

Abstract
In this work, we develop a drug-mimicking nanofibrous peptide hydrogel that shows long-term bioactivity comparable to a small-molecule inhibitor of inducible nitric oxide synthase (iNOS). The iNOS inhibitor, N6-(1-iminoethyl)-l-lysine (l-NIL), is a positively charged amino acid whose structure could be readily integrated into the framework of a positively charged multidomain peptide (MDP) through the modification of lysine side chains. This new l-NIL-MDP maintains the self-assembling properties of the base peptide, forming β-sheet nanofibers, which entangle into a thixotropic hydrogel. The l-NIL-MDP hydrogel supports cell growth in vitro and allows syringe-directed delivery that persists in a targeted location in vivo for several weeks. Multiple characterization assays demonstrate the bioactivity of the l-NIL-MDP hydrogel to be comparable to the l-NIL small molecule. This includes iNOS inhibition of macrophages in vitro, reduced nitrotyrosine immunostaining in murine subcutaneous histology, and reduced serum levels of vascular endothelial growth factor in vivo. This study expands the toolbox of available peptide hydrogel scaffold designs that can modify biological activity without the need for any additional small-molecule drugs, proteins, or cells.
Keywords: self-assembly, peptide, hydrogel, iNOS inhibition
INTRODUCTION
Biomaterials have attracted large interest in the past few decades for their versatility of design and unique biological applications. Biomaterial designs include polysaccharide-based scaffolds,1 polymer implants,2 mesoporous silica rods,3 and complex self-assembling structures based on polymers,4 nucleic acids,5 or peptides.6,7 Peptide-based biomaterials are particularly interesting as they possess easily customizable structures due to the modular nature of their amino acid building blocks, and they are inherently biocompatible and biodegradable.6,8 Based on rational sequence design and final material properties, peptide biomaterials can be developed to load and deliver practically any therapeutic,9,10 whether small molecules,11–13 biologics,14,15 or cells.16
Our lab has developed a versatile portfolio of multidomain peptide (MDP) biomaterials, which form syringe-injectable hydrogels.17 MDPs are a family of rationally designed peptides that self-assemble in the aqueous solution to create nanofibrous networks that resemble the extracellular matrix (ECM).18 MDP self-assembly is primarily driven by an amphiphilic core of alternating hydrophobic (often leucine) and hydrophilic (often serine) amino acids, resulting in a facial amphiphile that initially self-assembles to maximize hydrophobic packing of aliphatic residues. Further assembly occurs as the peptides form antiparallel β-sheet networks as a result of peptide-backbone hydrogen bonding (Figure 1C). Self-assembly is limited and controlled via molecular frustration or the introduction of competing supramolecular forces, which can be achieved through a variety of mechanisms.19 Generally, molecular frustration is introduced by incorporation of charged amino acids (often lysine) on the peptide termini, utilizing charge repulsion to prevent infinite self-assembly that would result in aggregation and poor solubility. This design also results in triggerable hydrogelation. Specifically, when buffers containing multivalent counterions [such as phosphate ions in phosphate-buffered saline (PBS)] are added to an MDP solution, charge shielding and side-chain crosslinking allow the peptides to form highly entangled nanofiber networks, resulting in soft ECM-like hydrogels ideal for use as injectable biomaterials (Figure 1B–D).17
Figure 1.
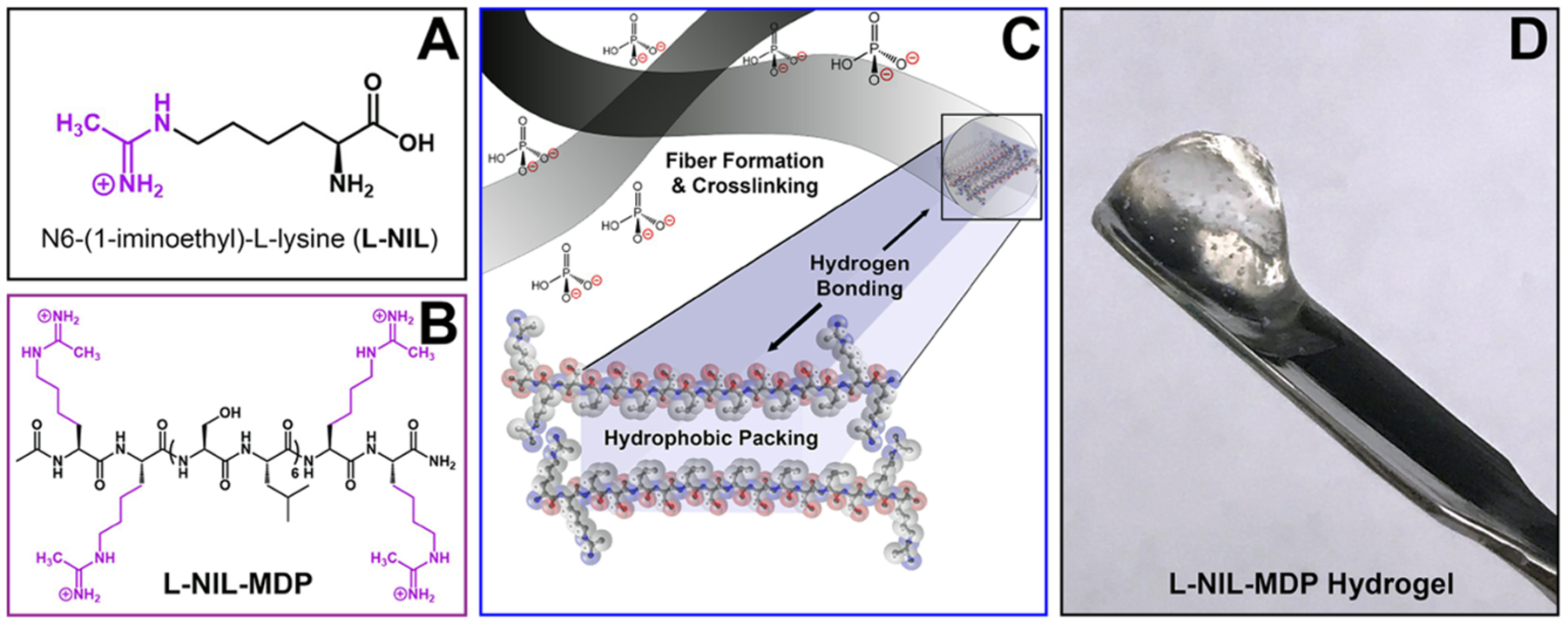
Chemical structures of the small-molecule drug N6-(1-iminoethyl)-l-lysine (l-NIL) (A) and the designed l-NIL multidomain peptide (B) chemically derived from the lysine-based peptide, K2(SL)6K2, by converting the starting material’s lysine side chains to l-NIL acetamidine functional groups. (C) Graphic depicting the nanofibers formed by self-assembly of multidomain peptides into antiparallel β-sheets, driven by hydrophobic packing and the formation of hydrogen-bonding networks between peptide backbones. Charged functional groups are displayed on the fiber surface and interact with phosphate counterions to extend and crosslink fibers. (D) Image of the easily manipulated hydrogel biomaterial that forms when the l-NIL-MDP is prepared at 1 wt % in phosphate-containing buffer.
MDPs have high sequence customizability, capable of incorporating a variety of residues while still resulting in nanofibers and hydrogels.19 Various agents can also be incorporated into the MDP or salt buffer solution prior to gelation, yielding loaded gel formulations evenly distributed with drugs, biologics, or cells.18 We have demonstrated in prior investigations that the chemistry and functionality of these nanofibrous peptide biomaterials can be tailored for extended drug delivery applications, including cancer therapy.20–23
In this study, our goal was to generate a new MDP biomaterial via chemical modification that would provide inherent bioactivity without exogenous drug loading. Such a material could be, subsequently, used in future combination therapy studies, where the material’s long-term pharmacological activity could be synergistically combined with time-released factors. Herein, we developed a peptide-based biomaterial to mimic the inhibitory action of a small-molecule inhibitor with immunomodulatory properties called N6-(1-iminoethyl)-l-lysine or l-NIL (Figure 1A). l-NIL and its derivatives are potent small-molecule drugs (molecular weight ≈ 190 g/mol) that selectively inhibit inducible nitric oxide synthase (iNOS).24,25 iNOS is one of the primary enzymes responsible for the in vivo synthesis of nitric oxide (NO) from l-arginine, with NO being a pervasive and multifunctional cell-signaling molecule that has many broad-ranging effects.26,27 The expression and activity of the enzyme iNOS are highly upregulated in many types of cancers, leading to increased synthesis of NO and promoting conditions conducive to tumor growth, such as increased angiogenesis,28,29 avoidance of apoptosis,30 and immunosuppression.27,31,32 The small-molecule drug l-NIL can be used to selectively inhibit iNOS and curtail aberrant NO production, thereby reducing various downstream effects that are conducive to tumor growth.
However, the use of l-NIL or other small-molecule iNOS inhibitors in preclinical research has been hindered by the need to repeatedly deliver large drug quantities over a long period of time to achieve treatment efficacy. This is usually achieved by the addition of l-NIL to the drinking water in murine models.25 While technically simple, this administration procedure results not only in excessive waste of the valuable material (as drug-doped water must be exchanged daily) but also results in inconsistent treatment results when administered “ad libitum” or “as much as desired.” Furthermore, reliance on the oral administration route must combat issues of absorption, bioavailability, and incomplete drug distribution.25 Therefore, an injectable, extended-activity system could reduce material waste, standardize drug intake, and increase the maximum dosage by bypassing the oral delivery route via direct intratumoral injection.
