

Abstract
Bone destruction in inflammatory osteolytic diseases including periodontitis is related to excessive activity of osteoclasts (OC), which originate from precursor cells of the myeloid lineage, termed osteoclast precursors (OCP). In contrast to ample knowledge that we currently have on mature OC, little is known about OCP and their regulation during bacterial infection. Therefore, this study aimed to identify and characterize OCP following chronic infection with a periodontal bacteria Porphyromonas gingivalis (Pg). We used a micro-osmotic pump to continually release Pg subcutaneously in a murine model. Two weeks after Pg infection, the frequency of CD11b+c-fms+Ly6Chi population is significantly elevated within the bone marrow, spleen and peripheral blood. In vitro and in vivo studies identified these cells as the OCP-containing population and Pg infection significantly enhanced the osteoclastogenic activity of these cells. Furthermore, mRNA sequencing analysis indicated a unique gene and pathway profile in CD11b+c-fms+Ly6Chi population following Pg infection, with changes in genes and pathways related to OC differentiation, cell proliferation and apoptosis, inflammatory response, phagocytosis and immunity, as well as antigen processing and presentation. Moreover, using IL-6 knockout mice, we found that IL-6 is important for Pg-induced accumulation of CD11b+c-fms+Ly6Chi population from the bone marrow and periphery. Our results provide new insights into the characterization and regulation of OCP following a chronic bacterial infection. This knowledge is relevant to the understanding of the pathogenesis of bacteria-induced bone loss, and to the identification of potential therapeutic targets of bone loss diseases.
Keywords: Porphyromonas gingivalis, osteoclast precursors, osteoclasts, osteoclastogenesis
Graphical Abstract
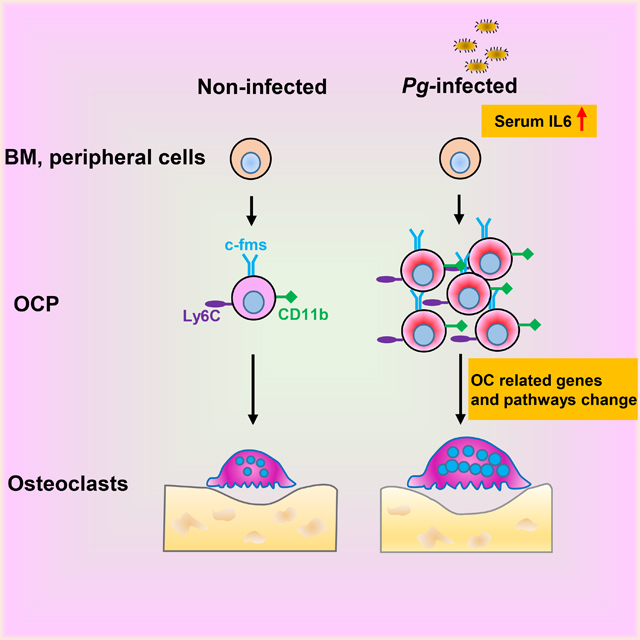
Summary sentence:
Chronic Porphyromonas gingivalis infection promotes CD11b+c-fms+Ly6Chi OCP accumulation in BM and periphery through elevated serum IL-6 and enhances their osteoclastogenic potential through changed gene signatures
1. INTRODUCTION
Excessive frequency and activity of osteoclasts (OC), the body’s exclusive bone resorbing cells, is a characteristic feature of pathological bone loss diseases such as periodontitis, rheumatoid arthritis (RA) and osteoporosis [1, 2]. OC are large, multinucleated cells differentiated from the myeloid/monocyte/macrophage lineage of the hematopoietic stem cells (HSC), the common precursors for macrophages and dendritic cells, following stimulation with the key factors, macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL) [3]. M-CSF and RANKL exert their effect by binding to c-fms (also known as CD115) and RANK, respectively, expressed on osteoclast precursors (OCP). In contrast to OC, which are tightly attached to bone matrix, OCP are present in bone marrow (BM) and periphery. And OC can be induced in vitro from BM, spleen (SPL) and peripheral blood (PB) cells in the presence of M-CSF and RANKL. It’s believed that the life span of individual HSC-derived OC is as short as a few weeks, and that a perpetual supply of OCP is required for the frequent renewal of OC for optimal postnatal maintenance and function [4, 5]. While it has been well known about osteoclasts, the identity of the osteoclast precursor population is still poorly defined, especially in bacterial infection.
