

CTD S2P mediates nutrient-dependent development.
Abstract
Serine 2 phosphorylation (S2P) within the CTD of RNA polymerase II is considered a Cdk9/Cdk12-dependent mark required for 3′-end processing. However, the relevance of CTD S2P in metazoan development is unknown. We show that cdk-12 lesions or a full-length CTD S2A substitution results in an identical phenotype in Caenorhabditis elegans. Embryogenesis occurs in the complete absence of S2P, but the hatched larvae arrest development, mimicking the diapause induced when hatching occurs in the absence of food. Genome-wide analyses indicate that when CTD S2P is inhibited, only a subset of growth-related genes is not properly expressed. These genes correspond to SL2 trans-spliced mRNAs located in position 2 and over within operons. We show that CDK-12 is required for maximal occupancy of cleavage stimulatory factor necessary for SL2 trans-splicing. We propose that CTD S2P functions as a gene-specific signaling mark ensuring the nutritional control of the C. elegans developmental program.
INTRODUCTION
Cloning of the gene encoding the largest subunit of polymerase II (Pol II), Rpb1 (RNA polymerase B), identified a unique repeated motif (YSPTSPS) forming the C-terminal end of the protein and referred to as the C-terminal domain (CTD). A large body of work next established a proposed universal model where hypophosphorylated Pol II is recruited to transcription units (TUs) and becomes phosphorylated first on serine 5 (S5) during the transition from initiation to early elongation and then on serine 2 (S2) during productive elongation (1–3). While the CTD can be modified in many additional ways on all of its residues, the anticorrelated gradients of S5 and S2 phosphorylations (S5P and S2P) are the most conserved and best characterized marks. Elegant work using fission yeast has shown that the only essential function of CTD S5P in this organism is to ensure the timely recruitment of the capping machinery to promote capping of the emerging transcript at the 5′-end of the TU where S5P peaks (4). This model is supported by other converging evidences (5, 6). The CTD S5P can therefore be seen as a generic scaffold required for maturation through both allosteric activation of processing factors and locally increasing their concentration at their site of action. By contrast, the profile of CTD S2P, which increases during elongation and peaks at the 3′-end, suggests that it plays a similar role in splicing and 3′-end processing, namely, cleavage and polyadenylation, and evidence supports the possibility that S2P enhances the recruitment of 3′-end factors (7, 8). mRNA 3′-end maturation consists of an endonucleolytic cleavage followed by polyadenylation of the free 3′-end. Termination is linked to 3′-end processing because the free 5′-end of the mRNA generated by cleavage is targeted by the nuclear 5′-3′ exoribonuclease Rat1/Xrn2, which acts as a “torpedo” and disengages Pol II from the template (9). Most of the components of both the cleavage and polyadenylation specificity factor (CPSF) and the cleavage stimulatory factor (CstF) (10–12) are conserved across species, despite differences in complex organization (13, 14).
However, while the 3′-end maturation machineries are essential in both budding and fission yeast, the S2P mark is dispensable in both species (15). Therefore, the dependency between CTD phosphorylation and mRNA processing may be less stringent for S2P at the 3′-end than it is for S5P at the 5′-end. Supporting this, when a yeast strain was engineered to express the T7 polymerase (that lacks a CTD) and a T7-dependent transcript, this mRNA was not capped but cleavage and polyadenylation were readily detected and dependent on the usual factors despite a reduced specificity for the poly-A site (16). In addition, experiments using the human β-globin gene have shown that inactivating the poly-A signal AAUAAA (17) or the CPSF subunit CPSF73 (18) inhibits downstream S2P, suggesting that the 3′-end peak of S2P may be a consequence, rather than the cause of 3′-end processing.
Two kinases, cyclin-dependent kinase 9 (Cdk9) and Cdk12, likely contribute to CTD S2P in vivo (19). Cdk12 acts closer to the 3′-end and is responsible for the bulk of S2P (20, 21). By contrast, the observed drop in CTD S2P may be an indirect consequence of decreased elongation following Cdk9 inhibition. Nevertheless, Cdk9 may phosphorylate CTD S2 directly closer to the 5′-end and be responsible for the remaining S2P observed upon Cdk12 inactivation (22). Like S2P and contrary to Cdk9, Cdk12 is not essential in either fission yeast (Lsk1) or budding yeast (Ctk1) but is associated with changes in poly-A site selection (7).
In fission yeast, despite being dispensable for vegetative growth, both Cdk12 and CTD S2P are absolutely required for mating and gametogenesis in a gene-specific manner (23, 24). The requirement of CTD S2P during metazoan development is an unexplored area.
An advantage of the nematode Caenorhabditis elegans to study the role and requirement of CTD S2P during development is the fact that about 15% of its genes are organized as 1255 multigene (up to 8) TU called operons that depend on a unique promoter. Contrary to bacterial operons, the single polycistronic mRNA is cotranscriptionally processed into mature independent mRNAs by splicing and 3′-end maturation, while peaks of CTD S2P are observed at every intergenic region in the absence of termination (25). This uncoupling of CTD S2P and termination, which allows an easier dissection of the role of this modification, results from the fact that genes in position 2 and over within operons undergo 5′-trans-splicing: the donation of a common spliced leader 2 (SL2) exon and its associated modified cap structure, which most likely impedes the torpedo termination process (26) as the 5′ extremity at the cleavage site is not accessible to Rat1/Xrn2. Monocistronic mRNAs and mRNAs in the first operon position are also trans-spliced by the related SL1.
SL2 trans-splicing has been shown to be associated with the 3′-end formation of the upstream gene because the SL2 small nuclear ribonucleoprotein (snRNP) is bound to CstF (27). CstF itself is thought to be recruited by direct binding to the mRNA through a U-rich region bound by its CstF64 subunit (28), while the CstF50 subunit has a CTD-binding domain (29).
Operons tend to contain housekeeping genes that are essential for growth (25) and were proposed to be adaptative by limiting the transcriptional resources required to induce a large set of genes simultaneously (30). C. elegans reversibly arrests development (diapause) when larvae at the first stage (L1, fig. S1A) hatch in the absence of food (31), and expression of operons increases markedly during recovery from the L1 arrest (30). Vertebrate diapause was also recently shown to help organisms to survive long periods of time in extreme environments without trade-off for subsequent growth and fertility (32).
Here, we inactivated CDK-12 by three different means to show that it is dispensable for embryogenesis but required to exit the L1 diapause. We further show that CDK-12 inhibition mimics a developmental arrest, displaying an “always starved” phenotype. A CTD S2A mutant also completes embryogenesis contrary to a full deletion of the CTD and similarly arrests at the L1 stage. Mechanistically, we show that CstF is recruited less efficiently when CDK-12 is inhibited, which is correlated with defective SL2 trans-splicing of mRNAs within operons and gradual loss of Pol II. In addition, during the physiological exit from diapause, both S2P and SL2 trans-splicing are strongly induced. On the basis of our data, we propose that CTD S2P is a developmentally regulated mark taking part in the signaling of nutrient availability.
RESULTS
The inhibition of CDK-12 results in the loss of CTD S2P and a developmental arrest at the L1 stage
We obtained a putative mutant allele (tm3846) of the C. elegans ortholog of cdk-12 from the National Bioresource Project. The mutation maintained as a heterozygote consists in a deletion of 314 base pairs (bp) (fig. S1, B and C), leading to a frameshift and resulting in a truncated protein lacking about half of the kinase domain. After two rounds of outcrossing, a pilot genotyping of individual adult worms within the population confirmed the presence of homozygous wild type (wt) (+/+), heterozygous (Δcdk-12/+), but no homozygous (Δcdk-12/Δcdk-12) mutant, suggesting that the mutation may be lethal. However, a quantitative analysis of the progeny of heterozygous worms revealed that 81% were wt and 19% appeared to be arrested at an early larval stage, likely L1 (fig. S1A), while no dead embryo was found (Fig. 1A). Genotyping of the arrested larvae indicated that these were homozygous for the Δcdk-12 allele, and 4′,6-diamidino-2-phenylindole (DAPI) staining confirmed that they arrested development at the late L1 stage, with no more than 10 germline cells (33). These results suggest that the CDK-12 protein may not be required for embryogenesis but is essential to exit the L1 stage. The L1 arrest of homozygous mutant worms is reminiscent of the L1 developmental diapause occurring when eggs hatch in the absence of food. Alternatively, cdk-12 mRNA may be provided maternally, which ensures the production of the protein during embryogenesis despite the homozygous null genotype. To test this, we knocked down cdk-12 by RNA interference (RNAi), which targets the mRNA, using cdk-9 as a control as RNAi against this gene was previously analyzed (34). Soaking young adults in double-stranded RNA (dsRNA) corresponding to cdk-9 mostly resulted in embryonic lethality, with about 24% of the worms developing past the L2 stage (Fig. 1B). Similarly, most cdk-12 RNAi-treated animals were arrested at an early larval stage, and 15% reached the L4 stage. As we were suspecting low penetrance on the basis of the fully penetrant embryonic lethality previously reported for cdk-9 (34), we combined RNAi by soaking and feeding, which resulted in full embryonic lethality in the case of cdk-9 and full larval arrest in the case of cdk-12 (Fig. 1B). We conclude that cdk-12 is only essential for postembryonic development in C. elegans and maternal contribution of the mRNA is unlikely. These data also confirmed the previously reported essential requirement of cdk-9 early during embryogenesis (34).
Fig. 1. The inactivation of cdk-12 results in the loss of detectable CTD S2P and a developmental arrest at the L1 stage.
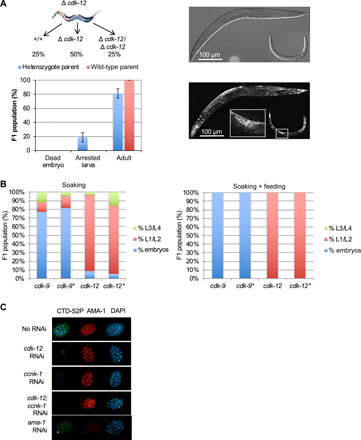
(A) Phenotypic analysis of the progeny of heterozygous wt/tm3846 worms (cdk-12 deletion). L4 larvae were allowed to grow for 24 hours and then removed and genotyped. The F1 progeny of six wt and six heterozygous worms was allowed to grow for 3 days, and the F1s were scored. Each column represents the averaged value ± SEM (n = 6). Heterozygote parent: arrested larva, 19%; adult, 81%. Wild-type parent: arrested larva, 0%; adult, 100%. Top: A schematic of the distribution of expected genotypes. Right: Differential interference contrast and DAPI staining of an adult and an arrested larva. The DAPI-stained germline is highlighted. (B) RNAi knockdown of cdk-9 and cdk-12 by soaking (left) or combined soaking and feeding (right). For each experiment, one L4 larva was allowed to grow for 24 hours on RNAi and then removed. The F1 progeny were allowed to grow for 4 days before scoring (the * is the replicate). (C) Immunostaining of early embryos collected after RNAi knockdown (soaking + feeding) of the indicated genes. Staining was performed with anti–CTD S2P, anti-AMA-1, or DAPI.
