

DNA methyltransferase 3B is identified as a protective target against pulmonary vascular remodeling.
Abstract
DNA methylation plays critical roles in vascular pathology of pulmonary hypertension (PH). The underlying mechanism, however, remains undetermined. Here, we demonstrate that global DNA methylation was elevated in the lungs of PH rat models after monocrotaline administration or hypobaric hypoxia exposure. We showed that DNA methyltransferase 3B (DNMT3B) was up-regulated in both PH patients and rodent models. Furthermore, Dnmt3b−/− rats exhibited more severe pulmonary vascular remodeling. Consistently, inhibition of DNMT3B promoted proliferation/migration of pulmonary artery smooth muscle cells (PASMCs) in response to platelet-derived growth factor–BB (PDGF-BB). In contrast, overexpressing DNMT3B in PASMCs attenuated PDGF-BB–induced proliferation/migration and ameliorated hypoxia-mediated PH and right ventricular hypertrophy in mice. We also showed that DNMT3B transcriptionally regulated inflammatory pathways. Our results reveal that DNMT3B is a previously undefined mediator in the pathogenesis of PH, which couples epigenetic regulations with vascular remodeling and represents a therapeutic target to tackle PH.
INTRODUCTION
Pulmonary hypertension (PH) is a progressive cardiopulmonary disorder characterized by high pulmonary artery pressure and elevation of pulmonary vascular resistance that usually succumbs to right heart failure and even death (1, 2). It has been well established that the predisposing gene BMPR2 leads to the development of PH and cellular signaling perturbations involved in PH pathogenesis (3, 4). Recently, a growing body of evidence highlights that PH is a complex disease mediated by the interplay of predisposed genetic background, epigenetic state, and injurious events.
Epigenetic modifications play a key role in establishing cell type–specific gene expression profile and patterns, which is fundamental for lineage commitment, differentiation, and proliferation (5). DNA methylation is one of the best characterized chemical modifications on chromatin linked to transcriptional silencing in physiology and pathophysiology (6). Archer et al. (7, 8) demonstrated that hypermethylation of Sod2 contributed to a proliferative, anti-apoptotic phenotype of pulmonary artery smooth muscle cell (PASMC). Moreover, an increase of DNA methylation in eNOS (endothelial nitric oxide synthase) promoter was identified in pulmonary artery endothelial cells in fetal lambs with persistent PH of the newborn (9). Notably, a predominantly different DNA methylation signature in pulmonary endothelial cells (10) and PASMC (11) was documented in PH patients compared to the corresponding controls. In addition, our previous study showed that BMPR2 promoter methylation was significantly higher in affected individuals than controls, accounting for the different penetration of heritable pulmonary arterial hypertension (PAH) (12). Together, these studies have explored certain genes regulated by DNA methylation in PH development. However, the role and mechanism of action by DNA methyltransferases (DNMTs), the most important enzyme regulating DNA methylation, are poorly understood.
Here, we found that global DNA methylation was significantly elevated in PH rat models, with an increase in DNMT3B expression in affected lungs. Furthermore, we showed that Dnmt3b−/− rats exhibited a deteriorated PH phenotype compared to wild-type (WT) littermates after monocrotaline (MCT) administration or hypobaric hypoxia exposure. On the other hand, augmenting DNMT3B rescued the pathological vascular remodeling. In addition, in vitro studies showed that Dnmt3b-specific inhibition, inactivation, or deficiency promoted PASMC proliferation and migration after platelet-derived growth factor–BB (PDGF-BB) stimulation, which was rescued by DNMT3B overexpression. Mechanistically, we also showed that DNMT3B transcriptionally regulated inflammatory pathways. Together, our studies reveal a critical role for DNMT3B in maintaining vascular homeostasis and provide a potential treatment strategy for devastating PH.
RESULTS
Global DNA methylation is increased in experimental rodent PH models
To determine changes in global DNA methylation pattern between normal and pulmonary hypertensive lungs, the percentage of 5-methylcytosine (5-mC) was examined in lung tissue of established PH rat models induced by MCT injection, which led to significant higher right ventricular systolic pressure (RVSP), mean pulmonary arterial pressure (mPAP), and right ventricular hypertrophy index (RVHI) 3 weeks later (fig. S1, A to C). DNA methylation analysis showed a 1.8-fold increase of global DNA methylation at day 21 in whole lungs from MCT-induced PH rats compared to controls (Fig. 1A).
Fig. 1. Global DNA methylation and Dnmt3b are elevated in lungs of PH rat models and pathological up-regulation of Dnmt3b in PAH.
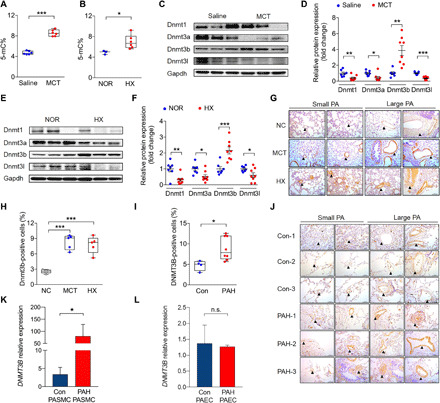
(A) Lungs of rats at week 3 after MCT injection (n = 6) or (B) under hypobaric hypoxia (HX) (n = 6) had higher levels of global DNA methylation [5-methylcytosine% (5-mC%)] than those of saline controls (n = 6) or normobaric normoxic controls (NOR) (n = 3). (C to F) Western blot analysis of Dnmt1, Dnmt3a, Dnmt3b, and Dnmt3l relative to Gapdh from PH rat lungs at week 3 after MCT injection or after hypobaric hypoxic exposure compared to its corresponding controls (n = 7 to 8 per group). (G and H) Representative images and quantification analysis of Dnmt3b-positive staining cell percentage showed increased Dnmt3b (brown) expression in the media and endothelium of pulmonary arteries of rats after MCT administration (n = 5) or under hypobaric hypoxic conditions (n = 5) compared to that of control rats (n = 5). Scale bars, 100 μm. (I and J) Quantification analysis and representative images of Dnmt3b-positive staining of four control patients (top three rows) and eight PAH lungs (bottom three rows). Scale bars, 100 μm. (K) DNMT3B mRNA expression in PASMCs from PAH patients or control subjects (n = 5 per group). (L) DNMT3B mRNA expression in PAECs from PAH patients or control subjects (n = 3 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 versus corresponding controls, Student’s t test (A, C, D, F, and I), Mann-Whitney test (K and L), and one-way analysis of variance (ANOVA) with Bonferroni correction for multiple comparisons (H). Data are presented as the mean ± SEM (D, F, K, and L) or box-and-whisker plots with scatter (A, B, H, and I). n.s., not significant.
Another PH model was generated by maintaining rats in a hypobaric hypoxia (50 kPa) chamber for 3 weeks (fig. S1, D to F). Likewise, rats with hypobaric hypoxia–induced PH had a 1.4-fold elevation in DNA methylation levels in lungs compared to normobaric normoxic animals (Fig. 1B). Together, these data suggest that global DNA methylation levels are up-regulated in PH rodent models.
DNMT3B is significantly increased in lungs from patients with PH and in experimental rodent PH models
To identify relevant DNA methyltransferase(s) in disease state, protein expression of the four DNA methyltransferases in two independent experimental PH models was determined. Among all the DNA methyltransferases detected—Dnmt1, Dnmt3a, Dnmt3b, and Dnmt3l—only Dnmt3b protein expression was up-regulated at day 21 in both PH models (Fig. 1, C to F). Consistently, PH induced by either MCT or hypoxia led to the increase of Dnmt3b and reduction of other Dnmts at mRNA levels (fig. S1, G to N).
Localization of Dnmt3b in the lung was next assessed. An elevation of Dnmt3b expression in vessel walls of lung tissue of both PH rodent models after MCT challenge or hypobaric hypoxia exposure was determined by immunohistochemistry (Fig. 1, G and H), consistent with an increase in global DNA methylation (Fig. 1, A and B). Furthermore, a significant elevation was seen in lung tissues of congenital heart disease (CHD)–PAH patients versus age- and gender-matched CHD patients without PAH (table S1 and Fig. 1, I and J). We also measured DNMT3B expression in PASMCs from patients with PAH and control subjects. A 24-fold of increase in DNMT3B was depicted in PASMCs from patients with PAH compared to those from control subjects (Fig. 1K), while no significant difference of DNMT3B was detected in pulmonary arterial endothelial cells (PAECs) from PAH patients compared to those from controls (Fig. 1L). Together, these results suggest that DNMT3B is up-regulated in PASMCs of patients with PAH and experimental PH models.
