

Abstract
In situ forming implants (ISFIs) allow for a high initial intratumoral concentration and sustained release of the chemotherapeutic. However, clinical translation is impeded primarily due to limited drug penetration from the tumor/boundary interface and poor intratumoral drug retention. Therapeutic ultrasound (TUS) has become a popular approach to improving drug penetration of transdermal devices and increased cellular drug uptake of nanoparticles. These effects are driven by the mechanical and thermal bioeffects associated with TUS. In this study, we characterize the released drug penetration, retention, and overall therapeutic response when exposing ISFI to the combination of the mechanical and thermal effects of TUS (C-TUS). ISFIs were intratumorally injected into subcutaneous murine tumors then exposed to C-TUS (exposure: 5 min, duty factor: 0.33, frequency: 3 MHz, intensity: 2.2 W/cm2, pulse duration: 2 ms, pulse repetition frequency: 165 Hz, effective radiating area: 5 cm2, energy delivered: 896 J, time average intensity: 0.88 W/cm2). Tumors treated with the combination of ISFI + C-TUS demonstrated a 2.5-fold increase in maximum drug penetration and a 3-fold increase in drug retention at 5- and 8-days post-injection, respectively, compared to ISFIs without US exposure. These improvements in drug penetration and retention translated into enhanced therapeutic responses. Tumors treated with ISFI + C-TUS showed a 62.6% reduction in tumor progression, a 50.0% increase in median survival time, and a 26.6% increase in necrotic percentage compared to ISFIs without US exposure. Combining intratumoral ISFIs with US may be beneficial for addressing some long-standing challenges with local drug delivery in cancer treatment and may serve as a viable noninvasive method to improve the poor clinical success of local drug delivery systems.
1. Introduction:
Systemic drug delivery systems (SDDS) have had limited clinical success as a neoadjuvant, standard, or adjuvant treatments for several types of cancers, such as breast, pancreatic, colorectal, and non-small-cell lung cancers[1–4]. This is attributed to the mononuclear phagocytic system, off-target toxicity leading to unpredictable side effects, and low intratumoral drug levels[5–8]. To combat these issues, local drug delivery systems (LDDS) allow for high initial drug levels at the tumor site without the associated toxicities seen with SDDS [9,10]. These systems have been used in treatments such as adjuvant chemotherapy following brain tumor resection[11], transarterial chemoembolization of liver cancer[12–14], intraperitoneally injected microparticles of abdominal cancers[10], and intratumoral immunotherapy[15]. Promising preclinical research has not translated into successful clinical trials mainly due to poor drug penetration from the implanttumor boundary and limited intratumoral drug retention[11,16–22]. The Gliadel® wafer, which is used as an adjuvant chemotherapy following brain tumor resection, and OncoGel™, which delivers paclitaxel locally to esophageal cancer, has been shown to have therapeutic drug levels ~2 mm away from the implant/tumor interface[11,17,23,24]. Preclinical research has shown local delivery systems used in brain and breast cancer result in limited drug retention (<10% after 2 weeks)[17,19]. Poor retention has been attributed to increased cellular drug metabolism, overexpression of drug efflux transporters, and removal of the drug through intratumoral vessels[8,16,20,25,26]. Overall, intratumoral implants have been overlooked as a treatment for solid tumors due to the lack of clinical success attributed to poor drug penetration and retention.
To overcome these challenges, this study characterized the effects of released drug penetration, retention, and overall therapeutic response when exposing an LDDS to therapeutic ultrasound (TUS). Pressure waves generated by TUS are known to induce several mechanical and thermal bioeffects[27–32] including enhanced drug uptake through cavitation[33,34], increased drug penetration via disruption of cellular junctions in transdermal drug delivery systems[35,36], and higher drug retention through inhibition of drug efflux transporters[37,38]. Moreover, the Acoustic Radiation Force (ARF) and the associated secondary waves, generated from the interaction of the TUS primary waves and ISFIs, may be coupled with an increase in tissue temperature due to the absorption of the waves. Here, we examined the mechanical and thermal bioeffects combined and separately to investigate their effect on drug distribution and retention released from the LDDS. To achieve this, we used a low duty cycle/ high exposure time TUS to produce predominately mechanical bioeffects (ISFI + M-TUS) and a hot pack to produce only thermal bioeffects (ISFI + HT). A high duty cycle/ low exposure time TUS was used to produce a combination of mechanical and thermal bioeffects (ISFI + C-TUS).
The LDDS used in this study is a phase-sensitive in situ forming implant (ISFI). In ISFI systems, the drug is dissolved in a biocompatible polymer – organic solvent solution. Once injected into an aqueous environment, the solution forms a solid drug depot via phase inversion (slow precipitation of insoluble polymer due to solvent exchange with surrounding water) allowing sustained release of the drug for months[39–46]. ISFIs offer several advantages including a minimally invasive injection (e.g. no surgical implantation at the tumor site), complete in situ degradation, and ease of manufacturing. Clinically used ISFIs are SDDS and include Lupron Depot® used for palliative treatment of prostate cancer, Zoladex® used for the treatment of breast cancer, and Somatuline® used for acromegaly[47]. Since the goal of these formulations is sustained delivery, they do not possess the drug penetration limitation discussed above. An example of an LDDS ISFI is OncoGel™ that delivers paclitaxel locally to esophageal cancer. The phase II trial that used OncoGel™ was terminated since it did not show any impact on overall tumor response[47]. This has been attributed, in part, to poor drug distribution[17,23].
The objective of this work was to use TUS to overcome this critical limitation of ISFIs. The ISFI used in this study is composed of poly(lactic-co-glycolic) acid (PLGA), a hydrolytically biodegradable polymer, and Doxorubicin (Dox), a commonly used chemotherapeutic, co-dissolved in 1-methyl-2-pyrrolidinone (NMP), a water-miscible organic solvent[48–51]. Here, we exposed the ISFI to TUS and investigated the released drug release, distribution and retention and the corresponding therapeutic response in a subcutaneous murine tumor model.
2. Materials and Methods:
2.1. Materials
1-N-methyl-2-pyrrolidinone (NMP) obtained from Sigma–Aldrich (St. Louis, MO). Doxorubicin HCl (Dox) was obtained from LC Laboratories (Woburn, MA). Poly(DL-lactic-co-glycolic) (PLGA, acid capped, 75:25, MW 28.8 kDa, inherent viscosity 0.28 dL/g) was obtained from Evonik Corp (Parsippany, NJ). A commercially available physical therapy US unit, Omnisound® 3000, was obtained from Accelerated Care Plus (Reno, NV). Thermocouples, with type K calibration 36-gauge wire, were obtained from Evolution Sensors and Controls, LLC (West Deptford, NJ). All materials were used as received.
2.2. ISFI solution preparation
ISFI solutions were composed of PLGA and Dox co-dissolved in NMP. Solutions of 39 wt.% PLGA and 1 wt.% Dox were added to 60 wt.% NMP in 4 mL glass scintillation vials. The components of the ISFI solution were mixed overnight inside an incubator shaker at 37°C. ISFI solutions were used within one day of mixing[48,52–57].
2.3. In vitro ISFI Dox release
ISFIs (45–50 mg) were injected into 70 mL of phosphate buffered saline (PBS). Implant weight was collected at the initiation of the experiment. The temperature of the PBS was recorded with a thermocouple for the duration of the experiment. Immediately after injection, the ISFI was exposed to TUS, which was placed 0.5 cm above the ISFI. A schematic of the experimental setup is shown in supplemental figure S1. Parameters for C-TUS were chosen based on prior experiments in which examined the effects of ultrasound on the distribution of dyes released from ISFIs injected into hydrogels and subcutaneously in mice [51,58]. Parameters for M-TUS were chosen to investigate the mechanical bioeffects seen with C-TUS. Details of these TUS exposures are seen in supplemental table S1. Control implants received a sham TUS, where the TUS was placed above the ISFI but not turned on. 1 mL of bath side solutions were collected at 2 hr, 5 hr, and 2 days post US exposure and replaced with 1 mL of fresh PBS. After 2 days, ISFIs were transferred into glass scintillation vials containing 10 mL of PBS. At 2, 3, 5,7, 11, 17, and 21 days, 1 mL of the bath side solution was collected and the rest of the bath side solution was aspirated and replaced with 10 mL of fresh PBS. Fluorescent intensity of collected samples was measured using a plate reader at an excitation/emission wavelength of 495/595 nm (Tecan Ltd, Infinite 200 series) to measure Dox released from the ISFI. Intensity values were compared to a standard curve of Dox in PBS to obtain the amount of Dox within a collected sample. These values were normalized to the actual Dox loading obtained from initial implant mass measurements to obtain a cumulative release[50,51,54].
2.4. Tumor cells
Human colorectal carcinoma, HCT-15 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD). The cells were maintained in RPMI-1640 media supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in an atmosphere of 5% CO2 at 37 °C. It is important to note that the HCT-15 cells overexpressed P-glycoprotein (Pgp), a drug efflux transporter, as documented by the National Cancer Insitutute[59].
2.5. Subcutaneous HCT-15 mouse tumor inoculation
All experiments involving mice were approved by the Case Western Reserve University Institutional Animal Care and Use Committee. 4-week-old BALB/c nude mice (n=5/group) were purchased from the Athymic Animal & Xenograft Core facility at CWRU. All animal experiments were performed in accordance with relevant guidelines and regulations of Case Western Reserve University Institutional Animal Care and Use Committee. Before the start of experiments, mice were kept in an aseptic environment for 7 days to acclimate to the new environment. To inoculate tumors, HCT-15 cells suspended in 100 μL of 1X PBS were subcutaneously injected into the dorsal right flank of each mouse at a concentration of 5 ×106 cells/mL. Tumors were allowed to grow for 10 days. ISFI solution (45–50 mg) was injected directly into the tumor via a 21-gauge needle (Figure 1A). The syringe and needle containing the ISFI solutions were measured before and after each injection to ensure equivalent amounts of ISFI were delivered for each treatment. Diagnostic US images (Siemens S3000, frequency: 17 MHz, gain: 3 dB, dynamic range: 60, mechanical index: 1.1) were taken post-injection to ensure injections were at the center of the tumor (Figure 1B). ISFIs not centrally injected were excluded from the study, which can be seen in supplemental figure S3.
Figure 1:
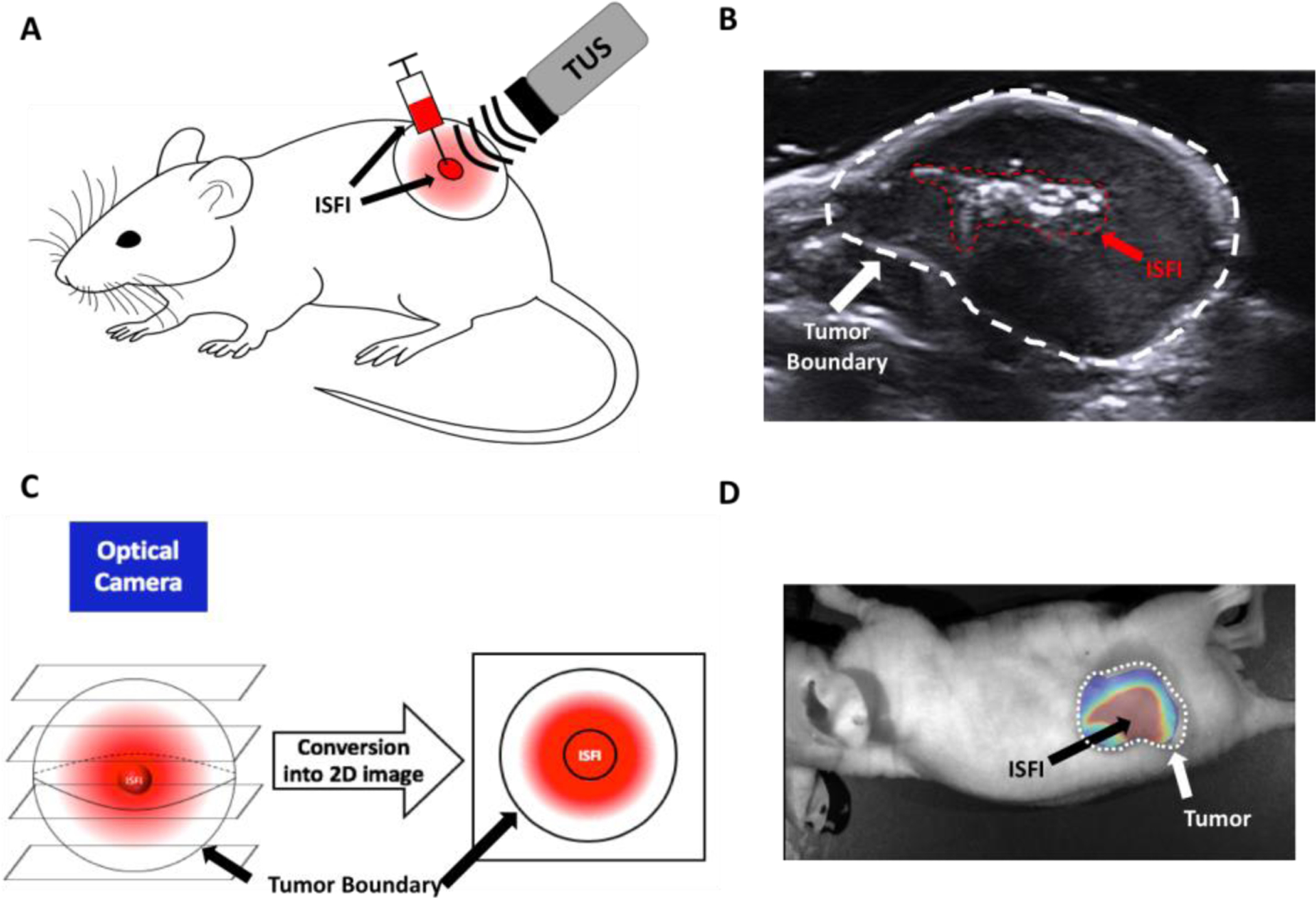
(A) Schematic of intratumoral ISFI injection then exposure to US treatment. (B) Diagnostic US image of the tumor after intratumoral ISFI injection to ensure central injection location. (C) Schematic of 3D distribution of ISFI becoming compressed into a 2D image for analysis. (D) Representative overlaid fluorescent image of intratumoral ISFI injection.
