

X-ray crystal structures of SARS-CoV-2 Mpro with dual inhibitors provide a new direction for the designing of COVID-19 antivirals.
Abstract
The main protease (Mpro) of SARS-CoV-2 is a key antiviral drug target. While most Mpro inhibitors have a γ-lactam glutamine surrogate at the P1 position, we recently found that several Mpro inhibitors have hydrophobic moieties at the P1 site, including calpain inhibitors II and XII, which are also active against human cathepsin L, a host protease that is important for viral entry. In this study, we solved x-ray crystal structures of Mpro in complex with calpain inhibitors II and XII and three analogs of GC-376. The structure of Mpro with calpain inhibitor II confirmed that the S1 pocket can accommodate a hydrophobic methionine side chain, challenging the idea that a hydrophilic residue is necessary at this position. The structure of calpain inhibitor XII revealed an unexpected, inverted binding pose. Together, the biochemical, computational, structural, and cellular data presented herein provide new directions for the development of dual inhibitors as SARS-CoV-2 antivirals.
INTRODUCTION
The COVID-19 (coronavirus disease 2019) pandemic emerged in late December 2019 in Wuhan, China and evolved to be one of the worst public health crises in modern history. The impact of COVID-19 on global public health and economy has been severe. The etiological agent of COVID-19 is severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which shares ~78% genetic similarity with SARS-CoV, the virus that led to the SARS outbreak in 2003. Although coronavirus outbreaks such as COVID-19 are not unpredicted, the high mortality rate and the ease of transmission of SARS-CoV-2 are unprecedented.
Currently, there are few antivirals and no vaccines available for SARS-CoV-2. Hence, it is imperative to identify drug targets that could lead to effective antivirals. Guided by research of similar coronaviruses, SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV), several viral proteins have been prioritized as SARS-CoV-2 antiviral drug targets: the spike protein, the RNA-dependent RNA polymerase (RdRp), the main protease (Mpro), and the papain-like protease (PLpro) (1, 2). The SARS-CoV-2 RdRp inhibitor remdesivir was granted emergency use authorization from the U.S. Food and Drug Administration on 1 May 2020. Remdesivir has broad-spectrum antiviral activity against SARS-CoV, SARS-CoV-2, and MERS-CoV in cell culture (3–5). The antiviral efficacy was further confirmed in MERS-CoV infection mouse and rhesus macaque models (6, 7). Additional RdRp inhibitors under investigation for SARS-CoV-2 include EIDD-2801, favipiravir (T-705), ribavirin, and galidesivir (8, 9). The fusion inhibitor EK1C4, which was designed on the basis of the H2 peptide in the S2 domain of the HCoV-OC43 spike protein, showed promising broad-spectrum antiviral activity against SARS-CoV-2, SARS-CoV, and MERS-CoV, as well as human coronaviruses HCoV-229E, HCoV-NL63, and HCoV-OC43 (10, 11). Meanwhile, Mpro has been extensively explored as a drug target for not only SARS-CoV-2 but also SARS-CoV and MERS-CoV, as well as enteroviruses, rhinoviruses, and noroviruses (12). Mpro is a viral-encoded cysteine protease that has a unique substrate preference for a glutamine residue at the P1 site, which was recently confirmed by substrate profiling for SARS-CoV-2 (13). Consequently, most of the designed Mpro inhibitors contain either 2-pyrrolidone or 2-piperidinone at the P1 site as a mimetic of the glutamine residue in the substrate (14). Examples include compounds N3, 13b, 11a, 11b, and our recently identified GC-376 (15–18)—all of which have potent enzymatic inhibition in biochemical assay and antiviral activity in cell culture. Their mechanism of action and mode of inhibition were revealed by the drug-bound x-ray crystal structures (15–18).
Our previous study found two unconventional SARS-CoV-2 Mpro inhibitors, calpain inhibitors II and XII, that are structurally dissimilar to the traditional Mpro inhibitors, such as GC-376 (15). Specifically, calpain inhibitors II and XII incorporate the hydrophobic methionine and norvaline side chains in the P1 position, respectively. This discovery challenges the idea that a hydrophilic glutamine mimetic is required at the P1 position. Furthermore, calpain inhibitor II is a potent inhibitor of human protease cathepsin L, with an inhibition constant Ki of 50 nM (19). Cathepsin L plays an important role in SARS-CoV-2 viral entry by activating the viral spike protein in the endosome or lysosome (20–22) and has a relatively broad substrate preference at the P1 position (23, 24). Studies have indicated that cathepsin L inhibitors such as MDL28170 can block or substantially decrease virus entry (20, 25). In this study, we explore two series of Mpro inhibitors, one is the dual inhibitors targeting both Mpro and cathepsin L such as calpain inhibitors II and XII, and the other is the Mpro-specific inhibitors such as GC-376 analogs UAWJ246, UAWJ247, and UAWJ248. To dissect the mechanism of action of dual inhibitors, we solved the high-resolution x-ray crystal structures of Mpro with calpain inhibitors II and XII. We found that calpain inhibitor II is bound to Mpro in the canonical, extended conformation, but calpain inhibitor XII adopts an unexpected binding mode, where it assumes an inverted, semi-helical conformation in which the P1′ pyridine ring is placed in the S1 pocket instead of the P1 norvaline side chain, as one would expect. The complex structures of calpain inhibitors II and XII, together with the structure-activity relationship studies, reveal that the S1 pocket of Mpro can accommodate both hydrophilic and hydrophobic substitutions, paving the way for the design of dual inhibitors that target both the viral Mpro and host cathepsin L. Last, guided by the x-ray crystal structure of SARS-CoV-2 Mpro with GC-376 [Protein Data Bank (PDB): 6WTT] (15), three Mpro-specific inhibitors UAWJ246, UAWJ247, and UAWJ248 were designed to profile the side-chain preferences of the S1′, S2, S3, and S4 pockets. Overall, the x-ray crystal structures and activity profile presented herein offer valuable insights into the substrate promiscuity of Mpro, as well as a new direction in the designing of dual inhibitors targeting both the SARS-CoV-2 Mpro and host cathepsin L.
RESULTS AND DISCUSSION
SARS-CoV-2 Mpro constructs used in this study
Three Mpro constructs were used in this study: the tag-free native Mpro (Mpro), Mpro with two extra residues histidine and methionine at the N terminus (HM-Mpro), and the Mpro with a native N terminus and a C-terminal his-tag (Mpro-His). The tag-free Mpro was used for all functional assays, while the other two constructs were used for structure determination due to ease of crystallization. The HM-Mpro construct was used for the complex structures of all five compounds including calpain inhibitors II and XII, UAWJ246, UAWJ247, and UAWJ248 (fig. S1). In addition, the Mpro-His construct was also cocrystallized with UAWJ246 as a control for the potential influence of extra HM residues at the N terminus on the Mpro structure and drug binding.
The Mpro-His and the native Mpro have similar enzymatic activity with kcat/Km (Michaelis constant) values of 6689 and 5748 s−1 M−1, respectively (fig. S2A). The HM-Mpro construct has significantly reduced enzymatic activity with a kcat/Km value of 214 s−1 M−1, which is about 3.7% of the Mpro (kcat/Km = 5748 s−1 M−1) (fig. S2A). This was expected as it has been shown that Mpro requires dimerization to be catalytically active, and the N-terminal finger plays an essential role in dimerization (26). Specifically, the first residue serine (Ser1) from one protomer interacts with the Glu166 of the adjacent protomer, a feature that is important for catalytic activity (fig. S2B). Nevertheless, the HM-Mpro turned out to be an excellent construct for crystallization, and we were able to determine several high-resolution drug-bound x-ray crystal structures. In contrast, efforts to obtain high-quality crystals with the Mpro-His construct was challenging, because of the localization of the disordered His-tag at the crystal packing interface (15). Several previous studies similarly used the enzymatically inactive Mpro with extra residues at the N terminus for the structural studies, and the ligand-binding poses were identical to those with tag-free Mpro (e.g., PDB 7BRP versus 6WNP and 6WTJ versus 6L70) (27–30). Therefore, the use of enzymatic inactive HM-Mpro construct for crystallographic study of inhibitor binding is justified.
X-ray crystal structures of SARS-CoV-2 Mpro in complex with calpain inhibitors II and XII
Previous studies have shown that SARS-CoV and SARS-CoV-2 Mpro cleave polyproteins at P2-P1 ↓ P1′ where P1′ is a residue with a small side chain (Ala, Ser, or Gly), P1 is glutamine, and P2 is a large, hydrophobic residue, such as leucine or phenylalanine (13, 31, 32). This consensus sequence has operated as the foundation for extensive inhibitor designs where a reactive warhead, usually an aldehyde, α,β-unsaturated ester, or α-ketoamide, is linked to a glutamine surrogate pyrrolidone that is connected to a hydrophobic residue via an amide bond (12, 33–35). This strategy has been largely successful, with the development of inhibitors such as GC-376 and 13b that have median inhibitory concentration (IC50) values in the low nanomolar range for SARS-CoV-2 Mpro (15, 17).
We recently reported that calpain inhibitors II and XII and boceprevir have low micromolar IC50 values against SARS-CoV-2 Mpro (15). These compounds have a hydrophobic side chain at the P1 position, challenging the notion that a hydrophilic moiety is required at this position. This has previously been demonstrated with SARS-CoV Mpro, where the aldehyde inhibitor Cm-FF-H binds with a Ki of 2.24 ± 0.58 μM despite having a phenylalanine at the P1 position (36). The observed substrate plasticity is in part attributed to the reactivity of the electrophilic warhead aldehyde with the catalytic cysteine, which offsets the requirement for favorable interactions with the hydrophilic S1 subsite. Furthermore, it introduces the prospect of modifying the P1 residue so that it interacts with multiple host or viral proteases that are essential for promoting SARS-CoV-2 viral entry or replication, which would increase antiviral spectrum and genetic barrier to drug resistance. To visualize the interactions between the P1 site and the hydrophobic S1 pocket, we solved the complex structures of SARS-CoV-2 Mpro with calpain inhibitor II and calpain inhibitor XII.
