

Liquid nitrogen–treated tumor cells serve as a drug carrier and cancer vaccine to facilitate chemo-immunotherapy.
Abstract
Live cells have been vastly engineered into drug delivery vehicles to leverage their targeting capability and cargo release behavior. Here, we describe a simple method to obtain therapeutics-containing “dead cells” by shocking live cancer cells in liquid nitrogen to eliminate pathogenicity while preserving their major structure and chemotaxis toward the lesion site. In an acute myeloid leukemia (AML) mouse model, we demonstrated that the liquid nitrogen–treated AML cells (LNT cells) can augment targeted delivery of doxorubicin (DOX) toward the bone marrow. Moreover, LNT cells serve as a cancer vaccine and promote antitumor immune responses that prolong the survival of tumor-bearing mice. Preimmunization with LNT cells along with an adjuvant also protected healthy mice from AML cell challenge.
INTRODUCTION
Acute myeloid leukemia (AML) is a hematological malignancy with a dismal prognosis and 5-year survival of only 30% (1, 2). The standard-of-care cytoreductive chemotherapy induces AML remission (3–5), but disease relapse frequently occurs (6, 7). Hematopoietic stem cell transplantation (HSCT) in patients who achieve remission after chemotherapy represents the only curative approach so far (8, 9). However, HSCT is associated with either the lack of suitable hematopoietic stem cell donors or the high risk of transplantation-related mortality (10). Hence, there is an urgent need to find further strategies for AML treatment.
AML originates in the bone marrow, and bone marrow creates leukemia-niches that promote leukemia survival (11). Furthermore, biodistribution of chemotherapeutics to the bone marrow is frequently poor (12, 13), and higher doses of chemotherapy required to ablate leukemia are toxic to normal tissues. Thus, developing targeting drug delivery to the bone marrow may not only enhance the therapeutic index of chemotherapy but also reduce its toxicity to nonhematopoietic tissues. Nevertheless, it is still challenging to engineer bone marrow–targeting moieties and bypass the blood–bone marrow barriers (14). Leveraging cells’ intrinsic properties offers solutions to overcome these limitations (15–17). Because AML cells naturally exhibit bone marrow homing capabilities (18–20), we developed an approach to use AML cells as drug carriers while eliminating their intrinsic pathogenicity.
Here, we used a liquid nitrogen–based cryo-shocking method to obtain therapeutic dead cells. These cells maintained the intact structure allowing for drug encapsulation, but lost their proliferation ability and pathogenicity. Specifically, cryo-shocked AML cells kept their bone marrow homing capability and served as a drug delivery vehicle of doxorubicin (DOX), which is a critical drug used in the induction chemotherapy in AML. Cryo-shocked AML cells stimulated an immune response that was in conjunction with chemotherapy to eradicate leukemia in tumor-bearing mice. Preimmunization with LNT cells together with an adjuvant protected healthy mice from AML cell challenge. We thus proposed a “dead cell”–based delivery vehicle that can be rapidly manufactured for clinical use compared with other live cell–based drug delivery systems (21).
RESULTS
Engineering and characterization of liquid nitrogen–treated cells
To obtain the liquid nitrogen–treated (LNT) cells, AML cells were suspended in the cell cryopreservation medium and immersed in liquid nitrogen for 12 hours. LNT cells were then thawed at 37°C and washed with phosphate-buffered saline (PBS) (Fig. 1A). When analyzed by confocal microscopy, LNT cells showed the same cellular structure as untreated live cells when assessed by nucleus and cytoskeleton staining (Fig. 1B). A slight decrease in cellular size was observed (Fig. 1C), with an average size of 11 μm for LNT cells and 12 μm for untreated live cells. The forward scatter (FSC) values measured by flow cytometry corroborated the cell size reduction of LNT cells, and similar side scatter (SSC) values suggested that the internal structure of LNT cells was maintained (Fig. 1D). Scanning electron microscopy (SEM) images revealed the sphere-like structure of LNT cells and the rougher cellular surface as compared with control live cells (Fig. 1E and fig. S1).
Fig. 1. Characterization of LNT cells.