Dnmt3b deficiency exacerbates PH progression in rodent models
To explore the role of Dnmt3b in PH, a Dnmt3b knockout rat model was generated (fig. S2A). At baseline, there was no significant difference in body weight between WT littermates and Dnmt3b homozygotes (fig. S2B). The susceptibility of Dnmt3b−/− rats to the MCT challenge (50 mg/kg) was then determined. After 4 weeks, Dnmt3b−/− rats demonstrated significantly higher RVSP, mPAP, RVHI, and Pcna expression than WT after MCT injection (Fig. 2, A to C, and fig. S2C). A similar pattern was observed in Dnmt3b−/− rats that exhibited thicker right ventricular free wall compared with WT rats in response to MCT (Fig. 2D and fig. S2D). However, we found no difference in tricuspid annular plane systolic excursion (TAPSE) between the two genotypes (fig. S2, E and F), suggesting that Dnmt3b is mainly involved in pulmonary vascular remodeling, while its effect on cardiac function is still uncertain. Dnmt3b−/− rats also displayed more profound pathological remodeling in the pulmonary vasculature, as measured by media to cross-sectional area (Fig. 2, E and F) and vessel muscularization (Fig. 2, G and H). These data collectively suggest that Dnmt3b deficiency exacerbates the development of MCT-induced PH.
Fig. 2. Dnmt3b deficiency exacerbates pulmonary vascular remodeling in MCT-induced PH rat model.
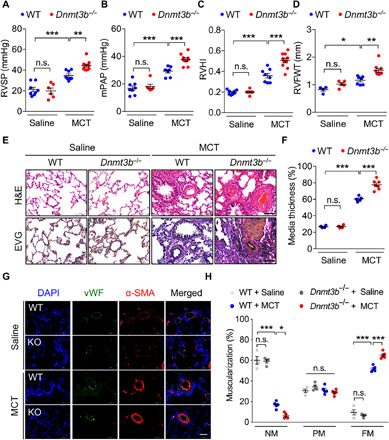
(A to D) Compared to WT saline group, Dnmt3b−/− homozygous [Dnmt3b−/−, knockout (KO)] Sprague-Dawley rats at week 4 after MCT injection exhibited an elevation in (A) RVSP, (B) mPAP, (C) RVHI, and (D) right ventricular free wall thickness (RVFWT) (A to C, n = 8 for WT saline group, n = 6 for KO group, n = 8 for WT MCT group, and n = 10 for KO MCT group; D, n = 4 for WT saline group, n = 5 for KO group, n = 7 for WT MCT group, and n = 10 for KO MCT group). (E) Representative photomicrographs of hematoxylin and eosin (H&E) staining and elastin–van Gieson (EVG) staining of lung tissue from MCT or saline-treated rats at day 28. Original magnification, ×400. Scale bars, 50 μm. (F) Pulmonary vascular remodeling by percentage of vascular medial thickness to total vessel size (n = 4 to 7 per group) and (G) quantification of vessel muscularization by immunofluorescence staining with anti–α-SMA (red, smooth muscle cells), anti-vWF (green, endothelial cells), and 4′,6-diamidino-2-phenylindole (DAPI) (blue, nuclei) for the PH model, demonstrating that Dnmt3b deficiency promoted a further elevation of pulmonary vascular wall thickness after MCT injection. Scale bars, 50 μm. (H) Proportion of nonmuscularized (NM), partially muscularized (PM), or fully muscularized (FM) pulmonary arterioles (20 to 50 μm in diameter) from MCT-treated rats, confirming that Dnmt3b deficiency significantly increased arteriole muscularization (n = 4 to 5 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT saline group or WT MCT group, one-way ANOVA with Bonferroni correction for multiple comparisons (A to D and F) and two-way ANOVA with Bonferroni’s post hoc analysis (H); mean ± SEM.
Whether disruption of Dnmt3b might accelerate disease progression was next tested in another experimental PH model. Because Sugen 5416/hypoxia rat PH model manifested a severe pulmonary vascular remodeling, it might be difficult to observe an additional deterioration in Dnmt3b−/− rats as hypothesized. WT and Dnmt3b deletion rats were then treated with chronic hypobaric (50 kPa) hypoxia for 3 weeks. Dnmt3b−/− rats displayed a significant increase in RVSP (42.2 ± 3.8 mmHg versus 34.8 ± 3.7 mmHg; P < 0.01) (Fig. 3A), mPAP (37.6 ± 4.8 mmHg versus 31.6 ± 2.3 mmHg, P < 0.05) (Fig. 3B), RVHI (0.37 ± 0.03 versus 0.31 ± 0.02, P < 0.001) (Fig. 3C), and right ventricular free wall thickness (1.5 ± 0.1 versus 1.2 ± 0.1, P < 0.01) (Fig. 3D and fig. S2G) compared with WT controls. Consistent with the MCT results, Dnmt3b−/− rats exhibited a similar TAPSE to that of WT littermates (fig. S2, H and I). Moreover, Dnmt3b deficiency aggravated pulmonary vascular remodeling induced by chronic hypobaric hypoxia through an elevation of pulmonary vascular wall thickness (Fig. 3, E and F) and muscularization (Fig. 3, G and H). Combined, these results provide the first evidence that Dnmt3b confers protection against PH development and progression.
Fig. 3. Dnmt3b deficiency exacerbates pulmonary vascular remodeling in hypobaric hypoxia–induced PH rat model.
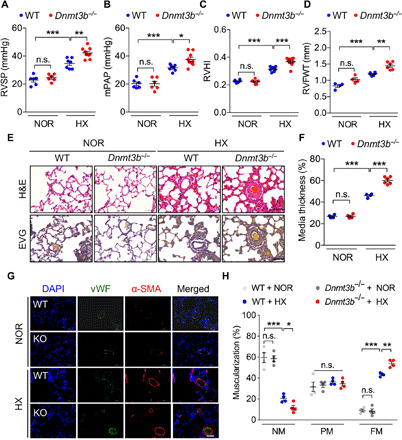
(A to D) Compared to WT normobaric normoxia group, Dnmt3b−/− (KO) Sprague-Dawley rats demonstrated an elevation in (A) RVSP, (B) mPAP, (C) RVHI, and (D) right ventricular free wall thickness (RVFWT) at day 21 after exposure to hypobaric hypoxia [A to C, n = 6 for WT normobaric normoxia (NOR) group, n = 6 for KO normobaric normoxia group, n = 7 for WT hypobaric hypoxia (HX) group, and n = 9 for KO hypobaric hypoxia group; D, n = 4 for WT or KO normobaric normoxia group and n = 6 for WT or KO hypobaric hypoxia group]. (E) Representative images of H&E staining and EVG staining of lung tissue from hypobaric hypoxia– or normobaric normoxia–treated rats at day 21. Original magnification, ×400. Scale bars, 50 μm. (F) Pulmonary vascular remodeling by percentage of vascular medial thickness to total vessel size (n = 4 to 6 per group) and (G) quantification of vessel muscularization by immunofluorescence staining with anti–α-SMA (red, smooth muscle cells), anti-vWF (green, endothelial cells), and DAPI (blue, nuclei) for the PH model, demonstrating that Dnmt3b deficiency promoted a further elevation of pulmonary vascular wall thickness under hypobaric hypoxic conditions. Scale bars, 50 μm. (H) Proportion of nonmuscularized (NM), partially muscularized (PM), or fully muscularized (FM) pulmonary arterioles (20 to 50 μm in diameter) from hypobaric hypoxia–treated rats, confirming that Dnmt3b deficiency significantly increased arteriole muscularization (n = 4 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 versus WT normobaric normoxia group or WT hypobaric hypoxia group, one-way ANOVA with Bonferroni correction for multiple comparisons (A to D and F) and two-way ANOVA with Bonferroni’s post hoc analysis (H); mean ± SEM.