The crystal structures of the SARS-CoV-2 HM-Mpro in complex with calpain inhibitors II (PDB: 6XA4) and XII (PDB: 6XFN) were solved in the C2 space group at 1.65 and 1.70 Å resolution with an Rwork/Rfree of 0.206/0.245 and 0.205/0.232, respectively, with one protomer per asymmetric unit (Fig. 1 and table S1). Like other peptidomimetic aldehyde inhibitors, the thiohemiacetal of calpain inhibitor II occupies the oxyanion hole formed by the backbone amide groups of Gly143, Ser144, and Cys145 (Fig. 1A). Here, it adopts the (S) configuration, which is typical for most Mpro aldehyde inhibitors, although the (R) configuration has also been observed (15, 37). Like previous Mpro and cathepsin L complex structures, the body of the inhibitor extends the length of the substrate-binding channel, with the side chains placed in their respective recognition pockets. The P1 methionine side chain projects into the S1 subsite where the sulfur forms a weak hydrogen bond with His163. The P2 leucine side chain forms hydrophobic interactions in the S2 pocket, while the P3 leucine occupies the solvent-accessible S3 position. Multiple hydrogen bonds form between the inhibitor amide backbone and the main chains of His164, Met165, and Glu166.
Fig. 1. X-ray crystal structures of SARS-CoV-2 Mpro in complex with calpain inhibitors II (PDB: 6XA4) and XII (PDB: 6XFN).
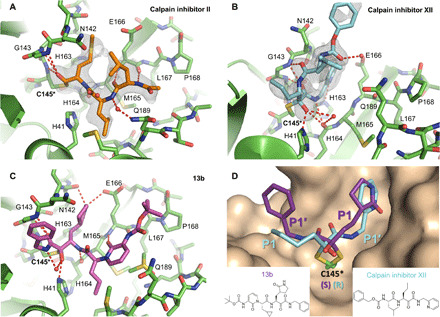
Unbiased Fo-Fc electron density map, shown in gray, is contoured at 2 σ. Hydrogen bonds are shown as red dashed lines. SARS-CoV-2 Mpro in complex with (A) calpain inhibitor II (orange) and (B) calpain inhibitor XII (cyan). (C) Binding mode and interactions of a previously reported α-ketoamide inhibitor 13b (purple, PDB ID: 6Y2F) with SARS-CoV-2 Mpro. (D) Close-up comparison of calpain inhibitor XII (blue) and 13b (purple) in the S1 and S1′ sites. The P1 pyrrolidinone ring and P1′ benzene of 13b occupy the S1 and S1′ sites, respectively. Conversely, the P1 norvaline and P1′ pyridine of calpain inhibitor XII adopt the S1′ and S1 sites, respectively. Accompanying the noncanonical positioning of the P1 and P1′ moieties, a stereochemical inversion of the thiohemiketal adduct with Cys145 is observed for calpain inhibitor XII. Like all other known α-ketoamide inhibitors, 13b adopts a (S) configuration. However, calpain inhibitor XII forms a covalent adduct in the (R) configuration.
In contrast to the pose of calpain inhibitor II, calpain inhibitor XII demonstrates an atypical binding mode where it adopts an inverted, semi-helical conformation that wraps around the catalytic core (PDB: 6XFN) (Fig. 1B). This is dissimilar to the extended configuration of previously published peptidomimetic inhibitors, including other α-ketoamide compounds such as 13b (Fig. 1, C and D) (18). For calpain inhibitor XII, the P1′ pyridine is placed in the S1 site while the P1 norvaline occupies the S1′ site. The P2 leucine projects outward toward the solvent near the TSEDMLN loop (residues 45 to 51), and the terminal carboxybenzyl (Cbz) group curls back toward the S1 site, forcing Asn142 upward while forming a water-mediated hydrogen bond with Glu166. Corresponding to this unique binding pose, we observed the (R) configuration of the thiohemiketal-Cys145 adduct among the known α-ketoamide inhibitors (Fig. 1D). From our recent x-ray crystal structure of the SARS-CoV-2 Mpro in complex with GC-376 (PDB: 6WTT) (15), it is known that the thiohemiacetal center of aldehyde-based inhibitors can assume either an (R) or (S) configuration, depending on which face of the aldehyde group undergoes nucleophilic attack from the thiolate of Cys145 during covalent bond formation (15). In contrast, the thiohemiketal group of all crystallographic solved α-ketoamide Mpro inhibitors such as N3 and 13b adopts the (S) configuration (Fig. 1, C and D) (16, 17). The new calpain inhibitor XII structure demonstrates that, similar to aldehyde-based inhibitors, the covalent adduct formed between α-ketoamide compounds and the catalytic cysteine can assume two different configurations as well.
In both the (R) and (S) configurations of the thiohemiketal adducts, the hydroxyl group is placed near His41, while the amide oxygen is positioned in the oxyanion hole. However, the exact locations of these two functional groups result in different hydrogen bond patterns. Compared to other α-ketoamide inhibitors, the unique binding mode of calpain inhibitor XII alters the hydrogen-bonding network of the catalytic core. The hydroxyl group forms a short hydrogen bond (2.5 Å in length) with the catalytic His41 and two weak hydrogen bonds (3.3 Å) with the main chain carbonyl of His164 and a water molecule in the central channel between the S1 and S2 pockets (Fig. 1B). The ketoamide amide oxygen establishes three hydrogen bonds in the oxyanion hole (2.9, 3.2, and 3.1 Å to the backbone ─NH of Gly143, Ser144, and Cys145, respectively), and its nitrogen forms a hydrogen bond (3.1 Å) with the mainchain carbonyl of His164 (Fig. 1B). In the canonical binding conformation for α-ketoamide inhibitors, such as 13b and those described herein, the hydroxyl group establishes one standard hydrogen bond (2.8 Å) with His41 and another hydrogen bond (3.3 Å) with a bulk water molecule (Fig. 1C). Meanwhile, the hydrogen bonds between the amide oxygen and the oxyanion hole now have distances of 3.3, 3.0, and 2.5 Å, respectively, while the amide nitrogen forms no hydrogen bond with the protein (Fig. 1C).
The S1 pocket recognizes the most conserved residue in the Mpro substrate, the P1 glutamine. Underscoring its importance for ligand binding, most specific Mpro inhibitors have a glutamine surrogate such as the pyrrolidone ring that occupies the S1 pocket. X-ray crystal structures of SARS-CoV-2 Mpro in complex with compounds like calpain inhibitor II, calpain inhibitor XII, and boceprevir (PDB: 6WNP and 7BRP) prove that a hydrophobic side chain can also be accommodated in the S1 pocket. Like previous inhibitors, hydrophobic interactions are observed between the newly identified inhibitors and the backbone atoms of Leu141/Asn142/Met165. However, these interactions are further enhanced in calpain inhibitor XII, where its aromatic pyridine ring is stacked and sandwiched between the two planar peptide bonds involving Asn142 and Met165 (Fig. 1B). Furthermore, a hydrogen bond is observed between His163 and the methionine sulfur and pyridine nitrogen for calpain inhibitors II and XII, respectively (Fig. 1, A and B).
To confirm the binding modes revealed by the x-ray crystal structures of calpain inhibitors II and XII, we designed two analogs, calpain inhibitor I and UAWJ257 (Fig. 2A). Specifically, to dissect the importance of the hydrogen bond between His163 and the sulfur atom from the methionine side chain of calpain inhibitor II, we designed the butyl analog, calpain inhibitor I (Fig. 2A). It was found that calpain inhibitor I has an IC50 that is ~10-fold weaker than calpain inhibitor II (Fig. 2, B versus C). Similarly, this hydrogen bond is important for calpain inhibitor XII binding, since the benzene counterpart of calpain inhibitor XII, compound UAWJ257, has a significantly reduced activity against SARS-CoV-2 Mpro (IC50 = 64.5 ± 15.4 μM) (Fig. 2, D versus E). Likewise, no binding was detected in the thermal shift assay [ΔTm (the shift of the unfolding temperature) = −0.07°C] for UAWJ257, while calpain inhibitor I showed reduced binding compared to calpain inhibitor II (Fig. 2F). In addition to abolishing the hydrogen bond, the marked loss of inhibition of UAWJ257 might be attributed to a clash between the proton on His163 Nε2 and the benzene hydrogen that replaces the lone pair on the pyridine nitrogen, which lies only 3.1 Å away from His163 Nε2. Overall, the structure-activity relationship results of calpain inhibitors II and XII are consistent with the binding poses shown in the x-ray crystal structures (Fig. 1).
Fig. 2. Biochemical and biophysical characterizations of calpain inhibitors II and XII and their analogs as SARS-CoV-2 Mpro inhibitors.
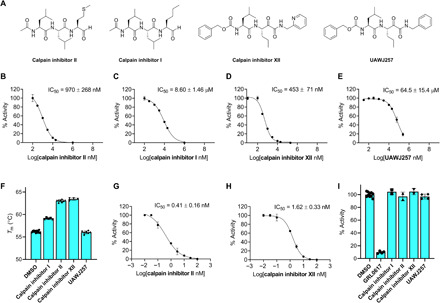
(A) Chemical structures of calpain inhibitors II, I, and XII and UAWJ257. (B to E) IC50 plots from in vitro FRET-based enzymatic assay against SARS-CoV-2 Mpro of calpain inhibitor II (B), calpain inhibitor I (C), calpain inhibitor XII (D), and UAWJ257 (E). (F) Thermal shift binding assay of calpain inhibitors and their analogs with 3 μM SARS-CoV-2 Mpro protein. Forty micromolar compounds were preincubated with Mpro protein for 30 min at 30°C. Tm was calculated as described in Materials and Methods. (G and H) IC50 plots from in vitro FRET-based enzymatic assay against human cathepsin L of calpain inhibitor II (G) and calpain inhibitor XII (H). (I) Counter screening of calpain inhibitors II, I, and XII and UAWJ257 against SARS-CoV-2 PLpro in the FRET-based enzymatic assay. GRL0617 was added as a positive control. The calculated enzymatic activity with each compound was normalized to dimethyl sulfoxide (DMSO) control. The results are average ± SD of two or more repeats.