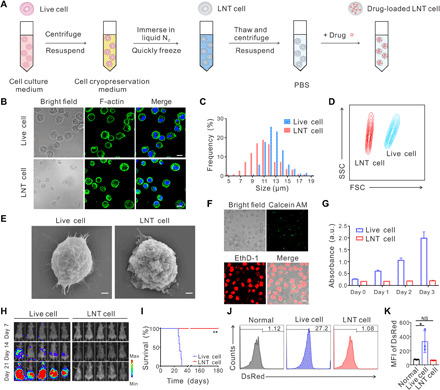
(A) Schematic of the procedure to prepare LNT cells. (B) Cellular structure of live and LNT C1498 cells. Cell nucleus was stained by Hoechst 33342 (blue), and cytoplasm F-actin was stained by AF488 phalloidin (green). Scale bars, 10 μm. (C) Cellular sizes of live and LNT C1498 cells. The cells were imaged by confocal microscopy, and cellular size was measured by the software Nano Measurer (cell numbers = 200). (D) Flow cytometry analysis of live and LNT C1498 cells under same voltages. FSC, forward scatter; SSC, side scatter. (E) SEM images of live and LNT cells. Scale bars, 1 μm. (F) Cell viability analysis of live and LNT cells by LIVE/DEAD viability kit. Calcein AM: live cells; EthD-1: dead cells. Scale bar, 10 μm. (G) Cell viability analysis of live and LNT cells by CCK8 assay (n = 6). a.u., arbitrary unit. (H) In vivo proliferation of 2 × 106 luciferase tagged live and LNT C1498 cells indicated by the bioluminescence signal (n = 5). (I) Survival of mice after challenge with 2 × 106 live and LNT tumor cells (n = 5). Typical flow cytometry images (J) and DsRed intensities (K) of peripheral blood 20 days after challenge with live and LNT DsRed tagged C1498 cells (n = 3). MFI, mean fluorescence intensity. Data are presented as means ± SD (G and K). Statistical significance was calculated via the log-rank (Mantel-Cox) test (I) and ordinary one-way analysis of variance (ANOVA) (K). *P < 0.05, **P < 0.01. NS, not significant.
Next, we evaluated the cell viability of LNT cells. As shown in Fig. 1F, nearly all the LNT cells were labeled with EthD-1 (indicating dead cells) and did not show intact fluorescence signal of calcein AM (indicating live cells). In addition, LNT cells did not show proliferative activity as compared with live cancer cells as measured with cell counting kit-8 (CCK8) assay (Fig. 1G). Furthermore, we confirmed the necrosis-dependent cell death of LNT cells by annexin-V–propidium iodide (PI) staining (fig. S2). We further verified the absence of pathogenicity of LNT cells in vivo. As shown in Fig. 1H, live C1498 AML cells quickly proliferated in mice and caused 100% death in 31 days, while mice receiving C1498 LNT cells exhibited no detectable bioluminescence signal, and all mice survived for at least 180 days (Fig. 1, H and I). Moreover, we quantitatively analyzed cancer cells in the peripheral blood at day 20 after injection. A notably higher DsRed signal was observed in mice injected with live C1498 cells, indicating a high portion of leukemia cells circulating in the blood, while the DsRed intensity for the mice challenged with LNT cells was similar to that of healthy mice (Fig. 1, J and K).
Leveraging LNT cells as the targeting drug carrier
Leukemia cells exhibit bone marrow homing and resident capabilities, which are at least in part associated with the expression of CXCR4 and CD44 chemokine, two typical adhesion receptors that interact with bone marrow (18, 22, 23). SDS–polyacrylamide gel electrophoresis (PAGE) showed that most of the proteins expressed by live C1498 cells were retained in LNT cells (fig. S3A). CXCR4 and CD44 were detected in both live and LNT cells as assessed by confocal imaging and flow cytometry (Fig. 2, A and B, and fig. S3, B and C). Despite some reduction in expression levels, Western blotting analysis indicated that CXCR4 and CD44 expression were 39 and 60%, respectively, in LNT cells compared with live cells (fig. S3, D and E). The bone marrow homing capacity of LNT cells was also evaluated. Upon intravenous infusion, LNT cells exhibited similar accumulation efficiency in bone barrow compared with live C1498 cells (Fig. 2, C and D, and fig. S4A). Cell signal was notably higher compared with paraformaldehyde-fixed cells, which reflects the loss of bioactivities upon paraformaldehyde fixation (Fig. 2, C and D). LNT cells also distributed in the liver, kidney, and spleen (fig. S4B), and were cleared from the bloodstream within 24 hours (fig. S5).