2.6. Dox release from intratumorally injected ISFI
After injection, the mice were divided randomly into four treatment groups: 1) no treatment control, 2) C-TUS 3) M-US or 4) 5 minutes of hyperthermia (HT). Each treatment was applied once at the beginning of the study. HT was applied using a nylon bag filled with ~70 g of jasmine rice, then placed on the tumor for 5 minutes. The bag was placed in a microwave and heated to approximately 60°C. This temperature was chosen to mimic a similar thermal profile rise seen with ISFI + C-TUS. Tumor temperatures were measured by a thermocouple inserted underneath the tumor for the duration of the treatment. To investigate the effects of each treatment on the release of Dox, ISFIs were removed from the tumor 1- and 8-days post-injection (n=3/group/time point). ISFIs were dissolved in 10 mL of NMP overnight. The fluorescence of Dox was read on a fluorescent microplate reader (Tecan Ltd, Infinite 200 series) at an excitation/emission wavelength of 495/595 nm. The fluorescent intensities were then compared to a standard curve of known Dox concentrations dissolved in NMP. The amount of Dox released was calculated from the actual loading of Dox during the injection. The standard curve for Dox in NMP can be seen in Figure S5.
2.7. Dox distribution within tumors
Fluorescent images of the tumors were taken on a fluorescent optical imaging system (CRI Maestro, Caliper Life Science) with a blue excitation (445–490 nm)/green emission (long pass 580 nm) filter exposed at 100 ms. Images were acquired immediately post ISFI injection, 1, 2, 3, 5, and 8 days after treatment. Group sizes are as followed: ISFI (n=10), ISFI + C-TUS (n=7), ISFI + HT (n=8), and ISFI + M-TUS (n=9). Images were analyzed in MATLAB (Mathworks) and ImageJ (NIH) to determine the total intratumoral Dox intensity, radial Dox distribution, and maximum Dox penetration. Images were exported into MATLAB and the background fluorescence, which was acquired from mice tumors with no ISFI injections was removed. These images were exported to ImageJ were a line intensity profile was used to determine the radial distribution from the center of the ISFI, where maximum Dox penetration was determined[50,58]. Radial distributions were normalized to the intensity at the center of the ISFI obtained directly after post-injection (time = 0 hr). Values below 0.1 normalized intensity in these radial distributions were disregarded due to poor signal to noise ratio. This is shown with a black line placed at the 0.1 value on the graphs seen in Figure 7. Penetration distances were determined from these radial distribution curves where the normalized Dox signal intensity reaches 0.1. Total intratumoral Dox intensity was determined by summing each intensity value in the radial distribution profiles. Total Dox intensity was normalized to the total intensity value obtained directly after ISFI injection (time = 0 hr). It is important to note that Dox distributes in all 3 dimensions from the ISFI. Accordingly, the 2D fluorescent images are obtained by compressing the z-plane Dox intensities (the Dox that distributes above and below the ISFI) along with the residual Dox in the ISFIs (Figure 1C). A representative 2D overlaid fluorescent image can be seen in figure 1D.
Figure 7:
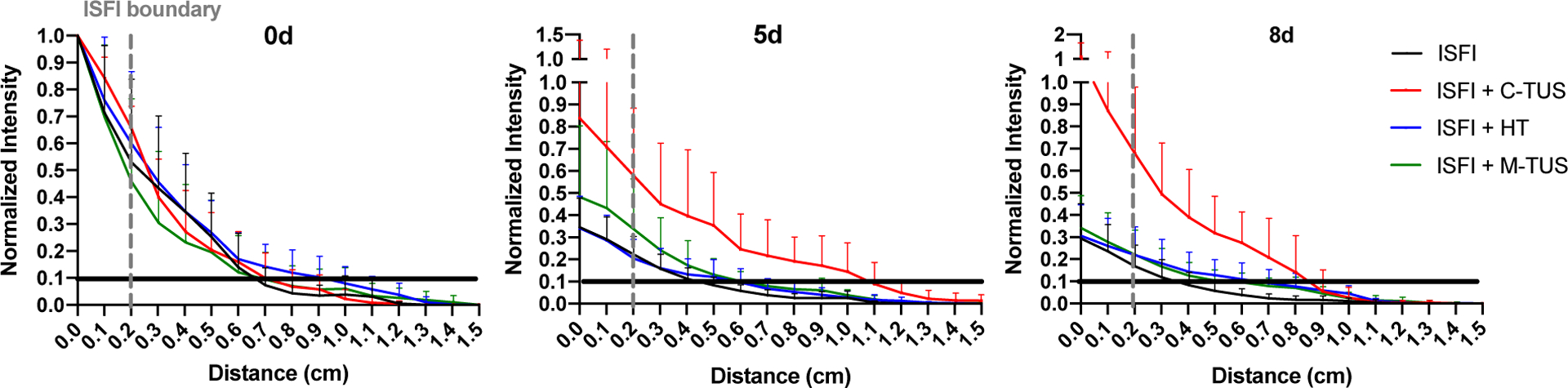
Radial Dox distribution from the center of intratumoral injected ISFI. ISFI + C-TUS showed an increase in maximum Dox penetration shown in Figure 7 at 5- and 8- days. Black lines were placed at 0.1 normalized intensity to ensure sufficient signal to noise ratio. The maximum penetration value was determined from this black line. The dashed gray line represents the edge of the intratumoral ISFI.
2.8. Therapeutic efficacy
Mouse tumor progression was measured using calipers and the volume was calculated using the formula: where, V = tumor volume in mm3, L = tumor length in mm and W = tumor width in mm. Group sizes are as follows: ISFI + C-TUS (n=9), ISFI alone (n=8), ISFI + M-TUS (n=7), ISFI + HT (n=5), Blank ISFI + C-TUS (n=4), C-TUS alone (n=5), HT alone (n=5). Tumor volumes were measured every other day after ISFI injection. For treatment groups where the tumor ulcerated, an adjusted tumor volume equation was used. Here, the tumor volume was calculated as the difference in the total tumor volume minus the volume of the ulcerated region. The ulcerated region, or necrotic tissue, was determined using calipers and calculated using the same volume formula described above. The survival of each mouse was evaluated over the course of 60 days after the ISFI injection. The endpoint for survival was chosen by a tumor volume cutoff of 2500 mm3 and a tumor burden score determined by the Institutional Animal Care and Use Committee (IACUC) at Case Western Reserve University. This score considers the appearance and well-being of the rodent, were above a certain score the mouse will be euthanized.
2.9. Histology
Tumors at the endpoint where harvested, fixed in paraformaldehyde, then frozen in OCT. Tumors were sliced at 10 μm thickness and stained with hematoxylin and eosin (n=4/group). Slices were imaged using a slide scanner (Olympus vs120). Images were exported into ImageJ, where the percentage of necrosis was calculated by dividing the area of necrosis by the total area of the slice.
2.10. Statistical analysis
Statistical analysis was performed using either a one-way or two-way ANOVA, assuming unequal variances between the two data sets in Prism 8. Significant differences among means of groups were evaluated using a Tukey multiple comparison test. The median survival for each animal was calculated from Prism. All data are reported as mean ± standard deviation except for survival data, where a single median survival number is shown.
3. Results:
3.1. In vitro Dox release from ISFI
The effects of US exposure were investigated on the modulation of Dox release in a PBS dissolution study over the course of 21 days. ISFI + C-TUS showed a 3.9 ± 0.1 °C increase in temperature from baseline (21.5 ± 0.2 °C) after 5 minutes and ISFI + M-TUSUS showed a 2.4 ± 0.3 °C increase in temperature from baseline (21.9 ± 0.1 °C) after 30 minutes (Figure 2A). ISFI, ISFI + C-TUS, ISFI + M-TUS showed Dox release of 37.5 ± 4.3%, 46.0 ± 3.0%, and 34.8 ± 2.0%, respectively, after 2 days. The average release after 21 days for ISFI, ISFI + C-TUS, ISFI + M-TUS were 53.4 ± 5.3%, 80.9 ± 5.3%, 56.5 ± 3.7%, respectively (Figure 2B). ISFI + C-TUS showed a significant (p < 0.05) increase in the release of Dox compared to ISFI + M-TUS at every time point over the 21 days.
Figure 2:
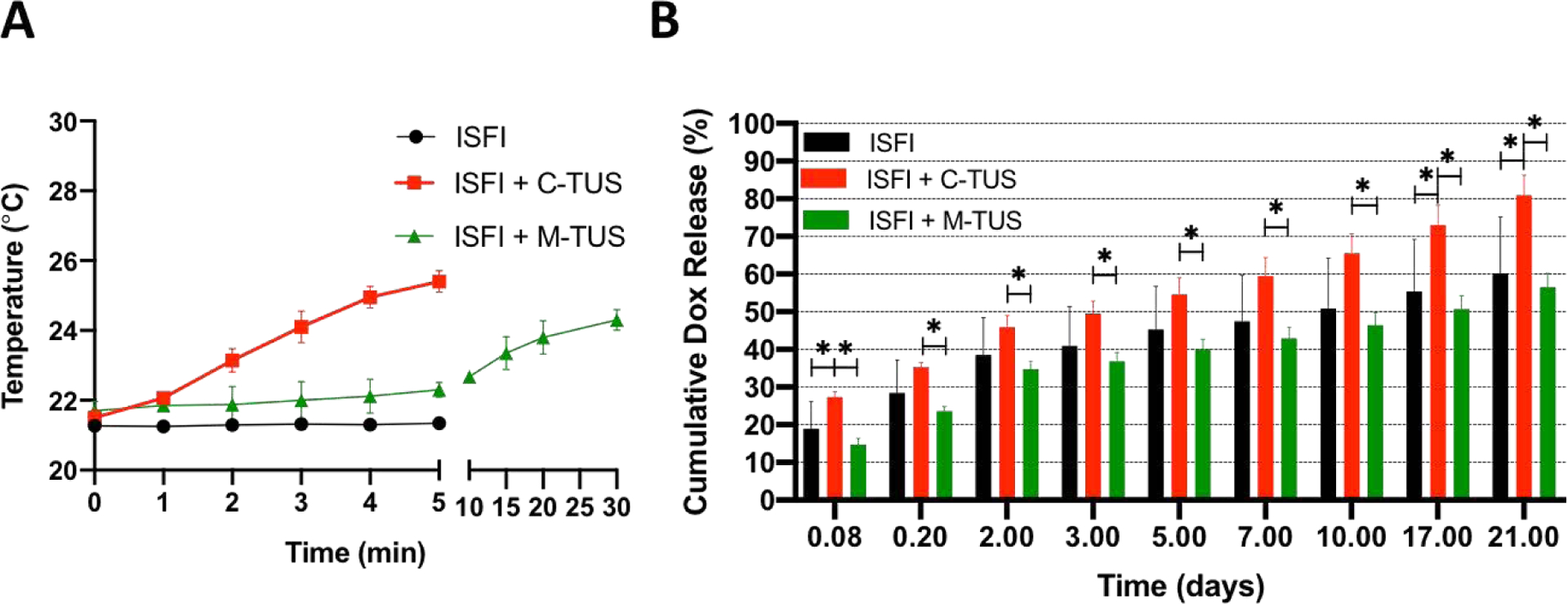
(A) PBS temperature changes due to TUS exposure of ISFI. (B) In vitro release of Dox with the application of US. ISFI + C-TUS showed an increased release of Dox over 21 days. N =7 for ISFI, n =4 for ISFI + C-TUS, and n=4 for ISFI + M-TUS. Error bars represent standard deviation * p < 0.05
3.2. Intratumoral Dox Release
Dox release from the ISFI was evaluated at 1- and 8-day post intratumoral injection as shown in Figure 3. Dox release from ISFI alone follows previously reported trends[60]. On day 1, there were no significant differences between any treatment groups compared to the ISFI alone. After 8 days, ISFI + HT and ISFI + M-TUS showed a non-significant decrease in the release of Dox compared to ISFI alone. At 8 days, ISFI + C-TUS showed a significant release of Dox compared to ISFI alone (96.3 ± 3.6% vs 67.2 ± 4.7%, respectively).
