

The molecular scissors Mus81 fixes or ruins chromosomes depending on the availability of DNA precursors in mitosis.
Abstract
Chromosome instability (CIN) underpins cancer evolution and is associated with drug resistance and poor prognosis. Understanding the mechanistic basis of CIN is thus a priority. The structure-specific endonuclease Mus81-Eme1 is known to prevent CIN. Intriguingly, however, here we show that the aberrant processing of late replication intermediates by Mus81-Eme1 is a source of CIN. Upon depletion of checkpoint kinase 1 (Chk1), Mus81-Eme1 cleaves under-replicated DNA engaged in mitotic DNA synthesis, leading to chromosome segregation defects. Supplementing cells with nucleosides allows the completion of mitotic DNA synthesis, restraining Mus81-Eme1–dependent DNA damage in mitosis and the ensuing CIN. We found no correlation between CIN arising from nucleotide shortage in mitosis and cell death, which were selectively linked to DNA damage load in mitosis and S phase, respectively. Our findings imply the possibility of optimizing Chk1-directed therapies by inducing cell death while curtailing CIN, a common side effect of chemotherapy.
INTRODUCTION
The DNA damage response (DDR), a complex network of interdependent signaling pathways activated upon DNA insults, assists the completion and fidelity of DNA replication. DDR defects are common across multiple cancers. Conventional anticancer therapy exploits this vulnerability by the use of chemicals or radiation that inflicts direct damage to the DNA. Along the same principle, DDR inhibitors have been introduced in clinical practice and have recently revolutionized the therapeutic landscape of cancer (1). One drawback of this strategy is that high levels of DNA damage and/or an inefficient DDR induce chromosome instability (CIN). CIN collectively refers to changes in chromosome number and structure, which can result from chromosome mis-segregation (2). The genetic diversity created by CIN provides tumor cells with growth advantages. Thus, contemporary anticancer therapy is a potential driver of malignancy. CIN is associated with poor prognosis and cancer relapse and correlates with resistance to antineoplastic treatments, both in tumor-derived cell lines and in clinical settings (2, 3). Optimizing therapies to suppress tumor growth while minimizing CIN is essential to addressing this clinical issue.
Checkpoint kinase 1 (Chk1) is a key mediator of the DDR that delays S phase progression, stabilizes replication forks, and promotes DNA repair upon replication stress (4). Chk1 inhibitors (Chk1i) are undergoing clinical evaluation in monotherapy or combination regimens (1). The current model prescribes that Chk1i unleash origin firing, slow down forks, and cause double-strand breaks (DSBs), perturbing the replication choreography and culminating in genomic instability and cell death (4–6). The link between altered replication dynamics, DSBs, and cell death has been unequivocally proven in Chk1-deficient cells (7–11). In particular, robust evidence has shown that Mus81-Eme2–dependent cleavage of stalled forks in S phase compromises cell survival upon Chk1 loss (8). Although surpassing a certain threshold of genomic instability is incompatible with cell survival (12), no unambiguous relationship has been established between genomic instability and cell death in Chk1-deficient cells (6).
The literature provides only scarce and isolated information on the contribution of Chk1 to genomic stability. A few reports have shown that Chk1 deficiency in cancer cells leads to the accumulation of CIN markers such as anaphase bridges, lagging chromosomes, and ultrafine bridges (UFBs) (13–15). These phenotypes are often the manifestation of under-replicated DNA (UR-DNA) being passed onto mitosis (16–19). DNA under-replication leads to nascent DNA synthesis in early mitosis, i.e., mitotic DNA synthesis (MiDAS). The inactivation of various DDR effectors induces UR-DNA and MiDAS (20–24). However, it remains unknown whether Chk1 loss induces UR-DNA and MiDAS and whether mitotic events prevent or promote CIN in Chk1-deficient cells.
MiDAS operates at common fragile sites and telomeres upon treatment with the DNA polymerase inhibitor aphidicolin (APH) (25). APH-induced MiDAS takes place in prophase to complete DNA duplication and thereby promote the proper segregation of sister chromatids in anaphase (22, 24, 26, 27). However, MiDAS is not necessarily restricted to origin-poor, late-replicating regions (28). Besides, MiDAS might not always fully complete DNA replication; instead, MiDAS might constitute the first step in the resolution of UR-DNA, which would ultimately take place during the next S phase within structures shielded by 53BP1 (16, 29). Our knowledge on the mechanistic details underlying MiDAS is similarly scarce and entirely restricted to studies with APH. APH-induced MiDAS is apparently not about the continuation of conventional, semiconservative replication that takes place in S phase (25). Instead, APH-induced MiDAS might represent a form of break-induced replication (BIR), a recombination-based pathway that repairs one-ended breaks in yeasts (24–26). During APH-induced MiDAS, the break that precedes replication is formed by a structure-specific endonuclease, Mus81-Eme1, or other nuclease functioning in complex with the SLX4 scaffold (22, 26). Together, much work still needs to be done to unveil the molecular details underlying MiDAS, its contribution to chromosome segregation, and its implications for cancer therapy.
Here, we demonstrate that Chk1 knockdown triggers the accumulation of UR-DNA, MiDAS intermediates, and aberrant anaphases. In contrast to APH-induced MiDAS, MiDAS in Chk1-depleted cells is not initiated by Mus81-Eme1–dependent cleavage of UR-DNA. Instead, Mus81-Eme1 functions downstream of MiDAS initiation in Chk1-depleted cells—it cleaves nascent DNA synthetized during mitosis. Such function of Mus81-Eme1 is activated because the nucleotide pool is insufficient to sustain MiDAS. This unprecedented role of Mus81-Eme1 propels CIN but does not compromise cell survival. Instead, cell death upon Chk1 inactivation is entirely dependent on Mus81-Eme2–mediated DSBs in S phase. Our data imply that treating cancer with Chk1i could unnecessarily lead to an enrichment of a chromosomally unstable subpopulation. We believe that our findings should be taken into consideration when designing Chk1-directed therapies.
RESULTS
Chk1 loss triggers replication catastrophe and chromosome mis-segregation by independent pathways
We have recently identified excess chromatin binding of the helicase cofactor CDC45 as the cause of increased origin firing and asymmetric fork slowdown in Chk1-deficient cells (9). Partial depletion of CDC45 does not interfere with cell proliferation (9, 30, 31), but it does nullify the massive accumulation of DNA damage and DSBs in Chk1-depleted U2OS cells (Fig. 1, A to C). These results are in good agreement with reports that have also partially inactivated CDC45 in Chk1-inhibited or Chk1-depleted cells (30–32). Hence, surplus CDC45 in Chk1-defective cells results in replication catastrophe, manifested as pan-nuclear γH2AX and a large number of DSBs per cell.
Fig. 1. Chk1 loss triggers replication catastrophe and chromosome mis-segregation by independent pathways.
