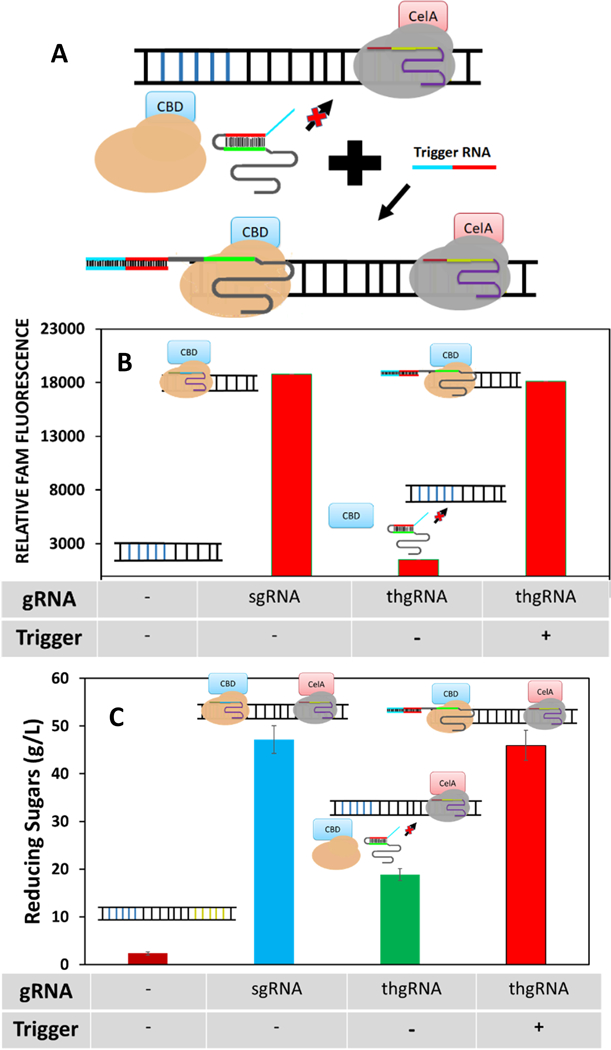
Conditional assembly of a two-component cellulosome. (A) Binding of SpdCas9-CBD is activated by a RNA trigger to unblock the spacer region of thgRNA by toehold-mediated strand displacement. (B) Conditional binding of SpdCas9-CBD using the Cas beacon assay. Binding was performed using either sgRNA or thgRNA. Activation was observed with the thgRNA only when a corresponding RNA trigger was added. (C) Cellulose hydrolysis by conditional cellulosome formation by toehold-mediated strand displacement. Cellulosomes assembled using sgRNA and the activated thgRNA produced similar reducing sugar titers while the inactivated thgRNA produced less reducing sugars comparable to the unassembled enzymes in Fig. 5.
To demonstrate dynamic cellulosome assembly, we designed a new thgRNA to modulate the conditional binding of SpdCas9-CBD. While the SpdCas9-CBD:sgRNA complex restored full fluorescence to the beacon, incubation with the SpdCas9-CBD:thgRNA complex exhibited only minimal background, reaffirming the ability of thgRNA to inhibit SpdCas9 binding. Full activation was achieved with the addition of a RNA trigger to a level similar to that with the unblocked sgRNA (Figure 5B). These data support the ability to gain conditional control over dCas9 binding using a simple RNA trigger.
We next assessed the ability to control cellulosome assembly. In this design, St1dCas9-CelA is always bound to the DNA scaffold while SpCas9-CBD binding is dynamically modulated (Fig. 5C). Simply by replacing the regular sgRNA with the thgRNA version, the level of reducing sugars reduced back to that of unassembled enzymes, consistent with the lack of binding by SpdCas9-CBD. Addition of the trigger RNA restored not only SpdCas9-CBD binding but also the enhancement in cellulose hydrolysis. The ability to control enzyme clustering dynamically using a simple RNA input provides a powerful platform to modulate the efficiency of many enzyme cascade reactions. The strategy is particularly attractive for dynamic control of in vivo enzyme assembly, as even a full-length mRNA has been shown to be capable of activating SpdCas9 binding. 25
In summary, we presented here a new approach to create enzyme cascades using dCas9-mediated assembly. By using orthogonal dCas9 pairs, site-specific assembly can be achieved. The high binding affinity affords by dCas9 proteins enables easy control over enzyme stoichiometry, order, and spacing without scarifying co-localization efficiency. The feasibility to modulate enzyme assembly dynamically using a simple RNA input should open up many in vivo opportunities for conditional activation of metabolic pathways.