DNMT3B induction alleviates pulmonary vascular remodeling in the PH mouse model
We next examined whether up-regulation of DNMT3B could arrest disease development. Human DNMT3B was engineered and packaged into adeno-associated virus 9 (AAV9). After intratracheal delivery, a robust increase in DNMT3B expression at the protein level was observed by immunofluorescence (Fig. 4A) and mRNA level by reverse transcription polymerase chain reaction (RT-PCR) (fig. S3A). In addition, the expression of DNMT3B in lung tissues was revealed to dominate over those in other organs from AAV9-DNMT3B recipients in ambient atmosphere (fig. S3, B and C). In contrast with Dnmt3b deficiency, overexpression of human DNMT3B elicited a remarkable reduction in RVSP (31.0 ± 0.7 mmHg versus 34.3 ± 0.8 mmHg; P < 0.01) (Fig. 4B) and RVHI (0.29 ± 0.01 versus 0.37 ± 0.02; P < 0.01) (Fig. 4C) compared to control mice receiving AAV9-null virus in response to hypoxia for another 2 weeks. Furthermore, we found that the protective effect of DNMT3B against vascular remodeling was consistent with suppression of media thickness (Fig. 4, D and E) and arteriolar muscularization (Fig. 4F), supporting a notion that DNMT3B may have a therapeutic potential to decelerate PH progression. In addition, Pcna+ PASMCs in lung tissues from mice that received recombinant AAV9-DNMT3B showed a decreased trend compared to mice that received AAV9-null under hypoxia condition by immunofluorescent colocalization staining (fig. S3, D and E).
Fig. 4. DNMT3B overexpression ameliorates hypoxia-induced PH in mice.
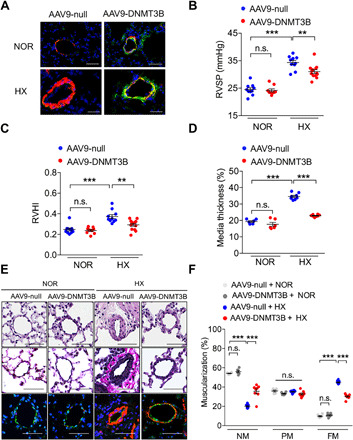
(A) Representative images of lung tissues from mice that received intratracheal delivery (AAV9-DNMT3B or AAV9-null) at day 28 under normoxia or hypoxia by immunofluorescence staining with anti–α-SMA (red), anti-DNMT3B (green), and DAPI (blue). Scale bars, 50 μm. (B and C) AAV9-DNMT3B–treated mice exhibited a reduction in (C) RVSP and (D) RVHI compared to AAV9-null–treated group at day 14 after exposure to hypoxia (C and D, n = 10 for AAV9-null–treated normoxia group, n = 9 for AAV9-DNMT3B–treated normoxia group, n = 10 for AAV9-null–treated hypoxia group, and n = 13 for AAV9-DNMT3B–treated hypoxia group). (D) Pulmonary vascular remodeling by percentage of vascular medial thickness to total vessel size (n = 6 for normoxia group and n = 8 for hypoxia group). (E) Representative images of H&E staining, EVG staining, and immunofluorescence staining with anti–α-SMA (red, smooth muscle cells), anti-vWF (green, endothelial cells), and DAPI (blue, nuclei) for the PH model of lung tissue from hypoxia- or normoxia-treated mice at day 14. Original magnification, ×400. Scale bars, 50 μm. (F) Quantification of vessel muscularization for the PH mouse model demonstrating that augmenting DNMT3B retarded pulmonary vascular remodeling under hypoxic conditions (n = 6 for normoxia group and n = 8 for hypoxia group). **P < 0.01 and ***P < 0.001 versus AAV9-null–treated normoxia group or AAV9-null–treated hypoxia group, one-way ANOVA with Bonferroni correction for multiple comparisons (B to D), and two-way ANOVA with Bonferroni’s post hoc analysis (F); mean ± SEM.
DNMT3B inhibition leads to a worse phenotype in PASMCs
Vascular smooth muscle cell proliferation is one of the underlying mechanisms of vascular remodeling in PH (13, 14). PDGF-BB is regarded as a potent mitogen for PASMC, and a higher PDGFB mRNA expression was demonstrated in PASMCs from patients with PAH compared to those from control subjects (fig. S4A). Thus, the role of DNMT3B in PASMC proliferation was assessed under the stimuli of PDGF-BB. It was found that DNMT3B expression was increased in PDGF-BB–induced human PASMCs (hPASMCs) (fig. S4B). When cells were treated with various concentrations of a DNMT3B inhibitor (nanaomycin A), proliferation was increased in a dose-dependent manner (Fig. 5A). To test whether DNMT3B inhibition affected hPASMC migration, a wound scratch assay was performed followed by PDGF-BB treatment. Nanaomycin A facilitated cell migration, as evidenced by a higher wound confluency in nanaomycin A–treated hPASMCs compared to control hPASMCs after PDGF-BB stimulation (Fig. 5, B and C). In accordance with the findings in hPASMCs, a DNMT3B inhibitor led to an increase in proliferation (fig. S4, C and D) and a stronger migration capacity (fig. S4, E and F) in rat PASMCs (rPASMCs) after PDGF-BB insult.
Fig. 5. Manipulation of DNMT3B alters PASMC capacity of proliferation and migration in vitro.
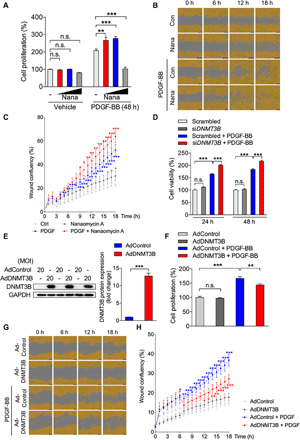
(A) hPASMCs treated with increasing concentrations (0.3, 1, or 3 μM) of DNMT3B inhibitor [nanaomycin A (Nana)] or vehicle in the presence or absence of PDGF-BB for 48 hours, demonstrating that nanaomycin A facilitated proliferation of hPASMCs in response to PDGF-BB. n = 3 independent experiments. (B) Representative images and (C) quantification analysis of wound confluency showed that nanaomycin A (0.3 μM) facilitated PDGF-BB–induced hPASMC migration. Red asterisk denotes the comparison to the PDGF-BB–treated group, and blue or gray asterisk denotes the comparison to the control (Con) group at examined time point. n = 3 independent experiments. (D) rPASMCs treated with siDnmt3b had higher proliferation compared to those with scrambled siRNA in response to PDGF-BB. n = 3 independent experiments. (E) An elevation in DNMT3B protein level in AdDNMT3B-treated hPASMCs was observed compared to AdControl-treated PASMCs at 48 hours after infection (MOI = 20). n = 3 independent experiments. (F) hPASMCs infected with either AdControl or AdDNMT3B (MOI = 20), demonstrating that Dnmt3b overexpression suppressed cell proliferation in response to PDGF-BB. n = 3 independent experiments. (G) Representative images and (H) quantification analysis of wound confluency showed that DNMT3B overexpression rescued PDGF-BB–induced hPASMC migration. Red asterisk denotes comparison to the PDGF-BB–treated group (AdControl + PDGF-BB), and blue asterisk denotes comparison to the control (AdControl) group at examined time points. n = 3 independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 versus corresponding controls, Student’s t test (E), one-way ANOVA with Bonferroni correction for multiple comparisons (A, D, and F), and two-way ANOVA with Bonferroni’s post hoc analysis (C and H). All data throughout the figure represent mean ± SEM.
Small interfering RNA (siRNA) was used against Dnmt3b to investigate whether Dnmt3b deficiency affected proliferation of rPASMCs. The knockdown efficiency was examined by Western blotting (fig. S4G). Dnmt3b silencing enhanced the proliferation and migration of rPASMCs in response to PDGF-BB at the indicated time points (Fig. 5D and fig. S4, H and I).
Moreover, a notable increase in proliferation was observed in Dnmt3b−/− rPASMCs compared to WT rPASMCs (fig. S4J). In aggregate, these data demonstrate that Dnmt3b deficiency causes a worse phenotype in PASMCs in the development of PH.
Overexpression of DNMT3B attenuates proliferation and migration of PASMCs
We next sought to determine whether overexpression of human DNMT3B by adenovirus could improve the phenotype of PASMCs. Cells infected with AdDNMT3B exhibited increases in DNMT3B expression at indicated multiplicity of infection (MOI), as evidenced by immunoblotting and immunofluorescence staining (Fig. 5E and fig. S5, A and B). Overexpression of human DNMT3B largely abolished the impact of PDGF-BB on proliferation of hPASMCs (Fig. 5F). In addition, DNMT3B augmentation resulted in a considerable reduction in migration ability in hPASMCs at each time point compared to the control group (Fig. 5, G and H). In accordance with the findings in hPASMCs, human DNMT3B overexpression resulted in a significant decline in cell viability and migration ability in rPASMCs in response to PDGF-BB (fig. S5, C to E). Collectively, these results suggest that DNMT3B acts against PDGF-BB–induced proliferation and migration of PASMCs in a protective manner.