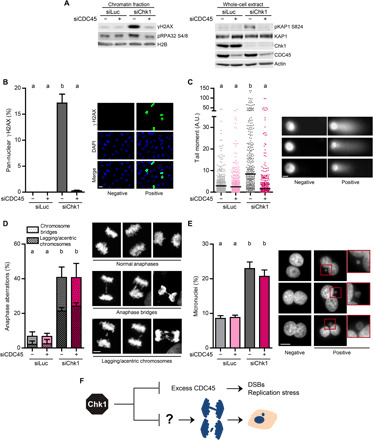
(A) Western blot of γH2AX, phospho-RPA32 Ser4/8, phospho-KAP1 Ser824, KAP1, Chk1, and CDC45 in U2OS cells, 48 hours after transfection. The left panel shows the chromatin fraction, obtained after an extraction with CSK buffer; the right panel shows whole-cell extracts. H2B and actin were used as loading controls. (B) Percentage (mean ± SD) and representative images of U2OS cells with pan-nuclear γH2AX staining. More than 2000 cells per sample were analyzed in three independent experiments. Scale bar, 20 μm. As in all graphs in the manuscript, different letters indicate significant differences (see Materials and Methods). (C) Quantification by neutral comet assay of DSB accumulation in U2OS cells (A.U., arbitrary units). The right panel shows representative images of DNA comets. Three hundred cells per sample were analyzed in three independent experiments. The bars on top of the distribution clouds indicate the median. Scale bar, 10 μm. (D) Percentage (mean ± SD) and representative Z-stack images of U2OS anaphase cells with aberrations. About 150 anaphases per sample were analyzed in three independent experiments. The total percentage of aberrant anaphases (bridges plus lagging chromosomes) was used to calculate the statistics. Scale bar, 5 μm. (E) Percentage (mean ± SD) and representative images of binucleated U2OS cells with micronuclei. About 750 binucleated cells per sample were analyzed in three independent experiments. Scale bar, 10 μm. (F) Excess CDC45 in Chk1-deficient cells provokes DSBs and acute replication stress but does not cause chromosome mis-segregation.
Replication stress often results in chromosome mis-segregation (6, 16, 23, 33). We then asked whether excess origin firing and fork slowdown result in chromosome mis-segregation in Chk1-deficient cells [phenotypes in S and M phases were determined 48 and 72 hours after transfection with small interfering RNA (siRNA), respectively; fig. S1A]. To this end, we measured the percentage of cells with anaphase aberrations and micronuclei after concomitant down-regulation of Chk1 and CDC45. Chk1-defective U2OS cells showed a steep increase in anaphase aberrations and micronuclei (Fig. 1, D and E, and fig. S1, B and C). However, CDC45 down-regulation failed to revert the chromosome segregation errors provoked by Chk1 loss (Fig. 1, D and E). This independency between altered replication dynamics and micronuclei accumulation was also observed in HCT116 and PANC-1 cells (fig. S2, A and B), whose replication speed was also fine-tuned by CDC45 expression levels (fig. S2, C and D). Thus, upon Chk1 loss, the replication catastrophe that follows origin usage and fork elongation defects is dissociated from chromosome mis-segregation (Fig. 1F).
Mus81 triggers CIN in Chk1-deficient cells
We next sought to identify the molecular triggers of chromosome mis-segregation in Chk1-depleted cells. Upon Chk1 loss, the unscheduled activation of the structure-specific endonuclease Mus81 leads to the accumulation of pan-nuclear γH2AX and DSBs in S phase (fig. S3, A to C) (7, 8, 10). Because chromosome segregation errors arise independently of the replication catastrophe elicited by Chk1 loss (Fig. 1, A to F), we predicted that Mus81 would not contribute to chromosome mis-segregation. Unexpectedly, however, increased rates of anaphase aberrations and micronuclei in Chk1-deficient cells did depend on Mus81 (fig. S3, D and E). In HCT116 and PANC-1 cells, Mus81 down-regulation also prevented the occurrence of micronuclei provoked by Chk1 inactivation (fig. S4, A and B). While these data indicate that Mus81-dependent DSBs precede the chromosome segregation errors elicited by Chk1 depletion, neutral comet assays did not reveal an association between DSB formation and chromosome mis-segregation (Fig. 1, A to F). This apparent contradiction might be explained by the fact that only cells with more than 50 DSBs generate a tail that can be detected by the neutral comet assay (34). Thus, after Chk1 depletion, chromosome mis-segregation might take place in cells that accumulate just a few Mus81-dependent DSBs.
Mus81-Eme1–dependent DSBs in mitosis trigger CIN in Chk1-deficient cells
Mus81 is the catalytic subunit of two human structure-selective endonucleases, Mus81-Eme1 and Mus81-Eme2 (35, 36). Unrestrained Mus81-Eme2–dependent cleavage of S phase replication intermediates leads to pan-nuclear γH2AX accumulation in cells deficient in Chk1 or WEE1, another DDR protein (37, 38). Notwithstanding this, our data suggest that Mus81 operates in an additional pathway upon Chk1 loss; this pathway is independent of quantifiable fork stalling in S phase and leads to chromosome mis-segregation. We hypothesized that the two functions of Mus81 in Chk1-depleted cells require either Eme1 or Eme2. Pan-nuclear γH2AX accumulation and DSBs, as measured by neutral comet assay, depended on Eme2 but not on Eme1 (Fig. 2, A to C, and fig. S5, A and B). In sharp contrast, the increase in anaphase aberrations and micronuclei depended on Eme1 but not on Eme2 (Fig. 2, D and E, and fig. S5C).
Fig. 2. Mus81-Eme1 triggers CIN in Chk1-deficient cells.
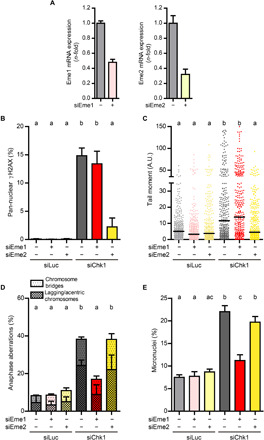
(A) Quantitative real-time PCR of Eme1 and Eme2 normalized to GAPDH in U2OS cells, 48 hours after transfection; error bars represent the SD of two technical replicates. (B) Percentage of U2OS cells with pan-nuclear γH2AX staining (mean ± SD). More than 1600 cells per sample were analyzed in two independent experiments. (C) Quantification by neutral comet assay of DSB accumulation in U2OS cells. Three hundred cells per sample were analyzed in three independent experiments. The bars on top of the distribution clouds indicate the median. (D) Percentage of U2OS anaphase cells with aberrations (mean ± SD). About 100 anaphases per sample were analyzed in two independent experiments. The total percentage of aberrant anaphases (bridges plus lagging chromosomes) was used to calculate the statistics. (E) Percentage of binucleated U2OS cells with micronuclei (mean ± SD). About 600 binucleated cells per sample were analyzed in three independent experiments.
Mus81-Eme2 and Mus81-Eme1 might function preferentially in S and M phases, respectively (39). The fact that Mus81-Eme1 leads to chromosome mis-segregation in Chk1-depleted cells points to mitotic DSBs as a prelude to such kind of CIN. Mitotic DSBs can be visualized directly as gaps/breaks in condensed, Giemsa-stained metaphase chromosomes (18, 19) or indirectly as γH2AX foci (40). In agreement with our hypothesis, Chk1 depletion increased the incidence of chromosomal gaps/breaks (Fig. 3A) and mitotic γH2AX foci (Fig. 3B). Moreover, the induction of mitotic DSBs depended on Mus81 and Eme1 but not on CDC45 or Eme2 (Fig. 3, A to C). So, contrary to the Mus81-Eme2–dependent, extensive chromosome pulverization observed in WEE1-inhibited cells (38), mitotic DNA damage in Chk1-depleted cells depends on Mus81-Eme1 and manifests as discrete DSBs. Together, our results provide strong evidence that Mus81-Eme1–dependent DSBs in mitosis conduce to chromosome mis-segregation, whereas Mus81-Eme2–dependent DSBs in S phase do not (Fig. 3D).
Fig. 3. Mus81-Eme1 triggers mitotic DSBs in Chk1-deficient cells.