OCP are identified by their expression markers. Regardless of many attempts to determine a specific precursor population for OC, it has been shown that multiple myeloid populations contain the potential of generating OC in vitro [6–10]. In physiological murine BM, OCP are identified as expressing B220−CD3−CD11b−/loCD115+ and either CD117hi, CD117int or CD117low, while in SPL and PB, OCP shared the same phenotype: B220−CD3−NK1.1−CD11b+Ly6Chi CD115+CCR2hiC×3CR1+ [11, 12]. In arthritis disease model, OCP are reported as CD11b–/loLy6Chi population, CD11b+CD115+Ly6Chi population or CD11b+CD115+Ly6C− population by different groups [9, 10, 13]. Overall, there are still controversial results about the cell surface markers of OCP, which mainly differ in the expression or lack of expression of CD11b and Ly6C, and if OCP markers are location-specific and/or disease-specific.
Under physiological conditions, only a small population of cells in BM, SPL or PB are able to differentiate into OC. However, given their precursor nature and wide distribution, OCP are highly plastic and dynamic, and are sensitive to environmental changes. Thus, it has been shown that inflammatory arthritis increases the number and function of circulating OCP [14], and that the number of OCP correlates with disease severity, as well as with the efficacy of therapies in arthritis [15, 16]. In addition, a significant upregulation in the frequency and osteoclastogenic activity of circulating OCP was also observed in patients with late-stage chronic kidney disease and on hemodialysis [17]. However, the nature and regulation of OCP during a chronic bacterial infection remain unclear.
Periodontitis is a dysbiotic inflammatory disease characterized by periodontal inflammation and alveolar bone loss, and is the leading cause of tooth loss in adults [18–20]. In addition, substantial epidemiological evidence suggest the association of periodontitis with systemic diseases/conditions, including rheumatoid arthritis (RA), osteoporosis, diabetes, preterm births, cardiovascular diseases, and Alzheimer’s disease [19, 21]. It is generally accepted that Porphyromonas gingivalis (Pg) is the keystone pathogen of periodontitis [22–24]. Importantly, animal and human studies have shown that Pg can disseminate from local infection sites to distal sites via the circulatory system and cause bacteremia [25–30]. Our previous in vitro studies have shown that Pg can inhibit OC differentiation from non-committed OCP, while enhancing the osteoclastogenesis of RANKL-committed OCP [31]. Recently, using an in vivo mouse calvarial infection model, our results showed that short term and localized Pg infection can upregulate the number and the osteoclastogenic activity CD11b+c-fms+ OCP population of BM and SPL [32]. Yet, the role of CD11b and Ly6C on Pg-induced OCP, their gene signature changes and their regulation remain to be established, especially in a chronic inflammation model.
In the present study, we sought to further address the effect of Pg on the regulation of the frequency and function of OCP in chronic infection, using an osmotic pump releasing system. We identified that the CD11b+c-fms+Ly6Chi population is increased in BM and periphery following Pg infection, and that this population is able to differentiate into OC in vitro as well as in vivo. In addition, Pg infection enhanced the osteoclastogenic activity of CD11b+c-fms+Ly6Chi populations from BM and periphery. Furthermore, analysis of mRNA sequencing of CD11b+c-fms+ Ly6Chi populations in control and Pg-infected mice demonstrated that these cells have unique genes and pathway characteristics after Pg infection, with changes in genes and pathways related to OC differentiation, cell proliferation and apoptosis, inflammation, phagocytosis and immunity, as well as antigen processing and presentation. Moreover, we found that IL-6 participates in the regulation of Pg-mediated accumulation of CD11b+c-fms+Ly6Chi population of BM and SPL in our model system. Our results provide new insight into the characterization and regulation of OCP following a chronic bacterial infection, which will be relevant for our understanding of the pathogenesis of bacteria-induced bone loss, as well as for the identification of therapeutic targets of bone loss diseases.
2. MATERIALS AND METHODS
2.1. Mice
C57BL/6 wild type (WT) and B6.129-IL6tm1kopf/J (IL-6−/−) mice were originally obtained from Jackson Laboratories. mT/mG;c-fms-Cre mice on a C57BL/g background were generated by crossing c-fms-Cre mice with a global-fluorescence Cre reported mouse line termed mT/mG mice[33, 34]. All mice were bred and maintained in an environmentally controlled, pathogen-free animal facility at the University of Alabama at Birmingham (UAB). All animal procedures were performed according to the National Institute of Health (NIH) guidelines, and protocols were approved by the UAB Institutional Animal Care and Use Committee.
2.2. Bacteria culture
Pg ATCC 33277 was cultured and maintained on enriched trypticase soy agar plates containing 1% yeast extract, 5% defibrinated sheep blood, 5 μg/ml hemin, and 1 μg/ml menadione, at 37°C in an anaerobic atmosphere of 10% H2, 5% CO2, and 85% N2 [32]. To prepare Pg for infection, the bacteria were grown in trypsin soy broth (BD Biosciences) containing 1% yeast extract, 5 μg/ml heme and 2.5 μg/ml menadione. The bacteria were collected by centrifugation and washed in PBS, and the number of bacteria (colony-forming units/ml) was determined by measuring the optical density at 600 nm and extrapolating using a standard curve, with a culture of OD600 of 1 equals 109 CFU/ml [32].