Our primary hypothesis was that l-NIL bioactivity could be successfully translated into a peptide biomaterial system, resulting in a unique material that would both retain the necessary supramolecular and mechanical properties to form a useful biomaterial, while also gaining new biological properties. In this study, we transformed the chemical structure of a peptide hydrogel so that it could possess the intrinsic functionality and bioactivity of the drug l-NIL. The chemical structure of l-NIL is simple and easily manipulatable, as it is derived from the natural amino acid lysine. Lysine is a residue our lab has extensive experience incorporating it into multidomain peptide hydrogels, and it is also an amino acid often used in the charged terminal regions of MDP designs.17,18,24 The l-NIL acetamidine functional group, like lysine’s amine, critically retains a positive charge at physiological pH. This cationic charge was predicted to allow for control of the self-assembly of a custom l-NIL acetamidine-based multidomain peptide via charge repulsion (Figures 1 and 2). In this report, we demonstrate that the l-NIL-MDP hydrogel possesses unique chemical and mechanical properties that make it a promising biomaterial, as it shows inherent drug bioactivity, remains in its implanted location for up to 3 weeks, and can create a durable effect observable in vitro and in vivo.
Figure 2.
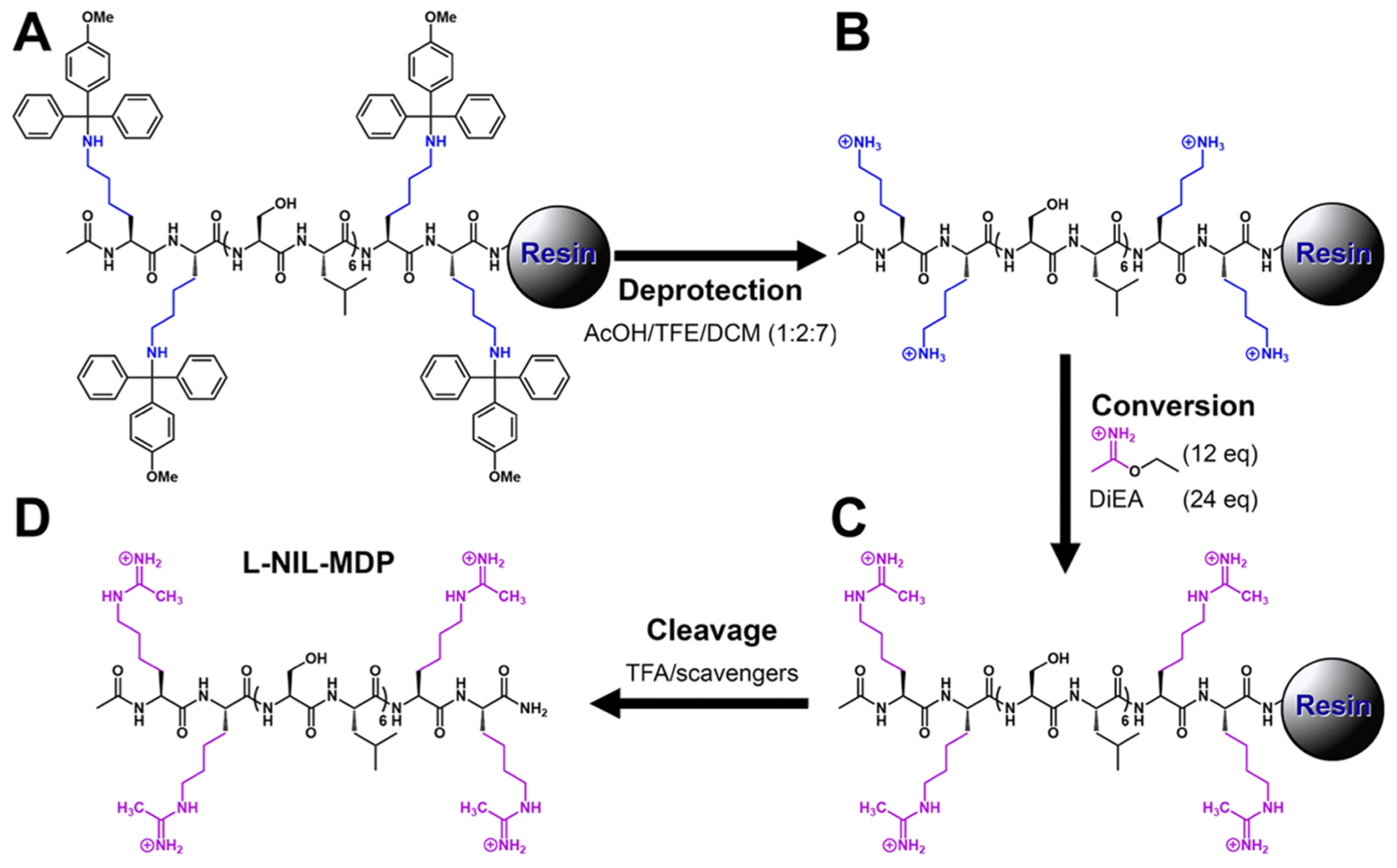
l-NIL-MDP solid-phase synthesis scheme. (A) K2Mmt(SL)6K2Mmt is synthesized on Rink Amide 4-methylbenzhydrylamine (MBHA) resin using standard solid-phase peptide chemistry. (B) Removal of the monomethoxytrityl (Mmt) protecting groups is performed using 10% acetic acid in trifluoroethanol (TFE) and dichloromethane (DCM) to yield deprotected K2(SL)6K2. (C) On resin, conversion to l-NIL-MDP is achieved by acetimidation reaction with excess ethyl acetimidate and N,N-diisopropylethylamine (DiEA), followed by (D) trifluoroacetic acid (TFA) cleavage from resin to yield free peptide.
EXPERIMENTAL METHODS
Peptide Synthesis, l-NIL-MDP Reaction, and Product Purification.
Peptide synthesis reagents were purchased from EMD Chemicals (Philadelphia, PA). A combination of manual synthesis and an Apex Focus XC (AAPPTec) automatic synthesizer was used to synthesize the multidomain peptides K2(SL)6K2 (K2-MDP), E2(SL)6E2 (E2-MDP), R2(SL)6R2 (R2-MDP), and the precursor to the l-NIL-MDP, K2Mmt(SL)6K2Mmt, where Mmt represents acid-labile monomethoxytrityl protecting groups on the lysine side chains. All peptides were synthesized according to standard F-MOC chemistry using solid-phase peptide synthesis methods previously published.33All peptides were N-terminally acetylated and C-terminally amidated.
The following method was used to convert K2Mmt(SL)6K2Mmt on low-loading Rink Amide MBHA resin to l-NIL-MDP before TFA cleavage (Figure 2). Deprotection of the lysine side chains was achieved by removal of the Mmt protecting groups with four successive 15 min washes of AcOH/TFE/DCM (1:2:7), washing until the yellow color of released trityl cation was no longer observed. After washing, a reaction mixture was added containing 12 equiv predissolved ethyl acetimidate–HCl and 24 equiv DiEA in DCM, and the mixture was shaken for 3 h. The reaction was then drained, a second reaction mixture added, shaken for 2 h, followed by a third fresh reaction mixture for 1–2 h. This sequence was done to ensure maximum conversion of the lysine side chains to l-NIL functional groups.
Cleavage of all peptides was then achieved by shaking for 3 h with TFA and protecting scavengers, in a 2:1:1:18 ratio of Milli-Q (MQ) H2O/triisopropylsilane/anisole/TFA. Rotary evaporation was used to remove excess TFA, and trituration with cold diethyl ether yielded crude peptide. The peptide was then dissolved in purified Milli-Q (MQ) water, pH adjusted to approximately 7.0, and dialyzed against MQ water for 5–7 days to remove small-molecule scavengers, TFA, and other contaminants using 100–500 Da MW cut-off dialysis tubing (Spectra/Por, Spectrum Laboratories Inc., Rancho Dominguez, CA). Dialyzed peptide solutions were adjusted to pH 7.2–7.4, after which they were sterile-filtered using 0.2 μm filters and lyophilized to powder for storage and use in experiments. All peptides were analyzed by Autoflex matrix-assisted laser desorption ionization (MALDI) time-of-flight mass spectrometry (MS) (Bruker Instruments, Billerica, MA) for confirmation of expected peptide mass (Figure S1).
Hydrogel Preparation and Loading.
All chemicals not otherwise specified were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO). For the preparation of all sterile K2-MDP, l-NIL-MDP, E2-MDP, and R2-MDP hydrogels, 2 wt % (20 mg/mL) peptide solutions were made by dissolving lyophilized peptide mass in sterile 298 mM sucrose. Sterile 1× Hank’s balanced salt solution (HBSS, Fisher Scientific, Hampton, NH) was used to initiate gelation in cationic MDPs (K2-MDP, l-NIL-MDP, and R2-MDP), while a separate solution of 45 mM Mg(Cl)2 in 1× HBSS was used with the anionic E2-MDP, calculated to achieve 22.5 mM (4 molar equiv) Mg2+ upon final dilution with peptide. To form all hydrogels, 2 wt % peptide solutions were diluted 1:1 with 1× HBSS or 1× HBSS + 45 mM Mg2+ depending on peptide gelation requirements, for final formulations containing 1 wt % peptide (10 mg/mL, approximately 5–6 mM), 0.5× HBSS, and 149 mM sucrose.
Circular Dichroism (CD).
Peptide samples were prepared at 0.1 wt % in MQ H2O (diluted from 1 wt % hydrogels in HBSS and sucrose) for CD analysis on a Jasco J-810 spectropolarimeter (Jasco Inc., Easton, MD). Scanning experiments were performed from 250 to 180 nm with a 0.1 nm pitch, using an accumulation of 5–10 scans to acquire averaged spectra with reduced noise. The 0.1 wt % peptide solutions were loaded into 0.1 or 0.01 mm glass cuvettes. Diagnostic peaks for the β-sheet secondary structure identification were as follows: minimum near 216 ± 2 nm and maxima near 196 ± 2 nm.
Attenuated Total Reflectance Fourier Transform Infrared (ATR-FT-IR) Spectroscopy.
Peptide samples (10 μL of 1 wt % peptide solution) were dried under nitrogen on a Golden Gate diamond window of an ATR stage for 4–24 h as necessary to remove water. IR spectra were then collected on a Jasco FT/IR-660 plus spectrometer (Jasco Inc., Easton, MD) at 1 cm−1 resolution with 64 scans accumulation, and the background was collected and subtracted to minimize the contribution of water in the resulting spectra. Relevant peaks in the 1750–1450 cm−1 range were plotted and analyzed to determine the secondary structure of the peptides.