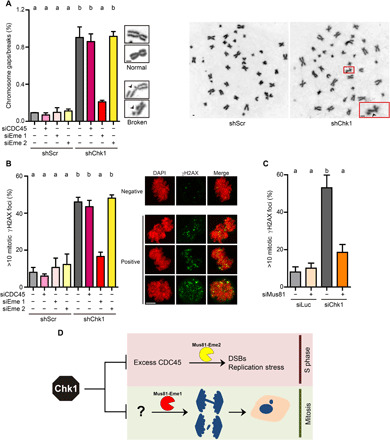
(A) Percentage of metaphase chromosomes with breaks/gaps (mean ± SD) and representative images of intact or broken chromosomes and whole metaphase spreads. HCT116 cells were transfected with the indicated siRNAs and transduced 5 hours later with nontargeting shRNA [shScramble (shScr)] or shRNA targeting Chk1 (shChk1). About 4500 chromosomes (from 100 metaphases) per sample were analyzed in two independent experiments. Scale bars, 1 μm. (B) Percentage (mean ± SD) and representative Z-stack images of mitotic U2OS cells with >10 γH2AX foci. About 150 metaphases per sample were analyzed in three independent experiments. Scale bar, 5 μm. (C) Percentage of mitotic U2OS cells with >10 γH2AX foci (mean ± SD). About 120 metaphases per sample were analyzed in three independent experiments. (D) Model in which Mus81-Eme1–dependent DSBs in mitosis trigger CIN in Chk1-deficient cells, independently of Mus81-Eme2–dependent DSBs in S phase.
MiDAS precedes Mus81-Eme1–dependent cleavage and CIN in Chk1-deficient cells
Mus81-Eme1–dependent DSBs in mitosis are well-known protectors of genome integrity (18, 19, 26). Our data indicate that Mus81-Eme1–dependent DSBs in mitosis are triggers of CIN as well; we then sought to understand the molecular basis of such an unprecedented role of Mus81-Eme1. Mus81-Eme1 cleaves DNA replication intermediates (35), and DNA synthesis may take place during early mitosis (21, 26). We thus hypothesized that late replication intermediates constitute the substrates for Mus81-Eme1–dependent cleavage. Chk1 depletion increased the percentage of cells that incorporated the nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU) in mitosis (Fig. 4A); EdU foci colocalized with FANCD2 (Fig. 4A), a marker of DNA synthesis in mitosis (21, 26). siChk1-induced MiDAS took place independently of Mus81 (fig. S6A), in sharp contrast with APH-induced MiDAS, which requires Mus81 [fig. S6B and (24, 26, 27)]. The depletion of the Mus81 scaffold SLX4, which phenocopies the depletion of Mus81, had also no effect on siChk1-induced MiDAS (fig. S6, C to F). These results reinforce the notion that Mus81-Eme1 acts downstream of MiDAS in Chk1-deficient cells.
Fig. 4. DNA synthesis in mitosis precedes Mus81-Eme1–dependent cleavage and CIN in Chk1-deficient cells.
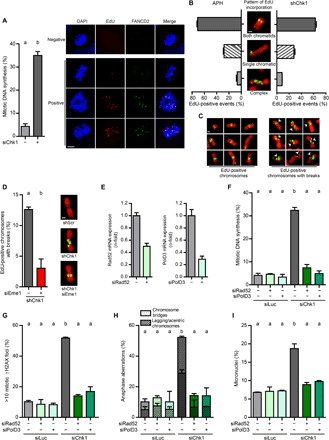
(A) Percentage (mean ± SD) and representative Z-stack images of mitotic U2OS cells with EdU/FANCD2 spots. About 100 metaphases per sample were analyzed in two independent experiments. Scale bar, 5 μm. (B) Percentage (mean ± SD) and representative images of HCT116 chromosomes (DAPI, red) with semiconservative, conservative, or complex patterns of EdU incorporation (green). APH (0.2 μM) was added as a control, 24 hours before EdU incorporation. Only shChk1-transduced and APH-treated samples are shown, because shScr-transduced and DMSO-treated samples did not exhibit EdU incorporation. Four hundred EdU-positive events per sample were analyzed in two independent experiments. Scale bar, 1 μm. (C) DAPI-negative breaks (white arrows) at sites of EdU incorporation in metaphase chromosomes from shChk1-transduced HCT116 cells. No breaks were detected in EdU-negative DNA. Scale bar, 1 μm. (D) Percentage of EdU-positive events with breaks (mean ± SD) in HCT116 metaphase chromosomes. Four hundred EdU-positive events per sample were analyzed in two independent experiments. The representative images depict Eme1-dependent chromosome breakage at sites of shChk1-induced EdU incorporation. Scale bar, 1 μm. (E) Quantitative real-time PCR of PolD3 and Rad52 normalized to GAPDH in U2OS cells, 48 hours after transfection; error bars represent the SD of two technical replicates. (F) Percentage of mitotic U2OS cells with EdU spots (mean ± SD). About 120 metaphases per sample were analyzed in three independent experiments. (G) Percentage of mitotic U2OS cells with >10 γH2AX foci (mean ± SD). About 100 metaphases per sample were analyzed in three independent experiments. (H) Percentage of U2OS anaphase cells with aberrations (mean ± SD). About 100 anaphases per sample were analyzed in two independent experiments. Total percentage of aberrant anaphases was used to perform the statistics. (I) Percentage of binucleated U2OS cells with micronuclei (mean ± SD). About 400 binucleated cells per sample were analyzed in two independent experiments.
To directly test this hypothesis, we visualized the pattern of EdU incorporation on DAPI (4′,6-diamidino-2-phenylindole)–stained metaphase spreads, which allows simultaneous detection of DNA synthesis in early mitosis and Mus81-dependent DSBs, visible as DAPI-negative gaps/breaks (26). EdU incorporation was only detectable in Chk1-depleted cells and not in control samples. The vast majority of metaphase chromosomes incorporating EdU did so in a pattern consistent with semiconservative replication (Fig. 4B, “both chromatids”). This might abrogate the need for a break before DNA synthesis; these results are thus in agreement with our observation that MiDAS in Chk1-defective cells is SLX4/Mus81-independent (fig. S6, A and D). EdU events were frequently detected at sites where no gap/break was visible, but all gaps/breaks coincided with sites of newly synthesized DNA tracks (representative images in Fig. 4C). The frequency of EdU-localized gaps/breaks was steeply reduced after Eme1 depletion (Fig. 4D). We propose that Chk1 deficiency prompts Mus81-Eme1–dependent cleavage at sites of semiconservative DNA synthesis in early mitosis.
To further assess the notion that MiDAS is a prelude to Mus81-Eme1–induced DNA damage, we abolished DNA synthesis in mitotic Chk1-deficient cells by depleting the Polδ subunit PolD3 and the repair protein Rad52, two factors involved in MiDAS (Fig. 4, E and F) (24, 26). When MiDAS was prevented, Mus81-Eme1–dependent accumulation of DNA damage in mitotic cells (Fig. 4G), anaphase aberrations (Fig. 4H), and micronuclei (Fig. 4I) was not observed. Replication fork slowdown and pan-nuclear γH2AX induced by Chk1 loss remained unaltered upon PolD3 or Rad52 depletion, ruling out any link between these S phase phenotypes and the damage observed in mitosis (fig. S7, A and B). In summary, Chk1 depletion induces PolD3- and Rad52-dependent MiDAS, whose intermediates are cleaved by Mus81-Eme1, ultimately causing CIN.
Nucleotide deficiency during MiDAS leads to CIN in Chk1-deficient cells
MiDAS provides a mechanism to completing DNA duplication beyond the S phase and hence safeguards chromosomal stability (21, 25, 26). In apparent contrast, our data show that MiDAS jeopardizes chromosome stability in a Chk1-deficient background. In cells deficient in the DDR components Chk1 and WEE1, the availability of nucleotides is restricted (8, 9, 38, 41, 42). In WEE1-inhibited, but not in Chk1-inhibited, cells, nucleotide scarcity triggers replication catastrophe and chromosome pulverization (8, 9, 37, 38). How nucleotide scarcity affects DNA duplication in a Chk1-deficient background remains unknown. We reasoned that if the nucleotide pool shortens, MiDAS might become suboptimal, generating replication intermediates that are prone to cleavage by Mus81-Eme1.