Fig. 2. LNT cells as the drug carrier.
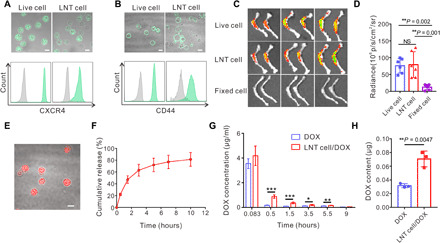
CXCR4 (A) and CD44 (B) expression of live and LNT C1498 cells analyzed by confocal microscopy (top) and flow cytometry (bottom). Scale bars, 10 μm. (C) Fluorescence images of bone isolated 6 hours after injection of cy5.5-labeled live C1498 cells, LNT C1498 cells, and paraformaldehyde-fixed C1498 cells. (D) Fluorescence intensities of the bone of indicated groups (n = 6). (E) Typical confocal image of DOX-loaded LNT cells. Scale bar, 10 μm. (F) Cumulative release profile of DOX from LNT cell/DOX (n = 3). (G) Plasma DOX concentration after intravenous injection of free DOX and LNT cell/DOX with DOX dose of 2.5 mg/kg (n = 4). (H) Bone marrow DOX content 3 hours after administration of the drug (n = 3). Data are presented as means ± SD (D and F to H). Statistical significance was calculated via ordinary one-way ANOVA (D) and Student’s t test (G and H). *P < 0.05, **P < 0.01, ***P < 0.001.
Because nuclear and cytoplasmic cellular structures are preserved in LNT cells (Fig. 1B), we assessed if these cells can be payload with DOX, via DNA intercalation and the electrostatic interactions between DOX and cytoplasm proteins (24–26), and deliver DOX to bone marrow. Briefly, DOX could be loaded into LNT cells via mixing and incubation with a loading capacity of 65 ± 16 μg per 1 × 107 LNT cells (Fig. 2E and fig. S6A). DOX was released from the drug-loaded LNT cells (LNT cell/DOX) in a sustained manner, and 81% of DOX was released within 10 hours (Fig. 2F). We then studied the in vitro cytotoxicity against C1498 cells of free DOX and LNT cell/DOX. The IC50 (median inhibitory concentration) values were 0.32 and 1.05 μg/ml, respectively (fig. S6B). Even though free DOX exhibited higher cytotoxicity against C1498 cells in vitro, LNT cell/DOX allowed longer detection of DOX in the blood and higher DOX accumulation within the bone marrow (Fig. 2, G and H). We used murine AML models to evaluate the therapeutic efficacy of LNT cell/DOX. In tumor-bearing C57BL/6J mice, tumor growth was monitored by bioluminescence signals upon treatment (fig. S7, A to C). In this leukemia model, although LNT cells alone exhibited no antitumor effects, LNT cell/DOX treatment reduced the tumor growth compared with control treatments (fig. S7, D to H).
Chemo-immunotherapy via LNT cells
Tumor cell lysates can function as cancer vaccines and initiate tumor-specific immune responses (27, 28). We hypothesized that LNT cells can enhance the antigen uptake and maturation of antigen-presenting cells (APCs). LNT cells cocultured with dendritic cells (DCs) caused their maturation as assessed by up-regulation of CD40, CD80, CD86, and major histocompatibility complex II (MHC-II) (fig. S8A). Moreover, CD4+ T cells and CD8+ T cells increased in the peripheral blood of the mice receiving LNT cells and the adjuvant of monophosphoryl lipid A (MPLA) (fig. S8B). DC maturation and T cell activation–related cytokines, including interferon-γ (IFN-γ), tumor necrosis factor–α (TNF-α), and interleukin-6 (IL-6), were also detected in mice treated with LNT cell and adjuvant (fig. S8C). We next evaluated the antitumor efficacy of LNT cell/DOX with adjuvant in leukemia-bearing mice. As demonstrated in Fig. 3 (A and B), bioluminescence of AML cancer cells increased rapidly in untreated mice, while AML had been partially inhibited after DOX or LNT cell and adjuvant treatment. AML cells were almost completely eliminated in mice treated with LNT cell/DOX and adjuvant up to 21 days after tumor inoculation (Fig. 3B). Quantitative analysis of tumor bioluminescence and survival analysis also demonstrated superior therapeutic activity of LNT cell/DOX combined with adjuvant (Fig. 3, C to E). Increased serum levels of IFN-γ and TNF-α (Fig. 3, F and G), as well as elevation of CD3+ T cell and CD8+ T cells, supported the occurrence of boosted immunity in the mice receiving LNT Cell/DOX and adjuvant treatment (Fig. 3, H and I).