To test the status of CTD S2P following cdk-12 knockdown, L4 larvae were treated by the soaking/feeding protocol targeting cdk-12 as above and embryos were collected from the uterus of treated mothers followed by immunostaining using anti–CTD S2P or anti–AMA-1 (ama-1 is the C. elegans rpb1 ortholog) (Fig. 1C). The RNAi knockdown of cdk-12 resulted in the loss of detectable CTD S2P signal in all nuclei. Similarly, targeting the ccnk-1 gene, which encodes the cyclin partner of CDK-12, alone or in combination with CDK-12 resulted in the loss of detectable CTD S2P signal, while the overall level of the largest subunit of Pol II, AMA-1, was maintained. As expected, targeting ama-1 resulted in the loss of both epitopes, supporting the specificity of the CTD S2P staining (Fig. 1C).
Together, these data suggest that CDK-12 is responsible for the bulk of CTD S2P during C. elegans embryogenesis but that this phosphorylation mark may be dispensable. In addition, the absence of CDK-12 unexpectedly mimicked the developmental diapause occurring when L1 larvae starve.
To dissect the specific requirement for CDK-12 after embryo hatching, we generated an adenosine 5′-triphosphate (ATP) analog-sensitive (-as) version of the kinase (35). On the basis of previous work on the fission and budding yeast orthologs of CDK-12, F397 (TTC codon) was identified as the gatekeeper residue (35) and CRISPR was used to generate a homozygous strain (cdk-12-as) where the phenylalanine is mutated to the much less bulky glycine residue (GGC codon) (Fig. 2A). Detailed analyses revealed a slight growth delay in the cdk-12-as strain in the absence of the inhibitor [the ATP analog 3MB–protein phosphatase 1 (PP1)], suggesting that the mutant allele may be slightly hypomorphic (fig. S2A). cdk-12-as and wt larvae at the L4 stage were cultured in the presence of the 3-MB-PP1 inhibitor, and the developmental stage of F1 progeny was scored after 3 days. Already at the 1 μM concentration of the inhibitor, all the progeny of the cdk-12-as line was arrested at an early larval stage, while the wt was not affected, therefore generating an asynchronous population of offspring (Fig. 2B).
Fig. 2. A cdk-12-as allele allows for the rapid depletion of CTD S2P and mimics the null mutant.
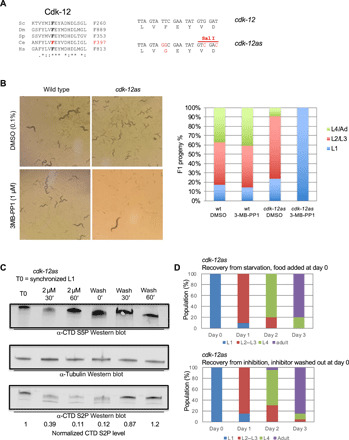
(A) Left: alignment of the residues flanking the gatekeeper phenylalanine (F) in the Cdk12 othologs from Sc (Saccharomyces cerevisae), Dm (Drosophila melanogaster), Sp (Schizosaccharomyces pombe), Ce (C. elegans), and Hs (Homo sapiens). Right: DNA sequence of the wt (TTC) and mutated (GGC) codons encoding the gatekeeper amino acid. Silent mutations introducing a Sal I restriction site used for genotyping are highlighted. (B) Left: Phenotypic analysis of wt and cdk-12-as lines cultured for 3 days in the presence or absence of 1 μM 3-MB-PP1 inhibitor. A single L4 larva was deposited at day 0. Note that the adult seen in the cdk-12-as/3-MB-PP1 condition is the parent. Right: Quantification of the phenotype of the F1 progeny grown in the indicated conditions (n = 3). (C) Western blot analysis measuring the levels of CTD S2P during inhibition of CDK-12-as and recovery. 3-MB-PP1 was added (2 μM) to synchronized L1 and later washed out at the indicated times. Total extracts were separated by polyacrylamide gel electrophoresis (PAGE) and probed with anti–CTD S5P, anti–CTD S2P, and anti-tubulin for normalization. The numbers indicate the level of tubulin-normalized CTD S2P as determined by ImageJ. (D) Comparison of the recovery from starvation (top) and recovery from CDK-12-as inhibition (bottom). Hatched L1 were added to NGM plates, and the number of individuals at the indicated developmental stages was determined every day for 3 days (n = 20). Hatched L1 were added to NGM plates containing 1 μM 3-MB-PP1 inhibitor for 24 hours and transferred to NGM plates without inhibitor on day 0. The number individuals at the indicated developmental stages was determined every day for 3 days (n = 20).
Immunostaining indicated that the level of CTD S2P dropped significantly (Wilcoxon-Mann-Whitney paired test P = 4.3 × 10−10) in all nuclei of L1 larvae after CDK-12-as inhibition, while levels of CTD S5P remained unaffected (Wilcoxon-Mann-Whitney paired test P = 0.38) (fig. S2, B to E). The very faint CTD S2P signal that persisted is unlikely to depend upon CDK-12, as increasing inhibitor concentration 10-fold did not result in a further reduction (fig. S2C).
Collectively, these data show that blocking CDK-12 using a chemical genetics strategy depletes the bulk of CTD S2P and results in a terminal phenotype identical to gene deletion or RNAi. They also exclude the idea that a maternal contribution ensures maintenance of S2P during embryogenesis because the CDK-12-as kinase is targeted by the inhibitor irrespectively of its maternal or zygotic origin.
To determine the kinetics of CDK-12-as inhibition, synchronized L1 larvae were supplemented with 2 μM inhibitor. Western blot analyses using anti–CTD S2P revealed that the signal dropped within 30 min of inhibition (Fig. 2C). When eggs were similarly treated, the drop of S2P took longer (about 60 min), suggesting that the eggshell is less permeable to the inhibitor (fig. S2F).
Removing the inhibitor from arrested larvae both restored the CTD S2P signal within 60 min and relieved the larval arrest, resulting in synchronous growth of the population until adulthood in the absence of noticeable difference compared to the release from diapause (Fig. 2D). These data indicate that rather than disorganizing transcription with consequent cell death, the inhibition of CDK-12 and the resulting loss of CTD S2P lead to a fully reversible developmental arrest reminiscent of the L1 diapause.
A full-length CTD S2A mutant displays an identical phenotype to CDK-12 inhibition
We reasoned that the residual CTD S2P signal detected by immunofluorescence after CDK-12 inactivation (fig. S2B) may be sufficient to ensure normal embryogenesis when CDK-12-as is inhibited. In addition, CTD-unrelated CDK-12 substrates may also contribute to the L1 arrest. These two issues were addressed by generating a mutant where all S2 within the CTD were replaced by alanine. We first attempted to use CRISPR to generate a strain harboring a synthetic CTD whose protein sequence is identical to the wt sequence but encoded by a shorter and optimized DNA sequence lacking the three introns located within the DNA sequence encoding the CTD and that collectively represent 1629 bp (fig. S3, A and B). The brood size of the synthetic genuine CTD (CTD S2S) was similar to the wt strain (fig. S3C), validating the strategy. We next used CRISPR to generate a heterozygous wt/S2A mutant. Approximately, 75% of progeny from these animals grew to adulthood (note that we can infer that about 25% of them are homozygous wt because they are not roller; see Materials and Methods), while about 25% of the progeny was arrested at an early larval stage (Fig. 3A). Some worms had undetectable CTD S2P signal (Fig. 3B). Genotyping of arrested larvae confirmed that these were homozygous for the CTD S2A allele (Fig. 3C). While generating the CTD S2A mutant, we isolated heterozygous animals containing a wt version of the CTD and a version lacking the entire CTD (CTD Δ), which was confirmed by sequencing. This occurred most likely when the two double-strand breaks were induced by Cas9 at the positions recognized by the two guides (and flanking the sequence encoding the CTD) and repair took place by nonhomologous end joining independently of the presence of the repair template. While 75% of the progeny of these worms reached adulthood (here, again, we can infer that about 25% of them are homozygous wt because they are not roller; see Materials and Methods), about 25% were embryonic lethal, indicating that the CTD Δ allele cannot sustain embryogenesis (Fig. 3D). The CTD Δ mutant strain is critical as it shows that any maternal contribution of either the ama-1 mRNA or the AMA-1 protein is not sufficient to ensure embryogenesis. This confirms previous work showing that RNAi targeting of ama-1 in embryos leads to no observable phenotype until the 28-cell stage when gastrulation begins and zygotic transcription is required (36). Therefore, these data collectively show that the CTD S2A mutant fulfils all the essential functions of the CTD required for embryogenesis.
Fig. 3. Homozygous CTD S2A mutant completes embryogenesis and arrests at the L1 stage while CTD Δ mutants are embryonic lethal.
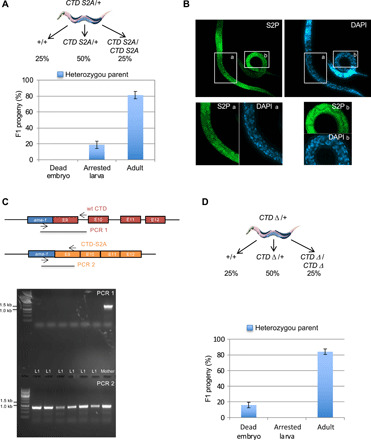
(A) Phenotypic analysis of the progeny from heterozygous wt/CTD S2A worms. Heterozygous roller L4 larvae were allowed to grow for 48 hours and then removed. F1 progeny of six heterozygous worms were allowed to grow for 3 days before scoring of individuals. Each column represents the averaged value ± SEM (n = 6). Arrested larva, 19%; adult, 81%. Top: A schematic of the distribution of expected genotypes. (B) Immunostaining of L1 progeny of a heterozygous CTD S2A/wt worm. Staining was performed with anti–CTD S2P and DAPI as indicated. Insets a and b correspond to enlarged regions of individuals positive (a) or negative (b) for the anti–CTD S2P staining. (C) Genotyping of the arrested L1 progeny of a heterozygous CTD S2A/wt worm. The mother is also shown as control. Primers used are indicated on the schematic. (D) Phenotypic analysis of the progeny of heterozygous wt/CTD Δworms. Heterozygous L4 larvae were allowed to lay for 24 hours and then removed and genotyped. F1 progeny of four heterozygous worms were allowed to grow for 3 days and individuals were scored. Each column represents the averaged value ± SEM (n = 4). Arrested larva, 16%; adult, 84%. Top: A schematic of the distribution of expected genotypes.
Together, this set of data is important as it supports that (i) CTD S2P is dispensable for embryogenesis, (ii) the L1 arrest observed when CDK-12-as is inhibited is attributable to the loss of CTD S2P, (iii) CDK-12 is the biologically relevant kinase of CTD S2P in vivo, and (iv) inhibiting CDK-12-as leads to the rapid loss of CTD S2P. We therefore used the -as version of the kinase to further analyze the role of CTD S2P following embryogenesis.