In agreement with our hypothesis, the percentage of Chk1-deficient cells showing MiDAS increased upon supplementation with nucleosides (Fig. 5A). These results suggest that, in mitotic Chk1-deficient cells, DNA is synthesized in a scenario of limited nucleotides, raising the intriguing possibility that such “limited” MiDAS leads to CIN. Nucleoside supplementation precluded the accumulation of mitotic γH2AX foci (Fig. 5B), anaphase aberrations (Fig. 5C), and micronuclei (Fig. 5D). Thus, if supplemented with nucleosides, MiDAS in Chk1-depleted cells resembles MiDAS in APH-treated cells in terms of its ability to prevent chromosome mis-segregation, but not in terms of the molecular event that initiates MiDAS—MiDAS in Chk1-depleted cells remains independent of Mus81, even if extra nucleosides are supplied (fig. S8A). We propose that, instead of fostering a mechanistic switch to a pathological type of MiDAS, nucleotide scarcity blocks the progression of replication forks in mitotic Chk1-deficient cells, thereby provoking Mus81-Eme1–dependent DSBs, which, in turn, generate CIN.
Fig. 5. Nucleotide deficiency during MiDAS leads to CIN in Chk1-deficient cells.
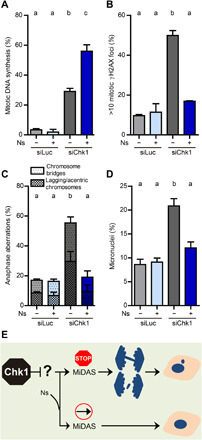
(A) Percentage of mitotic U2OS cells with EdU spots (mean ± SD). Nucleosides (Ns) were added 24 hours before fixation. About 120 metaphases per sample were analyzed in three independent experiments. (B) Percentage of mitotic U2OS cells with >10 γH2AX foci (mean ± SD). Cells were treated as in (A). About 100 metaphases per sample were analyzed in three independent experiments. (C) Percentage of U2OS anaphase cells with aberrations (mean ± SD). Cells were treated as in (A). About 100 anaphases per sample were analyzed in two independent experiments. The total percentage of aberrant anaphases (bridges plus lagging chromosomes) was used to calculate the statistics. (D) Percentage of binucleated U2OS cells with micronuclei (mean ± SD). Cells were treated as in (A). About 400 binucleated cells per sample were analyzed in two independent experiments. (E) Model in which limited nucleotide availability restrains the completion of DNA synthesis in mitosis and propels CIN.
To strengthen the link between nucleotide starvation during MiDAS and CIN, we mimicked Chk1 depletion by combining APH and hydroxyurea (HU), which inhibits nucleotide biosynthesis. In a context of limited nucleotides (HU), APH-induced MiDAS was no longer dependent on Mus81 (fig. S8B) and correlated with Mus81-induced chromosome segregation defects (fig. S8C). Thus, although MiDAS is generally regarded as a process that safeguards genomic stability, if incomplete, MiDAS may propel genomic instability (Fig. 5E).
Nucleotide deficiency during MiDAS leads to DNA under-replication in Chk1-deficient cells
Our data indicate that MiDAS is limited by low nucleotide availability in Chk1-deficient cells. Given the originality of this concept, we sought to explore it further. We determined the frequency of UFBs in anaphase and 53BP1-nuclear bodies (53BP1-NBs) in G1. UFBs and 53BP1-NBs reveal UR-DNA that escapes MiDAS and persists until anaphase and the next G1, respectively (16, 29, 43–46). In agreement with our data in Fig. 4A, Chk1 depletion induced UFBs and 53BP1-NBs, and the induction of both was prevented by supplementation with nucleosides (fig. S9, A and B). We conclude that the delivery of extra DNA precursors counteracts UR-DNA, probably by facilitating MiDAS.
We next depleted Rad52 to evaluate the effect of impeding MiDAS on the accumulation of UR-DNA. Rad52 depletion and nucleoside supplementation similarly attenuate chromosome segregation defects in Chk1-deficient cells (Figs. 4, H and I, and 5, C and D). However, because Rad52 depletion impedes, rather than exacerbates, MiDAS, we did not expect that it resolved UR-DNA, like extra DNA precursor supply did (fig. S9, A and B). Rad52 depletion did not prevent the accumulation of 53BP1-NBs and even augmented the frequency of UFBs in Chk1-depleted cells (fig. S9, A and B). Together, our results indicate that although MiDAS is required, it is insufficient to prevent the inheritance of DNA lesions by daughter cells. An adequate nucleotide supply is also needed, not to initiate MiDAS but to guarantee its completion and thereby avoid the aberrant activity of Mus81-Eme1.
CIN arising from nucleotide shortage during mitosis does not compromise survival of Chk1-deficient cells
We have described a molecular pathway leading to CIN in Chk1-depleted cells. Our data also show a dissection between chromosome mis-segregation and replication catastrophe. Replication catastrophe, which is characterized by pan-nuclear accumulation of γH2AX and single-stranded DNA, leads to cell death in S phase (7–9, 11, 30, 38, 41). So, we reasoned that chromosome mis-segregation might not precede cell death in Chk1-deficient cells. To directly test this hypothesis, we evaluated the impact of reverting the chromosome segregation defects stemming from the aberrant processing of late replication intermediates on cell death. Preventing the accumulation of micronuclei and anaphase aberrations by Rad52 depletion or exogenous nucleoside supply did not improve the survival of Chk1-deficient cells (Fig. 6A). Moreover, depletion of CDC45, which does not prevent chromosome mis-segregation, was sufficient to totally revert cell death upon Chk1 loss (Fig. 6B). In this regard, chromosome segregation errors correlate with the accumulation of γH2AX foci in mitosis, whereas cell death correlates with the accumulation of pan-nuclear γH2AX in S phase (Fig. 6B). These results demonstrate that chromosome mis-segregation is not the cause of cell death in Chk1-deficient cells.
Fig. 6. CIN arising from nucleotide shortage during mitosis does not compromise survival of Chk1-deficient cells.
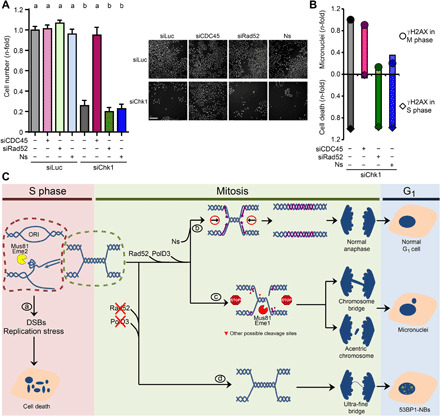
(A) Sensitivity of U2OS cells to Chk1 depletion and CDC45 or Rad52 depletion or nucleoside supplementation. Cell number was determined 6 days after transfection. Data represent the mean (±SD) of three independent experiments. The right panel shows representative images of the data. Scale bar, 500 μm. (B) Graph showing that chromosome mis-segregation and cell death are uncorrelated in Chk1-deficient cells. Chromosome mis-segregation correlates with the accumulation of γH2AX foci in mitosis, whereas cell death correlates with the accumulation of pan-nuclear γH2AX in S phase. Data correspond to Figs. 1E, 4I, and 5D (micronuclei); Fig. 6A (cell death); Figs. 3B, 4G, and 5B (γH2AX foci in mitosis); and Fig. 1B and fig. S7A (pan-nuclear γH2AX). The data on pan-nuclear γH2AX upon Ns addition shown here are not presented in any preceding figure. (C) Model in which Chk1 loss triggers chromosome segregation defects and cell death by independent pathways. During S phase, Chk1 deficiency prompts surplus origin firing, reduced and asymmetric fork elongation, and Mus81-Eme2–dependent DSBs, culminating in cell death (a). Independently of these S phase events, Chk1-deficient cells enter mitosis with UR-DNA, whose duplication is completed in mitosis only if extra DNA precursors are supplied (b). Otherwise, most replication intermediates in mitosis are cleaved by Mus81-Eme1, leading to chromosome mis-segregation (c). Restraining MiDAS results in persistent UR-DNA, manifested as UFBs and 53BP1-NBs in G1 (d).