While calpain inhibitor II was previously reported to inhibit cathepsin L with a Ki of 0.6 nM (19), which was confirmed by our data as well (IC50 = 0.41 nM) (Fig. 2G), the inhibition of cathepsin L by calpain inhibitor XII was unknown. We determined that calpain inhibitor XII is also a potent inhibitor of cathepsin L, with an IC50 value of 1.62 ± 0.33 nM (Fig. 2H). Because cathepsin L has been shown to activate the SARS-CoV-2 spike protein, cathepsin L inhibitors are known to block viral entry (20). This may provide an explanation for the superior antiviral activity of calpain inhibitors II and XII despite having inferior affinity for Mpro compared to the specific inhibitors GC-376, N3, UAWJ246, UAWJ247, and UAWJ248 (15, 18). Both calpain inhibitors II and XII had no detectable inhibition against the SARS-CoV-2 PLpro (IC50 > 20 μM) (Fig. 2I), suggesting that they are not nonspecific cysteine protease inhibitors. Collectively, the x-ray crystal structures of SARS-CoV-2 HM-Mpro in complex with calpain inhibitors II and XII, along with the enzymatic assay results, suggest that it is feasible to develop dual inhibitors that simultaneously targeting the SARS-CoV-2 Mpro and the host cathepsin L, both of which are validated antiviral drug targets for SARS-CoV-2 (20, 21).
Rational design of GC-376 analogs and the x-ray crystal structures of SARS-CoV-2 Mpro in complex with UAWJ246, UAWJ247, and UAWJ248
We recently demonstrated the inhibition of SARS-CoV-2 Mpro by GC-376 and solved the x-ray crystal structure of Mpro-His with GC-376 (PDB: 6WTT) (15). To profile the substrate spectrum of SARS-CoV-2 Mpro in the S1′, S2, S3, and S4 sites, several GC-376 analogs were designed (Fig. 3A). Specifically, compound UAWJ246 was designed to occupy the S1′ pocket according to the overlay structures of SARS-CoV-2 Mpro + GC-376 (PDB: 6WTT) and SARS-CoV Mpro (H41A) + substrate (PDB: 2Q6G) (fig. S3). UAWJ246 contains a pharmacological compliant α-ketoamide reactive warhead with a cyclopropyl substitution. UAWJ247 was designed to probe the substrate promiscuity in the S2 pocket. In the x-ray crystal structure of Mpro-His with GC-376, the TSEDMLN loop (residues 45 to 51) constituting the S2 pocket exhibits significant flexibility among the three protomers in the asymmetric unit, indicating that a variety of substitutions can be accommodated at this position. To test this hypothesis, UAWJ247 was designed with a benzyl substitution at the P2 position instead of the isopropyl in GC-376. UAWJ248 is a tripeptide and was designed to incorporate the P3 substitution. Enzyme kinetic studies showed that compounds UAWJ246 and UAWJ247 bound to Mpro reversibly with inhibition constant KI values of 0.036 ± 0.007 and 0.035 ± 0.008 μM, respectively (Fig. 3, B and C). In contrast, the enzyme kinetic curves for compound UAWJ248 was similar to that of GC-376, which showed a biphasic progression character (fig. S4), suggesting that UAWJ248 inhibits Mpro through a two-step process with an initial reversible binding followed by an irreversible inactivation. Fitting the progression curves with the two-step Morrison equation revealed the first step equilibrium dissociation constant KI and the second step reaction constant k2 as 13.20 nM and 0.001195 s−1, respectively, which corresponds to an overall k2/KI value of 9.05 × 104 M−1 s−1 (Fig. 3D). In comparison, the k2/KI value for GC-376 is 2.84 × 104 M−1 s−1 (Fig. 3E), suggesting that UAWJ248 is 3.2-fold more potent than GC-376. None of these compounds showed inhibition against the SARS-CoV-2 PLpro (IC50 > 20 μM) (Fig. 3F).
Fig. 3. Pharmacological characterization of the mechanism of action of GC-376 analogs UAWJ246, UAWJ247, and UAWJ248 in inhibiting SARS-CoV-2 Mpro.
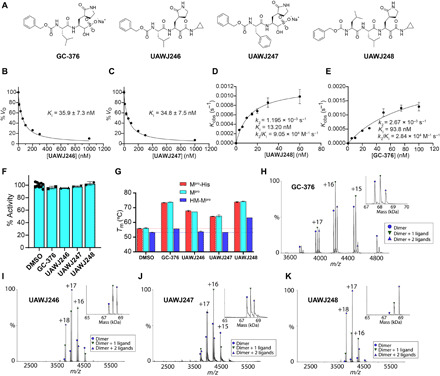
(A) Chemical structures of GC-376, UAWJ246, UAWJ247, and UAWJ248. (B to E) Morrison plot of the proteolytic reaction progression curves of SARS-CoV-2 Mpro in the presence or the absence of compounds. Original proteolytic reaction progression curves are in fig. S4. Detailed methods are described in Materials and Methods. UAWJ246 (B), UAWJ247 (C), UAWJ248 (D), and GC-376 (E). Kobs, observed pseudo first order rate constant. (F) Counter screening of GC-376, UAWJ246, UAWJ247, and UAWJ248 against SARS-CoV-2 PLpro in the FRET-based enzymatic assay. (G) Thermal shift binding assay of GC-376 analogs with different SARS-CoV-2 Mpro constructs. (H to K) Binding of inhibitors to SARS-CoV-2 Mpro using native mass spectrometry. Native mass spectra with the inset deconvolved spectra revealing ligand binding with (H) 10 μM GC-376 added, (I) 10 μM UAWJ246, (J) 10 μM UAWJ247 added, and (K) 10 μM UAWJ248 with 4 mM DTT added. The peaks are annotated with the blue circle as the dimer, green down triangle as the dimer with one ligand bound, and the purple up triangle as the dimer with two ligands bound. VO, initial velocity.
Next, we determined the mechanism of action with thermal shift binding assay and native mass spectrometry. As expected, binding of UAWJ246, UAWJ247, and UAWJ248 all stabilized SARS-CoV-2 Mpro, as shown by the ΔTm shift of 11.08°, 8.28°, and 18.10°C, respectively (Fig. 3G). All three compounds also stabilized the Mpro-His construct to the same degree as the tag-free Mpro, suggesting that these two constructs are functionally equivalent (Fig. 3G). In contrast, compounds UAWJ246 and UAWJ247 did not show stabilization for the HM-Mpro construct, while the more potent GC-376 and UAWJ248 stabilized this construct with ΔTm shift of 2.45° and 10.01°C, respectively (Fig. 3G).
The binding of all three compounds UAWJ246, UAWJ247, and UAWJ248 to Mpro was further confirmed by native mass spectrometry (Fig. 3, H to K). Like GC-376, addition of the ligands resulted in two new sets of peaks corresponding to one ligand per dimer and two ligands per dimer.
Most Mpro inhibitors with antiviral activity against SARS-CoV-2 use an α-ketoamide or aldehyde/bisulfite warhead to form a covalent adduct with the catalytic Cys145 (15, 17). We previously reported GC-376 as one of the most potent inhibitors of SARS-CoV-2 Mpro in vitro with an IC50 value of 0.033 μM and median effective concentration (EC50) value of 3.37 μM in the enzymatic assay and antiviral cytopathic effect assay (15). Here, we show that the α-ketoamide analog of GC-376, UAWJ246, has a comparable IC50 of 0.045 μM, suggesting that the α-ketoamide and the aldehyde are nearly equivalent in terms of inhibitory activity. We solved the complex structure of UAWJ246 with both SARS-CoV-2 HM-Mpro at 1.45 Å resolution with an Rwork/Rfree of 0.189/0.213 as a dimer (PDB: 6XBG) and Mpro-His at 2.35 Å as a trimer in the asymmetric unit (Fig. 4, A and B, and fig. S5). The binding pose of UAWJ246 in these two constructs was nearly superimposable (fig. S5), suggesting that the use of enzymatic inactive HM-Mpro construct for crystallographic study of inhibitor binding is justified. In the dimer structure of HM-Mpro with UAWJ246, the amide oxygen of the α-ketoamide occupies the oxyanion hole, while the thiohemiketal hydroxide forms a hydrogen bond with the catalytic histidine, His41 (Fig. 4A). The cyclopropyl group extends partly into the S1′ subpocket. The Cbz of UAWJ246 adopts a different conformation in each protomer (Fig. 4, A and B). In the A protomer of the 1.45-Å resolution structure crystallized in P21 space group, the Cbz moiety extends the length of the S3 and S4 subsites, forcing residues 189 to 191 to flip outward (Fig. 4A). In the B protomer, the Cbz moiety projects upward, where the S3 side chain is normally positioned (Fig. 4B). The A conformation resembles that of GC-376 in our previously solved structure (PDB: 6WTT) (fig. S6A), whereas the B conformation resembles GC-376 from PDB ID: 7BRR (fig. S6B) (28). The downward conformation establishes extensive interactions with the S4 pocket and may be one of the main reasons for the superior in vitro activity of GC-376 and its analogs. The upward conformation also forms favorable interactions with protein residues constituting the S3 site, including Glu166 and Gln189. In addition, it enables the formation of intramolecular interactions between the benzyl ring and the P1 side chain, similar to the hydrophobic intramolecular interactions formed between the P1 and P3 moieties in calpain inhibitor II and boceprevir, as well as, to some degree, the stacking between the pyridine and the Cbz linker of calpain inhibitor XII (Fig. 1B and fig. S7). It is likely that GC-376, UAWJ246, and other analogs exist in a dynamic equilibrium between these conformations, and the captured crystallographic poses are, in part, determined by the crystal-packing interface between protomers and/or differences in the pH or ionic strength of the crystallization solution.
Fig. 4. SARS-CoV-2 Mpro in complex with GC-376 analogs.
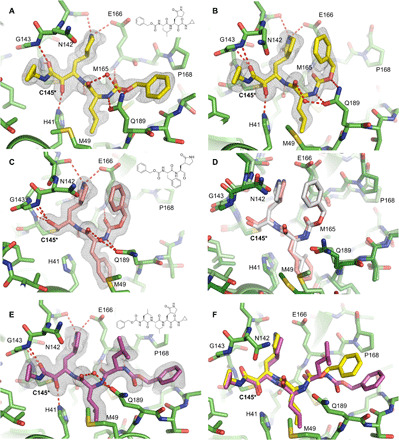
Unbiased Fo-Fc electron density map, shown in gray, is contoured at 2 σ. Hydrogen bonds are shown as red dashed lines. Solved as a dimer in the P21 spacegroup, two different conformations of the Cbz group of UAWJ246 were observed in the (A) protomer A and (B) protomer B. (C) The complex structure of UAWJ247, revealing that the P2 position can accommodate a Phe side chain. (D) Comparison of the binding poses of UAWJ247 (dark green/salmon) and GC-376 (light green/gray, PDB ID: 7BRR). (E) The complex structure of UAWJ248, solved as a dimer in the P1 space-group. Protomer A is shown here, and the inhibitor binding pose is identical in protomer B (fig. S8). (F) Comparison of the binding poses of UAWJ248 (purple) and UAWJ246 (yellow) in protomer A.