CDK-12 is essential for the expression of a subset of genes required to exit the L1 developmental diapause
Our results showing that both genetic and chemical inhibition of CTD S2P result in an L1 arrest following embryogenesis and hatching are reminiscent of the physiological diapause observed when embryos hatch in the absence of food. During this arrest, growth-related genes are down-regulated, and conversely, stress response genes are up-regulated. This tendency is reversed upon recovery within the first hours following addition of food (37). On the basis of these data, we designed an experimental setup where cdk-12-as or wt strains were synchronized in L1. Freshly hatched larvae were starved for 12 hours, and the populations were then divided into three groups: (S) 4 hours’ starvation; (R) 4 hours’ recovery in the presence of food; and (R + I) 4 hours’ recovery in the presence of food and inhibitor, all followed by total RNA sequencing (RNA-seq) (Fig. 4A). While the addition of food markedly affected the transcriptome, as previously reported (37), the inhibitor affected only the cdk-12-as strain with only minor effects on the wt (Fig. 4B). To provide an overview of the impact of CDK-12 inhibition on the transcriptome during exit from diapause, we selected the 6550 genes differentially expressed by the presence of “food” or “inhibitor.” Analyses of the resulting heatmap (Fig. 4C and fig. S4A) show that differentially expressed genes following the addition of food can roughly be divided into two categories: those that are up-regulated and encompass growth-related processes (including metabolism, translation, and replication) and those that are down-regulated and related to stress response (including catabolism and defense response) (fig. S4A). Unexpectedly, the inhibition of CDK-12 did not affect the steady-state expression levels of the majority of these genes corresponding roughly to clusters C and D and representing approximately 70% of the genes whose expression changes upon exit from the diapause. The smaller clusters B and E contain genes that behave similarly to the control but whose down-regulation (or repression) (cluster B) or overexpression (cluster E) during recovery is enhanced compared to the control. Cluster F contains genes that are normally down-regulated during recovery but maintain high expression when CDK-12 is inhibited. Last, cluster A represents growth-related genes (table S1) whose expression is normally induced during recovery but that fail to be induced or reach intermediate levels of expression, during recovery when CDK-12 is inhibited.
Fig. 4. CDK-12 is required for the expression of the subset of SL2 trans-spliced mRNAs.
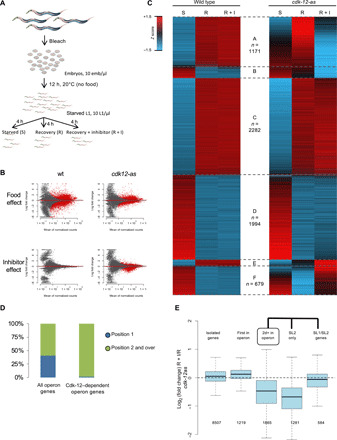
(A) Schematic of the experimental setup used. (B) MA plot of changes in the mean of normalized counts highlighting the effect of the addition of either food or inhibitor on either the wt or cdk-12-as lines. DESeq2 was used on the room temperature count to identify genes significantly (adjusted P < 0.05 and fold change >1.5) affected (in red) by the indicated parameters. (C) Heatmap of the Z score of the rlog-transformed expression score averaged over two replicates for the 6550 genes differentially expressed by the addition of food or inhibitor in the indicated lines. Six clusters (A to F) are defined according to the food and inhibitor effect. The number of genes in each cluster is indicated on the figure except for cluster B (271 genes) and cluster E (153 genes). (D) Distribution of the genes in position 1(blue) or in positions 2 and over (green) within all operon genes or operon genes whose induction requires CDK-12. (E) Boxplot of the log2 fold change due to CDK-12 inhibition during recovery for genes classified according to their genomic organization and their SL1 or SL2 trans-splicing status. The number of genes in each class is indicated below.
Together, these data reveal that approximately 30% of genes involved in recovery (1171 genes total) are not properly induced when CDK-12-as is inhibited. These genes may therefore explain the inability of the L1 larvae to exit the developmental arrest and were investigated further.
Genes in position 2 and over within operons specifically require CDK-12 for induction during recovery from the developmental diapause
The genes present in cluster A are not randomly distributed within the C. elegans genome but are highly enriched for genes located in positions 2 and over within operons (Fisher’s exact test P < 2.2 × 10−16) (Fig. 4D and fig. S4B). Metagene analyses confirmed that the presence of the inhibitor (R + I) does not globally affect the first gene in operons during recovery, while there is an approximately twofold global decrease in expression for downstream genes (fig. S4C). We note that in the cdk-12-as strain, those genes are expressed to an intermediate level in the absence of inhibitor, confirming that this allele is hypomorphic as suggested above (fig. S2A). This also suggests that the expression of these genes is affected in a dose-dependent manner by CDK-12 activity.
The metagene analysis also revealed that the genes in operons are globally up-regulated during exit from diapause in wt (fig. S4D). In sharp contrast, when CDK-12 is inhibited, while the first gene in operon is correctly induced, the expression of the downstream gene remains at its starvation level (Fig. 4D and fig. S4D).
For the first genes of the operon that are normally up-regulated in recovery from starvation, 94% (394 genes) of them are still induced upon Cdk12 inhibition and found in cluster C. In contrast, 79% (766 genes) of the downstream genes in the operon that are normally up-regulated in starvation recovery fail to be induced when Cdk12 is inhibited and are found in cluster A. As for the remaining 21% of the downstream genes in the operon that are still induced upon exit from diapause even when Cdk12 is inhibited, it appears that most of those genes (118 of 199 genes) can be expressed from an internal promoter.
A well-described discriminating feature between the genes located within operons is that the first genes undergo SL1 trans-splicing while most of the genes in position 2 and over are SL2 trans-spliced. Only those genes that are exclusively SL2 trans-spliced are affected upon CDK-12 inhibition, while genes that can also undergo SL1 trans-splicing owing to the presence of an auxiliary internal promoter are not affected (Fig. 4E). The trivial explanation that the level of the SL2 RNA precursors (also transcribed by Pol II) was itself down-regulated upon CDK-12 inhibition is not supported by quantitative reverse transcription polymerase chain reaction (qRT-PCR) measurements of 3 out of the 18 copies of the SL2 gene (fig. S4E).
Collectively, these results indicate that the gene-specific sensitivity to CDK-12 inhibition is not dictated by the position within operons but rather by the dependency to SL2 trans-splicing.
Inhibition of CDK-12 does not affect splicing or termination
A relatively small subset of genes (1211) was detected as differentially expressed upon CDK-12 inhibition. This is somewhat unexpected given that CTD S2P deposited by CDK-12 is a ubiquitous mark, present at most, if not all, expressed Pol II genes and that has been implicated in essential cotranscriptional processes such as splicing (8) and cleavage/polyadenylation (38) of the pre-mRNAs. However, we did not observe splicing defects (intron retention) (fig. S5A), or cleavage defects, and only minor readthrough defects (reads mapping beyond the gene boundaries) in the cdk-12-as strain in the R + I condition (fig. S5B). Quantification of the “readthrough” reads in the 500 nucleotides (nt) downstream the annotated transcription end sites revealed that only 53 genes (including 28 protein coding genes) have significantly more readthrough when CDK-12 is inhibited. In addition, visual inspection of those genes in a genome browser showed only minor differences. To further evaluate the effect of CDK-12 inhibition on polyadenylation, we used the extension of poly-A test (ePAT), an experimental assay to test the length of the poly-A tail on a set of individual mRNAs. The isolated gpd-4 gene and two genes located in position 1 of affected operons (ubxn-4 and rnf-21) were tested. We compared the length of the poly-A tail in the S, R, and R + I conditions after CDK-12 inhibition and observed no difference, indicating that the polyadenylation of these mRNAs is not affected by the loss of CDK-12 activity (fig. S5C). Therefore, CDK-12 activity appears to be only required for efficient SL2 trans-splicing of genes in operons, a process that is specifically induced upon exit from the developmental diapause.
Deficient SL2 trans-splicing results in gradual premature termination of pol II transcription within operons
Given the apparent inability of the CDK-12 inhibited strain to induce genes located in position 2 and over within operons, we next used chromatin immunoprecipitation sequencing (ChIP-seq) to monitor the presence of Pol II following CDK-12 inhibition during the R and R + I conditions in the cdk-12-as strain. ChIP at the L1 stage is challenging, and overall, the efficiency of the immunoprecipitation (IP) was poor. To obtain a robust ChIP signal, we selected the genes with a BEADS-normalized signal (see Materials and Methods) higher than a defined threshold allowing 1000 operon-contained genes to be selected. Meta-gene analysis of this set of genes revealed no difference in Pol II occupancy upon CDK-12 inhibition on the body of the first genes in operon (Fig. 5, A and B). In contrast, Pol II occupancy in the intergenic region—that is more enriched—between the first and the second genes decreases markedly, and the signal on the body of the second genes slightly decreased (Fig. 5, A and B). Overall, this analysis supports the hypothesis that the SL2 trans-splicing defect resulting from CDK-12 inhibition causes premature loss of the polymerase. A potential explanation is that the uncapped RNAs being generated activate Xrn2-dependent torpedo termination. Supporting this hypothesis, a gradual decrease in Pol II levels is observed over the operon length, the effect being amplified at each subsequent intergenic region, which is evident when the median of the log2 fold change value between the two conditions (R and R + I) is plotted at each position (Fig. 5B). At the end of the fourth genes in operons, the median log2 fold change reaches a minimum of about −0.6 (a difference of about 33%). Anti–CTD S2P ChIP-seq confirmed the robust loss of this mark when CDK-12 is inhibited (Fig. 5C).
Fig. 5. The inhibition of CDK-12 results in gradual premature termination of Pol II transcription within operons.
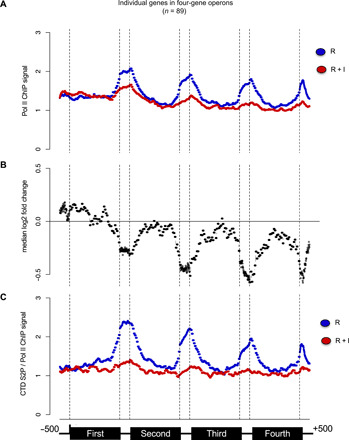
(A) Meta-operon profiles of BEADS-normalized Pol II occupancy data averaged over two replicates. The Pol II ChIP signal in the R (blue) and R + I (red) conditions is shown. The organization of the operons is shown at the bottom. Only the operons containing at least four genes were considered. Operons with any intergenic regions larger than 800 nt were also excluded from the analysis. (B) Meta-gene profiles of the median log2 fold change between Pol II ChIP signal in the R and R + I conditions as in (A). (C) Meta-operon profiles of S2 phosphorylated Pol II occupancy normalized on total Pol II occupancy data averaged over two replicates. The organization of the operons is shown at the bottom.
When lone genes are analyzed, a decreased occupancy of Pol II is also observed (fig. S5D), albeit weaker than in operon genes. These data suggest that the presence of CTD S2P may be globally required for robust 3′-end pausing, which may only be critical for efficient SL2 trans-splicing.
We next explored the correlation between the decreased occupancy of Pol II upon Cdk12 inhibition and gene length, but they appear uncorrelated (R2 ≈ 0.01) for both lone or operon genes (fig. S5E). Moreover, this analysis confirmed that the second genes in operons are, in general, more affected, clarifying that position in operon, rather than gene length, determines sensitivity to Cdk12 inhibition.
Because Pol II is not completely lost from the chromatin template upon CDK-12-as inhibition, it is likely that the mRNAs that are produced but not SL2-capped are targeted by nucleases, which could explain why the down-regulation at the mRNA level is higher than at the Pol II ChIP-seq level. In summary, two effects may coexist to explain the absence of induction of the mRNAs: a gradual premature termination of Pol II over the operon length and the degradation of the uncapped mRNAs. To gain more insights into these mechanisms, we set up a genetic screen for suppressors of the L1 arrest upon CDK-12 inhibition.