DISCUSSION
This report uncovers a mechanism by which aberrant processing of DNA synthesis intermediates in mitosis causes CIN. Rather than culminating in cell death, this mechanism results in the inheritance of damaged DNA by daughter cells. Another key finding of this study is that MiDAS does not necessarily occur as a consequence of fork elongation or origin usage defects. Moreover, our work predicts that nucleotide availability determines the fate of late replication intermediates.
Chk1 loss leads to CIN
Despite our vast understanding of how Chk1 loss affects the S phase, few studies have addressed how Chk1 deficiency translates into genomic instability. This study confirms previous reports showing that Chk1 inactivation induces chromatin bridges and laggards (13, 15); it also shows that Chk1 deficiency triggers micronuclei accumulation. Micronuclei and anaphase aberrations are mechanistically related (17, 47–49), and this is supported by our data, as we observed a tight correlation between these two variables. Checkpoint defects precipitate chromosome mis-segregation as the result of S phase deregulation or spindle assembly checkpoint failure (50). In Chk1-depleted cells, the fact that UR-DNA precedes chromosome mis-segregation favors S phase deregulation. However, the similarity in terms of chromosome segregation between siChk1 and siChk1-siCDC45 samples downplays the contribution of acute replication stress to CIN, although we cannot exclude the contribution of mild replication stress (minimal changes in fork speed or origin usage that escape detection by the established hallmark assays to monitor them).
Another key S phase parameter that could determine the success of chromosome segregation is timing of mitotic entry. That is, Chk1 deficiency might induce anaphase aberrations and micronuclei by exiting the S phase with levels of UR-DNA that cannot be dealt with in mitosis. Chk1-deficient cells show premature mitotic onset as a result of untimely activation of the mitotic master regulators CDK1 (cyclin-dependent kinase 1) and PLK1 (Polo-like kinase 1) (15, 51, 52). This is reminiscent of the faster progression through S phase observed after inhibition of the DDR proteins ATR and WEE1 (20, 38). We speculate that Chk1-depleted cells reach mitosis with UR-DNA because of untimely mitotic entry.
MiDAS is the source of CIN upon Chk1 loss
Our work points to incomplete DNA synthesis in mitosis as a source of CIN (Fig. 6C). Moreover, we identified Mus81-Eme1 as a molecular trigger of CIN. Mus81-Eme1 is known to contribute to UR-DNA resolution and genome maintenance upon low-APH treatment by participating in the initiation of MiDAS (18, 19, 26). In Chk1-depleted cells, Mus81-Eme1 not only is dispensable for MiDAS initiation but also aberrantly processes UR-DNA undergoing MiDAS. This unforeseen activity of Mus81-Eme1 leads to the accumulation of mitotic DSBs and the ensuing CIN. At the molecular level, the requirement of a Mus81-dependent initial break might be the sole distinction between the MiDAS described previously and herein. Earlier studies postulated that APH-induced MiDAS is conservative (24, 26), consistent with MiDAS being a BIR-like event (25); in agreement with a recent study (53), we show that both siChk1-induced MiDAS and APH-induced MiDAS are semiconservative. Thus, despite its requirement for Rad52 and PolD3, two known molecular effectors of BIR, MiDAS is probably not a conservative BIR-like process.
To our knowledge, our work is the first one to identify Mus81-Eme1–dependent cleavage of MiDAS intermediates as a source of CIN. Mus81-Eme1 normally initiates the repair of stalled replication intermediates, thereby avoiding the accumulation of aberrant anaphases (18, 19, 26, 39). Because homologous recombination (HR) might function suboptimally in Chk1-deficient cells (54), the cleavage products of Mus81-Eme1 might be processed by error-prone mechanisms or aberrant forms of HR, causing CIN. In checkpoint-deficient cells, Mus81-Eme2–dependent cleavage of stalled forks in S phase also fails to initiate repair and instead results in widespread DNA damage and cell death (7, 8, 38). This observation might explain why MiDAS, a salvage pathway that supposedly implies DNA replication of UR-DNA persisting until mitosis, culminates in CIN. However, toxic processing of UR-DNA undergoing MiDAS might not be necessarily circumscribed to Chk1-depleted cells. Every time the nucleotide supply shortens during mitosis, cells might exceed a threshold of repairable UR-DNA, even in HR-proficient scenarios. So, our work introduces a key concept: MiDAS might promote proper chromosome segregation only if DNA synthesis reaches completion within a single M phase.
Nucleotide deficiency in mitosis triggers CIN and persistence of UR-DNA
The ATR-Chk1 pathway adjusts the nucleotide pool via control of RRM2, a regulatory subunit of the ribonucleotide reductase, which catalyzes the rate-limiting step for deoxynucleoside triphosphate (dNTP) production (41). Chk1-inhibited cells show reduced dNTP levels (42), resulting in the slowdown of replication forks (8, 9). Notwithstanding this, we and others have been unable to detect an effect of such nucleotide deficiency on the DNA damage load in S phase (8, 9, 37). Unexpectedly, we show here that nucleotide shortage promotes a pathological form of MiDAS that fuels mitotic DSBs and CIN (Fig. 6C).
There is consensus that the delivery of extra DNA precursors alleviates DNA replication stress by promoting fork elongation. Thus, several studies have linked reduced fork speed to genomic instability and/or cell death based on experiments that normalize the nucleotide pools (33, 42, 49, 55–57). We propose that such experiments should be interpreted with care, as our findings indicate that nucleotide pools control DNA replication beyond the S phase. In this regard, it is important to highlight that the size of the dNTP pool peaks in S phase, in part due to tight control of RRM2 gene expression (58). Thus, nucleotide scarcity in mitosis is a typical scenario representing a threat to any cell that exits the S phase with high levels of UR-DNA. We conclude that cell cycle fluctuations of dNTP pools have the potential to challenge DNA replication in M phase and thereby induce anaphase anomalies.
CIN arising from nucleotide shortage in mitosis is not the cause of cell death in Chk1-deficient cells
Our study suggests that nucleotide starvation in Chk1-depleted cells impedes the accurate handling of UR-DNA, boosting genomic instability, manifested as anaphase aberrations, micronuclei, UFBs, and 53BP1-NBs. Distortion of dNTP pools, mitotic onset before completion of DNA duplication, and abnormal anaphases have been associated with cell death in cells lacking DDR proteins, either ATR, WEE1, or ETAA1 (20, 23, 55). Intriguingly, however, we found no such causal connection between genomic instability and cell death in Chk1-depleted cells (Fig. 6C). We consider two scenarios that might explain this observation. First, DNA lesions sequestered in 53BP1-NBs could be repaired during the next S phase (29), enabling cell survival. However, our model posits that not all UR-DNA in Chk1-depleted cells reaches the progeny bound to 53BP1. Second, chromosome segregation defects upon Chk1 loss might not be severe enough to impair the offspring’s fitness. In this case, genomic instability might get amplified across generations and so would the ensuing risk of acquiring selective growth advantages. We favor this possibility, as aberrant anaphases and micronuclei, which accumulate upon Chk1 inactivation, precede cellular transformation (17). Thus, our findings could be exploited therapeutically through selective mitigation of at least one source of genomic instability in Chk1-directed therapies. This could be achieved by avoiding mitotic onset with UR-DNA or enabling its resolution through enhanced MiDAS.