Supplementary Material
Acknowledgments
We would like to acknowledge the funding support from NSF (MCB1543838 and MCB1615731)
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interests
There are no conflicts to declare.
Notes and references
- 1.Pontrelli S, Chiu T-Y, Lan EI, Chen FY-H, Chang P, and Liao JC, Metab. Eng., 2018, 50, 16–46. [DOI] [PubMed] [Google Scholar]
- 2.Peralta-Yahya PP, Zhang F, del Cardayre SB and Keasling JD, Nature, 2012, 488, 320–328. [DOI] [PubMed] [Google Scholar]
- 3.Chen R, Chen Q, Kim H, Siu K-H, Sun Q, Tsai S-L and Chen W, Current opinion in biotechnology, 2014, 28, 59–68. [DOI] [PubMed] [Google Scholar]
- 4.Jørgensen K, Rasmussen AV, Morant M, Nielsen AH, Bjarnholt N, Zagrobelny M, Bak S and Møller BL, Current Opinion in Plant Biology, 2005, 8, 280–291. [DOI] [PubMed] [Google Scholar]
- 5.Garbett D and Bretscher A, Mol. Biol. Cell, 2014, 25, 2315–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smirnoff N, Plant Physiology, 2019, 179, 918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vélot C, Mixon MB, Teige M and Srere PA, Biochemistry, 1997, 36, 14271–14276. [DOI] [PubMed] [Google Scholar]
- 8.Good MC, Zalatan JG and Lim WA, Science, 2011, 332, 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conrado RJ, Wu GC, Boock JT, Xu H, Chen SY, Lebar T, Turnšek J, Tomšič N, Avbelj M, Gaber R, Koprivnjak T, Mori J, Glavnik V, Vovk I, Benčina M, Hodnik V, Anderluh G, Dueber JE, Jerala R and DeLisa MP, Nucleic Acids Res, 2012, 40, 1879–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P and Sigman M, Nat Chem, 2016, 8, 299–309. [DOI] [PubMed] [Google Scholar]
- 11.Sweetlove LJ and Fernie AR, Nature Communications, 2018, 9, 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Q and Chen W, Chem. Commun., 2016, 52, 6701–6704. [DOI] [PubMed] [Google Scholar]
- 13.Sun Q, Madan B, Tsai S-L, DeLisa MP and Chen W, Chem. Commun., 2014, 50, 1423–1425. [DOI] [PubMed] [Google Scholar]
- 14.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, V Ullal A, Prather KLJ and Keasling JD, Nat. Biotechnol., 2009, 27, 753. [DOI] [PubMed] [Google Scholar]
- 15.Moon TS, Dueber JE, Shiue E and Prather KLJ, Metab. Eng., 2010, 12, 298–305. [DOI] [PubMed] [Google Scholar]
- 16.V Pinheiro A, Han D, Shih WM and Yan H, Nat. Nanotechnol., 2011, 6, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu J, Liu M, Liu Y, Woodbury NW and Yan H, J. Am. Chem. Soc., 2012, 134, 5516–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adli M, Nat. Commun., 2018, 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang F, Zhou K, Ma L, Gressel S and Doudna JA, Science (80-. )., 2015, 348, 1477 LP-1481. [DOI] [PubMed] [Google Scholar]
- 20.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA and Charpentier E, Science (80-. )., 2012, 337, 816 LP-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, Esvelt KM and Church GM, Nat. Methods, 2013, 10, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ and Church GM, Nat. Methods, 2013, 10, 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semenova E, Kuznedelov K, Minakhin L, Mekler V and Severinov K, Nucleic Acids Res, 2016, 44, 2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Møller BL, Science (80-. )., 2010, 330, 1328 LP-1329. [Google Scholar]
- 25.Chen RP, Blackstock D, Sun Q and Chen W, Nature Chemistry, 2018, 10, 474–481. [DOI] [PubMed] [Google Scholar]
- 26.Siu K-H and Chen W, Nat. Chem. Biol., 2019, 15, 217–220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.