The knockdown of nucleases partially suppresses the developmental arrest resulting from CDK-12 inhibition
On the basis of the protocol of Saur et al. (39), we designed a genome-wide RNAi screen for suppressors of CDK-12-as inhibition (see Materials and Methods) (fig. S6). Starved synchronized cdk-12-as L1 larvae (future parents) were fed in 96-well plates with dsRNA-expressing bacteria, each well targeting a specific gene of C. elegans. The progenies were allowed to develop and screened for the ability to escape the L1 arrest induced when the inhibitor was added. Of the 19,000 tested candidates in a primary screen, 121 were selected, of which 33 were confirmed as robust suppressors in a secondary screen. Of these genes, panl-2 gene was identified on this basis and retained our attention as it encodes an ortholog of the human PAN2, a ribonuclease subunit that targets mRNAs to regulate their stability by shortening the poly-A tail (40), leading to their 3′-5′ degradation by the exosome or 5′-3′ degradation by the Xrn1 nuclease.
In light of our previous findings, and the possibility that XRN-2–dependent degradation is implicated in the loss of mRNAs in position 2 and over upon CDK-12-as inhibition, we decided to independently test the three well-described nucleases Xrn-1, Exo-1, and Xrn-2. While RNAi against xrn-1 and exo-1 had no effect on the larval arrest in the cdk-12-as strain, targeting xrn-2 or panl-2 resulted in about 30% of progeny developing past the L1 stage (fig. S6B). Analyses of the SL1/SL2 trans-splicing status of a gene in position 2 in individual worms revealed an increase in SL2 trans-spliced mRNAs (fig. S6C). These data suggest that a competition may exist between degradation and SL2 trans-splicing and that CTD S2P is required to tip the scales toward the second. In addition, the knockdown of xrn-2 restored mRNA level of genes in position 2 and over to a level close to wt (fig. S6D).
Collectively, these data bring support a model where inefficient SL2 trans-splicing and the resulting mRNA instability is likely the primary cause for the inability of the cdk-12-as strain to exit diapause upon CDK-12 inactivation.
CDK-12 is required for chromatin occupancy of the CstF complex
SL2 trans-splicing of mRNA from genes in position 2 and over within operons is coupled with the cleavage and polyadenylation of the gene in the first position. Mechanistically, this coupling results from the interaction of the CstF complex with both the CPSF complex and the SL2 trans-splicing complex (27). A recent ChIP-seq study reported overlapping occupancy of the CTD-S2 phosphorylated polymerase and both CstF50 (CPF-1) and CstF64 (CPF-2) subunits of CstF at the 3′-end of the C. elegans genes (26). Moreover, the CstF50 subunit contains a CTD-binding domain, which is required for 3′-end processing in human cells (29). On the basis of these observations, we reasoned that CDK-12–dependent CTD S2P could be required for the efficient recruitment or stabilization of a CstF/SL2snRNP complex, thereby ensuring coupling of the polyadenylation of an mRNA in position 1 (which is not affected by CDK-12) to trans-splicing of the downstream mRNA. To test this possibility, we first attempted to test for an in vivo link between S2-phosphorylated Pol II and CstF (Fig. 6A). Pol II was immunoprecipitated using anti–CTD S2P antibodies on nuclear extracts from a fed (4 hours) L1 larvae, followed by Western blot analysis using either an anti–CTD S2P or anti–CPF-2 [recognizing the 37-kDa CstF64 worm ortholog (27)] antibodies, while beads-only IPs were used as control. Next, the same experiment was performed with the cdk-12-as strain in the presence or absence of the inhibitor (Fig. 6A). These experiments support a coprecipitation between the CstF complex and the CTD S2P Pol II in vivo. We next performed CPF-1 ChIP-seq on cdk-12-as L1 larvae in identical conditions used previously (Fig. 4C) but failed to obtain reproducible results. This antibody has been used previously on L4 larvae (26), but as indicated above, L1 larvae constitute a more difficult material for ChIP, which may explain this failure. We attempted to construct a tagged version of the CstF-50 (CPF-1) worm ortholog, which turned out to show a strong synthetic negative interaction with cdk-12-as. While these data add further support to our previous finding that cdk-12-as may represent a hypomorphic allele, they also establish a genetic connection between CstF and CDK-12.
Fig. 6. CDK-12 is required for the efficient chromatin occupancy of the CstF complex.
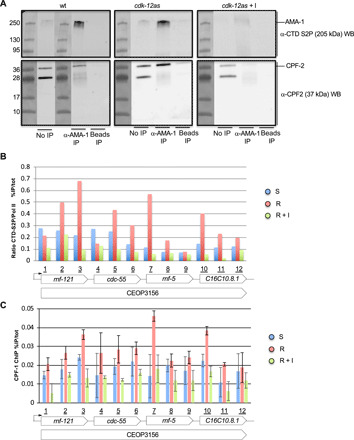
(A) Co-IP experiment of CTD S2P AMA-1 and CPF-2 (Csf64). Left: Nuclear extracts from fed L1 larvae were used for anti–CTD S2P IP and the precipitated material was separated on PAGE and submitted to anti–CTD S2P or anti–CPF-2 Western blotting (WB) as indicated. The ladder and corresponding size are shown. No IP refers to the crude nuclear extracts and beads only to IP where no antibodies were added. Middle: Same experiment performed on nuclear extracts from fed L1 larvae from the cdk-12-as strain. Right: Same experiment performed on nuclear extracts from fed L1 larvae from the cdk-12-as strain grown in the presence of the inhibitor. (B) ChIP experiments were performed on chromatin prepared from the indicated starvation (S), recovery (R), and recovery + inhibitor (R + I) conditions using anti–CTD S2P and anti–AMA-1 antibodies in triplicate. Amplicons marked from 1 to 12 allowed the scanning of the CEOP 3156 operon containing four genes. The CTD S2P signal was normalized on the total Pol II signal. (C) ChIP experiments were performed on chromatin prepared from the indicated starvation (S), recovery (R), and recovery + inhibitor (R + I) conditions using anti–CPF-1 (CstF50) antibodies in duplicate. Amplicons marked from 1 to 12 allowed the scanning of the CEOP 3156 operon containing four genes.
We next used ChIP-qPCR to look at CTD S2P and CPF-1 (CstF50) enrichment on a representative operon (CEOP 3156). We obtained robust data with the anti–CPF-1 (CstF50) antibody. Cdk-12-as L1 treated as before (S, R, and R + I) was used for anti–AMA-1, anti–CTD S2P, and anti–CPF-1 ChIP followed by qPCR (Fig. 6, B and C). We observed a marked (about fourfold at the S2P peak), CDK-12–dependent increase in normalized CTD S2P signal during recovery (R), peaking in the proximity of the intergenic regions as previously reported (26). The CstF subunit CPF-1 followed a similar trend, increasing concomitantly with CTD S2P during recovery (R). This effect was abolished when CDK-12 was inhibited (R + I) (Fig. 6C).
Together, these data indicate that the activity of CDK-12 is required for the efficient recruitment and/or the stabilization of CstF on chromatin. As CstF was shown to bring the SL2 particle necessary for trans-splicing, they also provide a molecular basis for the specific SL2 trans-splicing defect resulting form CDK-12 inhibition.
A specific CDK-12–dependent induction of SL2 trans-splicing upon exit from diapause
To better assess how CDK-12 affects the SL2 trans-splicing, we quantified from the RNA-seq dataset the SL1 and SL2 trans-splicing events in operon genes that can be trans-spliced by both SL1 and SL2. We used a bioinformatics pipeline (SL-quant) recently developed in the laboratory (41). Briefly, the SL sequences are identified with high specificity and are trimmed from the input reads, which are then remapped on the reference genome and quantified at the gene level. This analysis revealed two important features: (i) SL2 trans-splicing was statistically increased upon addition of food (S versus R) and (ii) this increase occurred in a manner dependent of CDK-12 activity (R versus R + I) (fig. S7A). However, the decrease in SL2 trans-splicing events when CDK-12 is inhibited could simply result from a reduction in the overall levels of the SL2 trans-spliced mRNA is reduced. To tease apart these two effects, we took advantage of the genes trans-spliced with either SL2 or SL1. In these so-called hybrid operons, a second internal promoter is present in addition to the upstream operon promoter. For these genes, the level of SL1 trans-splicing remains stable upon CDK-12 inhibition, while the level of SL2 trans-splicing significantly decreases (fig. S7A), demonstrating that CDK-12 activity is specifically required for the increase of SL2 trans-splicing upon exit from the diapause.
The global CDK-12–dependent increase of SL2-trans-splicing upon recovery from starvation is strong (+73%) (fig. S7B). This could simply reflect the fact that genes in operon are globally up-regulated during recovery. However, transcription from internal promoters (estimated from the level of SL1 trans-splicing for the same genes) is not induced in recovery, while polycistronic transcription (estimated from the level of SL2 trans-splicing) is significantly induced (fig. S7A).
When normalized on the total number of SL1 + SL2 events, it is clear that the proportion of SL2 trans-splicing increases in a CDK-12–dependent manner for genes in operons with auxiliary internal promoters (fig. S7C). We next wondered whether the efficiency of trans-splicing could be used by the cell to finely tune the expression of genes in operons during recovery from diapause. Consistent with this idea, the expression changes of the first and second genes in operon are poorly correlated during recovery (fig. S7D), with the second genes in operons significantly more overexpressed (fig. S7E).
Collectively, these data suggest that the expression of the polycistronic transcripts is uncoupled from expression of the operon promoter. This decoupling relies at least partly on different trans-splicing strategies. Upon exit from a starvation period leading to a developmental diapause in L1, CDK-12–dependent CTD S2P increases, leading to increased SL2 trans-splicing and mRNA production of growth genes. The inactivation of CDK-12 impedes this induction and results in an always-starved phenotype despite the presence of food.
DISCUSSION
Genetic dissection of the biological role of the CTD during cellular growth and differentiation has been hampered by both its repetitive nature and length that render manipulations difficult. An alternative strategy consisting in targeting the kinases responsible for CTD phosphorylation led to conflicting reports on the actual identity of the kinases because of relaxed in vitro specificity, low quality of the phospho-specific antibodies, or the opportunistic tendency of related kinases to take over territories left over when a genuine kinase is inactivated.
Improvement in gene synthesis and targeting has nevertheless allowed the precise replacement at the endogenous rpb1 locus of the authentic CTD by variants preserving its natural length in yeast (15, 42) and more recently in Drosophila (43), opening the way for genetic dissection of the requirement of CTD phosphorylation in vivo during growth and development. Unexpectedly, these experiments revealed that budding yeast or fission yeast CTD S2A are fully viable, a phenotype contrasting the lethality resulting from the deletion of most genes coding for 3′-end processors, including Pcf11, proposed to be recruited by the S2P mark. In addition, genome-wide analyses indicated that the transcriptomic signature of the CTD S2A mutant closely mirrors those resulting from deletion of CTK1 (44) or lsk1 (42), the respective orthologs of Cdk12 in the two yeasts, which also results in loss of the bulk, yet not all, of S2P. Furthermore, a more recent analysis of a full-length S2A mutant (albeit not expressed from the endogenous locus) in budding yeast showed that Pcf11 is still precisely recruited, though at lower efficiency (as analyzed by ChIP) that is inconsequential for cell viability (45). A modest average readthrough of 50 bp was noted for only 300 genes. These data indicate that the RNA binding capacities of the 3′-end processing complexes, CPSF that binds AAUAAA and CstF that binds a U-rich sequence downstream of the cleavage site, are sufficient to ensure their essential functions, and the CTD S2P is likely acting as a secondary layer of control to fine-tune the 3′-end maturation, thereby explaining its nonessentiality, at least in yeast. In addition, the above results suggest that the reason why, by contrast, S5P is absolutely required for capping may be that it acts early when the newly synthetized RNA is not long enough (about 30 nt) to allow the robust RNA-dependent recruitment of the 5′-end processing complexes.