Figure 3:
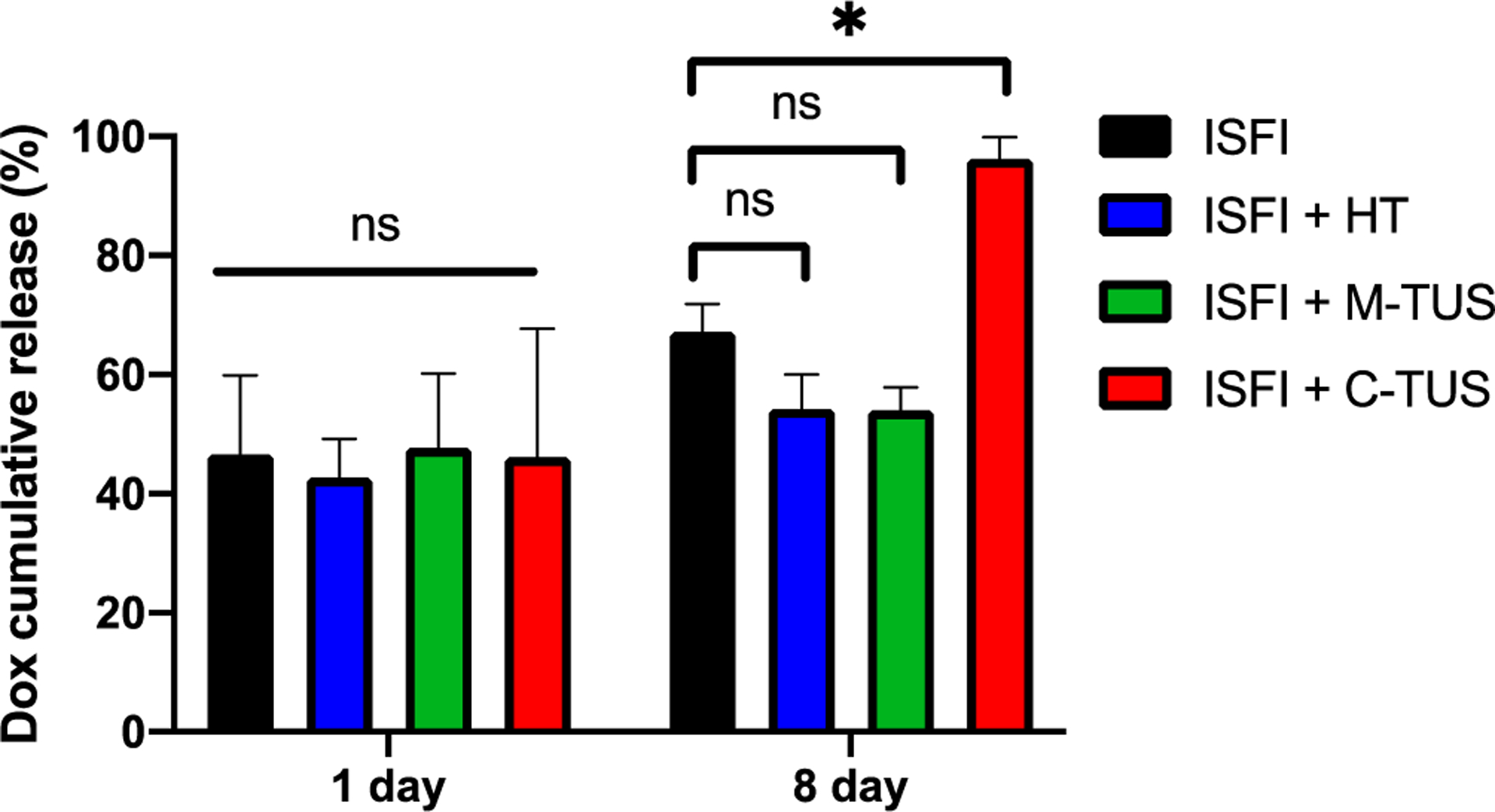
Intratumoral ISFI Dox release over 8 days. ISFI + C-TUS showed a significant increase in Dox release after 8 days. * p < 0.05
3.3. Tumor temperatures
Temperature changes from the various in vivo treatments are shown in Figure 4. All groups, except ISFI + M-TUS, showed equivalent final intratumoral temperatures after treatment. The change from the baseline temperature of the mice is shown in Table 1. Treatments with C-TUS showed greater changes in temperatures from baseline compared to HT treatment. This is due to a cold US gel being placed on the tumor prior to treatment, resulting in a reduced baseline temperature. As expected, ISFI + M-TUS showed the smallest change in intratumoral temperature with a final intratumoral temperature of 35.4 ± 1.2°C.
Figure 4:
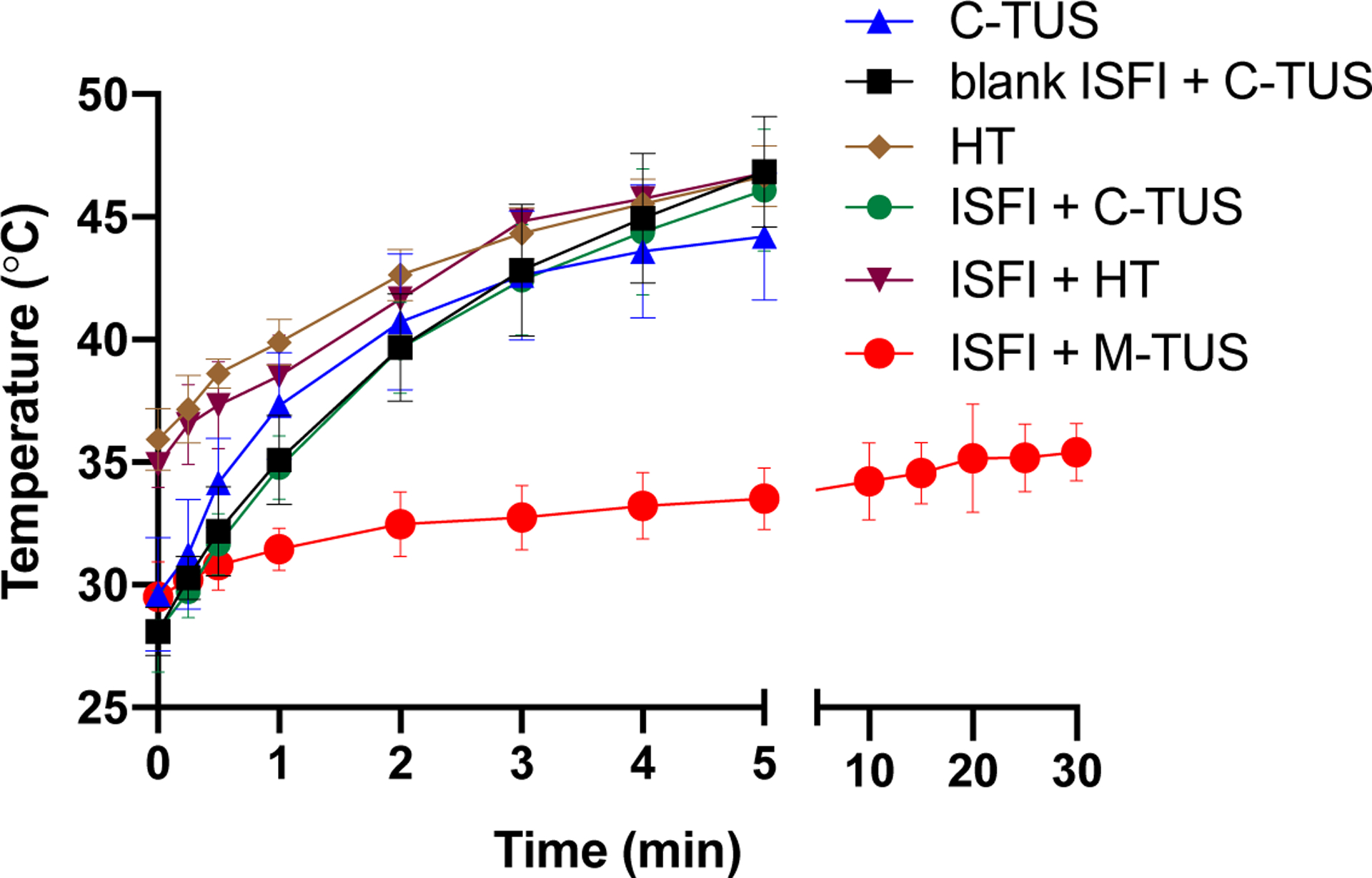
Intratumoral temperatures for all treatment groups. All groups except ISFI + M-TUS showed a final intratumoral temperature slightly above mild HT. The final temperature of ISFI + M-TUS was significantly below mild hyperthermia temperature (<43°C). C-TUS only and HT only are treatments done without ISFIs in the tumor.
Table 1:
Change in overall temperature from the baseline temperature from time = 0 to the end of the exposure.
| Treatment Group | ΔT from time = 0 |
|---|---|
| ISFI + C-TUS | 18.0 ± 3.2 |
| ISFI + HT | 11.9 ± 1.0 |
| ISFI + M-TUS | 5.9 ± 1.1 |
| Blank ISFI + C-TUS | 18.8 ± 1.5 |
| C-TUS | 14.6 ± 2.4 |
| HT | 10.7 ± 2.0 |
3.4. Dox distribution
Effects of US and HT exposure on the intratumoral Dox distribution were investigated through optical imaging. Representative fluorescent images and gross tumor images at 8 days are shown in figure 5. At 8 days, ISFI + C-TUS showed tumor ulcerations. ISFI + C-TUS showed a significantly (p < 0.05) greater total Dox intratumoral intensity over 8 days compared to all other groups as seen in figure 6. The greatest difference can be seen at 8 days with the ISFI + C-TUS having a normalized total intensity of 1.0 ± 0.4. ISFI alone, ISFI + HT, and ISFI + M-TUS showed a normalized total intensity of 0.3 ± 0.2, 0.4 ± 0.2, 0.4 ± 0.2, respectively. Radial Dox distribution can be seen in figure 7, where an equivalent distribution of Dox can be seen post-injection. ISFI + C-TUS showed an increased Dox distribution at 5 and 8 days leading to a significantly (p < 0.05) greater maximum Dox penetration compared to ISFI alone (figure 8). The greatest difference can be seen at 5 days, where ISFI + C-TUS showed a 2.5-fold increase in the maximum Dox penetration compared to ISFI alone.