The chemical structure of UAWJ247 is nearly identical to GC-376, except for the replacement of its S2 isobutyl moiety for a benzyl group, analogous to a Leu ➔ Phe exchange. To visualize the binding mode of UAWJ247, we solved the complex structure with SARS-CoV-2 Mpro at 1.60 Å with an Rwork/Rfree of 0.181/0.224 in the C2 space group with one protomer per asymmetric unit (PDB: 6XBH) (Fig. 4, C and D). Like their chemical structures, the binding poses between UAWJ247 and GC-376 are very similar (Fig. 4D), with minor differences observed for Gln189 and the catalytic histidine, His41, which swivels toward the S2 benzyl group to form face-to-face π-stacking interactions. As expected, the IC50 of 0.045 μM for UAWJ247 is very close to that of GC-376 and consistent with the preference for a hydrophobic residue at the S2 site. These data also suggest that replacing Leu for a larger Phe is tolerated and that aromaticity can be incorporated into the S2 site for the purpose of improving pharmacokinetic properties or broadening the spectrum of activity, with limited effect on Mpro inhibition.
UAWJ248 was designed to occupy the additional S4 pocket compared to UAWJ246. We solved the complex structure of UAWJ248 with SARS-CoV-2 HM-Mpro at 1.70 Å with an Rwork/Rfree of 0.181/0.219 as a dimer in the P1 monoclinic space group (PDB: 6XBI) (Fig. 4, E and F). The conformation is consistent in both protomers (fig. S8). The α-ketoamide warhead forms an adduct with Cys145 in the (S) conformation, like other cyclopropane α-ketoamide analogs described herein including UAWJ246 and previously published 13b (17). Similarly, the P1 γ-lactam and P2 isobutyl moieties occupy their respective S1 and S2 subsites. The P3 isobutyl orients upward into the S3 site where it forms no meaningful interactions. However, the insertion of this additional leucine into the UAWJ246 core ensures the formation of a hydrogen bond with the main chain amide oxygen of Glu166. The terminal Cbz is placed in the S4-S5 site, where nonpolar interactions occur between the benzene and side chain of Pro168 and Ala191 and π stacking with the main chain amides of Gln189, Thr190, and Ala191.
Molecular dynamics simulations of SARS-CoV-2 Mpro with inhibitors
The binding interactions between the covalently bound calpain inhibitor II, calpain inhibitor XII, UAWJ246, UAWJ247, and UAWJ248 with SARS-CoV-2 Mpro were further explored using 100-ns molecular dynamics (MD) simulations starting with the x-ray structures with PBD IDs 6XA4, 6XFN, 6XBG (dimer), 6XBH (monomer), and 6XBI (dimer). The MD simulations verified the stability of the interactions inside the binding cavity of SARS-CoV-2 Mpro observed in the x-ray structures, as inspected from the trajectories and shown in frequency interaction plots (Fig. 5, A, C, E, and G, and fig. S9, A, D, and G). The simulations further demonstrate that the complexes formed are stable, and the ligand positions do not deviate significantly from the crystallographic ones (Fig. 5, B, D, F, and H), with Cα root mean square deviation (RMSD) values less than 2.4 Å and an overall ligand RMSD less than 3.5 Å (Fig. 5, I to L, and fig. S9).
Fig. 5. MD simulations of SARS-CoV-2 Mpro with calpain inhibitors II and XII and UAWJ248.
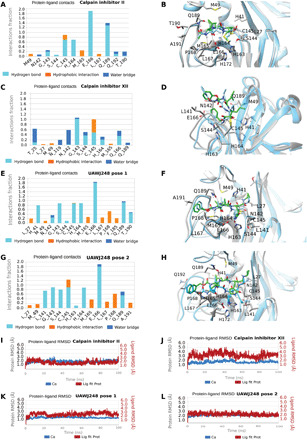
In (A), (C), (E), and (G), hydrogen bonding interactions bar is depicted in light blue, van der Waals in orange, and water bridges in blue. Interactions are plotted from 100-ns MD simulations for the complexes of the covalently bound calpain inhibitor II, calpain inhibitor XII, UAWJ248 (pose 1: first protomer), and UAWJ248 (pose 2: second protomer) in the active site of SARS-CoV-2 Mpro. They are considered important when the frequency bar is ≥0.2. In (B), (D), (F), and (H), the last snapshots of the abovementioned 100-ns MD-simulated complexes overlaid with experimental structures with PDB IDs 6XA4, 6XFN, 6XBI, respectively, are shown. In (I) to (L), the RMSD plots of Cα carbons (blue diagram, left axis) and of ligand (red diagram, right axis) of the abovementioned 100-ns MD-simulated complexes are shown.
Calpain inhibitor XII, UAWJ246, and UAWJ248 are ketoamides having a ketone carbonyl compared to the aldehyde group in calpain inhibitor II and UAWJ247. Calpain inhibitor XII forms hydrogen bond interactions with residues His41, Gly143, Ser144, Cys145, His164, and Glu166 (Fig. 5, C and D). The P1 pyridinyl group is positioned in the S1 region and forms a hydrogen bond with His163. Compared to calpain inhibitor XII, in UAWJ246, the pyrrolidone substitution occupies the S1 subsite instead of (2-pyridinyl)methyl in calpain inhibitor XII, leading to additional stabilizing hydrogen bonds as described previously. In addition, the small cyclopropyl group in UAWJ246 fits in the S1′ subsite, avoiding steric repulsions with S1′ subsite amino acids as seen in the MD simulation trajectory with calpain inhibitor XII. These changes resulted in a potency enhancement by 100-fold, i.e., from 0.45 μM for calpain inhibitor XII to 0.045 μM for UAWJ246. In UAWJ248, the length of the peptide was increased by adding a leucine between P2 Leu and Cbz group, and additional lipophilic contacts with P168 are observed (Fig. 5, E to H), but the activity remained unchanged. Two inhibitor-bound complexes are shown for UAWJ246 (poses 1 and 2) (fig. S9, A to C and D to F) and for UAWJ248 (poses 1 and 2) (Fig. 5, E to H), which correspond to different protomers. Minor differences are observed in the hydrogen bonding interactions between the two binding cavities in each protomer, reflecting the dynamic nature of the complexes. For example, a hydrogen bond with His41 was observed in UAWJ246 pose 1 (fig. S9A), while in UAWJ246 pose 2, a hydrogen bond with Asn142 was observed (fig. S9D). Similarly, hydrogen bonds with His41 and Thr190 were observed in UAWJ248 pose 1 (Fig. 5E), and a hydrogen bond with Ser144 was observed in UAWJ248 in pose 2 (Fig. 5G).
Compared to calpain inhibitor II (Fig. 5, A and B), in UAWJ247, which also has an aldehyde warhead, the methionine P1 substituent was changed to pyrrolidone, and additional hydrogen bonding interactions are formed. UAWJ247 forms important hydrogen bonding interactions between the P1 2-pyrrolidinone NH group and E166 side chain and peptidic carbonyl of Phe140 (fig. S9G), in addition to the hydrogen bonds with Gly143, Ser144, Cys145, His164, and Glu189. Furthermore, the P2 benzyl group in UAWJ247 fits better in the S2 subsite than the isobutyl from calpain inhibitor II, resulting in new van der Waals interactions with Met49, His41, and Met165 (fig. S9G). These additional stabilizing interactions reduce the IC50 against SARS-CoV-2 Mpro by 23-fold, i.e., from 0.97 μM in calpain inhibitor II to 0.042 μM in UAWJ247. The root-mean-square fluctuation (RMSF) values for the atoms in the P1 substitution of UAWJ246 (21 to 27), UAWJ247 (4 to 10), and UAWJ248 (29 to 35) were smaller than that from the corresponding atoms of calpain inhibitors II (1 to 4) and XII (27 to 34) (fig. S10), suggesting that the pyrrolidone substitution is preferred over methionine side chain and the (2-pyridinyl)methyl, although both of which are also tolerated at the S1 pocket.
Cellular antiviral activity and cytotoxicity of GC-376 analogs
To profile the antiviral activity of the GC-376 analogs UAWJ246, UAWJ247, and UAWJ248, we first tested their cellular cytotoxicity against multiple cell lines. All three compounds were not toxic to these cell lines with 50% cytotoxic concentration (CC50) values greater than 100 μM in most cases (table S2). Hence, we set the highest drug concentration as 30 or 100 μM in the antiviral assay. The antiviral activity of GC-376 analogs was tested in both the immunofluorescence assay and plaque assay using the wild-type SARS-CoV-2 virus. GC-376 was included as a positive control. In the immunofluorescence assay, GC-376, UAWJ246, UAWJ247, and UAWJ248 inhibited the viral replication in a dose-response manner with EC50 values of 1.50 ± 0.42, 15.13 ± 6.44, 6.81 ± 0.65, and 20.49 ± 3.71 μM, respectively (Fig. 6, A to D and I). In the plaque assay, GC-376, UAWJ246, UAWJ247, and UAWJ248 inhibited the viral replication with EC50 values of 0.48 ± 0.29, 4.61 ± 3.60, 2.06 ± 0.93, and 11.1 ± 4.2 μM, respectively (Fig. 6, E to H and J). Overall, all three GC-376 analogs UAWJ246, UAWJ247, and UAWJ248 had confirmed antiviral activity in cell culture. Comparing Mpro binding and antiviral potency among the GC-376 analogs, it appears that the aldehyde warhead may be more suitable for cell-based activities than the α-ketoamide. While the terminal groups of UAWJ248 may enhance the enzymatic inhibition potency of GC-376 by simultaneously occupying both S3 and S4 subpockets, the weaker cellular antiviral activity of UAWJ248 might be due to decreased cellular permeability or increased metabolic degradation. In addition, as shown by our previous study, the calpain inhibitors demonstrated more potent antiviral activity than GC-376 despite showing weaker binding affinity against Mpro in vitro (15), consistent with our hypothesis of synergistic inhibition of cathepsin L and Mpro.
Fig. 6. Antiviral activity of GC-376 analogs.