Fig. 3. Therapeutic efficacy of LNT cells in AML model.
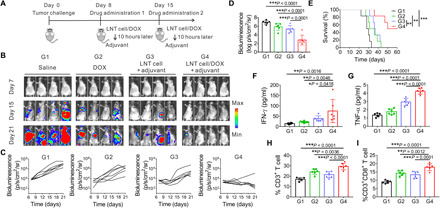
(A) Schematic of the treatment model. (B) AML progression in vivo as indicated by bioluminescence signal expressed by luciferase tagged C1498 cells during different treatments (G1, saline; G2, DOX; G3, LNT cell + adjuvant; G4, LNT cell/DOX + adjuvant). (C) Quantified bioluminescence of different treatment groups. (D) Bioluminescence intensity of treated mice on day 21 (n = 6). (E) Survival of the mice of different treatment groups (n = 6). Serum cytokine levels of IFN-γ (F), TNF-α (G), and proportion of peripheral CD3+ T cells (H) and CD8+ T cells (I) on day 13 (n = 6). Data are presented as means ± SD. (D and F to I). Statistical significance was calculated via ordinary one-way ANOVA (D and F to I) and log-rank (Mantel-Cox) test (E). *P < 0.05, **P < 0.01, ***P < 0.001.
Prophylactic efficiency of LNT tumor cells
We further evaluated the efficacy of LNT cells as a prophylactic cancer vaccine. Mice were first immunized at 21, 14, and 7 days before challenge with live C1498 cells. The onset of AML in mice was prevented in mice preimmunized with LNT cells and adjuvant (Fig. 4, A to C). Quantitative data also revealed that the tumor bioluminescence intensity of the group of LNT cells with adjuvant was substantially lower than control groups (Fig. 4D). Moreover, 71% of the mice treated with LNT cells and adjuvant were tumor free 90 days after tumor challenge, while all control mice died by day 34 (Fig. 4E). Serum levels of IFN-γ, TNF-α, IL-12, and IL-6 were significantly increased in mice treated with LNT cells and adjuvant (Fig. 4F), indicating that a prompt immune response was triggered upon tumor cell inoculation. In addition, CD3+ T cells and CD8+ T cells were significantly increased in the peripheral blood of mice vaccinated with LNT cells and adjuvant (Fig. 4, G and H, and fig. S9).
Fig. 4. In vivo prophylactic efficiency of LNT tumor cells.
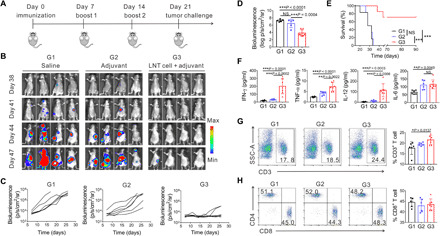
(A) Schematic of the treatment model. Bioluminescence images (B) and quantified bioluminescence (C) of the mice preimmunized with different treatment formulations (G1, saline; G2, adjuvant; G3, LNT cell + adjuvant). (D) Bioluminescence intensity of treated mice on day 47 (n = 5 for G1 and G2 for one mice died before day 47; n = 7 for G3). (E) Survival of the mice after tumor challenge (n = 6 for G1 and G2; n = 7 for G3). (F) Serum cytokine levels 3 days after challenge of live C1498 cells (n = 6 for G1 and G2; n = 7 for G3). (G) Representative flow cytometry images of CD3+ T cells (left) and proportion of peripheral CD3+ T cells (right) on day 24 (n = 6 for G1 and G2; n = 7 for G3). (H) Representative flow cytometry images of CD8+ T cells (left) and corresponding proportion of peripheral CD8+ T cell gating on CD3+ T cells (right) on day 24 (n = 6 for G1 and G2; n = 7 for G3). Data are presented as means ± SD. (D and F to H). Statistical significance was calculated via ordinary one-way ANOVA (D and F to H) and log-rank (Mantel-Cox) test (E), *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
In this study, we demonstrated the feasibility, efficacy, and safety of tumor dead cells used as a drug-targeting carrier and tumor vaccine for cancer therapy. Compared with the synthetic material–mediated delivery vehicles, cell-based carriers show unique targeting capacities and can bypass biological barriers (15, 29). AML cells originate in the bone marrow and naturally exhibit similar bone marrow homing capabilities as HSCs (22, 30, 31), rendering them suitable to be used as cellular drug carriers for AML therapy. However, it remains essential to develop strategies allowing the elimination of AML tumorigenicity while transiently preserving cellular integrity to deliver the payload at the tumor site. We therefore proposed to use the “dead” but “functional” AML cells as the drug carrier.