Figure 5:
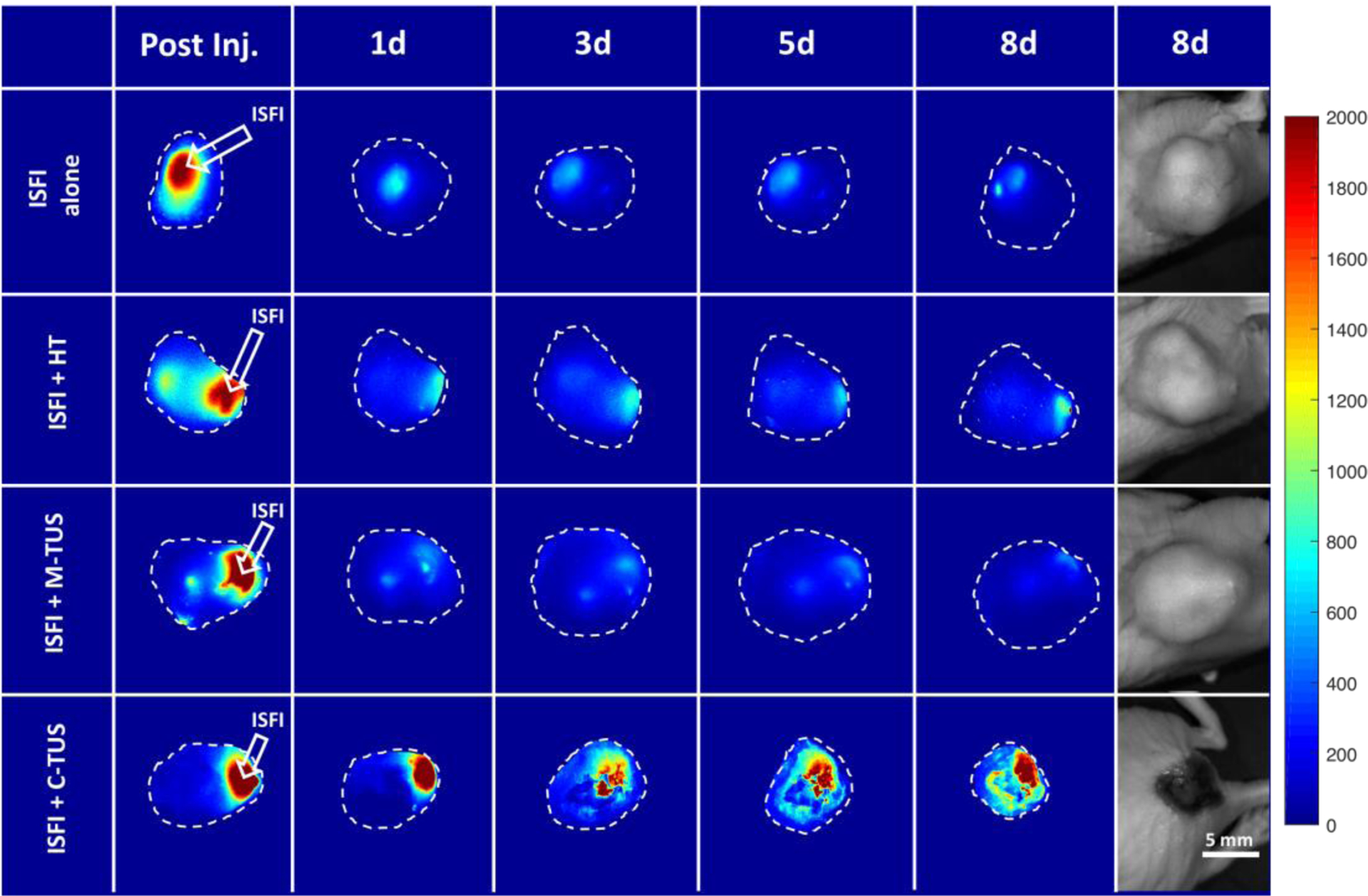
(A) Representative optical fluorescent images of intratumoral injected ISFIs over 8 days. ISFI + C-TUS shows significantly greater retention and maximum penetration of Dox over time. Total intensity and maximum penetration can be seen in the following three figures. (B) Gross representative images of tumors at 8 days. Tumor ulcerations can be seen with the ISFI + C-TUS treatment suggesting greater therapeutic response. Scale bar = 0.5 cm. The fluorescent intensity scale is arbitrary units.
Figure 6:
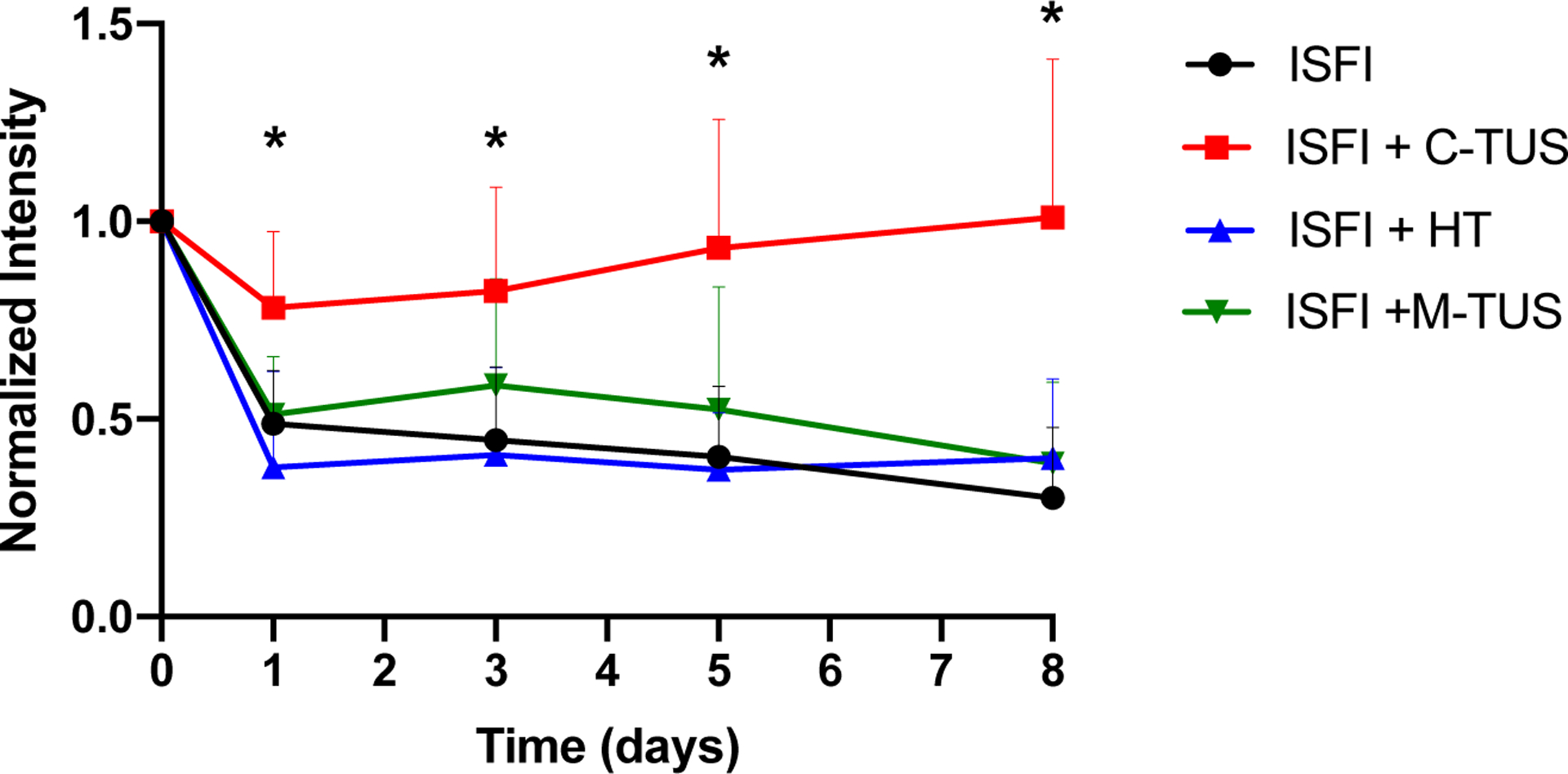
Total intratumoral Dox intensity over time. ISFI + C-TUS showed significant retention in the total Dox intensity over time compared to all other groups. * p < 0.05.
Figure 8:
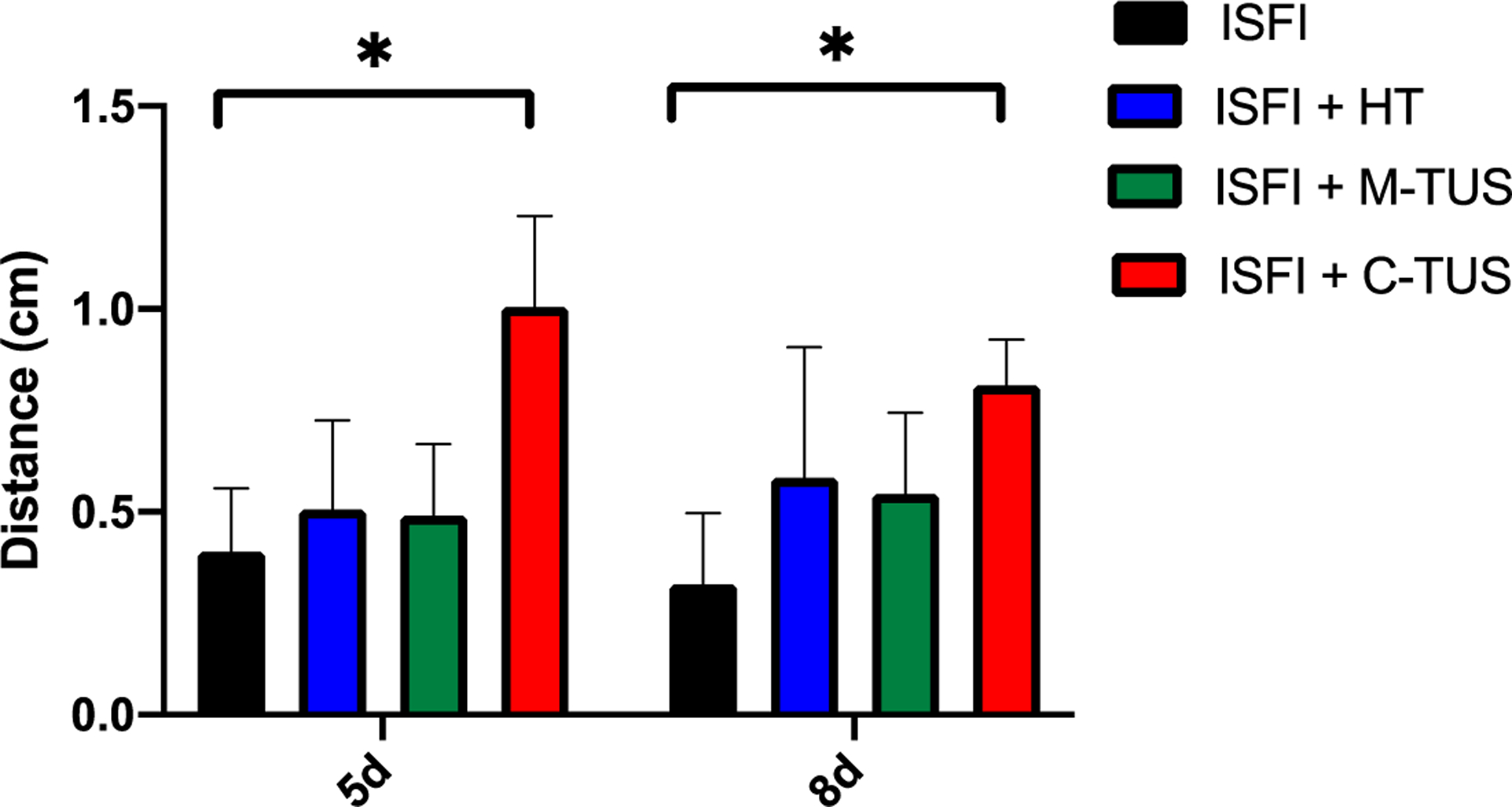
Maximum intratumoral Dox penetration at 5- and 8-days post-injection. ISFI + C-TUS showed a significant increase in penetration compared to ISFI alone. * p < 0.05
3.5. Tumor growth inhibition
Tumor growth rates for each individual treatment group over the course of 20 days can be seen in figure 9A. In figure 9B, the tumor volume at the 20-day time point is plotted to better visualize the differences between groups. The ISFI + C-TUS exposure showed the greatest inhibition of tumor growth, where after 20 days the final tumor volume was 485.2 ± 336.1 mm3. This was significantly smaller than all other treatment groups. Interestingly, the HT alone treatment group did not reach 20 days, and the ISFI + HT had the greatest increase in tumor volume, 2256.2 ± 229.4 mm3, over the 20 days.