Oscillatory Rheology.
Peptide hydrogels were analyzed for their mechanical properties by oscillatory rheology using a TA Instruments AR-G2 rheometer (TA Instruments, New Castle, DE). Samples of 150 μL of 1 wt % MDP hydrogels were prepared 24 h prior to use for rheology, to allow full equilibration of the material. Gel samples were then transferred from a cut syringe onto the rheometer stage equipped with a 12 mm stainless-steel parallel plate set to a 1000 μm gap height. The following program was used to monitor the storage modulus (G′) and loss modulus (G″) under various conditions, as has been previously published.34 Strain sweep analysis was performed using an applied strain of 0.01–200% at a frequency of 1 rad/s. Frequency sweep analysis was performed under 1% strain at 0.1–100 rad/s. Shear recovery analysis was performed by subjecting the peptide sample to 1% strain for 20 min, then 200% for 1 min, and finally 1% for 20 min, allowing for disruption of the hydrogel and monitoring of G′ and G″ recovery.
Transmission Electron Microscopy (TEM).
Peptide nanostructure was studied by TEM. All samples were prepared via negative staining on Quantifoil R1.2/1.3 holey carbon films on copper mesh grids. Peptide samples were prepared at concentrations ranging from0.1 to 0.01 wt %, and 10 μL from each solution was spotted onto individual grids. Excess peptide solution was blotted after being allowed to adsorb for 1 min. The sample grids were negative stained for 5 min using a 2 wt % phosphotungstic acid (PTA) solution in MQ H2O prepared at pH 7 and sterile-filtered. Excess PTA solution was wicked of the grids before drying overnight. Prepared samples were imaged at 120 kV and 40 000× magnification using a JEOL 2010 TEM microscope (JEOL USA Inc., Peabody, MA).
Cell Culture.
For in vitro experiments, mouse RAW 264.7 cells (ATCC TIB-71) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), l-glutamine, sodium pyruvate, and 1% penicillin–streptomycin. Cells were cultured at 37 °C with 5% CO2 and subcultured using scraping.
For two-dimensional (2-D) culture live–dead viability studies, peptide hydrogels were pipetted into 0.4 cm2 wells of Lab-Tek 16 well glass chamber slides (Thermo Fischer, Rochester, NY). Gels were pipetted to produce 70 μL pucks with a thickness of 1.75 mm. The gels were carefully pipetted into the bottom of each well and tapped before shear recovery to create flat gel profiles. After pipetting, the samples were allowed to shear recover for 5–10 min before adding 200 μL of cell media and 10 000 RAW 264.7 cells. For each experiment, time point (days 1 and 3) separate slides with duplicate gels were created. The cell media was changed every 2 days, taking care to not disturb or dislodge the gel material.
At each desired time point, the following procedure was used to stain the cells for live–dead analysis. Live–dead staining solution was prepared in Dulbecco’s phosphate-buffered saline (Thermo Fisher Scientific, Rockford, IL) with 2 μM Calcein AM (Life Technologies), 4 μM ethidium homodimer (Life Technologies), and 5 μg/mL of Hoechst 33342 (BD Biosciences, San Jose, CA). Cell media was removed from the top of the hydrogel samples, which were then washed with PBS. Samples were then stained with 100 μL of the prepared solution by incubating at 37 °C with 5% CO2 for 60 min while protected from light. After staining, the gels were removed from the glass chamber slides and placed in PBS before immediately imaging by confocal. Gels were analyzed by z-stack imaging using a Nikon A1 Confocal Microscope with a 40× water objective (405 nm blue channel laser, 488 nm green channel laser, 561 nm red channel laser). Image processing was done using NIS Elements, and the Imaris Cell Counting software was used to quantify cell viability and proliferation.
iNOS Knockdown in Vitro Greiss Assay.
To evaluate the iNOS knockdown efficacy of various materials, the following procedure was used to test % iNOS inhibition of RAW 264.7 cells stimulated with lipopolysaccharide (LPS, Thermo Fisher Scientific, Rockford, IL) and recombinant mouse interferon gamma (IFNγ) (Biolegend, San Diego, CA). RAW macrophages were plated at 2.5 × 105 cells per well in 24-well plates. For samples, testing the efficacy of 2-D surface cell culture, 200 μL of MDP hydrogels was applied to evenly coat the surface of each well before the addition of cells. Each experimental condition was performed in triplicate. The following controls were included in every experiment: (1) media alone, (2) stimulated cells alone, and (3) unstimulated cells alone. To each well, stimulation media or normal media was then added based on the experimental requirement. Stimulation media was prepared with LPS (final concentration 1 μg/mL) and IFN-γ (final concentration 100 U/mL). For samples, testing material efficacy in the solution (e.g., free l-NIL), after 6 h of incubation at 37 °C with 5% CO2, the desired concentration of the inhibitor was added to achieve a range of concentrations between 1 μM and 1 mM.
In all cases, after 24 h from the time of stimulation, samples of cell media were collected from each well after mixing media by pipetting before collection. Media samples were placed into a 96-well plate and centrifuged at 5000 rpm for 10 min. Supernatants were removed and placed in a new clear 96-well plate, and samples were immediately analyzed using a colorimetric Greiss Assay (Cayman Chemical, Ann Arbor, MI) to assess nitrite levels.35 The color was allowed to develop for 10 min following addition of Greiss reagents, and absorbance was read at 540 nm on a plate reader. Percent iNOS inhibition for each sample was calculated by first subtracting the absorbance of the media-only baseline and then converting to a percent of maximum nitrite signal as determined by the internal standard of stimulated cells alone. Data were normalized for cell viability on each hydrogel surface determined from the live–dead viability assay at day 1 time point.
Evaluation of Serum Vascular Endothelial Growth Factor (VEGF) Reduction in Vivo.
C57BL/6J male mice were purchased from The Jackson Laboratory. All experiments were performed with the approval of IACUC at Baylor College of Medicine and followed established protocols. B16-F0 melanoma cell line was purchased from American Type Culture Collection (ATCC) and maintained according to manufacturer instructions (DMEM high-glucose with 10% fetal bovine serum and 1% penicillin/streptomycin). Tumor inoculation was performed when mice reached 10–14 weeks of age. C57BL/6J mice were injected subcutaneously (s.c.) with 3 × 105 B16-F0 cells in a single flank. Once tumors became palpable (day 7 post-inoculation), mice were injected intratumorally (i.t.) with either buffer control (HBSS), free l-NIL dissolved in HBSS at 20 mM, K2-MDP hydrogel at 1 wt % (5.6 mM peptide), or l-NIL-MDP hydrogel at 1 wt % (5.2 mM peptide or approximately 20 mM l-NIL functional groups). All injections were done using 100 μL of total injection volume.
Mice were euthanized 5 days following i.t. injections, and the tumor/blood was collected. The blood was allowed to clot prior to centrifugation (2000g, 10 min, room temperature), and the serum was collected from the supernatant and stored at −80 °C prior to analysis. VEGF quantification of the undiluted serum was performed using a mouse VEGF Quantikine ELISA kit (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. All individual mouse samples were run in triplicate and averaged for the final reported value per mouse. Statistical differences were evaluated using Tukey’s multiple comparisons test.
Subcutaneous Experiments and Histology.
C57BL/6J female mice were purchased from The Jackson Laboratory at age 8–12 weeks for subcutaneous characterization experiments. Mice were housed under specific pathogen-free conditions in standard temperature and lighting conditions with free access to food and water, and experiments were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at Rice University and according to NIH guidelines. Mice were injected with 100 μL of MDP hydrogels in each of four separate sites in the subcutaneous space of the dorsal flank after the skin was shaved and sterilized with alcohol. At days 3, 7, 14, and 21, the mice were euthanized and the dorsal skin around the entire implant was removed, fixed overnight in 10% neutral buffered formalin, processed and paraffin embedded, and finally sectioned at 5 μm thickness for staining using standard Masson’s trichrome or hematoxylin and eosin procedures as well as immunohistochemical analyses [e.g., antinitrotyrosine immunohisto-chemistry (IHC) shown in Figure 7 or α-smooth muscle actin blood vessel analysis shown in Figure S4]. Antibodies used in all IHC experiments are listed in Table S1.
Figure 7.