Usually, the structure of the live cells can disintegrate upon dying with the loss of proteins and cytokines (32). In addition, external stimuli that could induce cell death, such as heat or radiation, will deactivate proteins as well (33, 34). Our data support the concept that cryo-shocked tumor cells obtained by rapid immersion of live cells in liquid nitrogen lose tumorigenicity while preserving transiently the integrity of the cell structure, which is critical for the drug loading and cargo release. Furthermore, certain critical functional proteins that include CD44 and CXCR4 were retained in LNT cells. CD44 can interact with hyaluronic acid that is highly expressed in the endosteum of bone marrow (18). CXCR4 enables cells to migrate toward the chemokine stromal cell–derived factor 1 (SDF-1) that is constitutively produced by the osteoblasts and stromal cells (23). CD44 and CXCR4 are two important adhesion receptors mediating AML cells homing toward bone marrow (35). Retention of both CD44 and CXCR4 in LNT cells, even if at reduced levels compared with live cells, is likely critical to promote their bone marrow homing. The proposed LNT-based strategy is simple and straightforward from a manufacturing point of view. Tumor cells in the case of liquid tumors can be readily collected in large quantity, for example, by leukapheresis. Similarly, for solid tumors, multiple devises are currently available to generate single cell suspension from resected tumors or tumor biopsies. The process of cell shocking in liquid nitrogen is also feasible to standardize in good manufacturing practice conditions.
We evaluated the proliferation and tumorigenicity of LNT tumor cells both in vitro and in vivo. The data that all mice treated with LNT cells exhibited no obvious side effects and no leukemia growth was recorded for 6 months after inoculation of LNT C1498 cells support at least in our mouse model the safety of the proposed strategy. After exposure to liquid nitrogen, the cellular membrane of LNT cells becomes permeable. While live cells require treatment with cell membrane detergent to obtain intracellular staining, LNT cells do not require this treatment, indicating the loss of long-term integrity of the cell membrane, which is essential to cell survival. However, our experiments demonstrate that liquid nitrogen treatment does not impair the capacity of LNT cells to function as drug carrier and tumor vaccine. The cryo-shocking technique could be a platform technology in cell bioengineering and could be applicable to various cell types. Here, we have further tested feasibility in 4T1 tumor cells (fig. S10). Regarding the potential impact in clinical use, the safety of LNT tumor cells, besides C1498 cells adopted in this work, should be evaluated thoroughly in other experimental animal models. In addition, the application of LNT cells to serve as drug carriers of other therapeutics, such as immune checkpoint inhibitors (36), is worth investigating.
In summary, we engineered LNT tumor cells to serve simultaneously as a drug delivery carrier and cancer vaccine. The simple liquid nitrogen treating process abrogates the tumorigenicity of tumor cells but preserves the integrity of their cellular structure. This in turn allows the possibility to load LNT cells with chemotherapy drugs and preserves the homing capacity of these cells to the tumor site. LNT cells in combination with adjuvant could elicit both therapeutic and protective immune antitumor responses and may avoid the complex quality control associated with isolated cells and synthesized material–based vehicles and enable large-scale production for clinical use.