Figure 9:
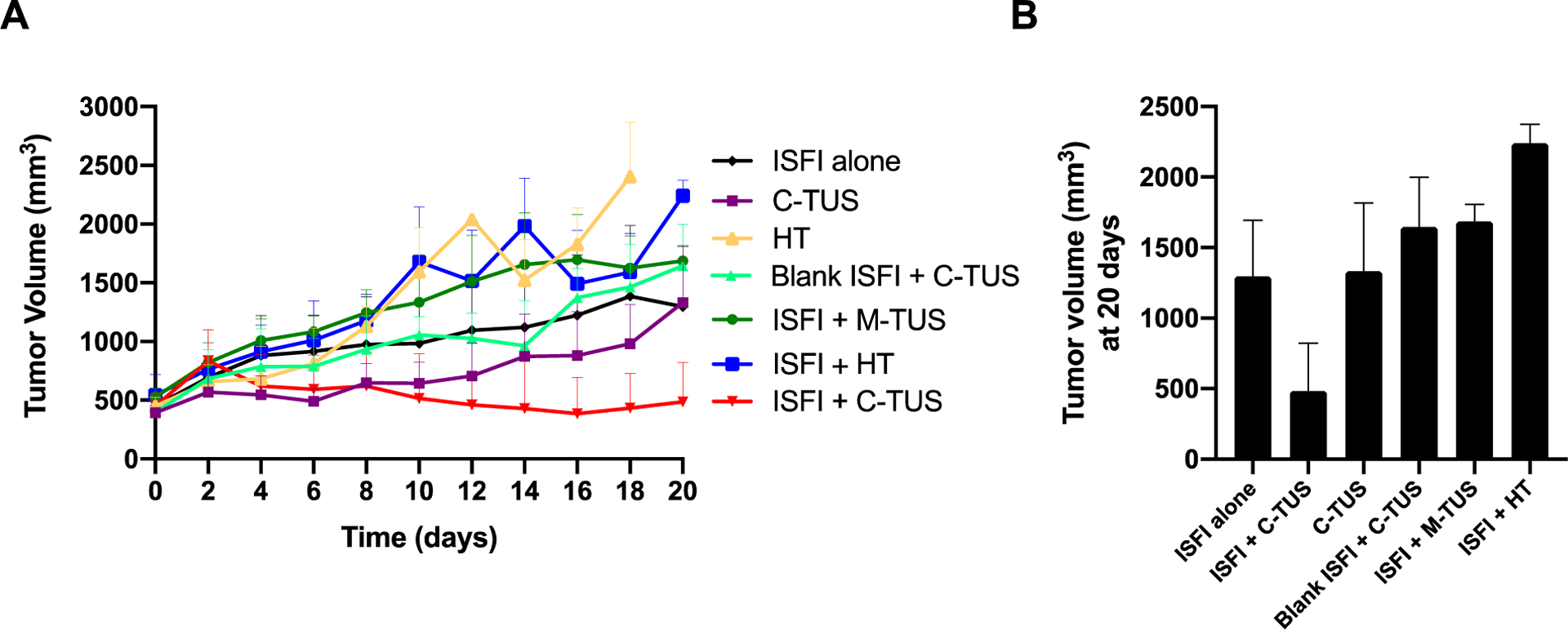
(A) Tumor growth curves for each treatment group over the course of 20 days. (B) End-point tumor volumes for each treatment group. ISFI + C-TUS demonstrated a significant (p < 0.05) inhibition of tumor growth compared to every other group at 20 days.
3.6. Survival
Each treatment group was evaluated over the course of 60 days, at which point the study was terminated. The survival curves can be seen in figure 10 for each treatment group and the median survival for each group in Table 2. ISFI + C-TUS showed the greatest median survival of 51 days, which was larger than all other treatment groups. Interestingly, all other treatment groups showed a reduction in the median survival compared to ISFI alone. The HT alone and ISFI + HT treatments showed the smallest median survival compared to all other groups.
Figure 10:
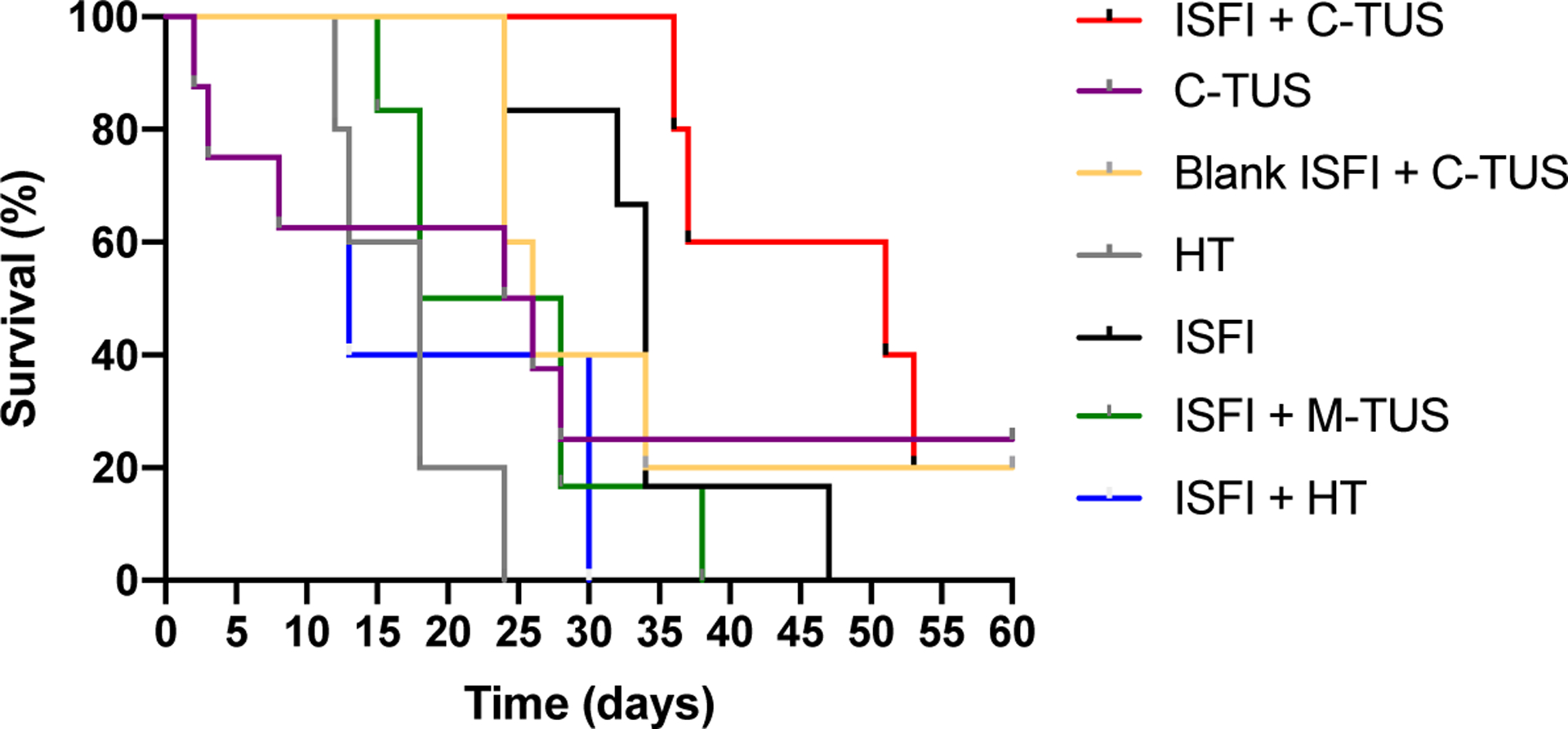
Survival curves of all treatment groups. ISFI + C-TUS showed extended survival compared to all other groups.
Table 2:
Median survival of all groups. ISFI + C-TUS showed an increase in median survival compared to all other groups.
| Treatment Group | Median Survival (days) |
|---|---|
| ISFI + HT | 13 |
| HT | 18 |
| ISFI + M-TUS | 23 |
| C-TUS | 25 |
| Blank ISFI + C-TUS | 26 |
| ISFI | 34 |
| ISFI + C-TUS | 51 |
3.7. Histology
Representative H&E stains are shown in figure 11 A–G. The percentage of necrosis was measured to determine if there were any correlations between survival/tumor growth patterns to the amount of necrosis shown in Figure 11 H. ISFI + C-TUS showed a significantly (p < 0.05) greater necrosis percentage compared to all other groups (73.5 ± 10.0%). No significant differences are seen between any of the other treatment groups.
Figure 11:
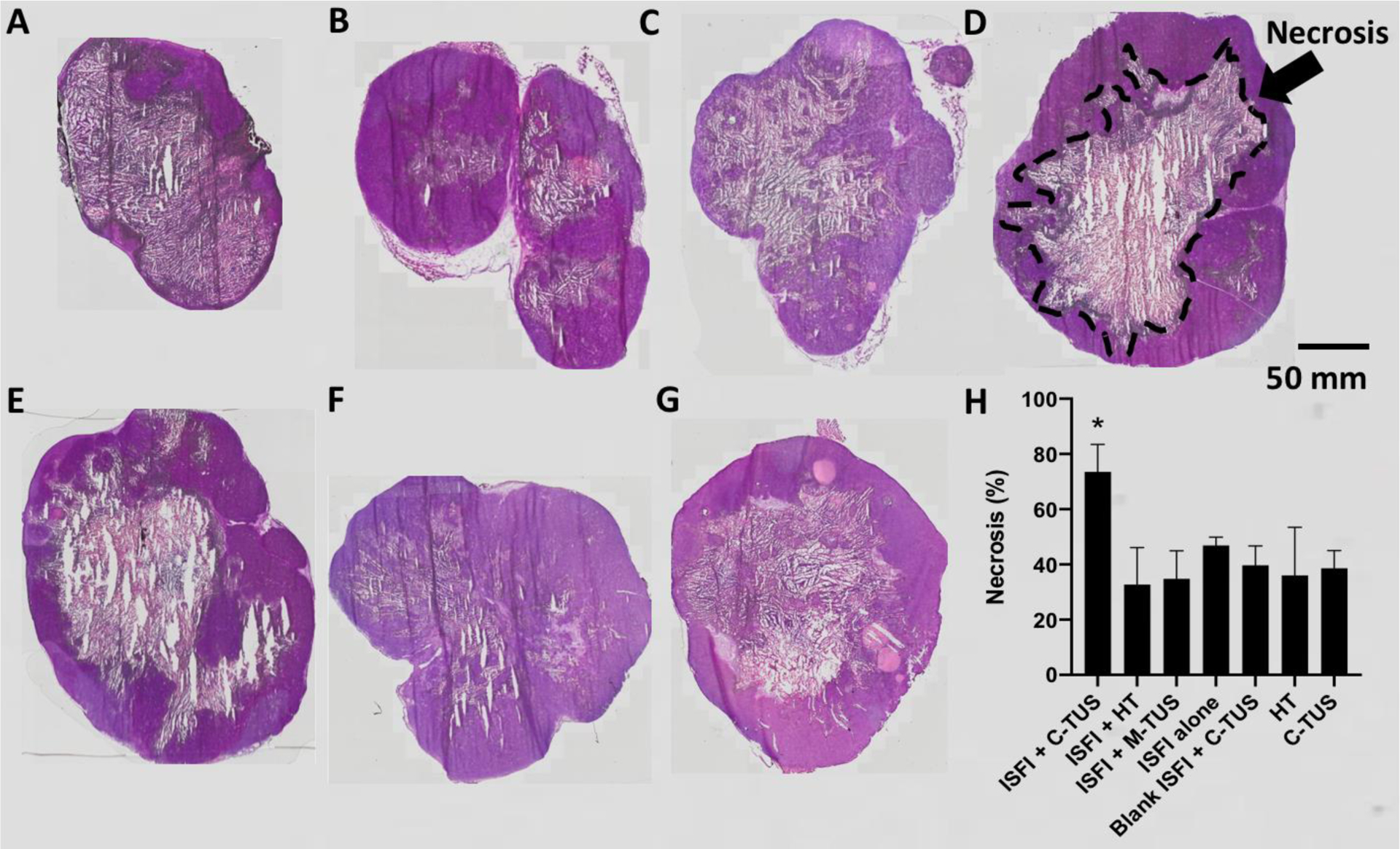
Representative H&E slices of each treatment group. (A) ISFI + C-TUS (B) ISFI + HT (C) ISFI + M-TUS (D) ISFI alone (E) Blank ISFI + C-TUS (F) HT (G) C-TUS. (H) Percentage of tumor necrosis for each treatment group. ISFI + C-TUS showed a significant increase in the necrosis percentage compared to all other groups. N=4 per treatment group.