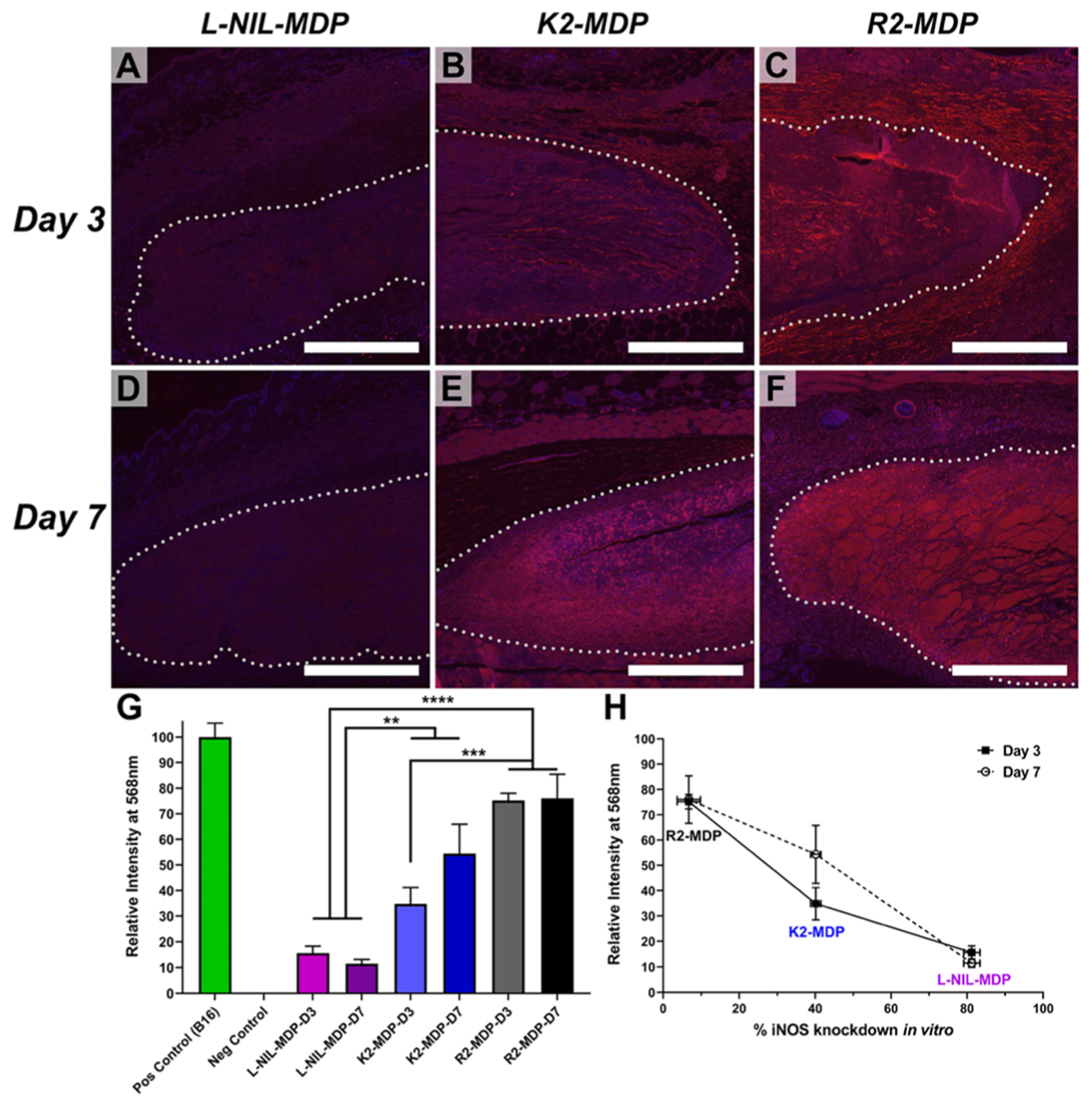
Antinitrotyrosine immunostaining of subcutaneously injected MDP hydrogels to evaluate the effects of relative inhibition of iNOS in vivo, using red 568 nm secondary antibody reactive to antinitrotyrosine primary and 4′,6-diamidino-2-phenylindole for nuclei counterstain. Hydrogel implant boundaries have been marked with dotted lines for additional clarity. (A–C) Immunostained implant images from left to right of l-NIL-MDP, K2-MDP, and R2-MDP, respectively, 3 days post-injection. (D–F) Images from left to right of l-NIL-MDP, K2-MDP, and R2-MDP 7 days post-injection. Scale bars = 0.5 mm. (G) Quantification of relative nitrotyrosine signaling by mean pixel intensity analysis of implant areas, and statistical significance is noted as **p value ≤0.049, ***p value ≤0.005, and ****p value ≤0.0002 [values are mean ± standard error of the mean (SEM) with n = 3]. The positive control was the staining intensity of an inflamed B16-F0 melanoma tumor section (Figure S6), and negative control was the background intensity of the unstained subcutaneous tissue from isotype controls that was subtracted from all datasets (Figures S7–S9). (H) Graph of nitrotyrosine staining intensity shown in (G) plotted vs % iNOS knockdown data shown in Figure 5 for all cationic MDPs, demonstrating a strong inverse correlation between the two separate datasets (values are mean ± SEM with n = 3).
RESULTS AND DISCUSSION
Material Synthesis and Chemical Characterization.
To synthesize a bioactive l-NIL-mimicking peptide nanofiber (or l-NIL-MDP), we used a modified solid-phase peptide synthesis method depicted in Figure 2 to maximize the chance for successful conversion of the large number of lysine side chains of the parent peptide. Since the parent K2-MDP is designed to aggregate and self-assemble into higher-ordered structures and nanofibers, the conversion reaction to the l-NIL-MDP would have been difficult to perform in the solution, due to concentration limitations and the likelihood for some of the key side-chain amines to be buried within the fibers and relatively inaccessible. A reaction on the solid-phase resin was, therefore, performed, and the low-loading MBHA resin was used to maximize interpeptide strand spacing and achieve theoretical “infinite dilution” to facilitate side-chain accessibility and a higher conversion ratio. The synthetic scheme (Figure 2) incorporates the use of a highly acid-labile monomethoxytrityl (Mmt) protecting group on the terminal lysine residues (Figure 2A), allowing for the on-resin deprotection of the lysine side chains prior to conversion. Thus, after first synthesizing K2Mmt(SL)6K2Mmt on the solid phase and acetylating the N-terminus, the lysine side chains were deprotected using the mild acidic conditions of 10% acetic acid (Figure 2B) and synthetically converted to the l-NIL moiety using an acetimidation reaction of the nucleophilic amines (Figure 2C), modified from literature procedures.24
After this on-resin conversion process, the modified peptide was cleaved from the solid-phase and recovered in the solution, with MALDI-MS analysis showing the successful conversion of the peptide to primarily the fully modified species (Figure 3A). Critical to the final design is that the l-NIL-MDP retains the cationic properties of the original lysine-based MDP, with the converted l-NIL acetamidine moieties bearing positive charges at physiological pH akin to canonical lysine or arginine amino acid side chains. This maintains the ability to control MDP self-assembly and hydrogel formation by charge repulsion and ionic crosslinking.
Figure 3.
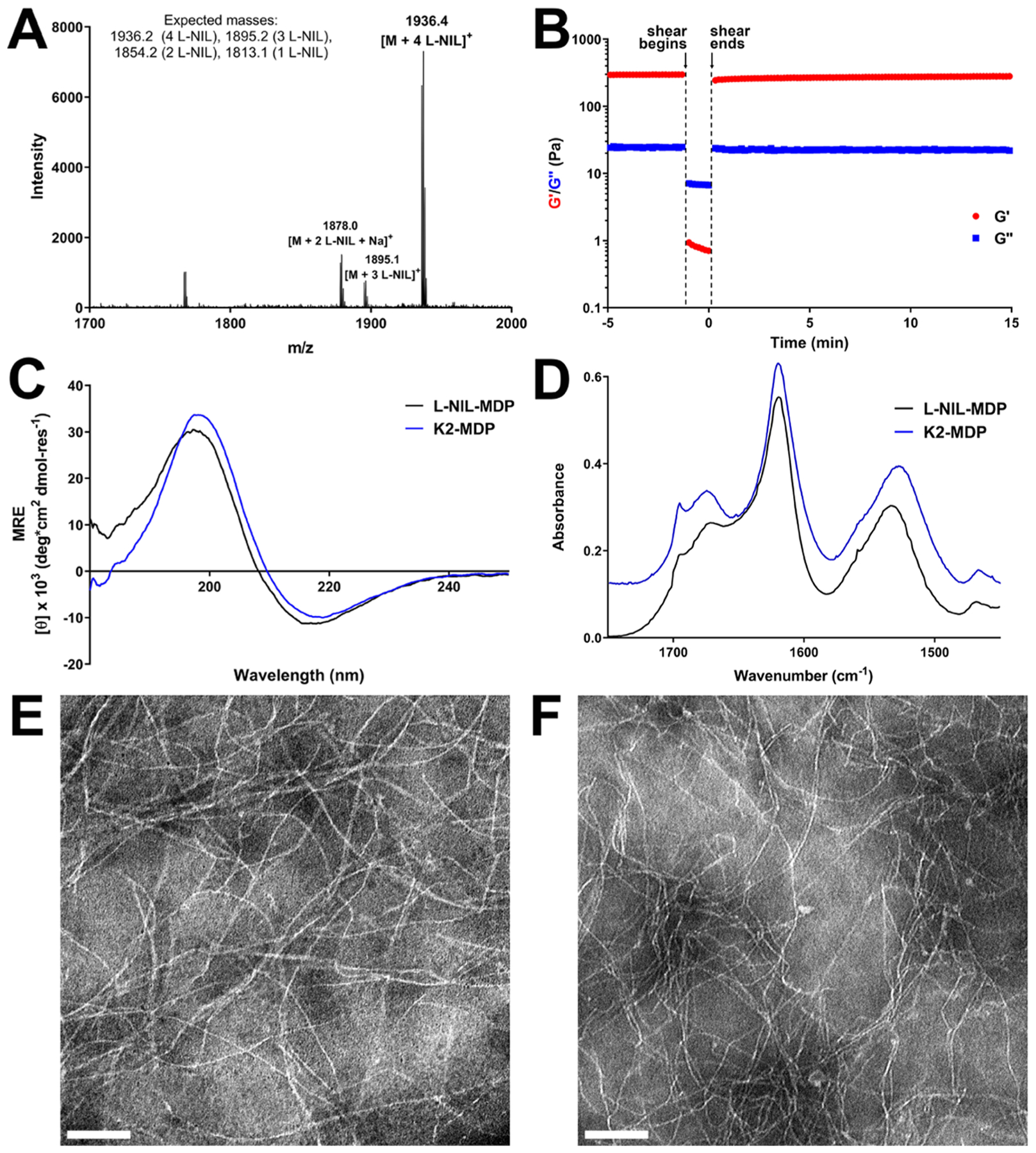
Chemical characterization data for the l-NIL-MDP biomaterial. (A) MALDI-mass spectrum showing the successful synthesis of l-NIL-MDP, primarily composed of a fully converted peptide (all four lysine side chains modified to l-NIL) with a small fraction possessing fewer modified side chains. (B) Oscillatory rheology showing the successful formation of l-NIL-MDP hydrogel with a storage modulus (G′) of ~300 Pa and loss modulus (G″) of ~25 Pa and with a shear recovery of ~86% 1 min after the shearing event. Circular dichroism (C) and attenuated total reflectance Fourier transform infrared spectroscopy (D) spectra (offset for clarity) confirmed the formation of the antiparallel β-sheet secondary structure that closely matches spectra for the parent peptide K2-MDP, demonstrating no significant change in the peptide structure upon conversion to l-NIL-MDP. Transmission electron microscopy images of (E) K2-MDP at 0.01 wt % and (F) l-NIL-MDP at 0.02 wt % shown at 40 000× magnification with scale bars = 100 nm.