This less stringent requirement of S2P for the core of transcriptional expression may have allowed for evolutionary selection of additional layers of regulation fundamentally contributing to gene expression in specific contexts, as typically shown in the present work. Supporting this, while S2P is dispensable during vegetative growth, it is absolutely required for gametogenesis in a gene-specific manner in fission yeast, and the inactivation of CDK12 in human specifically affects DNA damage response genes including the critical regulators of genomic stability BRCA1 and FANC1 (20, 21). These data plead for an analysis of the requirement of CTD S2P during metazoan development in a genetically tractable model organism.
Here, we show that a cdk-12 lesion in C. elegans through deletion, RNAi, or the use of an analog-sensitive version of the kinase expressed from the endogenous locus all result in a common terminal phenotype, namely, a developmental, postembryonic arrest at the L1 stage. We consider it unlikely that a maternal mRNA/protein contribution ensures persistent expression of CDK-12 during embryogenesis because RNAi targets maternal mRNAs and a maternally contributed protein is anyway inactivated in the cdk-12-as line. In addition, S2P is undetectable in the embryo (RNAi) or the arrested L1 (as) upon CDK-12 inactivation. These data suggest that Cdk12 is responsible for bulk S2P in C. elegans as in yeast. A previous study proposed that S2P is ensured by two distinct kinases (CDK9 and CDK12) in the soma and germline, respectively (46). Our current data do not support this model. A lower efficiency of cdk-12 depletion by RNAi may mask the terminal phenotype of cdk-9 and cdk-12 inactivation (Fig. 1B) and explain this discrepancy.
Because the L1 arrest phenotype that results from cdk-12 inactivation suggests that the S2P mark is dispensable for embryogenesis, we were concerned that a low level of persistent phosphorylation contributed by residual CDK-12 activity or CDK-9 could be sufficient to maintain and coordinate the high demand on transcription occurring during embryogenesis. We therefore created a full-length genuine S2A mutant expressed from the endogenous ama-1 locus. This mutant recapitulated the terminal phenotype of the cdk-12 null. While we cannot exclude the possibility that some embryonic lethality results from the absence of CTD S2P, our data indicate that it is a marginal effect. The possibility that embryogenesis occurs in the homozygous CTD S2A mutant due to maternal contribution is very unlikely as a mutant generated by the same strategy and completely lacking the CTD is embryonic lethal at an early stage and with complete penetrance. In addition, zygotic transcription was shown to be required from gastrulation (36). Therefore, we conclude that embryogenesis can occur in the complete absence of CTD S2P and that any resulting defects do not impede the complex and intricate regulation of gene expression typical of this phase of the life cycle.
Nevertheless, CTD S2P–depleted worms arrest development at the L1 larval stage, mimicking the diapause resulting from starvation. This always-starved phenotype is quickly and fully reversible when inhibition of Cdk-12 is relieved by washing out the inhibitor, restoring CTD S2P. This indicates that lowering CTD S2P at this stage does not simply lead to aberrant gene expression and cell death but is instead perceived as a physiologically integrated response. This points to a critical role for the control of S2P in coupling nutrient availability to development (see below).
The tight control of the cdk-12-as allele allowed us to precisely analyze the transcriptome upon L1 arrest after CDK12 inhibition. Similarly to what was observed in yeast, our analyses show that the steady-state level of most mRNAs is independent of S2P. In addition, we revealed a specific requirement for S2P in the induction of genes in position 2 and over within operons (Fig. 7). These genes are characterized by SL2 trans-splicing that requires the recruitment at short intercistronic regions of the SL2 snRNP, which interacts with the CstF complex through the CstF64 subunit (27).
Fig. 7. A schematic of the requirement of CDK-12 in trans-splicing.
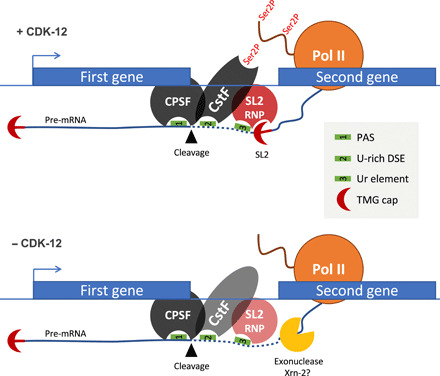
See Discussion for details. The fainter staining of CstF and SL2 RNP when CDK-12 is inactivated or absent represents a decreased occupancy of these complexes. PAS, polyadenylation signal; DSE, downstream element. TMG, trimethylated guanosine.
SL2 trans-splicing requires a specific sequence, the uracil-rich (Ur) element, that is not a CstF binding site (47) but likely base-pairs with the 5′ splice site on the SL2 snRNP. In addition, CstF also contains a CTD binding subunit, CstF50 (CPF-1), that was previously shown to bind the CTD S2P (48). Our data indicate that the CstF/Pol II interaction is conserved in C. elegans and that high-level CstF chromatin occupancy requires CDK-12–dependent CTD S2P. Therefore, the most likely explanation for the requirement of CDK-12 in the induction of SL2 modified mRNA is that the CstF-SL2 snRNP requires CTD S2P to be fully functional. We have not detected 3′-end defects associated with operon genes in position 1 or isolated genes, indicating that CTD S2P is not critically required for CstF activity. This most likely reflects the fact that CstF64 (CPF-2) has an RNA-binding domain that can target the U-rich at the 3′-end of processed mRNA.
Our data also indicate that the inactivation of CDK-12 and the failure to properly trans-splice SL2 result in the gradual disengagement of the polymerases, which tend to terminate early. As an unprotected 5′-end sensitizes the mRNA to degradation by XRN2, a likely explanation of this phenomenon is that a torpedo termination mechanism is activated (9). Supporting this possibility, RNAi inactivation of xrn-2 partially suppresses the L1 arrest. In addition, it has been reported that XRN-2 critically controls the level of expression of genes in position 2 and over within operons (49). Mechanistically, the Ur element bound by the SL2 snRNP was shown to block progression of the XRN-2 5′-3′ exonuclease attracted to the 5′-end generated by the cleavage (50). We therefore infer that the inability to induce growth genes from operons upon CDK-12 inactivation results from both premature transcription termination and the rapid degradation of unprotected full-length mRNAs produced by polymerases escaping termination (Fig. 7).
The panl-2 gene that encodes a 3′-5′ exonuclease (40) was also recovered as a suppressor, suggesting that the mRNAs expressed from operons may naturally be subject to deadenylation/degradation and that the down-regulation of PANL-2 balanced inefficient capping. Our data do not currently allow us to analyze this further.
We also noted that a small set of about 200 genes are down-regulated when cdk-12 is inactivated, despite not being expressed from operons. A longer than average length is the only common feature that we could highlight in this group of genes (Fig. 4). This is reminiscent of Drosophila or human cells where long genes seem to be particularly sensitive to Cdk12 activity (21, 51).
An important aspect of this work is our finding that CTD S2P is induced upon exit from starvation. This is not simply due to increased transcription, as we normalized CTD S2P signal to Pol II, and the steady-state levels of mRNA in position 2 and over is only poorly correlated to the first mRNA of the operon that belongs to the same TU. We propose that CDK-12–dependent CTD S2P is rate-limiting for exit from starvation because of its specific control of SL2 trans-splicing.
Previous work has already revealed that growth genes are regulated after recruitment during starvation to coordinate gene expression with nutrient availability, although the mechanism remains unclear. The present work reveals an additional, postinitiation layer of regulation ensuring that upon exit from starvation, premature termination within operons is proscribed and mRNA stability is maximized. CDK-12–dependent CTD S2P therefore optimizes the advantage of operon-organized growth genes, which is essential for the rapid recovery from the growth-arrested to fast-growing state (30). The transition from embryogenesis, where no net growth occurs, to larval development is archetypal of this situation (31), but we anticipate that CDK-12 may be required at every molt where growth takes place.
An outstanding open question is how the nutrient signal is communicated to CDK-12 to increase CTD S2P at operons. Previous studies in fission yeast have revealed that Cdk12 (Lsk1) in this species is directly regulated by a mitogen-activated protein kinase (MAPK) pathway during gametogenesis, when CTD S2P is essential (23, 24). As in fission yeast, gametogenesis only occurs during starvation, and the MAPK links the starvation signal to gene-specific CTD S2P. This occurs by phosphorylation of the long N-terminal extension of Cdk12 (Lsk1) that is absent from canonical CDKs (23). As this extension is conserved in all CDK-12 orthologs, it will be interesting to test if it plays an analogous role in C. elegans. The MAPK homolog, KGB-1, has been linked to nutrient availability (52), and the KGB-1 cascade is required to promote L1 recovery (53). Further work will clarify if this effect is mediated through CDK-12 phosphorylation.
The present work shows that beside its constitutive, likely nonessential role at the 3′-end of all RNA Pol II–transcribed genes, CTD S2P also functions as a gene-specific signaling mark that has evolved to allow rapid recovery from growth arrest during the metazoan life cycle.
MATERIALS AND METHODS
General C. elegans methods and antibody used
Worms were maintained at 20°C on Nematode Growth Media (NGM)–agar plates [51.3 mM NaCl, 1 mM CaCl2, 1 mM MgSO4, 25 mM KH2PO4, cholesterol (5 μg/ml), and 1.25 g of peptone with 8.5 g of agar for 500 ml] seeded with Escherichia coli OP50. In liquid medium, they were cultured on a shaker at 20°C in S-Basal [0.1 M NaCl, 50 mM KH2PO4, 3 mM MgSO4, 4 mM CaCl2, 1 mM potassium citrate, and cholesterol (5 μg/ml)] with E. coli HB101. The following were the strains used in this work:
WGM-002 cdk-12(tm3846)/+ III
WGM-0011 cdk-12(wdh1(cdk-12 F397G)) III
WGM-0057 ama-1 (wdh5 (ama-1 CTD S2A + loxP SEC loxP))/+ IV
WGM-0054 ama-1 (wdh3 (ama-1 CTD S2S))/IV
WGM-0055 ama-1 (wdh4 (ama-1 CTD Δ))/+ IV
The antibodies used were as follows:
Anti–CTD S2P: Millipore, 04-1571 (rat monoclonal), used for ChIP
Anti–CTD S2P: Abcam, ab5095 (rabbit polyclonal), used for IF (immunofluorescence)
Anti–CTD S5P: ChromoTek, 3E8 (rat monoclonal)
Anti–AMA-1: Novus, 38520002 (rabbit polyclonal)
Anti-tubulin: Sigma-Aldrich, T5168 (mouse monoclonal)
Anti–Cpf-1 (CstF50): a gift of T. Blumenthal (26) (rabbit polyclonal)
Anti–Cpf-2 (CstF64): a gift of T. Blumenthal (26) (rabbit polyclonal)
Brood size analysis and embryonic lethality
On day 0, one L4 worm of each genotype was transferred to individual plates. Every day, adults were transferred to a new plate for 6 days. Larvae and unhatched embryos were counted. The ratio of dead eggs to the total brood size is the embryonic lethality.