4. Discussion
ISFIs, an LDDS, may alleviate toxicities associated with SDDS, however, they struggle with intratumoral drug penetration and retention[61,62]. To overcome these challenges, this study investigated the exposure of TUS onto intratumoral injected ISFIs. A large body of scientific evidence supports that TUS exposure can improve tumor drug distribution[54,55], increase intracellular drug uptake in nanoparticle-based delivery systems[63], enhance drug penetration with transdermal delivery systems[35,36], and increase drug retention by overcoming drug efflux transporters[37,38], translating into a better therapeutic efficacy. These effects listed above are mainly due to either mechanical or thermal bioeffects produced by the US, such as heating, acoustic cavitation, microstreaming, and others[64]. Additionally, the generation of ARF can produce transverse (shear) waves which may potentially be another mechanism of drug transport[65,66]. We hypothesized that the combination of both mechanical and thermal bioeffects produced by the TUS (C-TUS group) will directly address the limitations of ISFIs. In this study, we examined the mechanical and thermal bioeffects separately and combined to investigate which underlying mechanism is responsible for the enhanced therapeutic efficacy.
In vitro, a significant increase in Dox release was observed within the first 2 hours when the ISFI was exposed to C-TUS (Figure 2) compared to the group without ultrasound. However, Dox release was reduced when the ISFI was exposed to M-TUS, although the difference was not significant (Figure 2). TUS has been shown to alter the conformation of polymer chains and increase crystallinity in sisal fibers and acrylamide gels[67,68]. Although these groups used higher intensity ultrasound, similar effects could be present in our study. The increase in crystallinity could reduce the release of Dox from the ISFI. These effects should also be present in the group exposed to ISFI + C-TUS. However, the increase in temperature with C-TUS may be a stronger driving force in increasing the release of Dox. After 17 days, a significant increase in Dox release was seen in the ISFI + C-TUS group compared to ISFI without ultrasound exposure (Figure 2). TUS has been shown to enhance the degradation and erosion of PLGA based systems due to increasing the energy state of the ester bonds making it more susceptible to hydrolysis[51,69,70]. This increase in degradation/erosion rate could lead to an enhanced release of Dox, which was observed both in vitro (Figure 2) and in vivo (Figure 3) in our experiments.
Optical fluorescent images of intratumorally injected implants showed a greater total Dox intensity with the ISFI + C-TUS treatment group over the 8-day period (Figure 5). Quantitative analysis of these images showed a significantly elevated total intratumoral Dox intensity (Figure 6) and maximum Dox penetration with the ISFI + C-TUS compared to ISFI alone (Figures 7 and 8). This enhancement can be attributed to changes in the Dox release profile and effects of the TUS on the tumor. Interestingly, both mechanical and thermal bioeffects alone have a slight increase in maximum Dox penetration at 8 days without an increase in the release of the drug. This led us to speculate that there were other intratumoral transport mechanisms involved which led to an increase in maximum penetration. The transport of intratumoral Dox is governed by the following first-order transport equation: , where D is the drug diffusivity, t is the time, C(r,t) is the tissue drug concentration, γ is the drug elimination, and ∇ is the gradient operator. To improve drug transport, one must increase the drug diffusivity (D) value and decrease the drug elimination (γ) value. Possible routes within the tumor that affect these values are: 1) through the extracellular space (ECS) due to a concentration gradient, 2) convective movement in the interstitial fluid (IF), 3) internalization by the cell, 4) metabolism within the cell, and 5) diffusion in the capillaries/blood vessel with subsequent elimination[16,71]. Studies have shown that the application of HT increases the drug diffusivity value in tissues through the decreased interstitial fluid pressure[72–76]. HT also causes damage to intratumoral blood vessels reducing the excretion of the drug form the tumor[27,28,74–76] thus decreasing the drug elimination term. Mechanical effects of US that have demonstrated transient intercellular space widening[77], which increases the drug diffusivity value. Additionally, the injection of ISFI is likely to have introduced air bubbles into the tumors. Hyperechoic regions dispersed throughout the ISFI solution can be seen in Supplemental Figure S4. These are likely to result from trapped air in the polymer solution. Under TUS insonation, cavitation of these air bubbles could allow for greater damage to the endothelial cells thus decreasing the drug elimination term[78,79].
Interestingly, the ISFI + M-TUS showed an increase in the total intratumoral Dox at 3 and 5 days (Figure 6). This could be due to the overexpression of P-glycoprotein (Pgp), a transmembrane drug efflux transporter, found in the HCT-15 cell line used in this study. Pgp can remove the intracellular drug into the extracellular space thus eliminating it from the system. Different reports have shown that US has the capability of reversing the expression of Pgp thus allowing greater retention of Dox within the cell over time[37,80,81].
Therapeutic efficacy was evaluated for all treatment groups in terms of tumor growth rates, survival, and tumor necrosis percentage. ISFI + C-TUS showed the greatest response in these three categories, where it inhibited tumor growth over 20 days (Figure 9), allowed for a 51-day median survival (Figure 10), and showed significantly greater tumor necrosis compared to all other groups (Figure 11). Interestingly, ISFI alone showed the second-best treatment response in terms of tumor growth inhibition, median survival, and necrosis percentage. All other groups, except for ISFI + C-TUS and ISFI alone, had a lower therapeutic efficacy. This might suggest that a small non-lethal exposure to either mechanical or thermal treatments induces a tumor shock allowing it to upregulate resistant pathways. We speculate this pathway might involve heat shock proteins (HSPs), an endogenous chaperone involved in protein folding, that is upregulated to promote cell proliferation under stressful environmental exposure, such as heat, cold, UV, and others[82–84]. Upregulation of HSPs due to our treatment exposures could be a possible mechanism into why several of the milder exposure regimes had treatment responses that were worse than the ISFI control but must be further investigated in the future.
The combination of ultrasound with the ISFI used in this manuscript has the potential to improve the therapeutic efficacy of several different solid tumors due to the minimally invasive approach. However, future studies will investigate the use of ISFI for intermediate-stage hepatocellular carcinoma (IS-HCC). IS-HCC is characterized as a large multinodular liver mass that is not responsive to most systemic chemotherapies. The current clinical treatment for IS-HCC is image guided transarterial chemoembolization (TACE)[85]. This procedure delivers and occludes the tumor vasculature with an embolic agent (typically beads or Lipiodol[86–88]) placed directly into the tumors using fluoroscopy. However, this treatment results in an overall survival of 10–20 months[88–90]. The poor treatment outcome is attributed, in part, to the poor drug distribution[91]. Injecting the ISFI into these tumors that have not metastasized and exposing it to ultrasound may allow for greater distribution of the drug leading potentially to a larger reduction in tumor size. The goal would be to downstage the tumor so the patient can receive either surgical resection of the tumor or liver transplantation (treatments for early-stage HCC) where overall survival is greater than 5 years[92,93].
The following are limitations of this study and possible future directions to address them. First, there was a single exposure of ISFI and US or HT at the beginning of the study. Since about 50% of the drug is released within 10 days and another 10% within the following 2 weeks, performing multiple injections and applying repeated US exposures throughout the study may elicit greater differences. Second, an unfocused transducer with a large surface area was used to sonicate the ISFIs. The unfocused nature of this probe could account for the high degree of variability shown in the treatments. A focused transducer will be used in future studies to minimize variability and concentrate the acoustic effects on the ISFI system to further decouple implant and tissue effects. Third, the thermal only bioeffect group likely had a different intratumoral temperature distribution and heating profile compared to the C-TUS group. This is because the heated bag superficially heated the tumor (conductive heating) while the US deposits energy throughout the entire tumor and heats in depth. Additionally, the use of the cold gel prior to the TUS group dropped the temperature of the tumor before treatment, which results in a different thermal dose between the ISFI + HT versus ISFI + C-TUS treatments. Although the ISFI + HT group had a greater thermal dose ( 106 equivalent minutes at 43 °C (CEM43)) compared to ISFI + C-TUS (53 equivalent minutes at 43 °C (CEM43)), it did not result in as large a Dox distribution/retention. This suggests both mechanical and thermal bioeffects are necessary to produce the results seen with ISFI + C-TUS. Fourth, variability in the drug distribution was introduced when injecting an ISFI into tumors, as the shape and surface area to volume ratio are difficult to control. Injecting pre-formed implants could be an alternative system that can eliminate this variability.
5. Conclusion
Local drug delivery systems, such as the ISFI, have been shown to alleviate off-target toxicity issues associated with systemic chemotherapy. Intratumoral released drug penetration and retention from ISFIs within the tumors still remain barriers to clinical application. In this study, we investigated both the mechanical and thermal properties of the TUS on ISFI drug penetration and retention. A C-TUS exposure on the ISFI showed greater drug penetration and retention compared to other exposure regimes. This translated into an improved treatment response, where the ISFI + C-TUS exposure inhibited tumor growth, showed greater survival, and tumor necrosis compared to all other groups. Overall, exposing ISFI to TUS serves as an excellent non-invasive method to improve the poor clinical success of local drug delivery systems.
Supplementary Material
Highlights.
Ultrasound increases penetration of drugs delivered by intratumoral in-situ forming implants.
Drug retention within tumor cells is enhanced with the application of ultrasound.