In characterizing the material, we compared its self-assembly, secondary structure, nanostructure, and viscoelastic properties to the parent material K2-MDP, with our hypothesis being that the process of chemical conversion to l-NIL-MDP should not significantly affect the final peptide material’s physical properties (Figure 3). Similar to previously designed cationic MDPs, hydrogelation of the l-NIL-MDP is controlled by the addition of multivalent negative ions (e.g., phosphate anions in PBS or HBSS buffers) or pH variation. When prepared at 1 wt % (5.2 mM) in an aqueous buffer containing sucrose for cell compatibility and phosphate anions for crosslinking (HBSS), the l-NIL-MDP forms a compliant hydrogel with a storage modulus (G′) of approximately 260–300 Pa, a loss modulus (G″) of approximately 20–25 Pa, and a shear recovery of ~86% of its initial storage modulus 1 min after a high strain event such as injection through a needle (Figure 3B). For comparison, the parent K2-MDP hydrogel has been reported to have a G′ in the range of approximately 200–500 Pa with a shear recovery of ~82% after 1 min.17 These results demonstrate that the converted material has retained thixotropic mechanical properties ideal for hydrogel syringe injectability. The l-NIL-MDP was also shown to possess a β-sheet secondary structure highly similar to the parent peptide, as confirmed by minima near 216 ± 2 nm and maxima near 196 ± 2 nm in the circular dichroism (CD) spectra for both K2-MDP and l-NIL-MDP (Figure 3C). Furthermore, infrared spectroscopy (ATR-FT-IR) confirmed the secondary structure to be an antiparallel β-sheet, with characteristic amide I absorbances observed at 1695–1696 and 1618–1620 cm−1 for both parent and converted peptides (Figure 3D). Transmission electron microscopy (TEM) analysis of the self-assembled nanostructure showed the formation of highly similar nanofibers for both MDPs, with the l-NIL-MDP nanofibers potentially showing a slightly reduced persistence length (Figure 3E,F). Fiber widths were also observed to be similar between both peptides and consistent with expectations based on previous studies.17,36 Thus, overall material characterization confirmed that the l-NIL-MDP retains the same secondary structure, nanostructure, and bulk material properties as the parent K2-MDP. Therefore, any resulting biological effects of the chemically modified peptide cannot be ascribed to differences in material properties but rather must be due to the new chemical functionality and bioactivity provided by the acetamidine conversion.
Evaluation of Material Bioactivity in Vitro.
As an essential step in the development of any new biomaterial, the l-NIL-MDP hydrogel was evaluated in various biological assays to determine how it interacts with living systems. First, the material’s cytocompatibility in vitro was compared to other MDP hydrogels developed by our lab [e.g., K2(SL)6K2, R2(SL)6R2, or E2(SL)6E2 MDPs].18 RAW 264.7 macrophages were utilized in all in vitro studies as nitric oxide (NO) is a key macrophage product for various cellular functions and activities, and RAW 264.7 is a murine cell line widely used in iNOS-related experiments in the literature.37–39 Live–dead analysis of RAW macrophages cultured for 1–3 days on 1 wt % hydrogel surfaces showed that the l-NIL-MDP was compatible for cell growth (approx. 96% cell survival after 3 days), facilitating the attachment and proliferation of macrophages on the hydrogel comparable to K2-MDP, R2-MDP, and E2-MDP (Figures 4 and S2). It was noted that the highest degrees of cell attachment and proliferation were observed for the cationic MDPs (l-NIL-MDP, K2-MDP, and R2-MDP), while the anionic E2-MDP showed poorer cell survival and proliferation rates likely due to inhibition of cell attachment by the negative carboxylic acid functional groups displayed by the material (Figure S2). This is consistent with various other studies that have demonstrated the ability of cationic moieties or materials (such as polylysine) to facilitate cell attachment, while negative materials often do not.40–42
Figure 4.
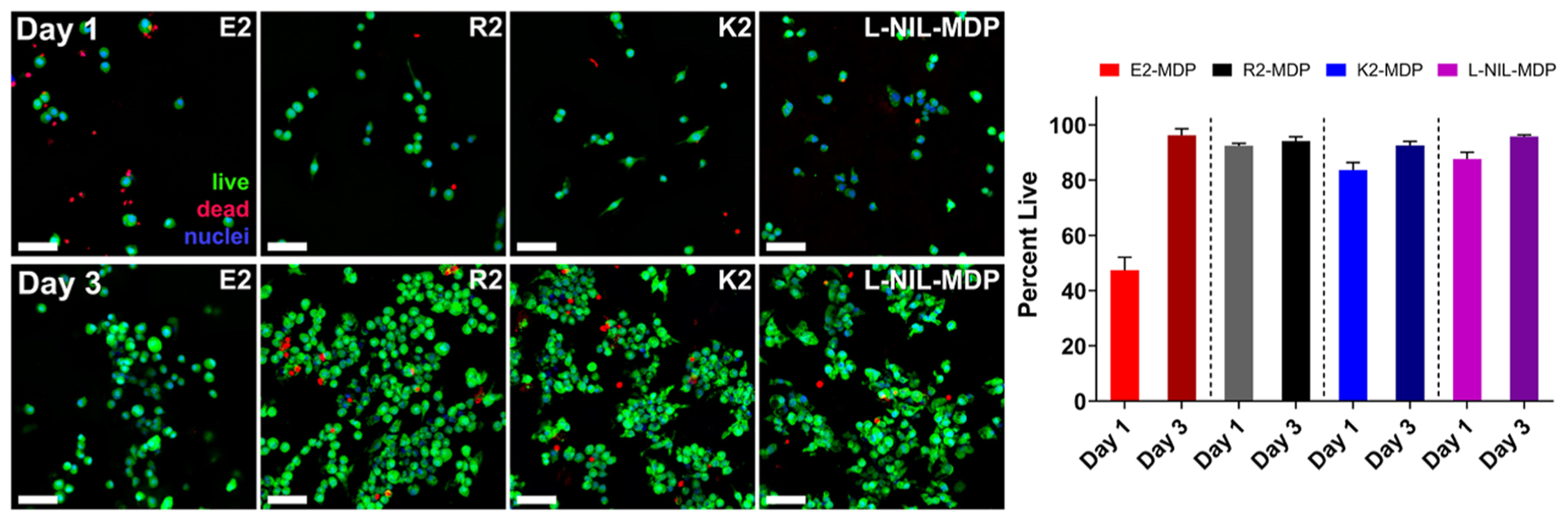
Viability of cells grown on MDP hydrogels. RAW 264.7 cells were seeded on top of 70 μL of hydrogel pucks under 200 μL of media (changed every 2 days) and processed under live–dead viability assays (green Calcein AM: live cells; red ethidium homodimer I: dead cells; blue Hoechst 33342: nuclei). Cells are shown to be able to attach and proliferate on all materials, though to a lesser degree on the anionic MDP hydrogel E2 compared to the cationic MDPs R2, K2, and l-NIL-MDP. Day 1 images are shown on the top row, day 3 images are shown on the bottom row, and quantification of cell viability is shown on the right [values are mean ± standard deviation (SD) with n = 3 images]. All scale bars are 50 μm.
Evaluating bioactivity is critical to studying any system that has changed the structure or availability of a drug. The conversion of a small-molecule drug to a chemical motif displayed on a larger scaffold in no way guarantees superior drug persistence (or even any drug activity at all), as changing the size or chemical structure of a molecule can fundamentally change how it interacts with its biological environment. To begin evaluating the bioactivity of the l-NIL-MDP, an in vitro iNOS knockdown assay was performed to compare the efficacy of different MDP hydrogels to free l-NIL drug in the solution as a positive control. RAW 264.7 macrophages were cultured on various material surfaces, comparing the effects of different MDP hydrogels to tissue culture plastic (TCP) alone or free l-NIL in the solution in 24-well plates. All cells were stimulated with LPS and IFN-γ to upregulate iNOS activity and promote the release of detectable iNOS products into the media (i.e., nitrites), and media was removed 24 h from the start of the experiment and assessed using a colorimetric Griess assay to determine the extent of relative iNOS inhibition by each material. The l-NIL-MDP was shown to be the most effective surface-culture material tested compared to TCP, K2-MDP, R2-MDP, and E2-MDP, exhibiting over 80% iNOS inhibition of RAW macrophages cultured on the hydrogel (Figure 5). This is comparable to the level of inhibition achieved when using 100 μM of free l-NIL in the solution (Figure 5B). However, it must be stated that in this static in vitro system, which tests only the first 24 h of activity, fluid flow and free diffusion into larger environments that are present in vivo are not occurring, and, therefore, the true advantages of an implantable and long-acting hydrogel (compared to a freely diffusible small molecule) are not fully realized. Furthermore, while we attempt to draw comparisons between material activities and soluble l-NIL concentrations, a direct concentration-based comparison of a discrete biomaterial to a freely diffusible small molecule is not truly appropriate, as cells will interact with each in a unique fashion.
Figure 5.