DNMT3B mediated inflammation at transcriptional level
The underlying mechanism by which DNMT3B confers protection against PH development was next determined. RNA-sequencing (RNA-seq) was performed to investigate the transcriptional profile in hPASMCs with adenovirus-mediated overexpression of DNMT3B (AdDNMT3B) compared to empty vector (AdControl). By quantitative analysis, 222 transcripts were differentially expressed in response to DNMT3B overexpression [q < 0.001; fold change (FC) ≥ 2], with 113 up- and 109 down-regulated (Fig. 6A). A total of 37 up-regulated and 34 down-regulated unique genes were displayed in a heatmap (Fig. 6B). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the most differentially expressed genes (DEGs) highlighted inflammation-related process as the most changed pathway that might act as a protector against vascular remodeling (Fig. 6C). To further explore interactions among the 637 overlapped transcripts, a strong connectivity of proinflammatory genes (i.e., CCL5 and CXCL6) stood out in protein-protein interaction (PPI) analysis (fig. S6, A and B). In addition, we also performed RNA-seq in PDGF-BB–treated hPASMCs infected with AdDNMT3B or AdControl and identified a list of DEGs. Next, we selected the DEGs with similar trend in response to excessive DNMT3B in PDGF-BB–treated hPASMCs as in those without PDGF-BB treatment. A heatmap showing the selected DEGs was generated (fig. S6C), in which proinflammatory genes were still documented. These transcriptome and protein analyses suggest that inflammatory regulation may be one of the underlying mechanisms of DNMT3B-mediated protection against PH progression.
Fig. 6. Mechanism of protective role of Dnmt3b in vascular remodeling in PAH.
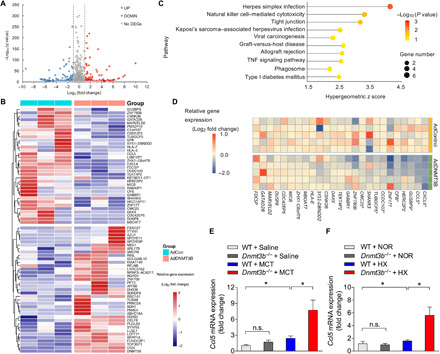
(A) Volcano plot of DEGs in hPASMCs infected with AdDNMT3B (MOI = 20) versus AdControl indicating up-regulated genes, which are highlighted in red, and down-regulated genes, which are highlighted in green. The thresholds are shown as dashed lines (horizontal: q < 0.001; vertical: absolute log2 FC ≥ 1). (B) Heatmap illustrating DEGs for hierarchical clustering. (C) KEGG pathway enrichment analysis of the identified DEGs between AdDNMT3B and AdControl groups. The 10 most significantly enriched pathways (P < 0.05 by Fisher’s exact test) are shown. TNF, tumor necrosis factor. (D) mRNA expression of selected down-regulated genes out of RNA-seq data was analyzed by RT-PCR. The relative FC, compared with those from AdControl-infected hPASMCs, was log-transformed at the base of 2 and shown in the heatmap. *P < 0.05 versus AdControl-infected hPASMCs, Student’s t test, n = 5 per group. (E and F) RT-PCR analysis of expression of Ccl5 in lungs of WT and Dnmt3b−/− PH rats after MCT injection (E) or under hypobaric hypoxic conditions (F), demonstrating a higher Ccl5 expression in Dnmt3b−/− PH rats (n = 4 to 8 per group). *P < 0.05 versus WT control group or WT MCT or WT hypoxia group as indicated, one-way ANOVA with Bonferroni correction for multiple comparisons; mean ± SEM.
Because of the inhibitory effect of methylation, we mainly focused on the down-regulated genes [q < 0.001; FC ≥ 2; fragments per kilobase per million mapped reads (FPKM) > 0.01] in hPASMCs overexpressing DNMT3B (table S2). Because a proliferative, promigratory, and proinflammatory phenotype in PASMCs is the key to pathologic vascular remodeling in PH, proinflammatory genes were reduced after DNMT3B overexpression by the RNA-seq data analysis. We next validated the candidate genes by quantitative PCR and demonstrated a reduction of the proinflammatory gene CCL5 (Fig. 6D and fig. S6D) as well as several other down-regulated genes (Fig. 6D and fig. S6, E to J) at transcriptional levels in hPASMCs overexpressing DNMT3B. Moreover, an additional increase in Ccl5 expression was observed in Dnmt3b−/− rats compared to WT littermates after MCT administration or hypoxia exposure (Fig. 6, E and F). In addition, a significant increase of Ccl5 expression was depicted in hypoxia-induced PH mice with AAV9-null infection compared to normoxia control mice, and a trend toward a down-regulation was displayed in AAV9-DNMT3B–infected mice after hypoxia exposure (fig. S6K). These findings demonstrated that inflammatory regulation might contribute to the protective role of Dnmt3b in vascular remodeling (fig. S7).
DISCUSSION
The acquisition and influence of DNA methylation in PH progression and establishment have largely been unexplored. The present study identifies that (i) global DNA methylation level is up-regulated in lungs from rodent models after MCT injection or under hypobaric hypoxic condition; (ii) DNMT3B expression increased in individuals with CHD-PAH, in two experimental PAH models, and in PASMCs of PAH patients as well as in PASMCs stimulated by PDGF-BB, the most potent mitogen of PASMCs involved in pathogenesis of PH; (iii) Dnmt3b-deficient (Dnmt3b−/−) rats have a markedly deteriorated hemodynamic response to hypobaric hypoxia or MCT that is associated with a worse vascular remodeling and more severe RVH; (iv) overexpression of DNMT3B leads to mitigation of PH development in a rodent model; (v) pharmacological inhibition, inactivation, or genetic ablation of Dnmt3b enhances PASMC proliferation and migration, which can be restored by overexpression of DNMT3B; (vi) DNMT3B may have a therapeutic potential against pulmonary vasculature remodeling via inflammatory regulation.
Archer et al. (8) showed that hypermethylation of a CpG island of Sod2 favors a proliferative, apoptosis-resistant state in fawn-hooded rats (FHRs) with spontaneous PH, which is associated with significantly higher Dnmt3b in lungs and Dnmt3b in isolated PASMCs. These findings are in line with our observations that Dnmt3b expression levels increased in lungs from two classical PH rat models and in PASMCs stimulated by growth factor. A reduction in Dnmt1 and Dnmt3a expression level after MCT administration and hypoxia exposure was observed in our study, but Archer et al. showed a slight increase in Dnmt1 and no change in Dnmt3a in lungs from FHRs (spontaneously develop PAH) compared to Sprague-Dawley rat lungs but not FH-BN1 consomic rats (identical to FHRs except that they have introgression of a normal chromosome 1 and do not develop PAH). One of the possibilities responsible for this discrepancy might be that Dnmt1 and Dnmt3a expression patterns vary from different PH experimental models. The expression of Dnmt1, Dnmt3a, and Dnmt3l decreased in our PH rodent models, while global DNA methylation and expression of Dnmt3b increased. It suggests that Dnmt3b might act as the main driver in DNA methylation in PAH. In addition, a demethylation agent, 5-aza-2′-deoxycytidine (5-AZA), inhibits PASMC proliferation through up-regulation of Sod2 expression, but whether 5-AZA can regress pulmonary vascular remodeling in vivo still warrants further investigation.
Apart from the role in pulmonary vascular remodeling, Dnmt3b deficiency enhanced RVH in rats, and in a complementary approach, overexpression of human DNMT3B via the intratracheal delivery of AAV-DNMT3B in mice alleviated hypoxia-induced RVH. In contrast to our findings, a site-specific higher DNA methylation was associated with an up-regulation of Dnmt3a and Dnmt3b expression levels, which contributed to endothelial cell dysfunction and led to right ventricular decompensation (15). However, our findings showed no increase of DNMT3B expression levels in PAECs in PAH patients compared to control subjects. One potential explanation to the divergent role of DNMT3B in PH could be that right ventricles in our PAH model were still at the compensation stage, which were different from decompensated right ventricles in the previous report.