Acoustically-driven modulation of intratumoral drug kinetics inhibits tumor growth and improves overall survival
Acknowledgments
This work was supported by the National Institute of Biomedical Imaging and Bioengineering and National Cancer Institute of the National Institutes of Health under award numbers R21EB026324-01, R01EB016960, T32-EB007509, and F31CA200373. The authors would like to acknowledge Dr. Christopher Hernandez for his insightful discussions on experimental technique and analysis. We would also like to acknowledge the CWRU SOM Light Microscope Core Facility and NIH Grant S10-RR021228. Views and opinions of and endorsements by the author(s) do not reflect those of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials, Lancet. 365 (2005) 1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- [2].Neoptolemos JP, Stocken DD, Bassi C, Ghaneh P, Cunningham D, Goldstein D, Padbury R, Moore MJ, Gallinger S, Mariette C, Wente MN, Izbicki JR, Friess H, Lerch MM, Dervenis C, Oláh A, Butturini G, Doi R, Lind PA, Smith D, Valle JW, Palmer DH, Buckels JA, Thompson J, McKay CJ, Rawcliffe CL, Büchler MW, for the European Study Group for Pancreatic Cancer, Adjuvant Chemotherapy With Fluorouracil Plus Folinic Acid vs Gemcitabine Following Pancreatic Cancer Resection, JAMA. 304 (2010) 1073. doi: 10.1001/jama.2010.1275. [DOI] [PubMed] [Google Scholar]
- [3].Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJM, Schrama JG, Erdkamp FLG, Vos AH, van Groeningen CJ, Sinnige HAM, Richel DJ, Voest EE, Dijkstra JR, Vink-Börger ME, Antonini NF, Mol L, van Krieken JHJM, Dalesio O, Punt CJA, Chemotherapy, Bevacizumab, and Cetuximab in Metastatic Colorectal Cancer, N. Engl. J. Med 360 (2009) 563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- [4].Yusuf S, Peto R, Lewis J, Collins R, Sleight P, Beta blockade during and after myocardial infarction: An overview of the randomized trials, Prog. Cardiovasc. Dis 27 (1985) 335–371. doi: 10.1016/S0033-0620(85)80003-7. [DOI] [PubMed] [Google Scholar]
- [5].Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll H-J, Tveit K-M, Gibson F, A Review of the Evolution of Systemic Chemotherapy in the Management of Colorectal Cancer, Clin. Colorectal Cancer 14 (2015) 1–10. doi: 10.1016/J.CLCC.2014.11.002. [DOI] [PubMed] [Google Scholar]
- [6].Jain RK, Martin JD, Stylianopoulos T, The Role of Mechanical Forces in Tumor Growth and Therapy, Annu. Rev. Biomed. Eng 16 (2014) 321–346. doi: 10.1146/annurev-bioeng-071813-105259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chauhan VP, Jain RK, Strategies for advancing cancer nanomedicine, Nat. Mater 12 (2013) 958–962. doi: 10.1038/nmat3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sriraman SK, Aryasomayajula B, Torchilin VP, Barriers to drug delivery in solid tumors, Tissue Barriers. 2 (2014) e29528. doi: 10.4161/TISB.29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gao L, Xia L, Zhang R, Duan D, Liu X, Xu J, Luo L, Enhanced antitumor efficacy of poly(D,L-lactide-co-glycolide)-based methotrexate-loaded implants on sarcoma 180 tumor-bearing mice., Drug Des. Devel. Ther 11 (2017) 3065–3075. doi: 10.2147/DDDT.S143942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ramazani F, van Nostrum CF, Storm G, Kiessling F, Lammers T, Hennink WE, Kok RJ, Locoregional cancer therapy using polymer-based drug depots, Drug Discov. Today 21 (2016) 640–647. doi: 10.1016/J.DRUDIS.2016.02.014. [DOI] [PubMed] [Google Scholar]
- [11].Westphal M, Ram Z, Riddle V, Hilt D, Bortey E, O. behalf of the E.C. of the G.S. Group, Gliadel® wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial, Acta Neurochir. (Wien) 148 (2006) 269–275. doi: 10.1007/s00701-005-0707-z. [DOI] [PubMed] [Google Scholar]
- [12].Lewis AL, Dreher MR, Locoregional drug delivery using image-guided intra-arterial drug eluting bead therapy, J. Control. Release 161 (2012) 338–350. doi: 10.1016/j.jconrel.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Trevisani F, Renzulli M, Bolondi L, Golfieri R, Cucchetti A, Bartolozzi C, Bargellini I, Breatta AD, Cioni R, Nani R, Gandini G, Giampalma E, Gasparini D, Randomised controlled trial of doxorubicin-eluting beads vs conventional chemoembolisation for hepatocellular carcinoma, Br. J. Cancer 111 (2014) 255–264. doi: 10.1038/bjc.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Du L, Huang Y, Zhang Q, Zhou Y, Huang J, Yan L, Yu Z, Qin A, Yang H, Chen M, Liang L, Bian B, Li X, Fu J, Synthesis and assessment of drug-eluting microspheres for transcatheter arterial chemoembolization, Acta Biomater. (2019). doi: 10.1016/J.ACTBIO.2019.02.035. [DOI] [PubMed] [Google Scholar]
- [15].Chua CYX, Jain P, Susnjar A, Rhudy J, Folci M, Ballerini A, Gilbert A, Singh S, Bruno G, Filgueira CS, Yee C, Butler EB, Grattoni A, Nanofluidic drug-eluting seed for sustained intratumoral immunotherapy in triple negative breast cancer, J. Control. Release 285 (2018) 23–34. doi: 10.1016/j.jconrel.2018.06.035. [DOI] [PubMed] [Google Scholar]
- [16].Weiser JR, Saltzman WM, Controlled release for local delivery of drugs: barriers and models, J. Control. Release 190 (2014) 664–673. doi: 10.1016/J.JCONREL.2014.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fung LK, Ewend MG, Sills A, Sipos EP, Thompson R, Watts M, Colvin OM, Brem H, Saltzman WM, Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain., Cancer Res. 58 (1998) 672–84. http://www.ncbi.nlm.nih.gov/pubmed/9485020 (accessed May 17, 2019). [PubMed] [Google Scholar]
- [18].Fung LK, Shin M, Tyler B, Brem H, Saltzman WM, Chemotherapeutic Drugs Released from Polymers: Distribution of 1,3-bis(2-chloroethyl)-l-nitrosourea in the Rat Brain, Pharm. Res 13 (1996) 671–682. doi: 10.1023/A:1016083113123. [DOI] [PubMed] [Google Scholar]
- [19].Lin Z, Gao W, Hu H, Ma K, He B, Dai W, Wang X, Wang J, Zhang X, Zhang Q, Novel thermo-sensitive hydrogel system with paclitaxel nanocrystals: High drug-loading, sustained drug release and extended local retention guaranteeing better efficacy and lower toxicity, J. Control. Release 174 (2014) 161–170. doi: 10.1016/J.JCONREL.2013.10.026. [DOI] [PubMed] [Google Scholar]
- [20].Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM, Revisiting the role of ABC transporters in multidrug-resistant cancer, Nat. Rev. Cancer 18 (2018) 452–464. doi: 10.1038/s41568-018-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gottesman MM, Pastan I, BIOCHEMISTRY OF MULTIDRUG RESISTANCE MEDIATED BY THE MULTIDRUG TRANSPORTER l * **, n.d. www.annualreviews.org (accessed May 9, 2019). [DOI] [PubMed] [Google Scholar]
- [22].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP–dependent transporters, Nat. Rev. Cancer 2 (2002) 48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- [23].Torres AJ, Zhu C, Shuler ML, Pannullo S, Paclitaxel delivery to brain tumors from hydrogels: A computational study, Biotechnol. Prog 27 (2011) 1478–1487. doi: 10.1002/btpr.665. [DOI] [PubMed] [Google Scholar]
- [24].Prasad A, Loh XJ, From Bench to Bedside—An Example of an In Situ Hydrogel in In Vivo Applications, in: Springer, Singapore, 2015: pp. 215–226. doi: 10.1007/978-981-287-152-7_9. [DOI] [Google Scholar]
- [25].Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC, Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues, Proc. Natl. Acad. Sci 84 (1987) 7735–7738. doi: 10.1073/PNAS.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM, Targeting multidrug resistance in cancer, Nat. Rev. Drug Discov 5 (2006) 219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- [27].Uchida T, Nakano M, Hongo S, Shoji S, Nagata Y, Satoh T, Baba S, Usui Y, Terachi T, High-intensity focused ultrasound therapy for prostate cancer, Int. J. Urol 19 (2012) 187–201. doi: 10.1111/j.1442-2042.2011.02936.x. [DOI] [PubMed] [Google Scholar]
- [28].Jang HJ, Lee J-Y, Lee D-H, Kim W-H, Hwang JH, Current and Future Clinical Applications of High-Intensity Focused Ultrasound (HIFU) for Pancreatic Cancer, Gut Liver. 4 (2010) S57. doi: 10.5009/gnl.2010.4.S1.S57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang Y-N, Khokhlova T, Bailey M, Hwang JH, Khokhlova V, Histological and Biochemical Analysis of Mechanical and Thermal Bioeffects in Boiling Histotripsy Lesions Induced by High Intensity Focused Ultrasound, Ultrasound Med. Biol 39 (2013) 424–438. doi: 10.1016/J.ULTRASMEDBIO.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shibata T, Shimada K, Isoda H, Hirokawa Y, Togashi K, Arizono S, Small Hepatocellular Carcinoma: Is Radiofrequency Ablation Combined with Transcatheter Arterial Chemoembolization More Effective than Radiofrequency Ablation Alone for Treatment?, Radiology. 252 (2009) 905–913. doi: 10.1148/radiol.2523081676. [DOI] [PubMed] [Google Scholar]
- [31].Nittayacharn P, Yuan H-X, Hernandez C, Bielecki P, Zhou H, Exner AA, Enhancing Tumor Drug Distribution with Ultrasound-Triggered Nanobubbles, J. Pharm. Sci 0 (n.d.). doi: 10.1016/J.XPHS.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stride E, Saffari N, Microbubble ultrasound contrast agents: A review, Proc. Inst. Mech. Eng. Part H J. Eng. Med 217 (2003) 429–447. doi: 10.1243/09544110360729072. [DOI] [PubMed] [Google Scholar]
- [33].Bouakaz A, Zeghimi A, Doinikov AA, Sonoporation: Concept and Mechanisms, in: Springer, Cham, 2016: pp. 175–189. doi: 10.1007/978-3-319-22536-4_10. [DOI] [PubMed] [Google Scholar]
- [34].Qin J, Wang T-Y, Willmann JK, Sonoporation: Applications for Cancer Therapy, in: Springer, Cham, 2016: pp. 263–291. doi: 10.1007/978-3-319-22536-4_15. [DOI] [PubMed] [Google Scholar]
- [35].Seah BC-Q, Teo BM, Recent advances in ultrasound-based transdermal drug delivery., Int. J. Nanomedicine 13 (2018) 7749–7763. doi: 10.2147/IJN.S174759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Levy D, Kost J, Meshulam Y, Langer R, Effect of ultrasound on transdermal drug delivery to rats and guinea pigs., J. Clin. Invest 83 (1989) 2074–8. doi: 10.1172/JCI114119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wan CPL, Jackson JK, (Nazly) Pirmoradi F, Chiao M, Burt HM, Increased Accumulation and Retention of Micellar Paclitaxel in Drug-Sensitive and P-Glycoprotein–Expressing Cell Lines Following Ultrasound Exposure, Ultrasound Med. Biol 38 (2012) 736–744. doi: 10.1016/J.ULTRASMEDBIO.2012.01.023. [DOI] [PubMed] [Google Scholar]
- [38].Wu F, Shao Z-Y, Zhai B-J, Zhao C-L, Shen D-M, Ultrasound Reverses Multidrug Resistance in Human Cancer Cells by Altering Gene Expression of ABC Transporter Proteins and Bax Protein, Ultrasound Med. Biol 37 (2011) 151–159. doi: 10.1016/J.ULTRASMEDBIO.2010.10.009. [DOI] [PubMed] [Google Scholar]
- [39].Berges R, Bello U, Effect of a new leuprorelin formulation on testosterone levels in patients with advanced prostate cancer, Curr. Med. Res. Opin 22 (2006) 649–655. doi: 10.1185/030079906X96425. [DOI] [PubMed] [Google Scholar]
- [40].Sartor O, Eligard: leuprolide acetate in a novel sustained-release delivery system, Urology. 61 (2003) 25–31. doi: 10.1016/S0090-4295(02)02396-8. [DOI] [PubMed] [Google Scholar]
- [41].Schulman C, Alcaraz A, Berges R, Montorsi F, Teillac P, Tombal B, Expert opinion on 6-monthly luteinizing hormone-releasing hormone agonist treatment with the single-sphere depot system for prostate cancer, n.d. http://www.acfw.com.br/crio1/prostata/british journal of urology/2007/julho/10.pdf (accessed July 2, 2019). [DOI] [PubMed] [Google Scholar]
- [42].Astaneh R, Nafissi-Varcheh N, Erfan M, Zinc–leuprolide complex: preparation, physicochemical characterization and release behaviour fromin situ forming implant, J. Pept. Sci 13 (2007) 649–654. doi: 10.1002/psc.894. [DOI] [PubMed] [Google Scholar]
- [43].Hatefi A, Amsden B, Biodegradable injectable in situ forming drug delivery systems, J. Control. Release 80 (2002) 9–28. doi: 10.1016/S0168-3659(02)00008-1. [DOI] [PubMed] [Google Scholar]
- [44].McHugh AJ, Miller DC, The dynamics of diffusion and gel growth during nonsolvent-induced phase inversion of polyethersulfone, J. Memb. Sci 105 (1995) 121–136. doi: 10.1016/0376-7388(95)00046-F. [DOI] [Google Scholar]
- [45].Graham PD, Brodbeck KJ, McHugh AJ, Phase inversion dynamics of PLGA solutions related to drug delivery, J. Control. Release 58 (1999) 233–245. doi: 10.1016/S0168-3659(98)00158-8. [DOI] [PubMed] [Google Scholar]
- [46].Brodbeck KJ, DesNoyer JR, McHugh AJ, Phase inversion dynamics of PLGA solutions related to drug delivery: Part II. The role of solution thermodynamics and bathside mass transfer, J. Control. Release 62 (1999) 333–344. doi: 10.1016/S0168-3659(99)00159-5. [DOI] [PubMed] [Google Scholar]