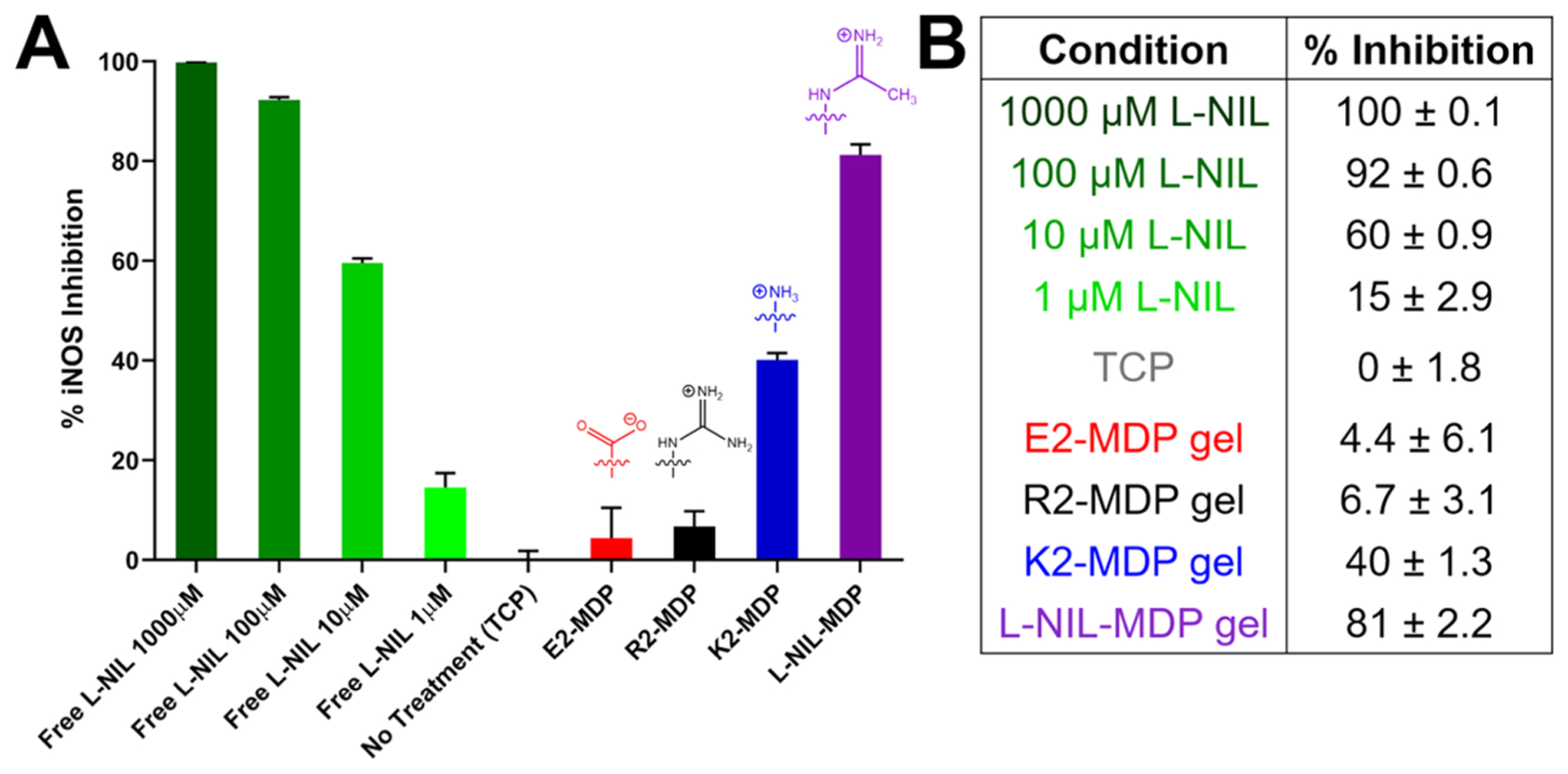
Greiss assay results used to evaluate iNOS knockdown efficacy of various materials. (A) The plot of % iNOS inhibition of RAW 264.7 cells stimulated with lipopolysaccharide (LPS) and IFN-γ cultured in different surface conditions, showing chemical structures of key surface-exposed functional groups. Data have been normalized for cell viability on each hydrogel surface determined from the viability assay shown in Figure 4. (B) Quantification of relative inhibition activity; values are from two experiments and are mean ± SD with three replicates tested per condition.
In comparing the activity of the l-NIL-MDP to other MDP hydrogels tested, it is interesting to note that the parent K2-MDP displays some inherent level of iNOS inhibition comparable to <10 μM free l-NIL (Figure 5B). This is likely because N6-[1-iminoethyl]-l-lysine (l-NIL) is chemically derived from lysine based on the originally published synthesis.24 Therefore, the lysine-rich K2-MDP was observed to display basal levels of activity due to its close chemical relation to l-NIL, though this activity is nearly 10-fold less than the l-NIL-MDP in terms of comparison to free l-NIL concentration. This only further reinforces the strengths of displaying the l-NIL drug moiety on a lysine-based peptide scaffold, suggesting that even the few unreacted lysine residues within the l-NIL-MDP material (Figure 3A) can still display basal inhibitory activity. For the R2-MDP, the simple structural comparison would suggest that the arginine guanidinium side chain would show relatively similar iNOS inhibition to l-NIL or the l-NIL-MDP, due to its similarity to the drug’s acetamidine moiety. Yet in contrast to l-NIL-MDP or even K2-MDP, the R2-MDP showed little to no iNOS inhibition. This can be attributed to the fact that arginine is the natural substrate for iNOS in the synthesis of nitric oxide (NO), and, therefore, in this system, it likely acts as a substrate for the enzyme.26 Finally, the glutamic acid-based E2-MDP similarly shows little to no iNOS inhibition likely due to complete structural mismatch. Most importantly, this assay was the first to demonstrate that the l-NIL-MDP does not eliminate the bioactivity of l-NIL, but rather it displays an ability to inhibit iNOS activity and nitrite production in cell populations that interact with the hydrogel material.
Histological Material Evaluation and Host Response.
Evaluation of material biocompatibility, degradation, and host response in vivo was performed by histological analysis of hydrogel implants injected subcutaneously in healthy mice. l-NIL-MDP hydrogel implants were injected as 100 μL boluses underneath the skin and extracted 3, 7, 14, and 21 days later to study material persistence and degradation as well as the host tissue response (Figure 6). At the day 3 time point (Figure 6A–C), a high density of cellular infiltration, mostly in the periphery of the hydrogel implant, was observed by cells that morphologically resemble innate immune cell populations such as monocytes, macrophages, and granulocytic cells. This represents an initial acute inflammatory reaction by the host to the foreign material that is consistent with previous immune cell profiling of other subcutaneously injected MDP hydrogels. However, over the course of 7–21 days, this initially large and uneven infiltrating cell population resolves into a homogenous and less dense infiltrate that is observed deeper into the core of the hydrogel (Figure 6D–L). Quantification of the local density of cell nuclei per mm2 within the hydrogel bolus shows a clear decrease in density over 2 weeks of implantation, likely as the initial acute inflammatory reaction resolves (Figure S3). This demonstrates that the material does not cause a chronic inflammatory reaction after injection but is biocompatible and facilitates cellular infiltration and remodeling of the hydrogel implant over 1–3 weeks.
Figure 6.
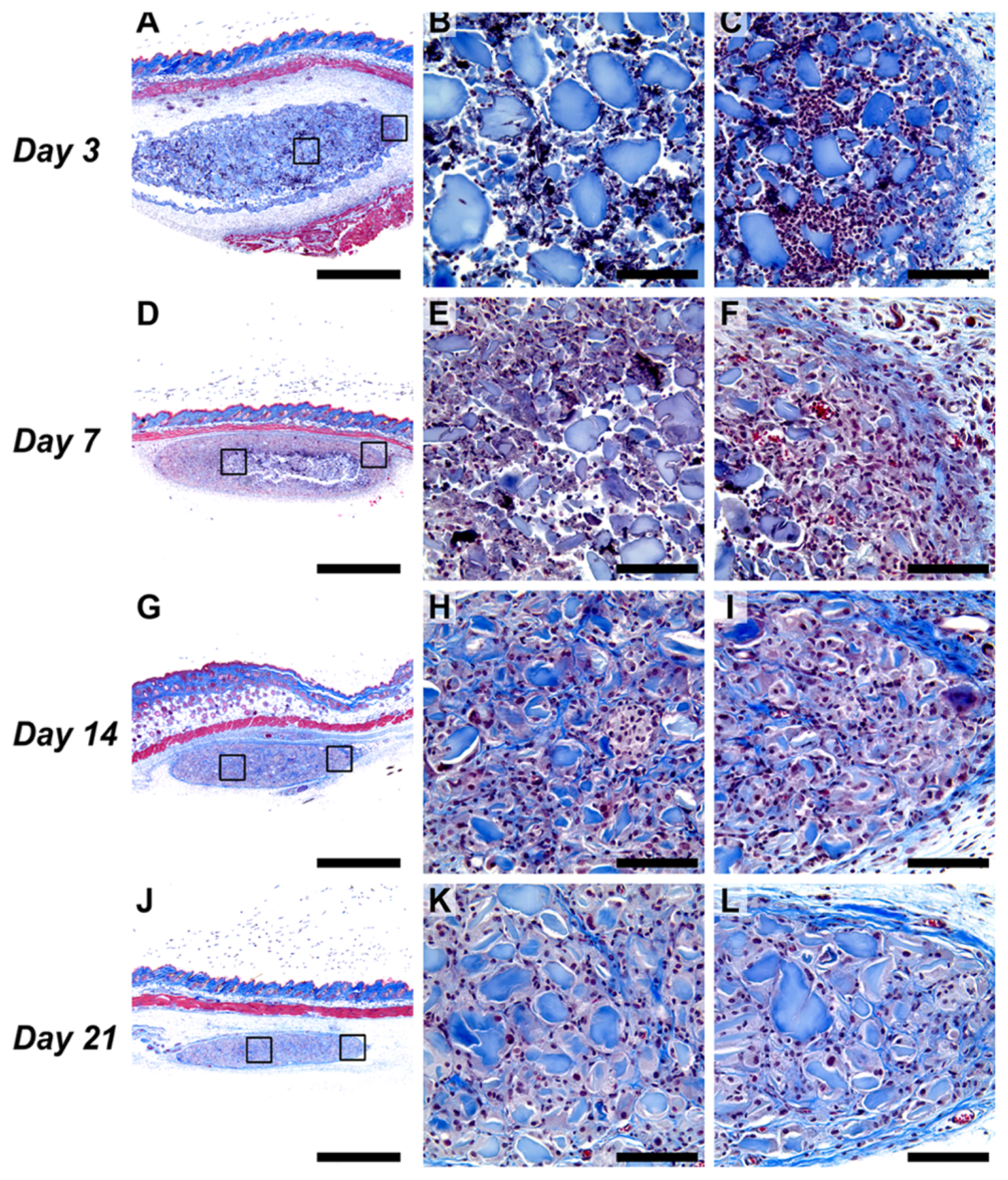
Histological analysis of l-NIL-MDP hydrogel implants extracted 3–21 days post-subcutaneous injection in mice (Masson’s trichrome stained). (A–C) l-NIL-MDP hydrogel 3 days post-injection, with (A) showing a 4× panorama and (B) and (C) showing representative zoomed 40× images of the core (B) followed by the periphery of the implant (C), denoted by the black boxes on the main image (same pattern follows in all series). (D–F) hydrogel 7 days post-injection; (G–I) hydrogel 14 days post-injection; (J–L) hydrogel 21 days post-injection. Scale bars are 1 mm in all 4× panoramas and 0.1 mm in all 40× images.