MATERIALS AND METHODS
Experimental design
The aim of this study was to use the cryo-shocked tumor cells as a kind of drug-targeting carrier and tumor vaccine for chemo-immunotherapy in the treatment of AML. After treating the live cells in liquid nitrogen, the cellular structure of the cryo-shocked cells was observed. The proliferation behavior, in vivo tumorigenicity, and targeting capability toward the bone marrow of the cryo-shocked cells were assessed. In vivo antitumor efficacy was analyzed in an AML model by intravenously injecting C1498 cells in C57BL/6J mice. Mice were randomly assigned to groups based on body weights. After different treatments, the mice were captured by in vivo imaging system (IVIS) to evaluate in vivo tumor progression. Survival curves, immune cell proportions, and cytokine levels were determined according to previous experimental experience. Specific information about treatment groups, sample numbers, and data analysis was denoted in the figure captions.
Materials, cell lines, and animals
Doxorubicin hydrochloride was purchased from Fisher Scientific Co. (D4193; purity, >95%). Noncontrolled-rate cell cryopreservation medium was bought from Cyagen Co. (NCRC-10001-50). AML cell line C1498 was purchased from the American Type Culture Collection (ATCC). Luciferase and DsRed tagged C1498 cell line was provided by B. Blazar of the University of Minnesota. The cells were cultured in 90% Dulbecco’s modified Eagle’s medium (Gibco) and 10% fetal bovine serum (Gibco) with penicillin (200 U ml−1) and streptomycin (200 U ml−1) (Gibco). The cells were passaged every 1 to 2 days. C57BL/6J mice (4 to 6 weeks, female) were purchased from the Jackson laboratory. All animal tests complied with the animal protocol approved by the Institutional Animal Care and Use Committee of the University of California, Los Angeles.
Preparation of LNT cells and drug-loaded LNT cells
C1498 cells were centrifuged at 250g for 3 min and suspended in noncontrolled-rate cell cryopreservation medium at a cell density of 1 × 106 to 1 × 107 ml−1. The cell-containing medium was immersed in liquid nitrogen for 12 hours. Before use, the medium was thawed at 37°C and LNT cells were pelleted at 500g for 3 min. After washing with PBS solution (pH 7.4), LNT cells were suspended in PBS and kept at 4°C. For preparation of DOX-loaded LNT cells, the LNT cells were suspended in DOX containing PBS. After incubation for 2 hours, the medium was centrifuged at 500g for 5 min and the pellets were DOX-loaded LNT cells.
In vivo treatment of AML
The AML model was established by intravenous injection of 5 × 106 C1498 cells on day 0. On day 8 and day 15, saline, LNT cell + adjuvant, free DOX, and LNT cell/DOX + adjuvant were administrated intravenously with DOX dose of 5 mg/kg and adjuvant (MPLA) 20 μg per mouse. Specifically, MPLA was intravenously injected 10 hours after injection of LNT cell or LNT cell/DOX. The bioluminescence images of mice were captured every 3 days. The exposure time was 2 min. On day 13, 400 μl of blood was collected via the orbital vein. Blood (200 μl) was treated with ammonium-chloride-potassium (ACK) buffer and centrifuged at 800g for 8 min to obtain pellets of white blood cells. After staining with BV421-CD3, PE-CD4, and APC-CD8, the samples were analyzed by flow cytometry. Another 200 μl of blood in blood serum collection tubes (BD Microtainer 365967) was centrifuged at 3000 rpm for 10 min. The upper serum was detected with the following enzyme-linked immunosorbent assay kits: IFN-γ (BioLegend 430804) and TNF-α (BioLegend 430904).
Statistical analysis
The results were presented as means ± SD or mean ± standard error of the mean (means ± SEM) as indicated. The data were compared by Student’s t test between two groups and ordinary one-way analysis of variance (ANOVA) for three or more groups. The survival curves were analyzed via the log-rank (Mantel-Cox) test. All statistical analyses were conducted by the GraphPad Prism software. The threshold of a statistically significant difference was defined as P < 0.05.