Recently, one study showed diminished TET2 in peripheral blood cells of PAH patients, which engenders the epigenetic link of DNA demethylation to PAH progression (16). However, DNA methylation regulated by DNMTs and TETs is strain, species, organ, and stimuli dependent. In our study, we found that higher DNMT3B was displayed in lungs from PAH patients, different PH rodent models, and PASMCs from patients with PAH compared to corresponding controls. All these can reflect local DNA methylation signature in the setting of PAH. The other study by Joshi et al. (17) unveiled the mechanisms that link reprogrammed metabolism to aberrant expression of functional phenotype-associated genes in PH. They suggest that functional TET2 is required for G6PD inhibition to reverse PH in mice. In contrast to our finding of up-regulation of Dnmt3b in hypoxia condition, Dnmts were not altered significantly in hypoxia versus normoxia and in hypoxia + NEOU (G6PD inhibitor) versus hypoxia, and we assume that the difference lies in that their finding was based on RNA-seq data with limited sample size. Here, the role of DNMT3B in the scenario of PAH warrants our further investigation.
The excessive proliferation and apoptosis resistance of PASMC have emerged as essential mechanisms in the remodeling of the pulmonary vasculature associated with PH (18, 19). Growing evidence shows that DNA methylation is a key regulator for smooth muscle cell phenotype switch and vascular remodeling (20). Although Dnmt3b was shown to functionally inactivate p53 in vascular smooth muscle cells induced by homocysteine, thereby indirectly promoting cell growth, our results reveal that dysfunction of Dnmt3b upsets the balance between PASMC growth and death and migration ability, thus favoring increased muscularization of the vasculature. Collectively, these results suggest that Dnmt3b may modulate smooth muscle cells through different pathways and exhibit opposite effects under various conditions.
Emerging evidence indicates strong similarities between the dysregulated growth of vascular and tumor cells (21, 22). Recent studies show that epigenetic disruptions caused by Dnmt3b are also involved in tumor transformation and progression. Altered promoter DNA methylation is identified as one of the most important epigenetic abnormalities in human cancers (23), contributing to some of the hallmarks of cancer (24). Dnmt3b can be negatively modulated by Ang and activates Mmp2, which contribute to bladder cancer oncogenesis (25). A more recent study on the effect of Dnmt3b overexpression in HaCaT cells on global gene expression was carried out to identify targets of Dnmt3b (26). They found that the Dnmt3b overexpression modulates the expression of genes related to cancer, down-regulating the expression of 151 genes with CpG islands; some of which promote tumorigenesis (27, 28), while some are in favor of inhibition of proliferation, tumorigenicity, or invasion (29, 30). The phenomenon that one active molecule exerts opposite effects in different tumors is not unusual, and the discrepancy may be largely attributable to the differences in tumor microenvironment or cell lines, and these results highlight the importance of Dnmt3b in gene expression and neoplastic transformation.
Dnmt3b was demonstrated to be posttranscriptionally regulated by multiple microRNAs (miR-29c, miR-26a, miR-26b, miR-203, etc.), and loss of expression of these regulatory microRNAs contributes to Dnmt3b overexpression in hypermethylation cell lines (31). Among them, the miR-29 family is up-regulated in heritable PAH and antagonism of miR-29 attenuates PAH in transgenic mouse models of Bmpr2 mutation (32). We speculate that up-regulation of miR-29 might contribute to a lower expression of Dnmt3b, thus promoting vascular remodeling and PAH. Dnmt3b, however, is not unique in this regard, as other molecules could also be regulated by miR-29.
Our current findings demonstrated enrichment of inflammation-related pathways in response to DNMT3B overexpression in hPASMCs, with an involvement of proinflammatory genes, such as CCL5. Chemokine CCL5 exerts a multitude of generally proinflammatory effects (33, 34), and the vascular smooth muscle cells are reported to orchestrate arterial inflammatory response via acute Ccl5 production and subsequent inflammatory cell recruitment, which may, in part, explain the development of adventitia remodeling in PH (35). Our observation of higher CCL5 in experimental PH models compared to control rats was consistent with previous studies that CCL5 was elevated in pulmonary adventitial fibroblasts from chronically hypoxic hypertensive calves (36) and lungs of patients displaying severe PAH (37). Although a further increase of Ccl5 expression was observed in Dnmt3b−/− rats after stress (MCT or hypoxia) compared to WT littermates, and CCL5 was reduced after DNMT3B overexpression in vitro, the functional study whether the inhibition or knockdown of CCL5 could prevent the adverse effect of DNMT3B silencing or vice versa was not investigated in our study, which could be one of our limitations. As therapeutic interventions targeting either PASMC proliferation (38) or lung inflammation (18) could ameliorate experimental PH, the modulation of DNMT3B to maintain balanced inflammatory signaling and PASMC proliferation might emerge as a therapeutic target for pulmonary vascular homeostasis.
Using PAECs from PAH patients, no difference of DNMT3B expression levels was detected in PAECs from PAH patients compared to those from controls. However, we did not investigate the role of Dnmt3b in endothelial function. At this point, we cannot exclude the possibility that effects of Dnmt3b on endothelial cells in response to stress differentially or synergistically affect PAH in a systemic knockout. This limitation precludes any conclusion regarding the cell-specific role of Dnmt3b in the progression of PAH. As for animal experiments, the efficiency of decreased DNMT3B activity and the rescue in rat models had not been explored, which can be considered as limitations of this study.
Although various pro-proliferative and inflammatory pathways in vascular remodeling have been reported in PH, the contribution of each remains to be fully understood. Here, we demonstrate that a link may exist between Dnmt3b-mediated proliferation and inflammatory response in the pathogenesis of PH and offer insights for future therapeutic gains.
MATERIALS AND METHODS
Human lung samples and patient characteristics
To mirror female predominance of clinical prevalence, human lung tissue from subjects with CHD-associated PAH (CHD-PAH) (n = 8) or CHD (n = 4) were obtained during lung biopsy. Subjects with CHD-PAH had a mean age (years ± SD) of 26 ± 2. Control CHD had a mean age (years ± SD) of 30 ± 2. The study protocol for tissue biopsy was approved by the ethics committee of Fuwai Hospital and in accordance with the Declaration of Helsinki. Written informed consent was obtained from each individual patient or the patient’s next of kin. All the detailed clinical data from lung tissues are listed in table S1. hPASMCs for detection of DNMT3B expression levels were isolated from five donors and five patients with PAH. PAECS used in this study were from three control subjects and patients with PAH.
Animal models
All animal protocols were approved by the Ethics and Animal Care and Use Committee of Fuwai Hospital and Beijing Shijitan Hospital or the University of Illinois at Chicago. All animal work was carried out in accordance with the relevant guidelines and regulations of these bodies. To test the DNA methyltransferase profiling in PH rodent models, male Sprague-Dawley rats (~250 to 300 g) were purchased from Charles River (Beijing, China) and kept either in hypobaric hypoxic chamber (50 kPa) or in room air (21% oxygen) for 3 weeks (n = 7 to 8 per group) for the hypoxia study. One-dose subcutaneous injection of MCT or saline was given to male Sprague-Dawley rats for the MCT study (n = 7 to 8 per group). On day 21, both hemodynamics and RVHI were performed, and lung tissues were harvested and stored in liquid nitrogen or formalin for subsequent analysis. Dnmt3b−/− rats were obtained as a gift from L. Zhang from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (39). Eight-week-old male Dnmt3b−/− and WT littermates were either put into the hypobaric hypoxic chamber for 3 weeks or received one-dose MCT injection to develop PH. At day 21 after hypobaric hypoxic exposure or day 28 after MCT injection, hemodynamics and RVHI were assessed as described above. Lung tissue were fixed in formalin or stored at −80°C. To investigate the effect of DNMT3B overexpression on the development of PH, 6- to 8-week-old C57BL/6 mice were delivered intratracheally with 2 × 1011 genome copies of either AAV9-DNMT3B or AAV9-null in a final solution of 100 μl at day 14 under hypoxia or normoxia condition; the mouse was maintained at a 45° angle for 1 min. Mice were kept in a hypoxia chamber or in normoxia for additional 2 weeks. At day 28, animals were sacrificed for analysis. Details are provided in Supplementary Materials and Methods.