Protein extraction for Western blotting
Worms (±18,000 L4 in liquid culture) were pelleted, washed twice in M9 (42.2 mM Na2HPO4, 22 mM KH2PO4, 85.7 mM NaCl, and 1 mM MgSO4), and frozen. Thawed worms were resuspended in 4× Laemmli Sample Buffer [200 mM tris (pH 8.5), 8% SDS, and 40% glycerol], boiled for 5 min at 95°C, vortexed, and then sonicated on a Diagenode Bioruptor Sonicator for 10 cycles (30 s on and 30 s off). Protein concentration was determined by a Pierce assay, and equal amounts of protein are loaded on Bio-Rad Mini-PROTEAN TGX gels 4 to 15%. After transfer on nitrocellulose, the membrane was blocked with Skin Milk Powder (5%) (Sigma-Aldrich) and exposed to primary antibodies overnight at 4°C [anti–α-tubulin 1/1000 (Sigma-Aldrich, T5168), anti–CTD 8WG16 1/1000 (Eurogentec, MMS-126P-050), anti–CTD Ser2P 3E10 1/1000 (Millipore, 04-1571), and anti-FLAG 1/1000 (Sigma-Aldrich, F3165)], washed three times in phosphate-buffered saline (PBS)–Tween (PBS-T; 0.05%), and then exposed to secondary antibody [anti-mouse immunoglobulin G (IgG) Perox (1/5000; GE, NA931) and anti-rat IgG Perox (1/5000; Dako, P0450)] and washed twice with PBS-T (0.05%). PerkinElmer Western Lightning Plus-ECL (enhanced chemiluminesence) was used for ECL and detection occurred on a GE ImageQuant LAS 4000 machine.
For Western blot on L1, L1s were grown for 4 hours and collected, washed once in M9, and frozen. Thawed worms were resuspended in 4× Laemmli Buffer [200 mM tris (pH 8.5), 8% SDS, and 40% glycerol] and then boiled for 5 min before being thoroughly vortexed and sonicated for 10 cycles (30 s on and 30 s off) on a Diagenode Bioruptor Sonicator. Protein concentration was measured by Bradford (Bio-Rad), and 15 μg of total protein was loaded on a Bio-Rad Mini-PROTEAN TGX gel 4 to 15%. Proteins were transferred to a nitrocellulose membrane using the Bio-Rad Trans-Blot Turbo transfer system, set up for high–molecular weight proteins (1.3A, up to 25 V). The membrane was then blocked in LI-COR Odyssey Blocking Buffer for at least 1 hour. Primary antibodies were incubated overnight at 4°C: anti–CTD S2P 3E10 1/1000 (Merck Millipore, 04-1571), anti–CTD S5P 3E8 1/1000 (ChromoTek), and anti–AMA-1 1/1000 (Novus Biologicals, 38520002). The membrane was washed three times in PBS-T and incubated for 1 hour with secondary antibodies coupled to infrared fluorophores (IRDye 800CW Goat anti-rat IgG, IRDye 800CW goat anti-mouse, and IRDye 680RD goat anti-rabbit) and then washed again two more times in PBS-T and one last time in PBS. Last, the membranes were dried out at room temperature, in the dark, overnight before the fluorescence was measured using the LI-COR Odyssey scanner and quantified using Image Studio (54).
Single-worm qRT-PCR
RNA extraction, genomic DNA digestion, cDNA synthesis, and qRT-PCR were performed as previously described (55). The Thermo Maxima H Minus First Strand cDNA Synthesis kit with dsDNase (#K1681) was used.
Immunofluorescence
Worms were killed by NaN3 (50 mM) on polylysine-coated slides and cut with a 22-gauge needle (BD Microlance 3). They were next frozen between slide and coverslip, for at least 1 hour, at −80°C on a metal plate, before being freeze-cracked by flipping the coverslip from the slide. The slides were fixed using MetOH 100% (20 min at room temperature) and a 3.7% formaldehyde solution (30 min at room temperature) [10 ml of formaldehyde solution 37% (Sigma-Aldrich, 252549) in 30 ml of PBS], washed twice with PBS and once with PBS-T (0.05%), and incubated with primary antibodies in wet chambers overnight at 4°C [anti–CTD 8WG16 (1/2000; Eurogentec, MMS-126P-050) and anti–CTD Ser2P 3E10 (1/2000; Millipore, 04-1571)]. The next day, the slides were washed three times with PBS-T (0.05%) and incubated with secondary antibodies 1 hour at room temperature in wet chambers [anti-rat IgG Alexa 488 (1/2000; Invitrogen, A21208) and anti-mouse IgG Alexa 594 (1/2000; Invitrogen, A11005)], washed twice with PBS-T (0.05%) and once with PBS, and mounted using Fluoroshield (containing DAPI) (Sigma-Aldrich, F6057).
Slides are observed on a Zeiss Axio Imager Z1 microscope with a Hamamatsu Digital Camera C11440 using the Zeiss software ZEN. The exposure was 3 s for Alexa 488, 1 s for Alexa 594, and 5 ms for DAPI.
For quantification, images were exported in TIFF format and analyzed in ImageJ with the following protocol: The background was removed from each picture using a sliding paraboloid of 2000 pixels in radius, the three channels (DAPI, Alexa 488, and Alexa 594) were stacked together, and the worm was detoured using the DAPI channel. The integrated value of fluorescent signal in the worm was measured for both Alexa 488 and Alexa 594 channels. The selection was then inversed to measure the background fluorescent signal in the rest of the slide. In Excel, these numbers were used to make a ratio of Alexa 488 (worm − background)/Alexa 594 (worm − background). All worms for which a picture was taken were measured, but those that had less than 200 integrated value for Alexa 594 in the worm were excluded from subsequent analysis as they appeared to have been less efficiently stained by AMA-1 primary antibodies and/or Alexa594 secondary antibody and the staining efficiency of Ser2P/Ser5P or Alexa 488 could not be trusted. The Ser2P/AMA-1 and Ser5P/AMA-1 ratio of more than 25 worms from at least two separate experiments were then plotted using R “boxplot” function. The P values are from Mann-Whitney test using the “wilcox.test” R function.
Single-worm PCR for genotyping
Worms (L1 to adults) were digested with proteinase K (1 mg/ml, final) (Roche Diagnostics, 03 115 828 001) in 1× Colorless GoTaq Reaction Buffer (Promega, M792A), first by snap-freezing for 15 min and then by incubating for 1 hour at 65°C to obtain a worm lysate. The proteinase K was inactivated 15 min at 95°C.
PCR was performed using GoTaq polymerase (GoTaq G2 DNA Polymerase, Promega, M748B) with 1.5 μl of worm lysate from proteinase K–treated worms, 5 μl of 5× Green GoTaq Reaction Buffer (Promega, M791A), 2 μl of primers at 10 μM, 0.25 μl of deoxynucleoside triphosphate at 20 mM, 0.125 μl of GoTaq polymerase, and 14.1 μl of water.
Genome editing by CRISPR
The CDK-12-as line was constructed using a dpy-10 co-CRISPR approach with a rol-6 selection and plasmid-borne Cas9 (56). After injection, 95 F1 animals having the roll and dumpy phenotypes were screened. The guide used targeted the CTATTTAGTATTCGAATATGngg where the underlined TTC corresponds to the codon encoding the gatekeeper F to be mutated into G and ngg indicates a point accepted mutation (PAM). The repair template consisted of a plasmid containing a genomic CDK-12 fragment harboring the mutated site flanked by 1.2 kb of homology on both sides and a mutated PAM. A silent Sal I site was added (see Fig. 2A) to facilitate the screening.
The ΔCTD, S2S, and S2A lines were constructed using a self-excising drug selection cassette (SEC) protocol (57) with the following modifications. A preassembled Cas9 RNP was prepared by incubating 10 min at 37°C, the protein Cas9 [IDT (Integrated DNA Technologies), 5 μg], the tracrRNA (2 μg), and both crRNAs targeting ama-1 (0.56 μg of each). The plasmid repair template (100 ng/μl, final), the plasmid coding for myo-3::m-Cherry (5 ng/μl, final), the plasmid coding for myo-2::m-Chery (2.5 ng/μl, final), and water to obtain a final volume of 20 μl are then added. To avoid needle clogging, the mix is spun at 14,000 rpm for 2 min and about 17 μl is transferred to a fresh tube. The RNA guides used were CAAUGAAGGAGGAUGGUCUCngg and UAUGAAUUUGGAUCAUAAGUngg (where ngg indicates the PAM) that targeted regions just upstream and downstream of the DNA coding for the CTD. The repair templates for S2S and S2A were constructed by Gibson Assembly to generate vectors where the fluorescent tag from Dickinson et al. (57) was replaced by the sequence coding a synthetic, codon optimized and intronless CTD S2S or CTD S2A (generated by Eurofins Genomics). In addition, two 1-kb homology regions were added upstream of the CTD and downstream of the SEC and PAM were mutated.
After the injection (on day 0), parental worms were allowed to lay eggs for 4 days. On day 4, 250 μl of hygromycin (10 mg/ml) (Hygromycin B from Streptomyces hygroscopicus; Sigma-Aldrich, H9773) was added per plate. From days 4 to 11, plates were checked to find roller and hygromycin-resistant F1 worms. These potential mutant worms were isolated into new plates (one worm per plate) and checked for the expression of the red comarkers using the Modular Stereo Microscope for Fluorescent Imaging Leica MZ10 F with the Leica Filter set ET mCherry (10450098). Only the fluorescence negative worms were kept because they likely integrated the repair plasmid rather than forming extra-chromosomal arrays. The progeny (F2) of these F1 roller and hygromycin-resistant worms were genotyped.
Gibson Assembly
The Gibson Assembly reactions required to prepare the repair constructs were performed using the Gibson Assembly Cloning Kit of New England Biolabs (E5510S) following their protocols [Gibson Assembly Protocol (E5510) and Gibson Assembly Chemical Transformation Protocol (E2611)]. The primers were designed with the NEBuilder Assembly Tool.
Q5 Site-Directed Mutagenesis
The Q5 Site-Directed Mutagenesis was made using the Q5 Site-Directed Mutagenesis Kit of New England Biolabs (E0554S) following the instructions of the manufacturer (Q5 Site-Directed Mutagenesis Kit Quick Protocol, E0554). The primers were designed with NEBaseChanger.
RNA interference
All the RNAi experiments were performed as described in (58), with the following precisions.
RNAi by injection
cDNA fragments were obtained by PCR amplification of a correspondent RNAi feeding plasmid. dsRNA was synthetized in vitro with the RiboMAXTM Large Scale RNA Production Systems T7 (Promega, P1300) and injected into L4 gonads at ~1 μg/μl. Injected worms are placed onto NGM plates at 20°C, and the development of the progeny was observed.