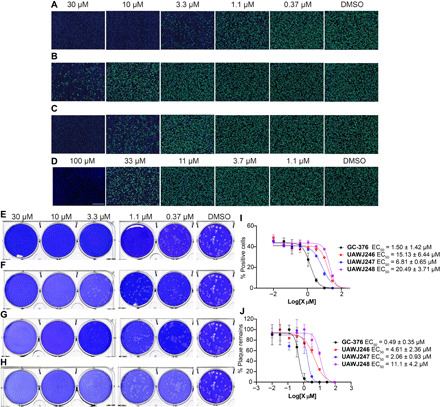
(A to D) Antiviral activity of GC-376 analogs against SARS-CoV-2 in the immunofluorescence assay. (A) GC-376; (B) UAWJ246; (C) UAWJ247; (D) UAWJ248. Vero E6 cells in a 96-well plate were infected with SARS-CoV-2 (USA-WA1/2020 isolate) at an MOI of 0.05 in the presence of the indicated concentrations of the tested compounds. At 48 hours post infection (hpi), the cells were fixed and stained with a rabbit monoclonal antibody against the SARS-CoV-2 NP and a secondary antibody conjugated with Alexa 488 (green). The nuclei were counterstained with Hoechst dye (blue). For each well, fluorescence images of approximately 10,000 cells were acquired and shown. The images are representatives of three repeats. (E to H) Antiviral activity of GC-376 analogs against SARS-CoV-2 in the plaque assay. (E) GC-376; (F) UAWJ246; (G) UAWJ247; (H) UAWJ248. Vero E6 cells in six-well plates were infected with approximately 40 plaque-forming units per well of SARS-CoV-2 (USA-WA1/2020 isolate). After 1 hour, the inoculum was removed, and the cells were overlaid with medium containing the indicated concentrations of the tested compounds and 1.2% Avicel RC-591. At 3 days post infection, the overlay was removed, and the cells were stained with 0.2% crystal violet. The images are representatives of two repeats. Data fitting of the antiviral activity of GC-376 analogs against SARS-CoV-2 in the immunofluorescence assay (I) and the plaque assay (J).
The ongoing COVID-19 pandemic needs an immediate intervention. If the previous SARS-CoV and MERS-CoV outbreaks are not severe enough to attract the attention from the scientific community, the current COVID-19 outbreak is a timely reminder of the threat of coronavirus. Encouraging progress has been made in developing antivirals and vaccines against SARS-CoV-2, such as remdesivir. However, despite the proof-reading function of the SARS-CoV-2 RdRp, SARS-CoV-2 continues to mutate, which will inevitably lead to resistance development. Drug resistance has evolved in cell culture against remdesivir using a model coronavirus, the murine hepatitis virus (38), raising concerns for the monotherapy of remdesivir. Hence, new drugs with distinct mechanisms of action are needed.
The coronavirus Mpro (3CLpro) has long been pursued as a promising antiviral drug target (12). The unique feature of Mpro is its stringent preference for the glutamine residue at the P1 position, while no known host protease has such preference. Accordingly, most Mpro inhibitors are designed to contain a glutamine mimetic at the P1 position such as pyrrolidone or 2-piperidinone. One of the most advanced lead compounds in this class is GC-376, an investigational veterinary drug that is currently being developed to treat feline infectious peritonitis. GC-376 has optimal in vivo pharmacokinetic properties and in vivo antiviral efficacy in feline infectious peritonitis virus (FIPV) infection cat model (39, 40). Our earlier study, coupled with an independent study from Vuong et al. (28), showed that GC-376 can similarly inhibit the enzymatic activity of SARS-CoV-2 Mpro and the viral replication of SARS-CoV-2 in cell culture. While this result is expected, our study also identified three additional nonconventional hits, boceprevir and calpain inhibitors II and XII (15). These three compounds differ from known Mpro inhibitors in that they contain hydrophobic substitutions at the P1 site, challenging the notion that hydrophilic glutamine mimetics are required for potent inhibition. Intrigued by this finding, we pursued to solve the x-ray crystal structures of SARS-CoV-2 Mpro with boceprevir and calpain inhibitors II and XII. During this process, the x-ray crystal structures of SARS-CoV-2 Mpro in complex with boceprevir were released in PDB (PDB ID: 7BRP and 6WNP), and we therefore shifted our focus to calpain inhibitors II and XII. The binding pose of calpain inhibitor II in the active site of Mpro is consistent with other peptidomimetic inhibitors, where the methionine and leucine side chains occupy the S1 and S2 pockets, respectively. The methionine sulfur atom forms a hydrogen bond with the side chain imidazole of His163. In contrast, calpain inhibitor XII binds to the Mpro active site in an inverted conformation, projecting the pyridine instead of the norvaline residue in the S1 pocket. Again, the nitrogen from the pyridine forms a hydrogen bond with the side chain imidazole of His163. Collectively, these two structures suggest that the His163 residue at the S1 pocket represents a binding hot spot for Mpro inhibitors and that the pyridine ring is a suitable side chain to engineer potent interactions with the S1 subsite. The ligand plasticity of the P1 site suggests that it is feasible to design dual inhibitors targeting both the viral Mpro and other important proteases, such as cathepsin L. Cathepsin L has been identified as a critical host protease for the SARS-CoV-2 cell entry. It plays an essential role in mediating the activation of the viral spike protein, thereby triggering membrane fusion and viral RNA release (20, 22). Although the antiviral potency of cathepsin L inhibitors has been demonstrated against coronaviruses including SARS-CoV-2 in several studies, the in vivo antiviral efficacy of cathepsin L inhibitors is not known. One potential concern is that the coronavirus spike protein can also be activated by other host proteases including trypsin, calpain, and TMPRSS2. Therefore, it is not clear whether inhibition of cathepsin L alone will be sufficient to completely stop viral replication in vivo. In this regard, a dual inhibitor design strategy that targets both the viral Mpro and host cathepsin L has certain advantages. First, compared to monospecific Mpro inhibitors, dual inhibitors might have a higher genetic barrier to drug resistance as they also target the host cathepsin L. Second, compared to monospecific cathepsin L inhibitors, dual inhibitors can lead to complete inhibition of viral replication as it targets the essential viral protein Mpro.
Comparing x-ray crystal structures of SARS-CoV-2 Mpro in complex with UAWJ246, UAWJ247, and UAWJ248 to the complex structure of GC-376, we can conclude that (i) the P1′ substitution does not contribute significantly to the potency of drug binding,and (ii) the P2 position prefers hydrophobic substitutions such as the leucine and phenylalanine side chains. Previous studies also demonstrate that the cyclopropyl and cyclohexyl groups can be similarly accommodated (17, 18); (iii) the P3 and P4 positions prefer to be hydrophobic substitutions. However, these two positions, especially P3, may not be as important as the P1 and P2 substitutions, but favorable interactions with S3 and S4 subpockets can still contribute to the potency of inhibitor binding; (iv) while the unique conformation of calpain inhibitor XII demonstrates the versatility of the α-ketoamide adduct formation, comparing the potent antiviral activities of the GC-376 analogs with different warheads indicate that aldehyde-based compounds may be better suited for cell-based activity.
In summary, the structure-activity relationship of Mpro inhibitors revealed by the x-ray crystal structures and enzymatic assays described herein can be used to guide lead optimization. P1 substitution and the reactive warhead contribute significantly to the drug binding potency, followed by P2 substitution. While P1′, P3, and P4 substitutions are less essential, they can be optimized for inhibition against other proteases important for SARS-CoV-2 entry/replication and to improve their pharmacokinetic properties.
MATERIALS AND METHODS
Cell lines and viruses
Human rhabdomyosarcoma (RD), Madin-Darby canine kidney, Vero, Huh-7, and HCT-8 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM); Caco-2 and MRC-5 cell lines were maintained in Eagle’s minimum essential medium. Both media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin antibiotics. Cells were kept at 37°C in a 5% CO2 atmosphere. VERO E6 cells (American Type Culture Collection, CRL-1586) were cultured in DMEM, supplemented with 5% heat-inactivated FBS in a 37°C incubator with 5% CO2. SARS-CoV-2, isolate USA-WA1/2020 (NR-52281), was obtained through BEI Resources and propagated once on VERO E6 cells before it was used for this study. Studies involving the SARS-CoV-2 were performed at the University of Texas Health Science Center at San Antonio (UTHSCSA) biosafety level-3 laboratory by personnel wearing powered air-purifying respirators.
Protein expression and purification
SARS-CoV-2 Mpro (Mpro or 3CLpro) gene from strain BetaCoV/Wuhan/WIV04/2019 was ordered from GenScript (Piscataway, NJ) in the pET29a(+) vector with Escherichia coli codon optimization. The expression and purification of His-tagged SARS-CoV-2 Mpro (Mpro-His) were described previously (15). Briefly, the Mpro gene was inserted into pET29a(+) with Nde I/Xho I digestion sites. The N-terminal methionine was removed by E. coli methionine aminopeptidase. There are extra LEHHHHHH residues at the C terminus. The protein sequence for the C-terminal His-tagged SARS-CoV-2 Mpro is SGFRKMAFPS GKVEGCMVQV TCGTTTLNGL WLDDVVYCPR HVICTSEDML NPNYEDLLIR KSNHNFLVQA GNVQLRVIGH SMQNCVLKLK VDTANPKTPKYKFVRIQPGQ TFSVLACYNG SPSGVYQCAM RPNFTIKGSF LNGSCGSVGF NIDYDCVSFC YMHHMELPTG VHAGTDLEGN FYGPFVDRQT AQAAGTDTTITVNVLAWLYA AVINGDRWFL NRFTTTLNDF NLVAMKYNYE PLTQDHVDIL GPLSAQTGIA VLDMCASLKE LLQNGMNGRT ILGSALLEDE FTPFDVVRQC SGVTFQLEHHHHHH.