Hemodynamic measurement, tissue collection, and histopathological analysis
For measurement of RVSP and mPAP in rats, a fluid-filled catheter (PE 50 tubing) was inserted into the right ventricle through the right jugular vein and connected to a force transducer. Rats were then sacrificed, and the hearts and lungs were harvested. RVH was determined as the ratio of right ventricular to left ventricular and septal weight (RV/LV + S). The right lung was snap-frozen in liquid nitrogen.
A fraction of the excised lungs was fixed in 4% paraformaldehyde, embedded in paraffin, and then sliced into 4-μm-thick sections. Some sections were stained with hematoxylin and eosin (H&E) and elastin–van Gieson (EVG) following the standard protocol for morphological examination as in previous studies, and the external diameter and internal diameter of these arteries were measured using Image-Pro Plus software (Media Cybernetics, USA). The arterial wall thickness was calculated as follows: percentage wall thickness = [(external diameter − internal diameter)/external diameter] × 100.
For the assessment of pulmonary arteriolar muscularization, immunofluorescent staining of rat lung sections was performed using a mouse monoclonal anti–α-SMA (smooth muscle actin) antibody (Sigma-Aldrich, USA) at 1:1000 in 5% bovine serum albumin (BSA), followed by Alexa Fluor 594–conjugated anti-mouse secondary antibody (ZSGB-BIO, China) to visualize PASMCs, as well as using a rabbit polyclonal anti–von Willebrand factor (vWF) antibody (Abcam, USA) at 1:1500 in 5% BSA, followed by Alexa Fluor 488–conjugated anti-rabbit secondary antibody (ZSGB-BIO, China), to visualize endothelial cells. Images were taken at ×400 magnification. The degree of pulmonary arteriolar muscularization and size was assessed in a blinded fashion. Sixty to 80 intra-acinar vessels per rat accompanying either alveolar ducts or alveoli were evaluated. Each vessel was categorized as nonmuscularized (i.e., no apparent muscle), partially muscularized (i.e., with only a crescent of muscle), or fully muscularized (i.e., with a complete medial coat of muscle). The percentages of nonmuscularized and partially or fully muscularized vessels were calculated by dividing the number of vessels in each category by the total number of blood vessels counted per slide.
To evaluate proliferation of PASMCs in vivo, immunofluorescence staining of mice lung sections was performed with primary antibodies specific to proliferating cell nuclear antigen (PCNA; diluted 1:1000, Cell Signaling Technology) and anti–α-SMA antibody overnight at 4°C. Slides were then washed with phosphate-buffered saline (PBS) three times and incubated with secondary antibodies conjugated with Alexa Fluor 488 and Alexa Fluor 594 for 1 to 2 hours at room temperature. Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) was used to mount and counterstain the slides. All of the fluorescence images were captured under a fluorescence microscope at 200× and 400×. All images were analyzed with ImageJ (National Institutes of Health).
To localize DNMT3B protein expression within pulmonary vascular lesions, following antigen retrieval, the lung sections were treated with 3% H2O2, permeabilized with 0.3%Triton X-100, blocked with 5% BSA, and incubated with anti-DNMT3B antibody (catalog no. ab79822, 1:250 dilution; Abcam, Cambridge, UK) overnight at 4°C, followed by serial incubations with biotin-conjugated goat anti-rabbit immunoglobulin G (IgG) secondary antibody and horseradish peroxidase (HRP)–labeled streptavidin (ZSGB-BIO, China). Diaminobenzidine (ZSGB-BIO, China) was added to initiate the chromogenic reaction, and hematoxylin was used to stain the nuclei. The sections were mounted and examined under a microscope (DM6000B, Leica, Germany) at ×200 magnification.
Echocardiography
Transthoracic echocardiography was performed with the VEVO2100 system equipped with a 13- to 24-MHz (MS250, rat cardiovascular) transducer (VisualSonics). To determine TAPSE, the M-mode cursor was oriented to the junction of the tricuspid valve plane. Right ventricular free wall thickness was measured at parasternal short-axis view of a right ventricular outflow level as previously described (40).
Global methylation analysis
Global DNA methylation status in lungs of MCT- or hypoxia-induced PH rodents was performed using the MethylFlash Methylated DNA Quantification Kit (EpiGentek, Farmingdale, NY, USA). Briefly, sample DNA is bound to high DNA affinity strip wells. Methylated DNA is detected using capture and detection antibodies to 5-mC and then quantified colorimetrically by reading absorbance at 450 nm using a microplate reader (Tecan, Sweden). The amount of methylated DNA is proportional to the OD (optical density) intensity measured. The absolute amount of methylated DNA was quantified as per protocol using a standard curve, plotting the OD values versus five serial dilutions of control methylated DNA (0.5 to 10 ng).
Cell culture
rPASMCs were isolated from adult Sprague-Dawley rats. hPASMC were purchased from ScienCell (San Diego, CA). Cells were maintained in complete Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium. Passages 3 and 4 were used for subsequent experiments.
Inhibition of DNMT3B
DNMT3B was inhibited either genetically by siRNA oligonucleotides using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions or pharmaceutically by nanaomycin A. Transfection conditions are available in Supplementary Materials and Methods.
Overexpression of DNMT3B
PASMCs were infected with pHBAd-MCMV-GFP adenovirus encoding the complementary DNA (cDNA) green fluorescent protein (GFP) (AdControl) and DNMT3B (AdDNMT3B). Detailed methods are available in Supplementary Materials and Methods.
Cell viability and proliferation assays
To test cell viability, culture supernatant was discarded and replaced with Cell Counting Kit-8 (CCK-8) solution (Dojindo Laboratories, China). Cells were incubated at 37°C, 5% CO2 for 1.5 hours. Absorbance values (OD) were determined with a microplate reader (Infinite M200 Pro, Tecan, Switzerland) at a wavelength of 450 nm. To test cell proliferation, culture supernatant was discarded and incubated with 0.1% crystal violet for 10 min at room temperature. The plate was washed three times, and 100 μl of 0.1% SDS was added. OD values were determined at a wavelength of 570 nm.
Cell migration
For nanaomycin A experiments, 1.5 × 104 hPASMCs per well or 1 × 105 rPASMCs per well were seeded in 96-well plates. A scratch was made using a 96-pin WoundMaker in human cells and with a P200 pipette tip in rat cells. Cells were left untreated or treated with nanaomycin A (0.3 μM) or vehicle in response to human PDGF-BB (20 ng/ml) or rat PDGF-BB (50 ng/ml) for the indicated time. For adenovirus infection experiments, 1 × 104 hPASMCs per well or 1 × 105 rPASMCs per well were infected with AdDNMT3B or AdControl for 24 hours, followed by a scratch as mentioned above, and then treated with PDGF-BB or vehicle for 48 hours. Human cell confluence was imaged by phase-contrast using the IncuCyte HD system (IncuCyte live cell). Data were processed and analyzed using IncuCyte Zoom 96-Well Cell Invasion Software Application Module (all from Essen BioScience Inc., Ann Arbor, MI, USA). Six phase-contrast images of each individual scratch of rat cells were obtained at ×100 magnification at indicated time points after scratching. Transwell assay was also performed to assess cell migration capacity. rPASMCs were transfected with siRNA as detailed in Supplementary Materials and Methods. Cells were then seeded into the upper chamber of 24-well 8-μm transwell plates (Corning Costar, Corning, NY, USA), with the lower chamber in the presence or absence of PDGF-BB. Eighteen hours later, cells were fixed and dyed with 0.1% crystal violet dye for 5 min. Detailed method is provided in Supplementary Materials and Methods.
Western blotting
Tissues and cells after indicated duration of treatment were treated with radioimmunoprecipitation assay (RIPA) lysis buffer (Applygen, Beijing, China), complete protease inhibitor cocktail, phosphatase inhibitor cocktail (both from Roche, Basel, Switzerland), and loading buffer (Applygen, Beijing, China). Lysates were frozen at −80°C until used. Cell lysates (20 μg total protein) were separated on reducing SDS–polyacrylamide gel electrophoresis (PAGE) gels, and proteins were transferred to polyvinylidene fluoride membranes. Membranes were then blocked and probed with rabbit monoclonal antibodies against DNMT3A (clone D23G1, catalog no. 3598, 1:1000 dilution), DNMT1 (clone D63A6, catalog no. 5032, 1:1000 dilution; all Cell Signaling Technology, Danvers, MA), or DNMT3B (catalog no. ab79822, 1:1000 dilution; Abcam, Cambridge, UK) and rabbit polyclonal antibodies against DNMT3L (catalog no. A-1005-050, 1:1000 dilution; EpiGentek, Farmingdale, NY). As a loading control, all blots were reprobed with a monoclonal antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalog no. 2118, 1:5000 dilution; Cell Signaling Technology, Danvers, MA). Densitometry was performed using ImageJ software.