RNAi by feeding
RNAi was performed by feeding L4 or synchronized L1 larvae with HT115 bacteria transformed with a plasmid expressing dsRNA targeting the corresponding gene. Synchronized L1s were obtained by bleaching treatment of adult worms. Embryos were collected and washed with M9 and incubated overnight at 20°C in M9 without food. Synchronized L1s were transferred on an NGM/Amp (ampicillin)/isopropyl-β-d-thiogalactopyranoside (IPTG) plate seeded with HT-115 bacteria transformed with a dsRNA-expressing plasmid.
RNAi by soaking and feeding
L4 larvae were soaked in dsRNA (1 μg/μl) for 24 hours at 20°C. The animals were then transferred onto NGM/Amp/IPTG plates seeded with HT-115 bacteria that had been transformed with a dsRNA expression plasmid or control L4440 plasmid. The feeding RNAi was performed at 20°C for ~55 hours, during which the feeding plate was exchanged once after the first 24 hours.
RNAi suppression screen
Genome-wide RNAi screen was performed as described in (39) with the following modifications. Each RNAi culture was grown overnight at 37°C in 500 μl of LB with ampicillin (100 mg/ml). The cultures were induced for 4 hours the following day in 1 ml of LB containing ampicillin (100 mg/ml) and IPTG (1 mM) and incubated for 4 hours at 37°C. Transfer 200 μl of the induced cultures to 96-well plates and centrifuge at 3000 rpm for 2 min. Discard the supernatant and resuspend the pellets in 150 μl of S medium + IPTG (1 mM) + Amp (50 μg/ml).
Obtain an L1 synchronized culture and resuspend them at one L1/μl in S medium. Add 5 μl of the solution in each well (around five L1s per well). Incubate for 3 days at 20°C until the worms reach the adult stage. Add 1.5 μl of 3-MB-PP1 (100 μM) and 10% dimethyl sulfoxide (DMSO) (final 1 μM) in each well. Incubate for three more days at 20°C. The progeny is screened to identify suppressors of the L1 arrest. The complete list of suppressors will be published elsewhere.
Analog inhibition assay
The CDK-12-as inhibition test protocol is based on previous work (59). OP50 E. coli (uracil auxotrophs) were cultured overnight in LB at 37°C. Pellet 500 μl, discard the supernatant, and resuspend the pellet in 650 μl of S medium + 3-MB-PP1 (Sigma-Aldrich) at the desired concentration or just DMSO for control. In a 96-well plate, add 150 μl of the resuspended OP50 per well and add one L4. Incubate for 3 days at 20°C and observe the progeny.
For assays on plate, 3-MB-PP1 was added at the desired concentration before pouring the NGM plates. These plates seeded with OP50 could be stored at 4°C for at least 1 week with no loss of inhibitor efficiency.
Preparation of L1 larvae culture for ChIP or RNA
Embryos were collected by bleaching treatment and resuspended in S medium (no food) at 10 embryos per microliter. Embryos were incubated at 20°C for at least 12 hours to obtain synchronized starved L1s (S0). The culture was divided into three conditions: 1) Starvation (S): DMSO added to a final concentration of 0.1%. 2) Recovery (R): DMSO added to a final concentration of 0.1% and a 50-ml pellet of OP50. 3) Recovery plus inhibitor (R + I): 3-MB-PP1 (in DMSO) added to the desired final concentration and a 50-ml pellet of OP50.
Proceed to ChIP, RNA extraction, or other techniques.
Total RNA extraction and RNA-seq
An L1 liquid culture of 40 ml was pelleted, 2 min at 2000 rpm and 4°C, and washed three times with 50 ml of M9. One milliliter of Tri Reagent was added and the tube was mixed by vortexing 30 s followed by snap-freezing. Upon thawing in a 42°C bath, 200 μl of chloroform was added and the tube was mixed by vortexing for 30 s, incubated for 3 min at room temperature, and centrifuged for 15 min at 12,000 rpm and 4°C. The aqueous phase was transferred (500 μl) and 500 μl of isopropanol was added, incubated for 1 hour at −20°C, and centrifuged for 15 min at 12,000 rpm and 4°C. The supernatant was discarded; 1 ml of EtOH 70% was added and centrifuged for 5 min at 7500 rpm and 4°C; and the pellet was dried out at room temperature and dissolved in 100 μl of nuclease-free water by heating 5 min at 65°C. RNA-seq library preparation was made using the Illumina TruSeq stranded total RNA library preparation kit following the instructions of the manufacturer with the notable exception that the quantity of AMPure XP beads used for cleanups was scaled up by a ratio of 1.1 to retrieve smaller RNA species such as tRNAs.
ChIPs and ChIP-seq
L1s were pelleted for 1 min at 1200 rpm and washed three times with M9. L1s were resuspended in 70 ml of M9 and 10 ml of formaldehyde 16% (final 2%) to cross-link the L1s. After 30 min at 20°C, 4.25 ml of glycine 2.5 M (125 mM final) was added and incubated for 15 min at 20°C. The worms were pelleted and washed with tris (pH 8.0) 20 mM (4°C), pelleted, and snap-frozen.
From here on, the protocol is based on our published ChIP protocol (60) with some modifications. The pellet was thawed on ice and washed with FA/SDS/protease inhibitors [50 mM Hepes-KOH (pH 7.5), 150 mM NaCl, 1 mM EDTA, 0.1% Na deoxycholate, 1% Triton X-100, 0.1% SDS, and 1 mM phenylmethylsulfonyl fluoride], resuspended in 1.6 ml of FA/SDS/PI, and incubated for 15 min on ice. The sample was sonicated with a Bioruptor (Diagenode) (cycles of 30 s on and 60 s off for 21 min). After transferring to a 1.5-ml microcentrifuge tube and centrifugation for 30 min at 14,000 rpm and 4°C, the supernatant was transferred to a new tube.
For each IP, an extract volume containing the equivalent of 400,000 L1 (around 750 μl, 10% left as total extract at −20°C) was used. Three microliters of the appropriate antibody [anti-Pol II 8WG16 (ab817) and anti-S2P 3E10] was added overnight at 4°C. Per IP, 50 μl of appropriate magnetic beads (Dynal/Invitrogen) was washed four times with PBS–bovine serum albumin (BSA) (1 mg/ml) and resuspended in PBS-BSA (10 mg/ml) and then added to each IP and incubated for 4 hours at 21°C. The beads were washed three times with 1 ml of FA/SDS + 500 mM NaCl, one time with 500 μl of IP buffer, and one time with 500 μl of TE (tris-EDTA) and resuspended in 125 μl of Pronase buffer 1× and incubated for 20 min at 65°C. The total extract (100 μl) was similarly treated by adding 25 μl of Pronase buffer 5× and 6.25 μl of Pronase and incubated for 1 hour at 37°C and then overnight at 65°C. Ribonuclease A was added for 1 hour at 37°C, and the DNA sample was purified on Rapace columns.
For qPCR, DNA from total (input) extracts were diluted 40×, 200×, 1000×, 5000×, and 25,000× for the standard curve, while DNA from IPs was diluted 10×. For ChIP-Seq, the resulting DNA was sequenced, along with total extracts, using the Illumina TruSeq ChIP-seq library preparation protocol according to the instructions of the manufacturer.
Extension of poly-2A assay
Total RNA was obtained as described above. The cDNA synthesis and ePAT were performed as previously described (61).
Nuclear extracts and co-IP
Worms were collected and frozen as droplets in liquid nitrogen. Using a mortar and pestle, the frozen worms were processed into a fine powder resuspended in NIB [nuclear isolation buffer; 25 mM Hepes (pH 7.5), 0.025% NP-40, 50 mM KCl, and 5 mM MgCl2] and dounced. The resulting material was centrifuged for 6 min at 100g and 4°C, and both pellet and supernatant were checked by DAPI for intact nuclei enrichment in the supernatant. The nuclei were pelleted by centrifugation for 10 min at 2000g and 4°C and resuspended in the following buffers in the following order: 500 μl of NIB + 300 mM NaCl, 500 μl of NIB + 300 mM NaCl + 0.01% SDS, 500 of μl NIB + 300 mM NaCl, 1 ml of FA lysis buffer, and 2 of ml NIB + 300 mM NaCl + 0.1% SDS. The mix was resuspended by pipetting up and down gently for 5 min followed by centrifugation for 10 min at 1000g and 4°C. Protein concentration was determined, and 1 mg was used for IP using anti–CTD S2P or a corresponding volume of buffer, while 5% were left for the no IP sample. Fifty microliters of appropriate magnetic beads (Dynal/Invitrogen) was used for each IP and incubated for 4 hours at 4°C. The beads were washed three times and eluted in sample buffer before Western blotting.
Bioinformatics analyses
For all high-throughput sequencing experiments, the reads quality was assessed using FastQC. Quality and adaptor clipping was performed using trimmomatic (62). Statistical analysis and figure drawing were performed within the R programming environment (R Core Team, 2017).
RNA-seq analyses
Reads were mapped on the C. elegans genome using HISAT2 (63) as previously described (41). The number of reads by region of interest was quantified using featureCounts (64). Differential expression analysis was carried out with DESeq2 (65). Criteria for a differentially expressed feature are a false discovery rate <0.01 and an absolute fold change >1.5.
The quantification of trans-splicing events from the RNA-seq data was performed using SL-quant (41) in sensitive mode.
ChIP-seq analyses
Reads were mapped on the C. elegans genome using bowtie2 (66). The C. elegans profiles were normalized using the BEADS algorithm (67).