For HM-Mpro expression and purification, the SARS-CoV-2 Mpro gene from strain BetaCoV/Wuhan/WIV04/2019 GenScript (Piscataway, NJ, USA) was inserted into pETGSTSUMO vector. The plasmid was transformed into Rosetta(DE3) pLysS Competent Cells (Novagen). A single colony was picked for overnight growth to inoculate 50 ml of LB broth with kanamycin (50 μg/ml) and chloramphenicol (35 μg/ml). Ten milliliters of the overnight culture was used to inoculate 1 liter of LB broth with kanamycin (50 μg/ml) and chloramphenicol (35 μg/ml). The 1-liter culture was grown at 250 rpm, 37°C until optical density (OD) reached 0.6 to 0.8. Expression was then induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 250 rpm, 20°C overnight. The culture was centrifuged at 5000g for 20 min, and the resulting pellet was resuspended in 30 ml of the lysis buffer [20 mM tris-HCl (pH 8.4), 300 mM NaCl, 10% glycerol, and 20 mM imidazole]. These cells were lysed by sonication on a 10-s sonication/15-s rest cycle for a total of 15 min at an amplitude of 6. The lysate was centrifuged at 40,000g for 45 min at 4°C, and the supernatant was filtered and then loaded onto a HiTrap HP column. The column was washed with lysate buffer, and the protein was then eluted by linear gradient of imidazole. The peak of the protein was pooled and concentrated. The protein was then diluted in ULP1 cleavage buffer [20 mM tris (pH 8.0), 100 mM NaCl, and 10% glycerol]. The protease ULP1 was added at 1:20 ratio with incubation at 20°C overnight. The sample was loaded to a HisTrap HP column, and the flowthrough containing the HM-Mpro was collected. The HM-Mpro was concentrated and loaded to Superdex 200/16 equilibrated with 20 mM tris (pH 8.0) and 250 mM NaCl. The peak fractions were pooled and concentrated to 10 mg/ml and flash-frozen with liquid nitrogen. The purity of the protein was evaluated by SDS–polyacrylamide gel electrophoresis (PAGE). The protein sequence for the SARS-CoV-2 HM-Mpro is HMSGFRKMAFPS GKVEGCMVQV TCGTTTLNGL WLDDVVYCPR HVICTSEDML NPNYEDLLIR KSNHNFLVQA GNVQLRVIGH SMQNCVLKLK VDTANPKTPKYKFVRIQPGQ TFSVLACYNG SPSGVYQCAM RPNFTIKGSF LNGSCGSVGF NIDYDCVSFC YMHHMELPTG VHAGTDLEGN FYGPFVDRQT AQAAGTDTTITVNVLAWLYA AVINGDRWFL NRFTTTLNDF NLVAMKYNYE PLTQDHVDIL GPLSAQTGIA VLDMCASLKE LLQNGMNGRT ILGSALLEDE FTPFDVVRQCSGVTFQ.
For the expression and purification of SARS-CoV-2 Mpro with unmodified N and C termini (Mpro), the SARS-CoV-2 Mpro gene was subcloned from pET29a(+) to the pE-SUMO vector according to the manufacturer’s protocol (LifeSensors Inc., Malvern, PA). The forward primer with the Bsa I site is GCGGTCTCAAGGTTCAGGATTTAGGAAGATGGCATTTCC; the reverse primer with Xba I site is GCTCTAGATTACTGAAAGGTCACGCCGCTGCATTGACG. After removal of the SUMO tag with SUMO protease, there are no extra residues at either the N or C terminus. The pE-SUMO plasmid with SARS-CoV-2 Mpro gene was transformed into BL21(DE3) cells with kanamycin selection. A single colony was picked to inoculate 10 ml of LB media and was cultured at 37°C overnight. This 10-ml culture was added to 1 liter of LB media and grown to around OD600 of 0.8. This culture was cooled on ice for 15 min and then induced with 0.5 mM IPTG. Induced cultures were incubated at 18°C for an additional 24 hours and then harvested, lysed the same way as His-tagged Mpro protein (15). The supernatant was incubated with Ni-NTA (nickel-nitrilotriacetic acid) resin overnight at 4°C on a rotator. The Ni-NTA resin was thoroughly washed with 30 mM imidazole in wash buffer [50 mM tris (pH 7.0), 150 mM NaCl, and 2 mM dithiothreitol (DTT)], and SUMO-tagged Mpro was eluted from Ni-NTA with 300 mM imidazole. Eluted SUMO-tagged Mpro was dialyzed against 100-fold volume dialysis buffer [50 mM tris (pH 7.0), 150 mM NaCl, and 2 mM DTT] in a 10,000–molecular weight cutoff dialysis tubing. After dialysis, SUMO-tagged Mpro was incubated with SUMO protease 1 at 4°C overnight, and SUMO tag was removed by application of another round of Ni-NTA resin. The purity of the protein was confirmed with SDS-PAGE gel. The protein sequence for the native SARS-CoV-2 Mpro is SGFRKMAFPS GKVEGCMVQV TCGTTTLNGL WLDDVVYCPR HVICTSEDML NPNYEDLLIR KSNHNFLVQA GNVQLRVIGH SMQNCVLKLK VDTANPKTPKYKFVRIQPGQ TFSVLACYNG SPSGVYQCAM RPNFTIKGSF LNGSCGSVGF NIDYDCVSFC YMHHMELPTG VHAGTDLEGN FYGPFVDRQT AQAAGTDTTITVNVLAWLYA AVINGDRWFL NRFTTTLNDF NLVAMKYNYE PLTQDHVDIL GPLSAQTGIA VLDMCASLKE LLQNGMNGRT ILGSALLEDE FTPFDVVRQCSGVTFQ.
For the expression and purification of SARS-CoV-2 PLpro, SARS-CoV-2 PLpro gene (open reading frame 1ab 1564 to 1876) from strain BetaCoV/Wuhan/WIV04/2019 was ordered from GenScript (Piscataway, NJ) in the pET28b(+) vector with E. coli codon optimization. The SARS-CoV-2 PLpro gene was inserted into pET28b(+) with Nco I/Xho I sites. The expression and purification procedures are similar to that of Mpro-His protein as described above, except that the lysis buffer and Ni-NTA wash and elution buffer are in pH 7.5 [50 mM tris (pH 7.5), 150 mM NaCl, and 2 mM DTT]. The N-terminal methionine was removed by E. coli methionine aminopeptidase based on our mass spectrum result. There are extra LEHHHHHH residues at the C terminus. The final protein sequence for the SARS-CoV-2 PLpro is GEVRTIKVFTTVDNINLHTQVVDMSMTYGQQFGPTYLDGADVTKIKPHNSHEGKTFYVLPN DDTLRVEAFEYYHTTDPSFLGRYMSALNHTKKWKYPQVNGLTSIKWADNNCYLATALLTL QQIELKFNPPALQDAYYRARAGEAANFCALILAYCNKTVGELGDVRETMSYLFQHANLDS CKRVLNVVCKTCGQQQTTLKGVEAVMYMGTLSYEQFKKGVQIPCTCGKQATKYLVQQESP FVMMSAPPAQYELKHGTFTCASEYTGNYQCGHYKHITSKETLYCIDGALLTKSSEYKGPI TDVFYKENSYTTTIKLEHHHHHH. Human liver cathepsin L was purchased from EMD Millipore (catalog no. 219402).
Peptide synthesis
The SARS-CoV-2 Mpro fluorescence resonance energy transfer (FRET) substrate Dabcyl-KTSAVLQ/SGFRKME(Edans) was synthesized as described before (15). The SARS-CoV-2 PLpro FRET substrate Dabcyl-FTLRGG/APTKV(Edans) was synthesized by solid-phase synthesis through iterative cycles of coupling and deprotection using the previously optimized procedure (41). Specifically, ChemMatrix rink-amide resin was used. Typical coupling condition was 5 equivalents of amino acid, 5 equivalents of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), and 10 equivalents of N,N-diisopropylethylamine (DIEA) in N,N′-dimethylformamide for 5 min at 80°C. For deprotection, 5% piperazine and 0.1 M 1-Hydroxybenzotriazole (HOBt) were used, and the mixture was heated at 80°C for 5 min. The peptide was cleaved from the resin using 95% trifluoroacetic acid (TFA), 2.5% tris, and 2.5% H2O, and the crude peptide was precipitated from ether after the removal of TFA. The final peptide was purified by preparative high-performance liquid chromatography (HPLC). The purity and identity of the peptide were confirmed by analytical HPLC (>98% purity) and mass spectrometry ([M + 2]2+ calculated, 888.04; detected, 888.80).
Compound synthesis and characterization
Details for the synthesis procedure (fig. S1) and characterization for compounds UAWJ257, UAWJ246, UAWJ247, and UAWJ248 can be found in the Supplementary Materials.
Native mass spectrometry
Before analysis, the protein was buffer-exchanged into 0.2 M ammonium acetate (pH 6.8) and diluted to 10 μM. DTT was dissolved in water and prepared at a 400 mM stock. Each ligand was dissolved in ethanol and diluted to 10× stock concentrations. The final mixture was prepared by adding 4 μl of protein, 0.5 μl of DTT stock, and 0.5 μl of ligand stock for a final concentration of 4 mM DTT and 8 μM protein. Final ligand concentrations were 10 μM. The mixtures were then incubated for 10 min at room temperature before analysis. Each sample was mixed and analyzed in triplicate.
Native mass spectrometry was performed using a Q-Exactive HF quadrupole-Orbitrap mass spectrometer with the Ultra-High Mass Range research modifications (Thermo Fisher Scientific). Samples were ionized using nano–electrospray ionization in positive ion mode using 1.0-kV capillary voltage at a 150°C capillary temperature. The samples were all analyzed with a 1000 to 25,000 mass/charge ratio (m/z) range, the resolution set to 30,000, and a trapping gas pressure set to 3. Between 10 and 50 V of source fragmentation was applied to all samples to aid in desolvation. Data were deconvolved and analyzed with UniDec (42).
Enzymatic assays
The Mpro enzymatic assays were carried out exactly as previously described in pH 6.5 reaction buffer containing 20 mM Hepes (pH 6.5), 120 mM NaCl, 0.4 mM EDTA, 20% glycerol, and 4 mM DTT (15). The SARS-CoV-2 PLpro enzymatic assays were carried out as follows: The assay was assembled in 96-well plates with 100 μl of 200 nM PLPro protein in PLPro reaction buffer [50 mM Hepes (pH 7.5), 0.01% Triton X-100, and 5 mM DTT]. Then, 1 μl of testing compound at various concentrations was added to each well and incubated at 30°C for 30 min. The enzymatic reaction was initiated by adding 1 μl of 1 mM FRET substrate (the final substrate concentration is 10 μM). The reaction was monitored in a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30°C for 1 hour. The initial velocity of the enzymatic reaction with and without testing compounds was calculated by linear regression for the first 15 min of the kinetic progress curve. The IC50 values were calculated by plotting the initial velocity against various concentrations of testing compounds with a dose-response function in Prism 8 software.