In general, the hydrogel bolus is also observed to steadily decrease in overall size with each passing week (Figure 6A,D,G,J), marking the natural biodegradation and remodeling of the material that can also be observed by increasing local collagen deposition (shown in blue) within and around the implant over time. These histology data show a degradation profile that is slower than other classes of MDPs studied in prior investigations, such as fast-degrading anionic or neutral MDPs,19 as the l-NIL-MDP hydrogel implants are still observable even 3 weeks post-injection. A slow degradation rate suggests that the hydrogel, and its biological effect, will persist in its injected location for multiple weeks. This is supported by the observation that at every time point, including the longest time point studied (day 21), large islets of what appear to be pristine, noninfiltrated l-NIL-MDP hydrogel are still present and have not yet been degraded by interacting cells (Figure 6J–L).
Evaluation of iNOS Inhibitory Activity in Vivo.
Immunohistochemistry staining was performed on hydrogel implant tissue sections to further study in vivo properties of the l-NIL-MDP compared to related cationic MDPs. Antinitrotyrosine immunostaining was used to assess relative levels of nitrosylation of tyrosine within and around the subcutaneously injected hydrogel implants as an indirect marker of iNOS activity (Figure 7). It is known that the formation of nitric oxide (NO) and other nitrogen oxides in vivo can lead to observable post-translation modifications of peptides and proteins.43 For example, surface-exposed 3-nitrotyrosine can be used as a marker of cell or tissue inflammation and damage by reactive nitrogen and oxygen species, such as NO.44 The higher the local levels of iNOS activity and NO production, the greater should be the density of nitrosylated tyrosine. Therefore, visualization of nitrotyrosine levels by targeted antibodies can serve as a general indication of in vivo iNOS activity or in the case of the l-NIL-MDP, local iNOS inhibition.
As shown in Figure 7, nitrotyrosine staining intensity shown in red was significantly lower in the l-NIL-MDP hydrogel implants compared to other cationic MDPs such as K2-MDP and R2-MDP. This result was striking, as all three cationic hydrogel materials are known to result in high levels of cellular infiltration indicative of similar initial immune reactions by histology. Therefore, the nitrotyrosine intensity differences observed here cannot be attributed to differences in cellular density. This result is most dramatic in the day 7 post-injection images shown in Figure 7D–F, as nitrosylation of tyrosine residues is a cumulative phenomenon that is known to increase with time of exposure to nitrosylating species, consistent with our observations here. Thus, it is even more striking that the l-NIL-MDP maintains low nitrotyrosine levels in and around the implant for over 7 days, even into the skin and muscle layer above the hydrogel bolus (Figure 7A,D). These data suggest durable iNOS inhibition from a single subcutaneous injection of the l-NIL-MDP, significantly reducing reactive nitrogen species in the surrounding environment and protecting labile species such as tyrosine from nitrosylation over time. Isotype control tests using rabbit IgG in place of rabbit antinitrotyrosine primary showed little to no nonspecific staining of antirabbit 568 nm secondary antibody, validating the significant staining intensity differences observed between materials (Figures S7–S9). While histological nitrosylation can be caused or inhibited by factors other than the iNOS pathway (e.g., reactive oxygen species levels), the results we see here are highly suggestive that the l-NIL-MDP is operating via a similar mechanism of action to the selective iNOS inhibitor l-NIL.
Furthermore, staining intensity increased in inverse correlation to previously evaluated iNOS inhibition efficacy in vitro for each material (Figure 7G,H), showing excellent agreement between two separate studies. The R2-MDP showed the lowest iNOS inhibition in vitro (Figure 5) and conversely the highest nitrotyrosine staining in vivo (Figure 7), the K2-MDP showed both moderate iNOS inhibition and nitrotyrosine intensity, and finally the l-NIL-MDP showed both the greatest degree of iNOS inhibition among the tested cationic hydrogels as well as the lowest degree of nitrotyrosine staining in vivo (Figure 7H). These highly correlated results demonstrate that the l-NIL-MDP hydrogel displays the desired bioactivity in various biological systems, validating our system design and experimental hypothesis.
Finally, the systemic effect of the l-NIL-MDP in vivo was evaluated by serum vascular endothelial growth factor (VEGF) quantification in tumor-bearing mice. Aberrant iNOS activity and NO production observed in various cancer models are known to increase VEGF secretion, such that inhibition or genetic knockdown of iNOS can decrease VEGF levels.27,45,46 ELISA quantification of the undiluted serum from mice injected intratumorally with either HBSS buffer control, soluble l-NIL, K2-MDP hydrogel, or l-NIL-MDP hydrogel showed that only the l-NIL-MDP significantly reduced systemic VEGF levels 5 days post-injection, returning them to basal levels observed in naïve mice (Figure S5). These results further demonstrate that the l-NIL-MDP biomaterial possesses inherent bioactivity in vivo and can generate a systemic response that was not observed with either K2-MDP or l-NIL alone.
CONCLUSIONS
In this study, a peptide biomaterial (l-NIL-MDP) was synthesized and characterized for its ability to incorporate a druglike functionality via side-chain modification while maintaining its ability to self-assemble into nanofibers and form a hydrogel. This bioactive peptide hydrogel showed excellent mechanical properties ideal for syringe injection and can readily interface with biological systems, facilitating cell adhesion and proliferation in vitro and cellular infiltration upon injection in vivo. The hydrogel displayed strong bioactivity by the Greiss assay analysis of iNOS knockdown in vitro, protected against tyrosine nitrosylation in vivo as observed by immunostaining of subcutaneous implants, and reduced systemic VEGF levels in sera from tumor-bearing mice. These results confirmed our hypothesis that the chemical conversion of a peptide-based scaffold to mimic a charged drug moiety would neither interfere with the bulk supramolecular chemistry and material properties of the system nor destroy the bioactivity of the original small-molecule drug. Future questions, however, can be investigated concerning the biological mechanism of such peptide-based iNOS inhibition such as the mechanisms of in vivo material degradation and drug release, cell membrane interactions, or intracellular functions.
In future treatment efficacy tests, the l-NIL-MDP may show unique utility over current combination treatment strategies that utilize the small-molecule drug l-NIL, as the material can potentially be used to create an extended systemic response with only a single injection. We hope to combine the useful properties of the l-NIL-MDP as a bioactive carrier material with additional loaded agents, creating extended-release, multicomponent combination therapies. The material design remains cationic and, thus, allows for the noncovalent incorporation of other anionic agents within the hydrogel, exactly analogous to past hydrogels used by our lab to intratumorally deliver potent anionic cancer immunotherapy drugs such as STING agonists.23 Yet what is fundamentally different is that now, what was previously only a carrier vehicle for the loading of exogenous drugs can display its own inherent axis of bioactivity. Fundamentally, this study establishes the successful development of a novel bioactive material, where neither the material properties of the hydrogel nor the pharmacological functions of the drug were compromised by their combination.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grants DE021798 and DE024173) and the Welch Foundation (C1557). D.G.L. was supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1842494. J.M.N. acknowledges financial support from the National Institute of General Medical Sciences T32 predoctoral training grant (T32GM088129) and the National Institute of Dental & Craniofacial Research F31 NRSA training grant (F31DE026682) both of the National Institutes of Health. A.G.S. acknowledges support from the Caroline Weiss Law Endowment for Academic Excellence; the Owens Foundation; and grants from the Cancer Research Institute (Team Strategy Grant) and the National Institutes of Health (NCI/NIDCR 1U01DE028233-01). T.L.L.-S. was supported by the Mexican National Council for Science and Technology (CONACyT) Ph.D. Scholarship Program (678341). This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.9b01447.