Supplementary Material
Acknowledgments
We thank F. Steiner for many insightful discussions and O. Finet for critical reading of the manuscript. We thank T. Blumenthal for the anti–CPF-1 and anti–CPF-2 antibodies. Funding: C.C., C.Y.-S., and F.X.S. were supported by a FRIA fellowship. This work has benefited from the facilities and expertise of the NGS platform of Institut Curie, supported by the grants ANR-10-EQPX-03 and ANR10-INBS-09-08 and by the Canceropôle Ile-de-France. This work was supported by grants ANR-15-CE12-0007 and ERC consolidator “DARK” to A.M. and by grants MIS F.4523.11, PDR T.0012.14, and CDR J.0066.16 to D.H. D.H. is an FNRS Research Director. Competing interests: The authors declare that they have no competing interests. Author contributions: C.C., C.Y.-S., and F.B. designed, performed, and analyzed the experiments. V.M. performed the CstF/AMA-1 co-IP. P.P. generated the CTD S2A line. F.X.S. contributed to the generation of the CTD S2S. M.W. and A.M. contributed to the design and funding of the genome-wide datasets. F.P. and V.R. supervised the RNAi screen. C.Y.-S. performed all the statistical analyses. D.H. designed the study, acquired funding, and supervised the project. D.H. wrote the paper with input from other authors. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. ChiP-seq and RNA-seq raw and processed data are available on Gene Expression Omnibus under the accession numbers GSE145456 and GSE145457, respectively. The RNA-seq read coverage can be viewed on the legacy ce10 Washu epigenome browser using session ID qNwYUnhblv.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabc1450/DC1
REFERENCES AND NOTES
- 1.Buratowski S., The CTD code. Nat. Struct. Biol. 10, 679–680 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Buratowski S., Progression through the RNA polymerase II CTD cycle. Mol. Cell 36, 541–546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egloff S., Murphy S., Cracking the RNA polymerase II CTD code. Trends Genet. 24, 280–288 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Schwer B., Shuman S., Deciphering the RNA polymerase II CTD code in fission yeast. Mol. Cell 43, 311–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho E. J., Takagi T., Moore C. R., Buratowski S., mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 11, 3319–3326 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCracken S., Fong N., Rosonina E., Yankulov K., Brothers G., Siderovski D., Hessel A., Foster S., Program A. E., Shuman S., Bentley D. L., 5’-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 11, 3306–3318 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn S. H., Kim M., Buratowski S., Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell 13, 67–76 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Gu B., Eick D., Bensaude O., CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 41, 1591–1603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim M., Krogan N. J., Vasiljeva L., Rando O. J., Nedea E., Greenblatt J. F., Buratowski S., The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Gilmartin G. M., Nevins J. R., An ordered pathway of assembly of components required for polyadenylation site recognition and processing. Genes Dev. 3, 2180–2190 (1989). [DOI] [PubMed] [Google Scholar]
- 11.Takagaki Y., Ryner L. C., Manley J. L., Four factors are required for 3′-end cleavage of pre-mRNAs. Genes Dev. 3, 1711–1724 (1989). [DOI] [PubMed] [Google Scholar]
- 12.Takagaki Y., Ryner L. C., Manley J. L., Separation and characterization of a poly(A) polymerase and a cleavage/specificity factor required for pre-mRNA polyadenylation. Cell 52, 731–742 (1988). [DOI] [PubMed] [Google Scholar]
- 13.Proudfoot N., O’Sullivan J., Polyadenylation: A tail of two complexes. Curr. Biol. 12, R855–R857 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Millevoi S., Vagner S., Molecular mechanisms of eukaryotic pre-mRNA 3’ end processing regulation. Nucleic Acids Res. 38, 2757–2774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cassart C., Drogat J., Migeot V., Hermand D., Distinct requirement of RNA polymerase II CTD phosphorylations in budding and fission yeast. Transcription 3, 231–234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dower K., Rosbash M., T7 RNA polymerase-directed transcripts are processed in yeast and link 3’ end formation to mRNA nuclear export. RNA 8, 686–697 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mapendano C. K., Lykke-Andersen S., Kjems J., Bertrand E., Jensen T. H., Crosstalk between mRNA 3’ end processing and transcription initiation. Mol. Cell 40, 410–422 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Davidson L., Muniz L., West S., 3’ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 28, 342–356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartkowiak B., Greenleaf A. L., Phosphorylation of RNAPII: To P-TEFb or not to P-TEFb? Transcription 2, 115–119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartkowiak B., Liu P., Phatnani H. P., Fuda N. J., Cooper J. J., Price D. H., Adelman K., Lis J. T., Greenleaf A. L., CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24, 2303–2316 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., Peterlin B. M., The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25, 2158–2172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keogh M.-C., Podolny V., Buratowski S., Bur1 kinase is required for efficient transcription elongation by RNA polymerase II. Mol. Cell. Biol. 23, 7005–7018 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Materne P., Anandhakumar J., Migeot V., Soriano I., Yague-Sanz C., Hidalgo E., Mignion C., Quintales L., Antequera F., Hermand D., Promoter nucleosome dynamics regulated by signalling through the CTD code. eLife 4, e09008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Materne P., Vázquez E., Sánchez M., Yague-Sanz C., Anandhakumar J., Migeot V., Antequera F., Hermand D., Histone H2B ubiquitylation represses gametogenesis by opposing RSC-dependent chromatin remodeling at the ste11 master regulator locus. eLife 5, e13500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.T. Blumenthal, Trans-splicing and operons in C. elegans, in WormBook (The C. elegans Research Community, 2012), pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrido-Lecca A., Saldi T., Blumenthal T., Localization of RNAPII and 3’ end formation factor CstF subunits on C. elegans genes and operons. Transcription 7, 96–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans D., Perez I., MacMorris M., Leake D., Wilusz C. J., Blumenthal T., A complex containing CstF-64 and the SL2 snRNP connects mRNA 3’ end formation and trans-splicing in C. elegans operons. Genes Dev. 15, 2562–2571 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacDonald C. C., Wilusz J., Shenk T., The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNAs downstream of the cleavage site and influences cleavage site location. Mol. Cell. Biol. 14, 6647–6654 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fong N., Bentley D. L., Capping, splicing, and 3’ processing are independently stimulated by RNA polymerase II: Different functions for different segments of the CTD. Genes Dev. 15, 1783–1795 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaslaver A., Baugh L. R., Sternberg P. W., Metazoan operons accelerate recovery from growth-arrested states. Cell 145, 981–992 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baugh L. R., To grow or not to grow: Nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics 194, 539–555 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu C.-K., Wang W., Brind’Amour J., Singh P. P., Reeves G. A., Lorincz M. C., Alvarado A. S., Brunet A., Vertebrate diapause preserves organisms long term through Polycomb complex members. Science 367, 870–874 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strome S., Wood W. B., Immunofluorescence visualization of germ-line-specific cytoplasmic granules in embryos, larvae, and adults of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 79, 1558–1562 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shim E. Y., Walker A. K., Shi Y., Blackwell T. K., CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev. 16, 2135–2146 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bishop A. C., Ubersax J. A., Petsch D. T., Matheos D. P., Gray N. S., Blethrow J., Shimizu E., Tsien J. Z., Schultz P. G., Rose M. D., Wood J. L., Morgan D. O., Shokat K. M., A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Powell-Coffman J. A., Knight J., Wood W. B., Onset of C. elegans gastrulation is blocked by inhibition of embryonic transcription with an RNA polymerase antisense RNA. Dev. Biol. 178, 472–483 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Baugh L. R., Demodena J., Sternberg P. W., RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324, 92–94 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Lunde B. M., Reichow S. L., Kim M., Suh H., Leeper T. C., Yang F., Mutschler H., Buratowski S., Meinhart A., Varani G., Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat. Struct. Mol. Biol. 17, 1195–1201 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saur T., DeMarco S. E., Ortiz A., Sliwoski G. R., Hao L., Wang X., Cohen B. M., Buttner E. A., A genome-wide RNAi screen in Caenorhabditis elegans identifies the nicotinic acetylcholine receptor subunit ACR-7 as an antipsychotic drug target. PLOS Genet. 9, e1003313 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wahle E., Winkler G. S., RNA decay machines: Deadenylation by the Ccr4–not and Pan2–Pan3 complexes. Biochim. Biophys. Acta 1829, 561–570 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Yague-Sanz C., Hermand D., SL-quant: A fast and flexible pipeline to quantify spliced leader trans-splicing events from RNA-seq data. Gigascience 7, giy084 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coudreuse D., van Bakel H., Dewez M., Soutourina J., Parnell T., Vandenhaute J., Cairns B., Werner M., Hermand D., A gene-specific requirement of RNA polymerase II CTD phosphorylation for sexual differentiation in S. pombe. Curr. Biol. 20, 1053–1064 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Lu F., Portz B., Gilmour D. S., The C-terminal domain of RNA polymerase II is a multivalent targeting sequence that supports drosophila development with only consensus heptads. Mol. Cell 73, 1232–1242.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lenstra T. L., Tudek A., Clauder S., Xu Z., Pachis S. T., van Leenen D., Kemmeren P., Steinmetz L. M., Libri D., Holstege F. C. P., The role of Ctk1 kinase in termination of small non-coding RNAs. PLOS ONE 8, e80495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collin P., Jeronimo C., Poitras C., Robert F., RNA polymerase II CTD tyrosine 1 is required for efficient termination by the Nrd1-Nab3-Sen1 pathway. Mol. Cell 73, 6551–669.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Bowman E. A., Bowman C. R., Ahn J. H., Kelly W. G., Phosphorylation of RNA polymerase II is independent of P-TEFb in the C. elegans germline. Development 140, 3703–3713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graber J. H., Salisbury J., Hutchins L. N., Blumenthal T., C. elegans sequences that control trans-splicing and operon pre-mRNA processing. RNA 13, 1409–1426 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCracken S., Fong N., Yankulov K., Ballantyne S., Pan G., Greenblatt J., Patterson S. D., Wickens M., Bentley D. L., The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385, 357–361 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Miki T. S., Carl S. H., Stadler M. B., Großhans H., XRN2 autoregulation and control of polycistronic gene expresssion in Caenorhabditis elegans. PLOS Genet. 12, e1006313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y., Kuersten S., Huang T., Larsen A., MacMorris M., Blumenthal T., An uncapped RNA suggests a model for Caenorhabditis elegans polycistronic pre-mRNA processing. RNA 9, 677–687 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan L., Xie W., Li K.-L., Yang Z., Xu J., Zhang W., Liu L.-P., Ren X., He Z., Wu J., Sun J., Wei H.-M., Wang D., Xie W., Li W., Ni J.-Q., Sun F.-L., Heterochromatin remodeling by CDK12 contributes to learning in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 112, 13988–13993 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andrusiak M. G., Jin Y., Context specificity of stress-activated mitogen-activated protein (MAP) kinase signaling: The story as told by Caenorhabditis elegans. J. Biol. Chem. 291, 7796–7804 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roux A. E., Langhans K., Huynh W., Kenyon C., Reversible age-related phenotypes induced during larval quiescence in C. elegans. Cell Metab. 23, 1113–1126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bamps S., Westerling T., Pihlak A., Tafforeau L., Vandenhaute J., Mäkelä T. P., Hermand D., Mcs2 and a novel CAK subunit Pmh1 associate with Skp1 in fission yeast. Biochem. Biophys. Res. Commun. 325, 1424–1432 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Ly K., Reid S. J., Snell R. G., Rapid RNA analysis of individual Caenorhabditis elegans. MethodsX 2, 59–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H., Ishidate T., Ghanta K. S., Seth M., Conte D. Jr., Shirayama M., Mello C. C., A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 197, 1069–1080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dickinson D. J., Pani A. M., Heppert J. K., Higgins C. D., Goldstein B., Streamlined genome engineering with a self-excising drug selection cassette. Genetics 200, 1035–1049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Conte D. Jr., MacNeil L. T., Walhout A. J. M., Mello C. C., RNA interference in Caenorhabditis elegans. Curr. Protoc. Mol. Biol. 109, 26.3.1–26.3.30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J. S., Lilley B. N., Zhang C., Shokat K. M., Sanes J. R., Zhen M., A chemical-genetic strategy reveals distinct temporal requirements for SAD-1 kinase in neuronal polarization and synapse formation. Neural Dev. 3, 23 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Migeot V., Hermand D., Chromatin immunoprecipitation-polymerase chain reaction (ChIP-PCR) detects methylation, acetylation, and ubiquitylation in S. pombe. Methods Mol. Biol. 1721, 25–34 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Jänicke A., Vancuylenberg J., Boag P. R., Traven A., Beilharz T. H., ePAT: A simple method to tag adenylated RNA to measure poly(A)-tail length and other 3’ RACE applications. RNA 18, 1289–1295 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheung M. S., Down T. A., Latorre I., Ahringer J., Systematic bias in high-throughput sequencing data and its correction by BEADS. Nucleic Acids Res. 39, e103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabc1450/DC1