- [47].Jain A, Kunduru KR, Basu A, Mizrahi B, Domb AJ, Khan W, Injectable formulations of poly(lactic acid) and its copolymers in clinical use, Adv. Drug Deliv. Rev 107 (2016) 213–227. doi: 10.1016/j.addr.2016.07.002. [DOI] [PubMed] [Google Scholar]
- [48].Solorio L, Wu H, Hernandez C, Gangolli M, Exner AA, Ultrasound-guided intratumoral delivery of doxorubicin from in situ forming implants in a hepatocellular carcinoma model, Ther. Deliv 7 (2016) 201–212. doi: 10.4155/tde-2015-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Solorio L, Olear AM, Hamilton JI, Patel RB, Beiswenger AC, Wallace JE, Zhou H, Exner AA, Noninvasive characterization of the effect of varying PLGA molecular weight blends on in situ forming implant behavior using ultrasound imaging., Theranostics. 2 (2012) 1064–77. doi: 10.7150/thno.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jeganathan S, Budziszewski E, Hernandez C, Bielecki P, Kolios MC, Exner AA, Ultrasound-Enhanced Distribution and Treatment Efficacy of Dox-Loaded Intratumoral In Situ Forming Implants in Murine HCT-15 Tumors, in: 2018. IEEE Int. Ultrason. Symp., IEEE, 2018: pp. 1–4. doi: 10.1109/ULTSYM.2018.8580108. [DOI] [Google Scholar]
- [51].Manaspon C, Hernandez C, Nittayacharn P, Jeganathan S, Nasongkla N, Exner AA, Increasing Distribution of Drugs Released from In Situ Forming PLGA Implants Using Therapeutic Ultrasound, Ann. Biomed. Eng 45 (2017) 2879–2887. doi: 10.1007/s10439-017-1926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhou H, Gawlik A, Hernandez C, Goss M, Mansour J, Exner A, Nondestructive Characterization of Biodegradable Polymer Erosion in Vivo Using Ultrasound Elastography Imaging, ACS Biomater. Sci. Eng 2 (2016) 1005–1012. doi: 10.1021/acsbiomaterials.6b00128. [DOI] [PubMed] [Google Scholar]
- [53].Jeganathan S, Gilbert D, Hernandez C, Tavri S, Exner A, Ultrasound characterization of slow precipitating implants for vascular occlusion, in: IEEE Int. Ultrason. Symp. IUS, 2017. doi: 10.1109/ULTSYM.2017.8091843. [DOI] [Google Scholar]
- [54].Bielecki P, Hernandez C, Jeganathan S, Manaspon C, Kolios M, Exner A, Enhancing fluorescein release from in-situ forming PLGA implants using therapeutic ultrasound, in: IEEE Int. Ultrason. Symp. IUS, 2017. doi: 10.1109/ULTSYM.2017.8091565. [DOI] [Google Scholar]
- [55].Manaspon C, Hernandez C, Nittayacharn P, Jeganathan S, Nasongkla N, Exner AA, Increasing Distribution of Drugs Released from In Situ Forming PLGA Implants Using Therapeutic Ultrasound, Ann. Biomed. Eng 45 (2017). doi: 10.1007/s10439-017-1926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Solorio L, Babin BM, Patel RB, Mach J, Azar N, Exner AA, Noninvasive characterization of in situ forming implants using diagnostic ultrasound, J. Control. Release 143 (2010) 183–190. doi: 10.1016/J.JCONREL.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].solorio L, Sundarapandiyan D, Olear A, Exner AA, The Effect of Additives on the Behavior of Phase Sensitive In Situ Forming Implants, J. Pharm. Sci 104 (2015) 3471–3480. doi: 10.1002/JPS.24558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bielecki P, Hernandez C, Jeganathan S, Wiese M, Manaspon C, Kolios M, Exner AA, Enhancing fluorescein distribution from in situ forming PLGA implants using therapeutic ultrasound, in: IEEE Int. Ultrason. Symp. IUS, 2017. doi: 10.1109/ULTSYM.2017.8091587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE, Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen., Mol. Pharmacol 46 (1994). [PubMed] [Google Scholar]
- [60].Patel RB, Solorio L, Wu H, Krupka T, Exner AA, Effect of injection site on in situ implant formation and drug release in vivo, J. Control. Release 147 (2010) 350–358. doi: 10.1016/j.jconrel.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Weinberg BD, Patel RB, Exner AA, Saidel GM, Gao J, Modeling doxorubicin transport to improve intratumoral drug delivery to RF ablated tumors, J. Control. Release 124 (2007) 11–19. doi: 10.1016/J.JCONREL.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Weinberg BD, Ai H, Blanco E, Anderson JM, Gao J, Antitumor efficacy and local distribution of doxorubicin via intratumoral delivery from polymer millirods, J. Biomed. Mater. Res. Part A 81A (2007) 161–170. doi: 10.1002/jbm.a.30914. [DOI] [PubMed] [Google Scholar]
- [63].Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY, A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system., J. Pharmacol. Exp. Ther 317 (2006) 1372–81. doi: 10.1124/jpet.106.101154. [DOI] [PubMed] [Google Scholar]
- [64].Dalecki D, Mechanical Bioeffects of Ultrasound, Annu. Rev. Biomed. Eng 6 (2004) 229–248. doi: 10.1146/annurev.bioeng.6.040803.140126. [DOI] [PubMed] [Google Scholar]
- [65].Wu J, Nyborg WL, Ultrasound , cavitation bubbles and their interaction with cells ☆, 60 (2008) 1103–1116. doi: 10.1016/j.addr.2008.03.009. [DOI] [PubMed] [Google Scholar]
- [66].Nightingale K, Acoustic Radiation Force Impulse (ARFI) Imaging: A Review, Curr. Med. Imaging Rev 7 (2011) 328–339. doi: 10.2174/157340511798038657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Krishnaiah P, Ratnam CT, Manickam S, Enhancements in crystallinity, thermal stability, tensile modulus and strength of sisal fibres and their PP composites induced by the synergistic effects of alkali and high intensity ultrasound (HIU) treatments, Ultrason. Sonochem 34 (2017) 729–742. doi: 10.1016/j.ultsonch.2016.07.008. [DOI] [PubMed] [Google Scholar]
- [68].Ebrahimi R, Mohammadi HG, Kinetics of sonodegradation of poly(acrylic acid co acrylamide) hydrogels by viscometry, Macromol. An Indian J 7 (n.d.). [Google Scholar]
- [69].Agrawal CM, Kennedy ME, Micallef DM, The effects of ultrasound irradiation on a biodegradable 50–50% copolymer of polylactic and polyglycolic acids, J. Biomed. Mater. Res 28 (1994) 851–859. doi: 10.1002/jbm.820280803. [DOI] [PubMed] [Google Scholar]
- [70].Lu L, Peter SJ, Lyman MD, Lai HL, Leite SM, Tamada JA, Uyama S, Vacanti JP, Robert Langer AG Mikos, In vitro and in vivo degradation of porous poly(DL-lactic-co-glycolic acid) foams, Biomaterials. 21 (2000) 1837–1845. doi: 10.1016/S0142-9612(00)00047-8. [DOI] [PubMed] [Google Scholar]
- [71].Saltzman WM, Radomsky ML, Drugs released from polymers: diffusion and elimination in brain tissue, Chem. Eng. Sci 46 (1991) 2429–2444. doi: 10.1016/0009-2509(91)80036-X. [DOI] [Google Scholar]
- [72].El-Kareh AW, Secomb TW, A theoretical model for intraperitoneal delivery of cisplatin and the effect of hyperthermia on drug penetration distance., Neoplasia. 6 (2004) 117–27. doi: 10.1593/neo.03205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].González-Moreno S, González-Bayón LA, Ortega-Pérez G, Hyperthermic intraperitoneal chemotherapy: Rationale and technique., World J. Gastrointest. Oncol 2 (2010) 68–75. doi: 10.4251/wjgo.v2.i2.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Stylianopoulos T, Munn LL, Jain RK, Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside., Trends in Cancer. 4 (2018) 292–319. doi: 10.1016/j.trecan.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Baronzio G, Parmar G, Baronzio M, Overview of Methods for Overcoming Hindrance to Drug Delivery to Tumors, with Special Attention to Tumor Interstitial Fluid, Front. Oncol 5 (2015) 165. doi: 10.3389/fonc.2015.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kirui DK, Koay EJ, Guo X, Cristini V, Shen H, Ferrari M, Tumor vascular permeabilization using localized mild hyperthermia to improve macromolecule transport, Nanomedicine. 10 (2014) 1487. doi: 10.1016/J.NANO.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Frenkel V, Kimmel E, Iger Y, Ultrasound-induced intercellular space widening in fish epidermis, Ultrasound Med. Biol 26 (2000) 473–480. doi: 10.1016/S0301-5629(99)00164-7. [DOI] [PubMed] [Google Scholar]
- [78].Lai C-Y, Fite BZ, Ferrara KW, Ultrasonic Enhancement of Drug Penetration in Solid Tumors, Front. Oncol 3 (2013) 204. doi: 10.3389/fonc.2013.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lentacker I, De Cock I, Deckers R, De Smedt SC, Moonen CTW, Understanding ultrasound induced sonoporation : De fi nitions and underlying mechanisms ☆, Adv. Drug Deliv. Rev 72 (2014) 49–64. doi: 10.1016/j.addr.2013.11.008. [DOI] [PubMed] [Google Scholar]
- [80].Xia W, Ma X, Li X, Lu C, Yang X, Zhu Z, Yi J, Reversal effect of low-intensity ultrasound on adriamycin-resistant human hepatoma cells in vitro and in vivo, Int. J. Imaging Syst. Technol 24 (2014) 23–28. doi: 10.1002/ima.22075. [DOI] [Google Scholar]
- [81].Sheikov N, McDannold N, Sharma S, Hynynen K, Effect of Focused Ultrasound Applied With an Ultrasound Contrast Agent on the Tight Junctional Integrity of the Brain Microvascular Endothelium, Ultrasound Med. Biol 34 (2008) 1093–1104. doi: 10.1016/j.ultrasmedbio.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Calderwood SK, Gong J, Heat Shock Proteins Promote Cancer: It’s a Protection Racket, Trends Biochem. Sci 41 (2016) 311–323. doi: 10.1016/J.TIBS.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Richter K, Haslbeck M, Buchner J, The Heat Shock Response: Life on the Verge of Death, Mol. Cell 40 (2010) 253–266. doi: 10.1016/J.MOLCEL.2010.10.006. [DOI] [PubMed] [Google Scholar]
- [84].Sreedhar AS, Csermely P, Heat shock proteins in the regulation of apoptosis: new strategies in tumor therapy: A comprehensive review, Pharmacol. Ther 101 (2004) 227–257. doi: 10.1016/J.PHARMTHERA.2003.11.004. [DOI] [PubMed] [Google Scholar]
- [85].Forner A, Gilabert M, Bruix J, Raoul JL, Treatment of intermediate-stage hepatocellular carcinoma, Nat. Rev. Clin. Oncol 11 (2014) 525–535. doi: 10.1038/nrclinonc.2014.122. [DOI] [PubMed] [Google Scholar]
- [86].Golfieri R, Giampalma E, Renzulli M, Cioni R, Bargellini I, Bartolozzi C, Breatta AD, Gandini G, Nani R, Gasparini D, Cucchetti A, Bolondi L, Trevisani F, PRECISION ITALIA STUDY GROUP, Randomised controlled trial of doxorubicin-eluting beads vs conventional chemoembolisation for hepatocellular carcinoma, Br. J. Cancer 111 (2014) 255–264. doi: 10.1038/bjc.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lencioni R, Chemoembolization for Hepatocellular Carcinoma, Semin. Oncol 39 (2012) 503–509. doi: 10.1053/J.SEMINONCOL.2012.05.004. [DOI] [PubMed] [Google Scholar]
- [88].Kloeckner R, Weinmann A, Prinz F, Pinto dos Santos D, Ruckes C, Dueber C, Pitton MB, Conventional transarterial chemoembolization versus drug-eluting bead transarterial chemoembolization for the treatment of hepatocellular carcinoma, BMC Cancer. 15 (2015) 465. doi: 10.1186/s12885-015-1480-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pitton M, Sergent G, Mueller C, Chevallier P, Pfammatter T, Avajon Y, Lammer J, Pilleul F, Benhamou Y, Vogl T, Terraz S, Pomoni M, Denys A, Malagari K, Schuchmann M, Dumortier J, Gruenberger T, Lencioni R, Langenberger H, Watkinson A, Prospective Randomized Study of Doxorubicin-Eluting-Bead Embolization in the Treatment of Hepatocellular Carcinoma: Results of the PRECISION V Study, Cardiovasc. Intervent. Radiol 33 (2009) 41–52. doi: 10.1007/s00270-009-9711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].El Fouly A, Ertle J, El Dorry A, Shaker MK, Dechêne A, Abdella H, Mueller S, Barakat E, Lauenstein T, Bockisch A, Gerken G, Schlaak JF, In intermediate stage hepatocellular carcinoma: radioembolization with yttrium 90 or chemoembolization?, Liver Int. 35 (2015) 627–635. doi: 10.1111/liv.12637. [DOI] [PubMed] [Google Scholar]
- [91].Dreher MR, Sharma KV, Woods DL, Reddy G, Tang Y, Pritchard WF, Chiesa OA, Karanian JW, Esparza JA, Donahue D, Levy EB, Willis SL, Lewis AL, Wood BJ, Radiopaque drug-eluting beads for transcatheter embolotherapy: Experimental study of drug penetration and coverage in swine, J. Vasc. Interv. Radiol 23 (2012) 257–264.e4. doi: 10.1016/j.jvir.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Raza A, Sood GK, Hepatocellular carcinoma review: Current treatment, and evidence-based medicine, World J. Gastroenterol 20 (2014) 4115–4127. doi: 10.3748/wjg.v20.i15.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].O’Connor JK, Orr DW, Berger BD, Brown B, Meler JD, Dempster J, Walberg MW, Goldstein RM, Davis GL, Hepatocellular Carcinoma: Management of an Increasingly Common Problem, Baylor Univ. Med. Cent. Proc 21 (2017) 266–280. doi: 10.1080/08998280.2008.11928410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.