Supplementary Material
Acknowledgments
We acknowledge B. Blazar at the University of Minnesota for providing the luciferase and DsRed tagged C1498 cell line. Funding: This work was supported by the NIH (R01 CA234343-01A1) and grants from the start-up packages of UCLA. Author contributions: Z.G. and T.C. proposed the conception of the project. T.C. and H.L. performed all the experiments and collected the data with the help of G.C., Z.W., J.W., P.A., and Y.T. All authors analyzed the data and contributed to the writing of the manuscript, discussed the results and implications, and edited the manuscript at all stages. Competing interests: Z.G. and T.C. are inventors on a U.S. patent application related to this work filed by University of California, Los Angeles (no. 63/094,034, filed [Oct 20th, 2020]). Z.G. is a scientific cofounder of ZenCapsule Inc. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabc3013/DC1
REFERENCES AND NOTES
- 1.Döhner H., Weisdorf D. J., Bloomfield C. D., Acute myeloid leukemia. N. Engl. J. Med. 373, 1136–1152 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Shah N. J., Najibi A. J., Shih T.-Y., Mao A. S., Sharda A., Scadden D. T., Mooney D. J., A biomaterial-based vaccine eliciting durable tumour-specific responses against acute myeloid leukaemia. Nat. Biomed. Eng 4, 40–51 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Mayer R. J., Davis R. B., Schiffer C. A., Berg D. T., Powell B. L., Schulman P., Omura G. A., Moore J. O., McIntyre O. R., Frei E. III; Cancer and Leukemia Group B , Intensive postremission chemotherapy in adults with acute myeloid leukemia. N. Engl. J. Med. 331, 896–903 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Woods W. G., Neudorf S., Gold S., Sanders J., Buckley J. D., Barnard D. R., Dusenbery K., DeSwarte J., Arthur D. C., Lange B. J., Kobrinsky N. L., A comparison of allogeneic bone marrow transplantation, autologous bone marrow transplantation, and aggressive chemotherapy in children with acute myeloid leukemia in remission: A report from the Children’s Cancer Group. Blood 97, 56–62 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Fernandez H. F., Sun Z., Yao X., Litzow M. R., Luger S. M., Paietta E. M., Racevskis J., Dewald G. W., Ketterling R. P., Bennett J. M., Rowe J. M., Lazarus H. M., Tallman M. S., Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med. 361, 1249–1259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konopleva M., Konoplev S., Hu W., Zaritskey A. Y., Afanasiev B. V., Andreeff M., Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 16, 1713–1724 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Bradstock K. F., Gottlieb D. J., Interaction of acute leukemia cells with the bone marrow microenvironment: Implications for control of minimal residual disease. Leuk. Lymphoma 18, 1–16 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Copelan E. A., Hematopoietic stem-cell transplantation. N. Eng. J. Med. 354, 1813–1826 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Duval M., Klein J. P., He W., Cahn J.-Y., Cairo M., Camitta B. M., Kamble R., Copelan E., de Lima M., Gupta V., Keating A., Lazarus H. M., Litzow M. R., Marks D. I., Maziarz R. T., Rizzieri D. A., Schiller G., Schultz K. R., Tallman M. S., Weisdorf D., Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J. Clin. Oncol. 28, 3730–3738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai T., Li J., Sinclair A., Imren S., Merriam F., Sun F., O’Kelly M. B., Nourigat C., Jain P., Delrow J. J., Basom R. S., Hung H.-C., Zhang P., Li B., Heimfeld S., Jiang S., Delaney C., Expansion of primitive human hematopoietic stem cells by culture in a zwitterionic hydrogel. Nat. Med. 25, 1566–1575 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Emyr B., Qattan M., Mutti L., Demonacos C., Krstic-Demonacos M., The role of microenvironment and immunity in drug response in leukemia. Biochim. Biophys. Acta 1863, 414–426 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Adjei I. M., Sharma B., Peetla C., Labhasetwar V., Inhibition of bone loss with surface-modulated, drug-loaded nanoparticles in an intraosseous model of prostate cancer. J. Control. Release 232, 83–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karantanou C., Godavarthy P. S., Krause D. S., Targeting the bone marrow microenvironment in acute leukemia. Leuk. Lymphoma 59, 2535–2545 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Shadduck R. K., Waheed A., Wing E. J., Demonstration of a blood-bone marrow barrier to macrophage colony-stimulating factor. Blood 73, 68–73 (1989). [PubMed] [Google Scholar]