The cathepsin L enzymatic assay was carried out as follows: Human liver cathepsin L (EMD Millipore 219402) was activated by incubating in reaction buffer [20 mM sodium acetate, 1 mM EDTA, and 5 mM DTT (pH 5.5)] for 30 min at 30°C. Upon activation, the assay was assembled in 96-well plates with 100 μl of 300 pM cathepsin L protein in cathepsin L reaction buffer. Then, 1 μl of testing compound at various concentrations was added to each well and incubated at 30°C for 30 min. The enzymatic reaction was initiated by adding 1 μl of 100 μM FRET substrate Z-Phe-Arg-AMC (the final substrate concentration is about 1 μM). The reaction was monitored in a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30°C for 1 hour. The IC50 values were calculated as described in the above section.
Differential scanning fluorimetry
The thermal shift binding assay was carried out using a Thermo Fisher Scientific QuantStudio 5 Real-Time Polymerase Chain Reaction System as described previously (15). Briefly, 3 μM SARS-CoV-2 Mpro protein in Mpro reaction buffer [20 mM Hepes (pH 6.5), 120 mM NaCl, 0.4 mM EDTA, 4 mM DTT, and 20% glycerol] was incubated with testing compounds at 30°C for 30 min. SYPRO orange dye (1×) was added, and fluorescence of the well was monitored under a temperature gradient range from 20° to 90°C with 0.05°C/s incremental step.
Cytotoxicity measurement
Evaluation of the cytotoxicity of compounds was carried out using the neutral red uptake assay (43). Briefly, 80,000 cells/ml of the tested cell lines were dispensed into 96-well cell culture plates at 100 μl per well. Twenty-four hours later, the growth medium was removed and washed with 150 μl of PBS buffer. Two hundred microliters of fresh serum-free medium containing serial diluted compounds was added to each well. After incubating for 5 days at 37°C, the medium was removed and replaced with 100 μl of DMEM medium containing neutral red (40 μg/ml) and incubated for 2 to 4 hours at 37°C. The amount of neutral red taken up was determined by measuring the absorbance at 540 nm using a Multiskan FC Microplate Photometer (Thermo Fisher Scientific). The CC50 values were calculated from best-fit dose-response curves with variable slope in Prism 8.
MD simulations
MD simulations were carried out to the covalently bound calpain inhibitor II, calpain inhibitor XII, UAWJ246, UAWJ247, and UAWJ248 with SARS-CoV-2 Mpro corresponding to PDB ID: 6XA4 (monomer), PDB ID: 6XFN (monomer), PDB ID: 6XBG (dimer), PDB ID: 6XBH (monomer), and PDB ID: 6XBI (dimer) prepared as described previously (44). The most favored protonation states of ionizable residues (D, E, R, K, and H) at pH 7 were assigned using Maestro. The protonation states of the histidines in the binding region were set to δ position to contribute to the stabilization of complexes. Crystal waters were kept. All hydrogens atoms of the protein complex were minimized with the OPLS2005 force field (45) by means of Maestro/Macromodel (Schrodinger 2017-1) using a distance-dependent dielectric constant of 4.0. The molecular mechanics minimization was performed with a conjugate gradient method, and a root mean square of the energy gradient (threshold) value of 0.005 kJ Å−1 mol−1 was used as the convergence criterion.
Each complex was solvated using the TIP3P (46) water model. Using the “System Builder” utility of Schrodinger Desmond v.11.1, each complex was embedded in an orthorhombic water box extending beyond the solute 15 Å in x,y plane and z direction. Na+ and Cl− ions were placed in the water phase to neutralize the systems and to reach the experimental salt concentration of 0.150 M NaCl. Each complex consists of ca 305 amino acid residues and 4653 atoms and ~18,600 water residues (55,700 water atoms) or ca ~60,350 atoms for the monomer proteins and ~29,700 water residues (89,200 water atoms) for the dimer proteins, i.e., 98,000 atoms.
The OPLS2005 force field (47, 48) was used to model all protein and ligand interactions and lipids. The particle mesh Ewald method (49, 50) was used to calculate long-range electrostatic interactions with a grid spacing of 0.8 Å. Van der Waals and short-range electrostatic interactions were smoothly truncated at 9.0 Å. The Nose-Hoover thermostat was used to maintain a constant temperature in all simulations, and the Martyna-Tobias-Klein barostat was used to control the pressure. Periodic boundary conditions were applied (68 × 95 × 97) Å3 for the monomers and (105 × 106 × 96) Å3 for the dimers. The equations of motion were integrated using the multistep reversible reference system propagator algorithms (RESPA) integrator (51) with an inner time step of 2 fs for bonded interactions and nonbonded interactions within a cutoff of 9 Å. An outer time step of 6.0 fs was used for nonbonded interactions beyond the cutoff.
Each system was equilibrated in MD simulations with a default protocol for water-soluble proteins provided in Desmond, which consists of a series of restrained MD simulations designed to relax the system, while not deviating substantially from the initial coordinates. The first simulation was a Brownian dynamics run for 100 ps at a temperature of 10 K in the NVT (constant number of particles, volume, and temperature) ensemble with solute heavy atoms restrained with a force constant of 50 kcal mol Å−2. The Langevin thermostat (52) was applied in the NVT ensemble and a MD simulation for 12 ps with solute heavy atoms restrained with a force constant of 50 kcal mol Å−2. The velocities were randomized, and MD simulation for 12 ps was performed in the NPT ensemble and a Berendsen barostat (53) with solute heavy atoms equally restrained at 10 K and another one at 300 K. The velocities were again randomized, and unrestrained MD simulation for 24 ps was performed in the NPT ensemble. The abovementioned equilibration was followed by 100-ns simulation without restrains. Two MD simulations for each system were performed, one in workstation with GTX 970, and using the GPU implementation of the MD simulations code, and one in ARIS supercomputer system with CPU cores. The visualization of produced trajectories and structures was performed using the programs Chimera (54) and VMD.
Immunofluorescence assay
Vero E6 cells in 96-well plates (Corning) were infected with SARS-CoV-2 (USA-WA1/2020 isolate) at a multiplicity of infection (MOI) of 0.05 in DMEM supplemented with 1% FBS. Immediately before the viral inoculation, the tested compounds in a threefold dilution concentration series were also added to the wells in triplicate. The infection proceeded for 48 hours without the removal of the viruses or the compounds. The cells were then fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with DMEM containing 10% FBS, and stained with a rabbit monoclonal antibody against SARS-CoV-2 nucleoprotein (NP) (GeneTex, GTX635679) and an Alexa Fluor 488–conjugated goat anti-mouse secondary antibody (Thermo Fisher Scientific). Hoechst 33342 was added in the final step to counterstain the nuclei. Fluorescence images of approximately 10,000 cells were acquired per well with a 10× objective in a Cytation 5 (BioTek). The total number of cells, as indicated by the nuclei staining, and the fraction of the infected cells, as indicated by the NP staining, were quantified with the cellular analysis module of the Gen5 software (BioTek).
Plaque assay
Vero E6 cells in six-well plates (Corning) were infected with SARS-CoV-2 (USA-WA1/2020 isolate) at approximately 40 plaque-forming units per well. After 1 hour of incubation at 37°C, the inoculum was removed and replaced with DMEM containing 1% FBS, 1.2% Avicel RC-591 (Dupont), and the tested compounds at different concentrations, in duplicate. After 3 days of infection, the overlay was removed, and the cells were fixed with 4% paraformaldehyde and stained with 0.2% crystal violet.
Mpro crystallization and structure determination
SARS-CoV-2 Mpro was diluted to 5 mg/ml and incubated with 1.5 mM inhibitor at 4°C overnight. Samples were centrifuged at 13,000g for 1 min to remove precipitate. Crystals were grown by mixing the protein-inhibitor sample with an equal volume of crystallization buffer [20% PEG 3000 (polyethylene glycol, molecular weight 3000), 0.2 M Na citrate (pH 5.6)] in a vapor diffusion, hanging drop apparatus. A cryoprotectant solution of 35% PEG 3000 and 30% glycerol was added directly to the drop and soaked for 15 min. Crystals were then flash-frozen in liquid nitrogen for x-ray diffraction.
X-ray diffraction data for the SARS-CoV-2 Mpro structures were collected on the Structural Biology Center 19-ID beamline at the Advanced Photon Source in Argonne, IL and processed with the HKL3000 software suite (55). The CCP4 version of MOLREP was used for molecular replacement using a previously solved SARS-CoV-2 Mpro structure, PDB ID: 7BRR as a reference model for the dimeric P21 Mpro with UAWJ246 (56). PDB ID: 6YB7 was used as the reference model for the C2 monomeric Mpro with calpain inhibitors II and XII and UAWJ247, and the P1 dimeric structure with UAWJ248. PDB ID: 6WTT was used as the reference model for the P3221 trimer with UAWJ246. Rigid and restrained refinements were performed using REFMAC, and model building was performed with COOT (57, 58). Protein structure figures were made using PyMOL (Schrödinger LLC).