RNA isolation and RNA-seq library preparation, sequencing, and analysis
Total mRNA was isolated from frozen rat lung tissues and hPASMCs with TRIzol (Invitrogen). The purity, concentration, and integrity of RNA were determined using Agilent 2100 Bioanalyzer. RNA libraries were prepared for sequencing using the BGISEQ-500 sequencing platform [Beijing Genomics Institute (BGI), Wuhan, China] with a single-end read length of 50 base pairs (bp). After quality control with SOAPnuke (version 1.5.2), approximately 24.1 million clean reads were obtained and the average mapping ratio to the reference gene was about 90% (fig. S6A). The RNA-seq analyses were performed at the BGI. Briefly, high-quality reads were aligned to the human reference genome (GRCh38) using HISAT2 (version 2.0.4) and Bowtie2 (version 2.2.5). Normalized gene expression was calculated using the FPKM with RSEM (version 1.2.12). A Student’s t test algorithm was used to identify DEGs between the different sample groups, and P values were corrected using the Benjamini-Hochberg algorithm (q values). DEGs defined from the pairwise comparisons had to satisfy two selection criteria, including (i) FC ≥ 2 and (ii) corresponding q ≤ 0.001. The annotation information regarding genes involved in biological pathways was obtained from the KEGG database. A KEGG pathway enrichment analysis was performed using Fisher’s exact test, and only pathways with a corresponding P ≤ 0.05 were considered as significantly enriched. DIAMOND (version .0.8.31) was used to map the DEGs to the STRING (version 10) database to obtain the interaction between DEG-encoded proteins using homology with known proteins. The top 100 interaction networks were selected and imported into Cytoscape for network analysis.
cDNA synthesis and real-time PCR
Isolated RNA was subsequently transcribed into cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen) according to the manufacturer’s instructions. Real-time PCR was then performed with the FastStart Universal SYBR Green Master (Rox) Kit (Roche) or TaqMan Real-Time PCR Master Mixes in the ABI 7500 Real-Time Detection System. The threshold cycle (Ct) values of the target genes were normalized to that of the housekeeping gene (endogenous control) encoding GAPDH. All data were analyzed by adopting 2−ΔΔCt method, with 2−ΔΔCt demonstrating the relative expression ratios of the target gene of the case group to the control group (ΔΔCt = ΔCtcase group − ΔCtcontrol group, ΔCt = CtTarget gene − CtGAPDH). Relative mRNA expression is shown, with the average from control samples set to 1. Primers were purchased from AuGUT (Beijing, China), and sequences are shown in table S3. The TaqMan gene expression assays used in this study were as follows: Hs00966522_m1 (PDGFB), Hs00171876_m1 (DNMT3B), and Hs99999905_m1 (GAPDH).
Statistical analysis
Data are presented as the mean ± SEM or median (interquartile range). When only two groups were compared, statistical differences were assessed with unpaired two-tailed Student’s t test if normally distributed. Otherwise, Mann-Whitney U test was used. Statistical significance among three groups or over was determined using one-way analysis of variance (ANOVA) followed by Bonferroni multiple comparison test or Kruskal-Wallis test or two-way ANOVA with Bonferroni’s post hoc analysis as appropriate. The data were analyzed using GraphPad Prism version 5.0, and P < 0.05 was considered to indicate a statistically significant difference.
Supplementary Material
Acknowledgments
We thank the patients and families for their involvement in this study. We thank L. Huang (University of Illinois at Chicago) for technical support in cell experiments and Y. Ma (Key Laboratory of Human Disease Comparative Medicine, Ministry of Health, Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences) and X.-J. Wang (from FuWai Hospital, Academy of Medical Sciences) for technical support in cell experiments, thoughtful discussion and important comments. Funding: This work was supported by grants from the National Natural Science Foundation of China (81630003, 81670052), 13th Five-Year Plan—Precision Medicine—Key Research and Development Program—Clinical Cohort of Rare Disease (2016YFC0901500), Beijing Natural Science Foundation (7181009), and CAMS Innovation Fund for Medical Sciences (2016-I2M-1-002, 2017-I2M-1-004, 2017-I2M-1-011, 2017-I2M-2-001, 2017PT32016, and 2017-I2M-BR-02). Author contributions: Y.Y., Y.-Y.H., and X.J. contributed to the study’s design and conduction, data analyses, data interpretation, and manuscript preparation; Y.W. contributed to the study’s design and data interpretation; J.-W.C. performed mice experiments and DNA methylation detection assays and analyzed data; Y.Y., Y.-Y.H., and J.-H.Z. performed rat experiments, transcriptomic analyses, and real-time PCR; J.Y. and T.-Y.L. performed the proliferation and migration assay; Xu Zhang maintained the Dnmt3b−/− rats; R.-J.Z. performed staining experiments; D.L. assisted in data analysis and figure preparation; S.-S.G. performed Western blot experiments; X.-Q.X. collected clinical human lung tissues; K.S. contributed to statistical analyses; S.-Q.L. performed echocardiography; L.-F.Z. helped design the knockout rats; Xue Zhang and S.-Y.Z. participated in the study design. Z.-C.J. conceived and supervised the study, designed the experiments, and revised the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eaba2470/DC1
REFERENCES AND NOTES
- 1.Farber H. W., Loscalzo J., Pulmonary arterial hypertension. N. Engl. J. Med. 351, 1655–1665 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Hoeper M. M., Bogaard H. J., Condliffe R., Frantz R., Khanna D., Kurzyna M., Langleben D., Manes A., Satoh T., Torres F., Wilkins M. R., Badesch D. B., Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 62 (suppl. 25), D42–D50 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Hopper R. K., Moonen J.-R. A. J., Diebold I., Cao A., Rhodes C. J., Tojais N. F., Hennigs J. K., Gu M., Wang L., Rabinovitch M., In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133, 1783–1794 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwangbo C., Lee H.-W., Kang H., Ju H., Wiley D. S., Papangeli I., Han J., Kim J.-D., Dunworth W. P., Hu X., Lee S., El-Hely O., Sofer A., Pak B., Peterson L., Comhair S., Hwang E. M., Park J.-Y., Thomas J.-L., Bautch V. L., Erzurum S. C., Chun H. J., Jin S.-W., Modulation of endothelial bone morphogenetic protein receptor type 2 activity by vascular endothelial growth factor receptor 3 in pulmonary arterial hypertension. Circulation 135, 2288–2298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu L., Li Y., Tollefsbol T. O., Gene-environment interactions and epigenetic basis of human diseases. Curr. Issues Mol. Biol. 10, 25–36 (2008). [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrlich M., DNA hypomethylation in cancer cells. Epigenomics 1, 239–259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonnet S., Michelakis E. D., Porter C. J., Andrade-Navarro M. A., Thébaud B., Bonnet S., Haromy A., Harry G., Moudgil R., McMurtry M. S., Weir E. K., Archer S. L., An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 113, 2630–2641 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Archer S. L., Marsboom G., Kim G. H., Zhang H. J., Toth P. T., Svensson E. C., Dyck J. R. B., Gomberg-Maitland M., Thébaud B., Husain A. N., Cipriani N., Rehman J., Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 121, 2661–2671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ke X., Johnson H., Jing X., Michalkiewicz T., Huang Y.-W., Lane R. H., Konduri G. G., Persistent pulmonary hypertension alters the epigenetic characteristics of endothelial nitric oxide synthase gene in pulmonary artery endothelial cells in a fetal lamb model. Physiol. Genomics 50, 828–836 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hautefort A., Chesné J., Preussner J., Pullamsetti S. S., Tost J., Looso M., Antigny F., Girerd B., Riou M., Eddahibi S., Deleuze J.-F., Seeger W., Fadel E., Simonneau G., Montani D., Humbert M., Perros F., Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget 8, 52995–53016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., Huang X., Leng D., Li J., Wang L., Liang Y., Wang J., Miao R., Jiang T., DNA methylation signatures of pulmonary arterial smooth muscle cells in chronic thromboembolic pulmonary hypertension. Physiol. Genomics 50, 313–322 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Liu D., Yan Y., Chen J.-W., Yuan P., Wang X.-J., Jiang R., Wang L., Zhao Q.-H., Wu W.-H., Simonneau G., Qu J.-M., Jing Z.