Our results collectively challenge the simplistic view that defects in origin usage and/or fork elongation, mitotic abnormalities, and genomic alterations succeed each other before cell death. Given the clinical interest in developing therapeutic strategies that curtail genomic instability without compromising the killing efficiency, our study provides an important proof of concept for DDR-directed therapies.
MATERIALS AND METHODS
Cell culture and chemicals
U2OS (American Type Culture Collection), HCT116 (a gift from B. Vogelstein, Johns Hopkins University, Baltimore), and PANC-1 (a gift from T. Seufferlein, Department of Internal Medicine, University of Ulm) were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) with 10% fetal bovine serum (Natocor). EmbryoMax Nucleosides (1:1000; Millipore), APH (0.2 μM; Sigma-Aldrich), and HU (100 μM; Sigma-Aldrich) were added 24 hours before fixation; Gö6976 (1 μM; Calbiochem) was added 36 hours before fixation.
Small interfering RNAs
Transfections were performed using JetPRIME (Polyplus) according to the manufacturer’s instructions. Except from survival assays, cells were harvested 48 or 72 hours (γH2AX detection in mitotic cells and 53BP1 detection in G1 and micronuclei assays) after transfection. siRNAs were purchased from Dharmacon or Eurofins Genomics: siLuc (100 nM), 5′-CGUACGCGGAAUACUUCGA-3′ (59); siChk1 (100 nM), 5′-GAAGCAGUCGCAGUGAAGA-3′ (59); siCDC45 (10 nM in U2OS and PANC-1 and 5 nM in HCT116), 5′-GCAAGACAAGATCACTCAA-3′ (9); siMus81 (100 nM), 5′-CAGCCCUGGUGGAUCGAUA-3′ (39); siEme1 (100 nM), 5′-GCUAAGCAGUGAAAGUGA-3′ (18); siEme1#2 (100 nM), 5′-GCUCAAAGGCUUACAUGUA-3′ (18); siEme2 (100 nM), 5′-GCGAGCCAGUGGCAAGAG-3′ (39); siEme2#2 (50 nM), 5′-UGGAGCCCGAGGAGUUUCU-3′ (39); siRAD52 (100 nM), 5′-GGAGUGACUCAAGAAUUA-3′ (24); siPOLD3 (50 nM), 5′-GAUAGUGAAGAGGAGCUUA-3′ (60); siSLX4 (100 nM), 5′-GGAGAAGGAAGCAGAGAAU-3′ (61).
Lentiviral production and infection
The lentivirus production and infection were conducted exactly as previously described (9, 62).
Micronuclei assay
Twenty-four hours after transfection, cells were replated at low density. Twenty-four hours after replating, cytochalasin B (4.5 μg/ml; Sigma-Aldrich) was added to the media, and after 36 hours, cells were fixed with 2% paraformaldehyde (PFA)/sucrose for 20 min. DAPI (Sigma-Aldrich) staining served to visualize nuclei. About 200 binucleated cells were measured per sample per experiment. Representative images were acquired with a Zeiss Axio Observer 3 microscope.
Anaphase aberration assay
Asynchronous cells were fixed with 2% PFA/sucrose for 20 min. When required, cells were incubated with nucleosides, APH/DMSO (dimethyl sulfoxide), or HU 24 hours before harvesting. DAPI (Sigma-Aldrich) staining served to visualize anaphases. About 50 anaphases were measured per sample per experiment. Z-stacks were acquired with a Zeiss LSM 880 confocal microscope. Maximum intensity projections were generated using the Black ZEN Imaging Software (Zeiss).
Neutral comet assay
Neutral comet assay was conducted exactly as previously described (9).
Immunostaining and microscopy
Cells were fixed with 2% PFA/sucrose, and immunodetection of γH2AX (1:1000; Millipore, 05-636), FANCD2 (1:500; Novus, NB100-182), 53BP1 (1:1500; Santa Cruz Biotechnology, sc-22760), and PICH (1:100; Abnova, H00054821-B01P) was conducted exactly as previously described (9, 62, 63). Detection of S phase cells by EdU incorporation was performed exactly as previously described (62). Images of cells in interphase were acquired with a Zeiss Axio Observer 3 microscope. Images of mitotic cells were acquired with a Zeiss LSM 880 confocal microscope. γH2AX fluorescence intensity was quantified with the CellProfiler software (www.cellprofiler.org).
EdU labeling and detection in mitotic cells
Asynchronous cells were pulsed for 45 min with 20 μM EdU, which was detected with the Click-iT EdU Alexa Fluor 555 Imaging Kit (Life Technologies), following the manufacturer’s instructions. DAPI (Sigma-Aldrich) staining served to visualize metaphases. About 50 metaphases were measured per sample per experiment. Z-stacks were acquired with a Zeiss LSM 880 confocal microscope. Maximum intensity projections were generated using the Black ZEN Imaging Software (Zeiss).
Metaphase spreads and chromosome breakage
Chromosome aberration assays were conducted as previously described (64). Briefly, after a 24-hour treatment with colcemid (0.1 μg/ml; KaryoMAX, Invitrogen), mitotic cells were collected, pelleted, suspended in 75 mM KCl, incubated at 37°C for 5 min, and fixed using methanol:acetic acid (3:1). Cells were then dropped onto slides and aged for 24 hours before staining with 6% (w/v) Giemsa (Merck) for 2 min. Images of metaphase chromosomes were acquired using an automated CytoVision system (version 3.7, Applied Imaging). About 50 metaphases were analyzed per sample per experiment, and obvious chromosomal gaps/breaks were quantified.
EdU labeling and detection in chromosomes
As previously described in (26), cells were synchronized in late G2 by incubation with 10 μM RO-3306 (Calbiochem) for 20 hours, washed three times with phosphate-buffered saline (PBS) for 5 min, and released into fresh medium containing 20 μM EdU and colcemid (0.1 μg/ml; KaryoMAX, Invitrogen) for 60 min. Metaphase spreads and EdU detection were prepared/performed as described above. Chromosomes were stained with DAPI (Sigma-Aldrich). Images were acquired with a Zeiss LSM 880 confocal microscope and processed with the Black ZEN Imaging Software (Zeiss). About 200 EdU-positive events were analyzed per sample per experiment.
DNA fiber spreading
DNA fiber spreading was conducted exactly as previously described (9, 62, 63).
Flow cytometry
Flow cytometry analyses were performed exactly as previously described (64). Briefly, cells were fixed with ice-cold ethanol and resuspended in PBS containing ribonuclease I (100 mg/ml; Sigma-Aldrich) and propidium iodide (50 mg/ml; Sigma-Aldrich). Samples were subjected to fluorescence-activated cell sorting (Calibur, Becton Dickinson), and data were analyzed using the Summit 4.3 software (DAKO Cytomation). Ten thousand events were analyzed per sample per experiment.
Quantitative real-time PCR
Quantitative polymerase chain reaction (PCR) was conducted exactly as previously described (9, 63). Primer sequences were as follows: GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-AGCCTCCCGCTTCGCTCTCT-3′ (forward) and 5′-GAGCGATGTGGCTCGGCTGG-3′ (reverse) (63); EME1, 5′-CTCATCCCTGAGGGCTAGAA-3′ (forward) and 5′-AGTTGAAAGAGTGGCGGGA-3′ (reverse); EME2, 5′-AGGTGGAAGAGGCCCTGGTA-3′ (forward) and 5′-CCCTGCTGTGCAGAAGGAGA-3′ (reverse) (65); POLD3, 5′-ACCTCCTTCTGTCAAGAGCT-3′ (forward) and 5′-CAGGATTCACTCTCGTAGACT-3′ (reverse); RAD52, 5′-ACAGCGTTTGCCACCAGAA-3′ (forward) and 5′-ATGAGATTCCCAGTTTCCTGT-3′ (reverse); SLX4, 5′-AGTCGTGCTGTGTCACCTA-3′ (forward) and 5′-CCTGTAGTCCCAGCTATCT-3′ (reverse).