Supplementary Material
Acknowledgments
We thank M. Kemp for assistance with crystallization and x-ray diffraction data collection. We also thank the staff members of the Advanced Photon Source of Argonne National Laboratory, particularly those at the Structural Biology Center (SBC), for x-ray diffraction data collection. SBC-CAT is operated by UChicago Argonne LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Funding: This research was partially supported by the NIH (grants AI147325 and AI157046) and the Arizona Biomedical Research Centre Young Investigator grant (ADHS18-198859) to J.W. J.A.T. and M.T.M. were funded by the National Institute of General Medical Sciences and NIH (grant R35 GM128624 to M.T.M.). Y.X. was funded by the NIH (grant AI151638). The SARS-CoV-2 experiments were supported by a COVID-19 pilot grant from UTHSCSA to Y.X. Author contributions: J.W. and Y.C. conceived and designed the study; C.M. expressed the Mpro and PLpro; C.M. performed the IC50 determination, thermal shift binding assay, and enzymatic kinetic studies; M.D.S. carried out Mpro crystallization and structure determination with the assistance of X.Z. and analyzed the data with Y.C.; P.L. performed the MD simulations under the guidance of A.K.; A.G. and N.K. synthesized the compounds for cocrystallization; Y.H. performed the cytotoxicity assay; X.M. and P.D. performed the SARS-CoV-2 immunofluorescence assay and plaque assay under the guidance of Y.X. B.H. and B.T. performed the initial antiviral assay with SARS-CoV-2; J.A.T. performed the native mass spectrometry experiments with the guidance from M.T.M.; J.W. and Y.C. secured funding and supervised the study; J.W., Y.C., and M.D.S. wrote the manuscript with input from others. Competing interests: C.M. and J.W. are inventors on a provisional patent application related to this work filed by the Arizona Board of Regents on behalf of the University of Arizona, J.W. and C.M., 8 April 2020, “Small Molecule Inhibitors of SARS-COV-2 Viral Replication and Uses Thereof,” U.S. provisional patent application, pending. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The drug-bound complex structures for SARS-CoV-2 Mpro have been deposited in the PDB with accession numbers 6XA4 (SARS-CoV-2 HMPro + calpain inhibitor II), 6XFN (SARS-CoV-2 HMPro + calpain inhibitor XII), 6XBG (SARS-CoV-2 HMPro + UAWJ246), 6XBH (SARS-CoV-2 HMPro + UAWJ247), and 6XBI (SARS-CoV-2 HMPro + UAWJ248). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/sciadv.abe0751/DC1
REFERENCES AND NOTES
- 1.Liu C., Zhou Q., Li Y., Garner L. V., Watkins S. P., Carter L. J., Smoot J., Gregg A. C., Daniels A. D., Jervey S., Albaiu D., Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 6, 315–331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pillaiyar T., Meenakshisundaram S., Manickam M., Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 25, 668–688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30, 269–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eastman R. T., Roth J. S., Brimacombe K. R., Simeonov A., Shen M., Patnaik S., Hall M. D., Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 6, 672–683 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheahan T. P., Sims A. C., Graham R. L., Menachery V. D., Gralinski L. E., Case J. B., Leist S. R., Pyrc K., Feng J. Y., Trantcheva I., Bannister R., Park Y., Babusis D., Clarke M. O., Mackman R. L., Spahn J. E., Palmiotti C. A., Siegel D., Ray A. S., Cihlar T., Jordan R., Denison M. R., Baric R. S., Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 9, eaal3653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Wit E., Feldmann F., Cronin J., Jordan R., Okumura A., Thomas T., Scott D., Cihlar T., Feldmann H., Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. U.S.A. 117, 6771–6776 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheahan T. P., Sims A. C., Leist S. R., Schäfer A., Won J., Brown A. J., Montgomery S. A., Hogg A., Babusis D., Clarke M. O., Spahn J. E., Bauer L., Sellers S., Porter D., Feng J. Y., Cihlar T., Jordan R., Denison M. R., Baric R. S., Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 11, 222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Anirudhan V., Du R., Cui Q., Rong L., RNA-dependent RNA polymerase of SARS-CoV-2 as a therapeutic target. J. Med. Virol. 10.1002/jmv.26264 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Sheahan T. P., Sims A. C., Zhou S., Graham R. L., Pruijssers A. J., Agostini M. L., Leist S. R., Schäfer A., Dinnon K. H. III, Stevens L. J., Chappell J. D., Lu X., Hughes T. M., George A. S., Hill C. S., Montgomery S. A., Brown A. J., Bluemling G. R., Natchus M. G., Saindane M., Kolykhalov A. A., Painter G., Harcourt J., Tamin A., Thornburg N. J., Swanstrom R., Denison M. R., Baric R. S., An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 12, eabb5883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L., Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia S., Yan L., Xu W., Agrawal A. S., Algaissi A., Tseng C.-T. K., Wang Q., Du L., Tan W., Wilson I. A., Jiang S., Yang B., Lu L., A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 5, eaav4580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H., An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 59, 6595–6628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Hilgenfeld R., Drag M., Substrate specificity profiling of SARS-CoV-2 Mpro protease provides basis for anti-COVID-19 drug design. bioRxiv 2020.03.07.981928 (2020). [Google Scholar]
- 14.Ullrich S., Nitsche C., The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 30, 127377 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma C., Sacco M. D., Hurst B., Townsend J. A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M. T., Chen Y., Wang J., Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L. W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H., Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.-K., Xu Y., Yang H., Liu H., Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki T., Kishi M., Saito M., Tanaka T., Higuchi N., Kominami E., Katunuma N., Murachi T., Inhibitory effect of di- and tripeptidyl aldehydes on calpains and cathepsins. J. Enzyme Inhib. 3, 195–201 (1990). [DOI] [PubMed] [Google Scholar]
- 20.Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F., Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 117, 11727–11734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T., Luo S., Libby P., Shi G.-P., Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 213, 107587 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dana D., Pathak S. K., A review of small molecule inhibitors and functional probes of human cathepsin L. Molecules 25, 698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choe Y., Leonetti F., Greenbaum D. C., Lecaille F., Bogyo M., Brömme D., Ellman J. A., Craik C. S., Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 281, 12824–12832 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z., Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 11, 1620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia B., Kang X., Activation and maturation of SARS-CoV main protease. Protein Cell 2, 282–290 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye G., Wang X., Tong X., Shi Y., Fu Z. F., Peng G., Structural basis for inhibiting porcine epidemic diarrhea virus replication with the 3C-like protease inhibitor GC376. Viruses 12, 240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vuong W., Khan M. B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H. A., McKay R. T., van Belkum M. J., Joyce M. A., Young H. S., Tyrrell D. L., Vederas J. C., Lemieux M. J., Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 11, 4282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verschueren K. H. G., Pumpor K., Anemüller S., Chen S., Mesters J. R., Hilgenfeld R., A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 15, 597–606 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou C.-Y., Chang H.-C., Hsu W.-C., Lin T.-Z., Lin C.-H., Chang G.-G., Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry 43, 14958–14970 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Chuck C.-P., Chong L.-T., Chen C., Chow H.-F., Wan D. C.-C., Wong K.-B., Profiling of substrate specificity of SARS-CoV 3CLpro. PLOS ONE 5, e13197 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muramatsu T., Takemoto C., Kim Y.-T., Wang H., Nishii W., Terada T., Shirouzu M., Yokoyama S., SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. U.S.A. 113, 12997–13002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang S., Chen S.-J., Hsu M.-F., Wu J.-D., Tseng C.-T. K., Liu Y.-F., Chen H.-C., Kuo C.-W., Wu C.-S., Chang L.-W., Chen W.-C., Liao S.-Y., Chang T.-Y., Hung H.-H., Shr H.-L., Liu C.-Y., Huang Y.-A., Chang L.-Y., Hsu J.-C., Peters C. J., Wang A. H.-J., Hsu M.-C., Synthesis, crystal structure, structure–activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 49, 4971–4980 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Shie J.-J., Fang J.-M., Kuo T.-H., Kuo C.-J., Liang P.-H., Huang H.-J., Wu Y.-T., Jan J.-T., Cheng Y.-S. E., Wong C.-H., Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α,β-unsaturated esters. Bioorg. Med. Chem. 13, 5240–5252 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., Snijder E. J., Liu H., Hilgenfeld R., α-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 63, 4562–4578 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Zhu L., George S., Schmidt M. F., Al-Gharabli S. I., Rademann J., Hilgenfeld R., Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res. 92, 204–212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galasiti Kankanamalage A. C., Kim Y., Damalanka V. C., Rathnayake A. D., Fehr A. R., Mehzabeen N., Battaile K. P., Lovell S., Lushington G. H., Perlman S., Chang K.-O., Groutas W. C., Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 150, 334–346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agostini M. L., Andres E. L., Sims A. C., Graham R. L., Sheahan T. P., Lu X., Smith E. C., Case J. B., Feng J. Y., Jordan R., Ray A. S., Cihlar T., Siegel D., Mackman R. L., Clarke M. O., Baric R. S., Denison M. R., Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 9, e00221-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pedersen N. C., Kim Y., Liu H., Galasiti Kankanamalage A. C., Eckstrand C., Groutas W. C., Bannasch M., Meadows J. M., Chang K.-O., Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 20, 378–392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim Y., Liu H., Galasiti Kankanamalage A. C., Weerasekara S., Hua D. H., Groutas W. C., Chang K.-O., Pedersen N. C., Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLOS Pathog. 12, e1005531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cady S. D., Wang J., Wu Y., DeGrado W. F., Hong M., Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J. Am. Chem. Soc. 133, 4274–4284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marty M. T., Baldwin A. J., Marklund E. G., Hochberg G. K. A., Benesch J. L., Robinson C. V., Bayesian deconvolution of mass and ion mobility spectra: From binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Repetto G., del Peso A., Zurita J. L., Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 3, 1125–1131 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Musharrafieh R., Lagarias P., Ma C., Hau R., Romano A., Lambrinidis G., Kolocouris A., Wang J., Investigation of the drug resistance mechanism of M2-S31N channel blockers through biomolecular simulations and viral passage experiments. ACS Pharmacol. Transl. Sci. 3, 666–675 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaminski G. A., Friesner R. A., Tirado-Rives J., Jorgensen W. L., Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487 (2001). [Google Scholar]
- 46.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 47.Jorgensen W. L., Maxwell D. S., Tirado-Rives J., Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996). [Google Scholar]
- 48.Shivakumar D., Williams J., Wu Y. J., Damm W., Shelley J., Sherman W., Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 6, 1509–1519 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Darden T., York D., Pedersen L., Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- 50.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G., A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 51.Humphreys D. D., Friesner R. A., Berne B. J., A multiple-time-step molecular dynamics algorithm for macromolecules. J. Chem. Phys. 98, 6885–6892 (1994). [Google Scholar]
- 52.Izaguirre J. A., Catarello D. P., Wozniak J. M., Skeel R. D., Langevin stabilization of molecular dynamics. J. Chem. Phys. 114, 2090–2098 (2001). [Google Scholar]
- 53.Feller S. E., Zhang Y., Pastor R. W., Brooks B. R., Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 103, 4613–4621 (1995). [Google Scholar]
- 54.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Minor W., Cymborowski M., Otwinowski Z., Chruszcz M., HKL-3000: The integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62 ( Pt. 8), 859–866 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Vagin A., Teplyakov A., Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66 ( Pt. 1), 22–25 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67 ( Pt. 4), 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60 ( Pt. 12 Pt. 1), 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Mandadapu S. R., Gunnam M. R., Tiew K.-C., Uy R. A., Prior A. M., Alliston K. R., Hua D. H., Kim Y., Chang K.-O., Groutas W. C., Inhibition of norovirus 3CL protease by bisulfite adducts of transition state inhibitors. Bioorg. Med. Chem. Lett. 23, 62–65 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/sciadv.abe0751/DC1