- 15.Hu Q., Sun W., Wang J., Ruan H., Zhang X., Ye Y., Shen S., Wang C., Lu W., Cheng K., Dotti G., Zeidner J. F., Wang J., Gu Z., Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat. Biomed. Eng 2, 831–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z., Hu Q., Gu Z., Leveraging engineering of cells for drug delivery. Acc. Chem. Res. 51, 668–677 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Tang J., Hubbard-Lucey V. M., Pearce L., O’Donnell-Tormey J., Shalabi A., The global landscape of cancer cell therapy. Nat. Rev. Drug Discov. 17, 465–466 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Jin L., Hope K. J., Zhai Q., Smadja-Joffe F., Dick J. E., Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 12, 1167–1174 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Mishra A., Shiozawa Y., Pienta K. J., Taichman R. S., Homing of cancer cells to the bone. Cancer Microenviron. 4, 221–235 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marjon K. D., Termini C. M., Karlen K. L., Saito-Reis C., Soria C. E., Lidke K. A., Gillette J. M., Tetraspanin CD82 regulates bone marrow homing of acute myeloid leukemia by modulating the molecular organization of N-cadherin. Oncogene 35, 4132–4140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Q., Cheng H., Peng H., Zhou H., Li P. Y., Langer R., Non-genetic engineering of cells for drug delivery and cell-based therapy. Adv. Drug Deliv. Rev. 91, 125–140 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Konopleva M. Y., Jordan C. T., Leukemia stem cells and microenvironment: Biology and therapeutic targeting. J. Clin. Oncol. 29, 591–599 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavor S., Petit I., Porozov S., Avigdor A., Dar A., Leider-Trejo L., Shemtov N., Deutsch V., Naparstek E., Nagler A., Lapidot T., CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 64, 2817–2824 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Zunino F., Di Marco A., Zaccara A., Gambetta R. A., The interaction of daunorubicin and doxorubicin with DNA and chromatin. Biochim. Biophys. Acta 607, 206–214 (1980). [DOI] [PubMed] [Google Scholar]
- 25.Ferrant A., Hulhoven R., Bosly A., Cornu G., Michaux J. L., Sokal G., Clinical trials with daunorubincin-DNA and adriamycin-DNA in acute lymphoblastic leukemia of childhood, acute nonlymphoblastic leukemia, and bronchogenic carcinoma. Cancer Chemother. Pharmacol. 2, 67–71 (1979). [DOI] [PubMed] [Google Scholar]
- 26.Trouet A., Deprez-De Campeneere D., Daunorubicin-DNA and doxorubicin-DNA. A review of experimental and clinical data. Cancer Chemother. Pharmacol. 2, 77–79 (1979). [DOI] [PubMed] [Google Scholar]
- 27.Chiang C. L.-L., Benencia F., Coukos G., Whole tumor antigen vaccines. Semin. Immunol. 22, 132–143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye Y., Wang C., Zhang X., Hu Q., Zhang Y., Liu Q., Wen D., Milligan J., Bellotti A., Huang L., Dotti G., Gu Z., A melanin-mediated cancer immunotherapy patch. Sci. Immunol. 2, eaan5692 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Xue J., Zhao Z., Zhang L., Xue L., Shen S., Wen Y., Wei Z., Wang L., Kong L., Sun H., Ping Q., Mo R., Zhang C., Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 12, 692–700 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Sison E. A. R., Brown P., The bone marrow microenvironment and leukemia: Biology and therapeutic targeting. Expert Rev. Hematol. 4, 271–283 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnet D., Dick J. E., Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Green D. R., Llambi F., Cell death signaling. Cold Spring Harb. Perspect. Biol. 7, a006080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez Y., Lindquist S., HSP104 required for induced thermotolerance. Science 248, 1112–1115 (1990). [DOI] [PubMed] [Google Scholar]
- 34.Prise K. M., Schettino G., Folkard M., Held K. D., New insights on cell death from radiation exposure. Lancet Oncol. 6, 520–528 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Becker P. S., Dependence of acute myeloid leukemia on adhesion within the bone marrow microenvironment. ScientificWorldJournal 2012, 856467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C., Wang J., Zhang X., Yu S., Wen D., Hu Q., Ye Y., Bomba H., Hu X., Liu Z., Dotti G., Gu Z., In situ formed reactive oxygen species–responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 10, eaan3682 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabc3013/DC1