Western blot
Cells were lysed and harvested with Laemmli buffer, followed by 8 min of incubation at 99°C. The chromatin fraction was obtained after a 5-min extraction with ice-cold CSK buffer [10 mM Pipes (pH 7.5), 100 mM NaCl, 300 mM sucrose, 1 mM EGTA, 3 mM MgCl2, and 2% Triton X-100]. The following antibodies were used: α-Chk1 at 1:1000 (Santa Cruz Biotechnology, sc-8408), α-γH2AX at 1:4000 (Millipore, 05-636), α-phospho-KAP1 Ser824 at 1:4000 (Bethyl Laboratories, A300-767A), α-Mus81 at 1:1000 (Santa Cruz Biotechnology, sc-53382), α-CDC45 at 1:1000 (Santa Cruz Biotechnology, sc-20685), α-KAP1 at 1:4000 (Bethyl Laboratories, A300-274A), α-phospho-RPA Ser4/8 at 1:8000 (Bethyl Laboratories, A300-245A), α-H2B (histone 2B) at 1:2000 (Santa Cruz Biotechnology, sc-515808), and α-actin at 1:20,000 (Sigma-Aldrich, A2066). Incubations with secondary antibodies (Jackson ImmunoResearch) and enhanced chemiluminescence (ECL) detection (GE Healthcare) were performed according to the manufacturers’ instructions. Western blot images were acquired with ImageQuant LAS4000 (GE Healthcare), which allows the capture and the quantification of images within a linear range.
Cell survival assays
Twenty-four hours after transfection, 1000 cells per well were replated in 96-well plates. Five days after replating, cells were fixed with 2% PFA/sucrose for 20 min. DAPI staining served to visualize nuclei. INCell 2200 and INCell Analyzer WorkStation were used to image and count nuclei, respectively (9).
Statistical analysis
GraphPad Prism 5 was used for statistical analyses. Frequency distributions were analyzed with one-way analysis of variance (ANOVA) (followed by a Bonferroni posttest), and data shown as the mean (±SD) of independent experiments were analyzed with repeated-measures ANOVA (followed by a Newman-Keuls posttest). In all graphs, different letters indicate groups that are significantly different. Thus, if two samples share the same letter, they are not significantly different, while if two samples do not share any letter, they are significantly different. P < 0.001 or P < 0.01 was considered significant for frequency distribution or data shown as the mean of independent experiments, respectively.
Supplementary Material
Acknowledgments
We thank B. Vogelstein (Johns Hopkins University) and T. Seufferlein (University of Ulm) for the gift of cell lines. We thank A. Álvarez Julia and A. H. Rossi for technical support with tissue culture and microscopy. Funding: This work was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT; PICT 2016-1239) and the Instituto Nacional del Cáncer (INC; Asistencia Financiera IV) to V.G. M.A.G.B. and V.G. are researchers from the National Council of Scientific and Technological Research (CONICET). N.L.C. is supported by a fellowship from CONICET. Author contributions: M.A.G.B. and V.G. conceived the study; N.L.C. and M.A.G.B. designed and performed the experiments; N.L.C., M.A.G.B., and V.G. interpreted the data; N.L.C. designed the figures with the help of M.A.G.B. and V.G.; N.L.C. generated the figures; M.A.G.B. and N.L.C. wrote the manuscript, and all authors edited it; M.A.G.B. and V.G. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data and materials related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabc8257/DC1
REFERENCES AND NOTES
- 1.Pilié P. G., Tang C., Mills G. B., Yap T. A., State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 16, 81–104 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bakhoum S. F., Cantley L. C., The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 174, 1347–1360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Targa A., Rancati G., Cancer: A CINful evolution. Curr. Opin. Cell Biol. 52, 136–144 (2018). [DOI] [PubMed] [Google Scholar]
- 4.González Besteiro M. A., Gottifredi V., The fork and the kinase: A DNA replication tale from a CHK1 perspective. Mutat. Res. Rev. Mutat. Res. 763, 168–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorensen C. S., Syljuasen R. G., Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 40, 477–486 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Técher H., Koundrioukoff S., Nicolas A., Debatisse M., The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 18, 535–550 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Murfuni I., Basile G., Subramanyam S., Malacaria E., Bignami M., Spies M., Franchitto A., Pichierri P., Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLOS Genet. 9, e1003910 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Técher H., Koundrioukoff S., Carignon S., Wilhelm T., Millot G. A., Lopez B. S., Brison O., Debatisse M., Signaling from Mus81-Eme2-dependent DNA damage elicited by Chk1 deficiency modulates replication fork speed and origin usage. Cell Rep. 14, 1114–1127 (2016). [DOI] [PubMed] [Google Scholar]
- 9.González Besteiro M. A., Calzetta N. L., Loureiro S. M., Habif M., Bétous R., Pillaire M.-J., Maffia A., Sabbioneda S., Hoffmann J.-S., Gottifredi V., Chk1 loss creates replication barriers that compromise cell survival independently of excess origin firing. EMBO J. 38, e101284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forment J. V., Blasius M., Guerini I., Jackson S. P., Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLOS ONE 6, e23517 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakurikar N., Thompson R., Montano R., Eastman A., A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase. Oncotarget 7, 1380–1394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cahill D. P., Kinzler K. W., Vogelstein B., Lengauer C., Genetic instability and darwinian selection in tumours. Trends Cell Biol. 9, M57–M60 (1999). [PubMed] [Google Scholar]
- 13.Petsalaki E., Dandoulaki M., Morrice N., Zachos G., Chk1 protects against chromatin bridges by constitutively phosphorylating BLM serine 502 to inhibit BLM degradation. J. Cell Sci. 127 (pt. 18), 3902–3908 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Gemble S., Buhagiar-Labarchède G., Onclercq-Delic R., Biard D., Lambert S., Amor-Guéret M., A balanced pyrimidine pool is required for optimal Chk1 activation to prevent ultrafine anaphase bridge formation. J. Cell Sci. 129, 3167–3177 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Tang J., Erikson R. L., Liu X., Checkpoint kinase 1 (Chk1) is required for mitotic progression through negative regulation of polo-like kinase 1 (Plk1). Proc. Natl. Acad. Sci. U.S.A. 103, 11964–11969 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moreno A., Carrington J. T., Albergante L., Al Mamun M., Haagensen E. J., Komseli E.-S., Gorgoulis V. G., Newman T. J., Blow J. J., Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. U.S.A. 113, E5757–E5764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bizard A. H., Hickson I. D., Anaphase: A fortune-teller of genomic instability. Curr. Opin. Cell Biol. 52, 112–119 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Naim V., Wilhelm T., Debatisse M., Rosselli F., ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Ying S., Minocherhomji S., Chan K. L., Palmai-Pallag T., Chu W. K., Wass T., Mankouri H. W., Liu Y., Hickson I. D., MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Eykelenboom J. K., Harte E. C., Canavan L., Pastor-Peidro A., Calvo-Asensio I., Llorens-Agost M., Lowndes N. F., ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 5, 1095–1107 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Bergoglio V., Boyer A.-S., Walsh E., Naim V., Legube G., Lee M. Y. W. T., Rey L., Rosselli F., Cazaux C., Eckert K. A., Hoffmann J.-S., DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 201, 395–408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai X., Broderick R., Bergoglio V., Zimmer J., Badie S., Niedzwiedz W., Hoffmann J.-S., Tarsounas M., MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 8, 15983 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Achuthankutty D., Thakur R. S., Haahr P., Hoffmann S., Drainas A. P., Bizard A. H., Weischenfeldt J., Hickson I. D., Mailand N., Regulation of ETAA1-mediated ATR activation couples DNA replication fidelity and genome stability. J. Cell Biol. 218, 3943–3953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhowmick R., Minocherhomji S., Hickson I. D., RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 64, 1117–1126 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Özer Ö., Hickson I. D., Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol. 8, 180018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minocherhomji S., Ying S., Bjerregaard V. A., Bursomanno S., Aleliunaite A., Wu W., Mankouri H. W., Shen H., Liu Y., Hickson I. D., Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Di Marco S., Hasanova Z., Kanagaraj R., Chappidi N., Altmannova V., Menon S., Sedlackova H., Langhoff J., Surendranath K., Hühn D., Bhowmick R., Marini V., Ferrari S., Hickson I. D., Krejci L., Janscak P., RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol. Cell 66, 658–671.e8 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Teixeira L. K., Wang X., Li Y., Ekholm-Reed S., Wu X., Wang P., Reed S. I., Cyclin E deregulation promotes loss of specific genomic regions. Curr. Biol. 25, 1327–1333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spies J., Lukas C., Somyajit K., Rask M.