Expanded peptide synthesis and purification methods; MALDI-TOF mass spectroscopy spectra; quantification of live cell density; quantification of cell density; blood vessel immunostaining of L-NIL-MDP hydrogel implants; VEGF serum levels in mice bearing B16-F0 melanoma tumors; antinitrotyrosine immunostaining of inflamed B16-F0 tumor; day 3 L-NIL-MDP isotype control panorama; day 3 K2-MDP isotype control panorama; day 3 R2-MDP isotype control panorama; raw greiss assay results (Figures S1–S10); antibodies used for immunostaining protocols (Table S1) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Baldwin AD; Kiick KL Polysaccharide-Modified Synthetic Polymeric Biomaterials. Biopolymers 2010, 94, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ali OA; Huebsch N; Cao L; Dranoff G; Mooney DJ Infection-mimicking materials to program dendritic cells in situ. Nat. Mater 2009, 8, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dharmaraj N; Piotrowski SL; Huang C; Newton JM; Golfman LS; Hanoteau A; Koshy ST; Li AW; Pulikkathara MX; Zhang B; Burks JK; Mooney DJ; Lei YL; Sikora AG; Young S Anti-tumor immunity induced by ectopic expression of viral antigens is transient and limited by immune escape. OncoImmunology 2019, 8, No. e1568809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Alexander A; Ajazuddin; Khan J; Saraf S; Saraf S Poly(ethylene glycol)–poly(lactic-co-glycolic acid) based thermosensitive injectable hydrogels for biomedical applications. J. Controlled Release 2013, 172, 715–729. [DOI] [PubMed] [Google Scholar]
- (5).Cheng E; Xing Y; Chen P; Yang Y; Sun Y; Zhou D; Xu L; Fan Q; Liu D A pH-Triggered, Fast-Responding DNA Hydrogel. Angew. Chem., Int. Ed 2009, 48, 7660–7663. [DOI] [PubMed] [Google Scholar]
- (6).Cui H; Webber MJ; Stupp SI Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen J; Pompano RR; Santiago FW; Maillat L; Sciammas R; Sun T; Han H; Topham DJ; Chong AS; Collier JH The use of self-adjuvanting nanofiber vaccines to elicit high-affinity B cell responses to peptide antigens without inflammation. Biomaterials 2013, 34, 8776–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Collier JH; Segura T Evolving the use of peptides as components of biomaterials. Biomaterials 2011, 32, 4198–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bookstaver ML; Tsai SJ; Bromberg JS; Jewell CM Improving Vaccine and Immunotherapy Design Using Biomaterials. Trends Immunol. 2018, 39, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Leach DG; Young S; Hartgerink JD Advances in immunotherapy delivery from implantable and injectable biomaterials. Acta Biomater. 2019, 88, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Majumder P; Baxa U; Walsh STR; Schneider JP Design of a Multicompartment Hydrogel that Facilitates Time-Resolved Delivery of Combination Therapy and Synergized Killing of Glioblastoma. Angew. Chem., Int. Ed 2018, 57, 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Song H; Huang P; Niu J; Shi G; Zhang C; Kong D; Wang W Injectable polypeptide hydrogel for dual-delivery of antigen and TLR3 agonist to modulate dendritic cells in vivo and enhance potent cytotoxic T-lymphocyte response against melanoma. Biomaterials 2018, 159, 119–129. [DOI] [PubMed] [Google Scholar]
- (13).Nagai Y; Unsworth LD; Koutsopoulos S; Zhang S Slow release of molecules in self-assembling peptide nanofiber scaffold. J. Controlled Release 2006, 115, 18–25. [DOI] [PubMed] [Google Scholar]
- (14).Medina SH; Li S; Howard OMZ; Dunlap M; Trivett A; Schneider JP; Oppenheim JJ Enhanced immunostimulatory effects of DNA-encapsulated peptide hydrogels. Biomaterials 2015, 53, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nagy-Smith K; Yamada Y; Schneider JP Protein release from highly charged peptide hydrogel networks. J. Mater. Chem. B 2016, 4, 1999–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yamada Y; Patel NL; Kalen JD; Schneider JP Design of a Peptide-Based Electronegative Hydrogel for the Direct Encapsulation, 3D Culturing, in Vivo Syringe-Based Delivery, and Long-Term Tissue Engraftment of Cells. ACS Appl. Mater. Interfaces 2019, 11, 34688–34697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Aulisa L; Dong H; Hartgerink JD Self-assembly of multidomain peptides: sequence variation allows control over crosslinking and viscoelasticity. Biomacromolecules 2009, 10, 2694–2698. [DOI] [PubMed] [Google Scholar]
- (18).Moore AN; Hartgerink JD Self-Assembling Multidomain Peptide Nanofibers for Delivery of Bioactive Molecules and Tissue Regeneration. Acc. Chem. Res 2017, 50, 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lopez-Silva TL; Leach DG; Li IC; Wang X; Hartgerink JD Self-Assembling Multidomain Peptides: Design and Characterization of Neutral Peptide-Based Materials with pH and Ionic Strength Independent Self-Assembly. ACS Biomater. Sci. Eng 2019, 5, 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kumar VA; Shi S; Wang BK; Li I-C; Jalan AA; Sarkar B; Wickremasinghe NC; Hartgerink JD Drug-triggered and cross-linked self-assembling nanofibrous hydrogels. J. Am. Chem. Soc 2015, 137, 4823–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li IC; Moore AN; Hartgerink JD ″Missing Tooth″ Multidomain Peptide Nanofibers for Delivery of Small Molecule Drugs. Biomacromolecules 2016, 17, 2087–2095. [DOI] [PubMed] [Google Scholar]
- (22).Wickremasinghe NC; Kumar VA; Hartgerink JD Two-Step Self-Assembly of Liposome-Multidomain Peptide Nanofiber Hydrogel for Time-Controlled Release. Biomacromolecules 2014, 15, 3587–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Leach DG; Dharmaraj N; Piotrowski SL; Lopez-Silva TL; Lei YL; Sikora AG; Young S; Hartgerink JD STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials 2018, 163, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Moore WM; Webber RK; Jerome GM; Tjoeng FS; Misko TP; Currie MG L-N6-(1-Iminoethyl)lysine: A Selective Inhibitor of Inducible Nitric Oxide Synthase. J. Med. Chem 1994, 37, 3886–3888. [DOI] [PubMed] [Google Scholar]
- (25).Sikora AG; Gelbard A; Davies MA; Sano D; Ekmekcioglu S; Kwon J; Hailemichael Y; Jayaraman P; Myers JN; Grimm EA; Overwijk WW Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin. Cancer Res 2010, 16, 1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tang W; Li H; Poulos TL; Silverman RB Mechanistic studies of inactivation of inducible nitric oxide synthase by amidines. Biochemistry 2015, 54, 2530–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fukumura D; Kashiwagi S; Jain RK The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521. [DOI] [PubMed] [Google Scholar]
- (28).Jayaraman P; Parikh F; Lopez-Rivera E; Hailemichael Y; Clark A; Ma G; Cannan D; Ramacher M; Kato M; Overwijk WW; Chen S-H; Umansky VY; Sikora AG Tumor-Expressed Inducible Nitric Oxide Synthase Controls Induction of Functional Myeloid-Derived Suppressor Cells through Modulation of Vascular Endothelial Growth Factor Release. J. Immunol 2012, 188, 5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jenkins DC; Charles IG; Thomsen LL; Moss DW; Holmes LS; Baylis SA; Rhodes P; Westmore K; Emson PC; Moncada S Roles of nitric oxide in tumor growth. Proc. Natl. Acad. Sci. U.S.A 1995, 92, 4392–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mannick JB; Schonhoff C; Papeta N; Ghafourifar P; Szibor M; Fang K; Gaston B S-Nitrosylation of mitochondrial caspases. J. Cell Biol 2001, 154, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang XM; Xu Q Metastatic melanoma cells escape from immunosurveillance through the novel mechanism of releasing nitric oxide to induce dysfunction of immunocytes. Melanoma Res. 2001, 11, 559–567. [DOI] [PubMed] [Google Scholar]
- (32).Fukumura D; Gohongi T; Kadambi A; Izumi Y; Ang J; Yun C-O; Buerk DG; Huang PL; Jain RK Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dong H; Paramonov SE; Aulisa L; Bakota EL; Hartgerink JD Self-Assembly of Multidomain Peptides: Balancing Molecular Frustration Controls Conformation and Nanostructure. J. Am. Chem. Soc 2007, 129, 12468–12472. [DOI] [PubMed] [Google Scholar]
- (34).Li I-C; Hartgerink JD Covalent Capture of Aligned Self-Assembling Nanofibers. J. Am. Chem. Soc 2017, 139, 8044–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bryan NS; Grisham MB Methods to detect nitric oxide and its metabolites in biological samples. Free Radicals Biol. Med 2007, 43, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bakota EL; Sensoy O; Ozgur B; Sayar M; Hartgerink JD Self-Assembling Multidomain Peptide Fibers with Aromatic Cores. Biomacromolecules 2013, 14, 1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Nathan CF; Hibbs JB Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol 1991, 3, 65–70. [DOI] [PubMed] [Google Scholar]
- (38).MacMicking J; Xie Q -w.; Nathan, C. Nitric Oxide and Macrophage Function. Annu. Rev. Immunol 1997, 15, 323–350. [DOI] [PubMed] [Google Scholar]
- (39).Aldridge C; Razzak A; Babcock TA; Helton WS; Espat NJ Lipopolysaccharide-stimulated RAW 264.7 macrophage inducible nitric oxide synthase and nitric oxide production is decreased by an omega-3 fatty acid lipid emulsion. J. Surg. Res 2008, 149, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schneider GB; English A; Abraham M; Zaharias R; Stanford C; Keller J The effect of hydrogel charge density on cell attachment. Biomaterials 2004, 25, 3023–3028. [DOI] [PubMed] [Google Scholar]
- (41).Kim YH; Baek NS; Han YH; Chung M-A; Jung S-D Enhancement of neuronal cell adhesion by covalent binding of poly-d-lysine. J. Neurosci. Methods 2011, 202, 38–44. [DOI] [PubMed] [Google Scholar]
- (42).Lam AT-L; Li J; Chen AK-L; Reuveny S; Oh SK-W; Birch WR Cationic surface charge combined with either vitronectin or laminin dictates the evolution of human embryonic stem cells/microcarrier aggregates and cell growth in agitated cultures. Stem Cells Dev. 2014, 23, 1688–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ischiropoulos H Biological selectivity and functional aspects of protein tyrosine nitration. Biochem. Biophys. Res. Commun 2003, 305, 776–783. [DOI] [PubMed] [Google Scholar]
- (44).Ahsan H 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol 2013, 74, 1392–1399. [DOI] [PubMed] [Google Scholar]
- (45).Ambs S; Merriam WG; Ogunfusika MO; Bennett WP; Ishibe N; Hussain SP; Tzeng EE; Geller DA; Billiar TR; Harris CC p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat. Med 1998, 4, 1371. [DOI] [PubMed] [Google Scholar]
- (46).Song Z-J; Gong P; Wu Y-E Relationship between the expression of iNOS, VEGF, tumor angiogenesis and gastric cancer. World J. Gastroenterol 2002, 8, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.