-C., Hypermethylation of BMPR2 promoter occurs in patients with heritable pulmonary arterial hypertension and inhibits BMPR2 expression. Am. J. Respir. Crit. Care Med. 196, 925–928 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Dai J., Zhou Q., Chen J., Rexius-Hall M. L., Rehman J., Zhou G., Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat. Commun. 9, 3850 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boucherat O., Peterlini T., Bourgeois A., Nadeau V., Breuils-Bonnet S., Boilet-Molez S., Potus F., Meloche J., Chabot S., Lambert C., Tremblay E., Chae Y. C., Altieri D. C., Sutendra G., Michelakis E. D., Paulin R., Provencher S., Bonnet S., Mitochondrial HSP90 accumulation promotes vascular remodeling in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 198, 90–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potus F., Ruffenach G., Dahou A., Thebault C., Breuils-Bonnet S., Tremblay È., Nadeau V., Paradis R., Graydon C., Wong R., Johnson I., Paulin R., Lajoie A. C., Perron J., Charbonneau E., Joubert P., Pibarot P., Michelakis E. D., Provencher S., Bonnet S., Downregulation of MicroRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation 132, 932–943 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Potus F., Pauciulo M. W., Cook E. K., Zhu N., Hsieh A., Welch C. L., Shen Y., Tian L., Lima P., Mewburn J., D’Arsigny C. L., Lutz K. A., Coleman A. W., Damico R., Snetsinger B., Martin A. Y., Hassoun P. M., Nichols W. C., Chung W. K., Rauh M. J., Archer S. L., Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation 141, 1986–2000 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joshi S. R., Kitagawa A., Jacob C., Hashimoto R., Dhagia V., Ramesh A., Zheng C., Zhang H., Jordan A., Waddell I., Leopold J., Hu C.-J., McMurtry I. F., D’Alessandro A., Stenmark K. R., Gupte S. A., Hypoxic activation of glucose-6-phosphate dehydrogenase controls the expression of genes involved in the pathogenesis of pulmonary hypertension through the regulation of DNA methylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 318, L773–L786 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hurst L. A., Dunmore B. J., Long L., Crosby A., Al-Lamki R., Deighton J., Southwood M., Yang X., Nikolic M. Z., Herrera B., Inman G. J., Bradley J. R., Rana A. A., Upton P. D., Morrell N. W., TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 8, 14079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong Z., Chen K.-H., DasGupta A., Potus F., Dunham-Snary K., Bonnet S., Tian L., Fu J., Breuils-Bonnet S., Provencher S., Wu D., Mewburn J., Ormiston M. L., Archer S. L., MicroRNA-138 and MicroRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension cancer phenotype. Am. J. Respir. Crit. Care Med. 195, 515–529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuang J., Luan P., Li H., Wang K., Zhang P., Xu Y., Peng W., The Yin–Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 37, 84–97 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Rai P. R., Cool C. D., King J. A. C., Stevens T., Burns N., Winn R. A., Kasper M., Voelkel N. F., The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 178, 558–564 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voelkel N. F., Gomez-Arroyo J., Abbate A., Bogaard H. J., Nicolls M. R., Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur. Respir. J. 40, 1555–1565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baylin S. B., DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2 (suppl. 1), S4–S11 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D., Weinberg R. A., The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Peres R., Furuya H., Pagano I., Shimizu Y., Hokutan K., Rosser C. J., Angiogenin contributes to bladder cancer tumorigenesis by DNMT3b-mediated MMP2 activation. Oncotarget 7, 43109–43123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peralta-Arrieta I., Hernández-Sotelo D., Castro-Coronel Y., Leyva-Vázquez M. A., Illades-Aguiar B., DNMT3B modulates the expression of cancer-related genes and downregulates the expression of the gene VAV3 via methylation. Am. J. Cancer Res. 7, 77–87 (2017). [PMC free article] [PubMed] [Google Scholar]
- 27.Buchert M., Papin M., Bonnans C., Darido C., Raye W. S., Garambois V., Pelegrin A., Bourgaux J.-F., Pannequin J., Joubert D., Hollande F., Symplekin promotes tumorigenicity by up-regulating claudin-2 expression. Proc. Natl. Acad. Sci. U.S.A. 107, 2628–2633 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi Y. K., Woo S.-M., Cho S.-G., Moon H. E., Yun Y. J., Kim J. W., Noh D.-Y., Jang B. H., Shin Y. C., Kim J.-H., Shin H. D., Paek S. H., Ko S.-G., Brain-metastatic triple-negative breast cancer cells regain growth ability by altering gene expression patterns. Cancer Genomics Proteomics 10, 265–275 (2013). [PubMed] [Google Scholar]
- 29.Gkirtzimanaki K., Gkouskou K. K., Oleksiewicz U., Nikolaidis G., Vyrla D., Liontos M., Pelekanou V., Kanellis D. C., Evangelou K., Stathopoulos E. N., Field J. K., Tsichlis P. N., Gorgoulis V., Liloglou T., Eliopoulos A. G., TPL2 kinase is a suppressor of lung carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 110, E1470–E1479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan J.-Y., Huang X., Luo Y.-L., PSMA7 inhibits the tumorigenicity of A549 human lung adenocarcinoma cells. Mol. Cell. Biochem. 366, 131–137 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Sandhu R., Rivenbark A. G., Coleman W. B., Loss of post-transcriptional regulation of DNMT3b by microRNAs: A possible molecular mechanism for the hypermethylation defect observed in a subset of breast cancer cell lines. Int. J. Oncol. 41, 721–732 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X., Talati M., Fessel J. P., Hemnes A. R., Gladson S., French J., Shay S., Trammell A., Phillips J. A., Hamid R., Cogan J. D., Dawson E. P., Womble K. E., Hedges L. K., Martinez E. G., Wheeler L. A., Loyd J. E., Majka S. J., West J., Austin E. D., Estrogen metabolite 16α-hydroxyestrone exacerbates bone morphogenetic protein receptor type II–associated pulmonary arterial hypertension through MicroRNA-29–mediated modulation of cellular metabolism. Circulation 133, 82–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikolajczyk T. P., Nosalski R., Szczepaniak P., Budzyn K., Osmenda G., Skiba D., Sagan A., Wu J., Vinh A., Marvar P. J., Guzik B., Podolec J., Drummond G., Lob H. E., Harrison D. G., Guzik T. J., Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 30, 1987–1999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krohn R., Raffetseder U., Bot I., Zernecke A., Shagdarsuren E., Liehn E. A., van Santbrink P. J., Nelson P. J., Biessen E. A., Mertens P. R., Weber C., Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation 116, 1812–1820 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Kovacic J. C., Gupta R., Lee A. C., Ma M., Fang F., Tolbert C. N., Walts A. D., Beltran L. E., San H., Chen G., St. Hilaire C., Boehm M., Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J. Clin. Invest. 120, 303–314 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M., Riddle S. R., Frid M. G., El Kasmi K. C., McKinsey T. A., Sokol R. J., Strassheim D., Meyrick B., Yeager M. E., Flockton A. R., McKeon B. A., Lemon D. D., Horn T. R., Anwar A., Barajas C., Stenmark K. R., Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J. Immunol. 187, 2711–2722 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dorfmüller P., Zarka V., Durand-Gasselin I., Monti G., Balabanian K., Garcia G., Capron F., Coulomb-Lherminé A., Marfaing-Koka A., Simonneau G., Emilie D., Humbert M., Chemokine RANTES in severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 165, 534–539 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Rothman A. M. K., Arnold N. D., Pickworth J. A., Iremonger J., Ciuclan L., Allen R. M. H., Guth-Gundel S., Southwood M., Morrell N. W., Thomas M., Francis S. E., Rowlands D. J., Lawrie A., MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J. Clin. Invest. 126, 2495–2508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Y., Zhang X., Shen B., Lu Y., Chen W., Ma J., Bai L., Huang X., Zhang L., Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 24, 122–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koskenvuo J. W., Mirsky R., Zhang Y., Angeli F. S., Jahn S., Alastalo T.-P., Schiller N. B., Boyle A. J., Chatterjee K., De Marco T., Yeghiazarians Y., A comparison of echocardiography to invasive measurement in the evaluation of pulmonary arterial hypertension in a rat model. Int. J. Cardiovasc. Imaging 26, 509–518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eaba2470/DC1