-B., Lukas J., Neelsen K. J., 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 21, 487–497 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Toledo L. I., Altmeyer M., Rask M.-B., Lukas C., Larsen D. H., Povlsen L. K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J., ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Gagou M. E., Zuazua-Villar P., Meuth M., Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell 21, 739–752 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Syljuasen R. G., Sorensen C. S., Hansen L. T., Fugger K., Lundin C., Johansson F., Helleday T., Sehested M., Lukas J., Bartek J., Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 25, 3553–3562 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burrell R. A., McClelland S. E., Endesfelder D., Groth P., Weller M. C., Shaikh N., Domingo E., Kanu N., Dewhurst S. M., Gronroos E., Chew S. K., Rowan A. J., Schenk A., Sheffer M., Howell M., Kschischo M., Behrens A., Helleday T., Bartek J., Tomlinson I. P., Swanton C., Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olive P. L., Banáth J. P., The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 1, 23–29 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Ciccia A., Constantinou A., West S. C., Identification and characterization of the human Mus81-Eme1 endonuclease. J. Biol. Chem. 278, 25172–25178 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Ciccia A., Ling C., Coulthard R., Yan Z., Xue Y., Meetei A. R., Laghmani el H., Joenje H., McDonald N., de Winter J. P., Wang W., West S. C., Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 25, 331–343 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Beck H., Nahse-Kumpf V., Larsen M. S., O’Hanlon K. A., Patzke S., Holmberg C., Mejlvang J., Groth A., Nielsen O., Syljuasen R. G., Sørensen C. S., Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 32, 4226–4236 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duda H., Arter M., Gloggnitzer J., Teloni F., Wild P., Blanco M. G., Altmeyer M., Matos J., A mechanism for controlled breakage of under-replicated chromosomes during mitosis. Dev. Cell 39, 740–755 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Pepe A., West S. C., MUS81-EME2 promotes replication fork restart. Cell Rep. 7, 1048–1055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giunta S., Belotserkovskaya R., Jackson S. P., DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 190, 197–207 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buisson R., Boisvert J. L., Benes C. H., Zou L., Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol. Cell 59, 1011–1024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfister S. X., Markkanen E., Jiang Y., Sarkar S., Woodcock M., Orlando G., Mavrommati I., Pai C. C., Zalmas L. P., Drobnitzky N., Dianov G. L., Verrill C., Macaulay V. M., Ying S., La Thangue N. B., D’Angiolella V., Ryan A. J., Humphrey T. C., Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell 28, 557–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Pedersen R. S., Grofte M., Chan K. L., Hickson I. D., Bartek J., Lukas J., 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Harrigan J. A., Belotserkovskaya R., Coates J., Dimitrova D. S., Polo S. E., Bradshaw C. R., Fraser P., Jackson S. P., Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baumann C., Korner R., Hofmann K., Nigg E. A., PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell 128, 101–114 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Chan K. L., North P. S., Hickson I. D., BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffelder D. R., Luo L., Burke N. A., Watkins S. C., Gollin S. M., Saunders W. S., Resolution of anaphase bridges in cancer cells. Chromosoma 112, 389–397 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Utani K.-i., Kohno Y., Okamoto A., Shimizu N., Emergence of micronuclei and their effects on the fate of cells under replication stress. PLOS ONE 5, e10089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilhelm T., Olziersky A. M., Harry D., De Sousa F., Vassal H., Eskat A., Meraldi P., Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 10, 3585 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez Besteiro M. A., Gottifredi V., ETAA1 ensures proper chromosome segregation: A matter of S phase or mitosis? J. Cell Biol. 218, 3883–3884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lemmens B., Hegarat N., Akopyan K., Sala-Gaston J., Bartek J., Hochegger H., Lindqvist A., DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol. Cell 71, 117–128.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Enomoto M., Goto H., Tomono Y., Kasahara K., Tsujimura K., Kiyono T., Inagaki M., Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J. Biol. Chem. 284, 34223–34230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chappidi N., Nascakova Z., Boleslavska B., Zellweger R., Isik E., Andrs M., Menon S., Dobrovolna J., Balbo Pogliano C., Matos J., Porro A., Lopes M., Janscak P., Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at co-transcriptional R-loops. Mol. Cell 77, 528–541.e8 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Sorensen C. S., Hansen L. T., Dziegielewski J., Syljuasen R. G., Lundin C., Bartek J., Helleday T., The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 7, 195–201 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Lopez-Contreras A. J., Specks J., Barlow J. H., Ambrogio C., Desler C., Vikingsson S., Rodrigo-Perez S., Green H., Rasmussen L. J., Murga M., Nussenzweig A., Fernandez-Capetillo O., Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev. 29, 690–695 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bester A. C., Roniger M., Oren Y. S., Im M. M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D. S., Kerem B., Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruiz S., Lopez-Contreras A. J., Gabut M., Marion R. M., Gutierrez-Martinez P., Bua S., Ramirez O., Olalde I., Rodrigo-Perez S., Li H., Marques-Bonet T., Serrano M., Blasco M. A., Batada N. N., Fernandez-Capetillo O., Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat. Commun. 6, 8036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mathews C. K., Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 15, 528–539 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Speroni J., Federico M. B., Mansilla S. F., Soria G., Gottifredi V., Kinase-independent function of checkpoint kinase 1 (Chk1) in the replication of damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 109, 7344–7349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim M. S., Machida Y., Vashisht A. A., Wohlschlegel J. A., Pang Y. P., Machida Y. J., Regulation of error-prone translesion synthesis by Spartan/C1orf124. Nucleic Acids Res. 41, 1661–1668 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson J. S., Tejera A. M., Castor D., Toth R., Blasco M. A., Rouse J., Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 4, 853–860 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mansilla S. F., Bertolin A. P., Bergoglio V., Pillaire M. J., Gonzalez Besteiro M. A., Luzzani C., Miriuka S. G., Cazaux C., Hoffmann J.-S., Cyclin kinase-independent role of p21CDKN1A in the promotion of nascent DNA elongation in unstressed cells. eLife 5, e18020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vallerga M. B., Mansilla S. F., Federico M. B., Bertolin A. P., Gottifredi V., Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc. Natl. Acad. Sci. U.S.A. 112, E6624–E6633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Federico M. B., Vallerga M. B., Radl A., Paviolo N. S., Bocco J. L., Di Giorgio M., Soria G., Gottifredi V., Chromosomal integrity after UV irradiation requires FANCD2-mediated repair of double strand breaks. PLOS Genet. 12, e1005792 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lemacon D., Jackson J., Quinet A., Brickner J. R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N., Vindigni A., MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8, 860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/50/eabc8257/DC1