2.3. Infection model
Mice (8–10 weeks of age) were anesthetized and prepared for the dorsolumbar implantation of the micro-osmotic pumps (model 1002, Alzet Osmotic Pumps) as previously described [35]. This pump system has a reservoir volume of 100 μl and allows for the continuous delivery of capsule solutions for 14 days without the need for external connections or frequent handling of animals. Pg infected mice were implanted with pumps (one pump/mouse) containing a single dose of 100 μl of bacteria (2×1010 CFU/ml). Control mice were implanted with pumps containing PBS. Mice were sacrificed at day14.
2.4. FACS analysis and cell sorting
Single-cell suspensions were prepared from BM, SPL or PB, as previously described [31, 32, 35, 36]. Briefly, femur and tibiae were isolated and both ends of the bones were cut off and bone marrow was flushed with PBS using a 25-gauge needle. Bone marrow was then mechanically dispersed through a 100-μm cell strainer to prepare single-cell suspensions. Erythrocytes were removed using BD Pharm Lyse™ lysis buffer (BD biosciences) [31]. To harvest the SPL cells, spleens were isolated and minced through a 100-μm cell strainer in PBS and removed of erythrocytes with lysis buffer [35]. For the PB cells, blood (~500 μl) was collected via retro-orbital bleeding, followed by lysis of erythrocytes [36]. Subsequently, cells were suspended in FACS buffer (PBS containing 5% bovine serum albumin) and stained with CD11b (M1/70)-FITC (11-0112-82), CD115 (c-fms) (AFS98)-APC (17-1152-82) and Ly-6C (HK1.4)-PE-Cyanine7 (25-5932-82) (eBioscience). FACS analysis was done with a LSR II flow cytometer (Becton Dickinson), followed by data analysis with FlowJo (Tree Star). Cell sorting was done on a FACSAria II system (BD).
2.5. In vitro osteoclastogenesis assays
To induce OC differentiation, BM, SPL and PB cells were cultured in 24-well plates at a density of 1×105 cells/well (BM) or 2×105 cells/well (SPL and PB) in osteoclastogenic medium (α-MEM supplemented with 10% FBS, 5% M-CSF, and 100 ng/ml RANKL) for 5–7 days. Sorted cells from BM or SPL were cultured in 96-well plates (104 cells/well) for 4–6 days. Cells were stained for tartrate-resistant acid phosphatase (TRAP) activity using a leukocyte acid phosphatase kit (Sigma). TRAP+ multinucleated cells (MNCs, ≥ 3 nuclei) were considered as OC [32]. To evaluate F-actin ring formation of OC, differentiated cells were fixed, permeabilized, and stained with Rhodamine Phalloidin (Invitrogen) [32]. For in vitro bone resorption assay, OC differentiation was induced in 48-well plates (2 × 104 cells/well) containing bovine cortical bone slices. Bone slices (thickness: 0.25–0.5 mm) were prepared from bovine long bones as previously described [37, 38]. Bone slices were harvested on day 7 or day 9, and cells were removed by sonication in PBS, and then washed with 0.3% H2O2 for 30 min. Resorption pits were visualized by staining with wheat germ agglutinin (WGA) (Sigma) and 3,3′-diaminobenzidine (DAB) (Vector laboratories), and analyzed by Image J software (NIH).
2.6. Adoptive cell transfer
Micro-osmotic pumps loaded with Pg as described above were implanted in mT/mG;c-fms-Cre mice. These mice express membrane-targeted tandem dimer Tomato (mT) prior to Cre-mediated excision and green fluorescence protein (GFP) after Cre excision [33]. Fourteen days later, CD11b+c-fms+Ly6Chi population was sorted from the SPL of these Cre mice by flow cytometry, and suspended in PBS at a concentration of 5×106 cells/ml as the donor cells. Recipient C57BL/6 WT mice were first injected with Pg (1×108 CFU in 20 μl of PBS) into their cranial suture on day 0 to initiate osteoclastogenesis [9]. On day 1 and day 4, 100 μl of purified donor cells (5×105 cells) or PBS were transferred to the recipient mice via tail vein injection. Calvariae of the recipient mice were harvested on day 7 for histological analysis.
2.7. Histology and Immunofluorescent staining
Calvariae were decalcified with 10% EDTA and tissue sections (5 μm) were prepared as previously described [32]. Sections were incubated with the primary mouse anti-mouse GFP antibody (Abcam, ab1218), followed by secondary Alexa Fluor 488 goat anti-mouse IgG (Life Technologies, A-10680). Nuclei were labeled by DAPI (Sigma), and the fluorescence staining was observed under a fluorescent microscope (Olympus 1000, Nikon). OC differentiation was examined by TRAP staining as described above.
2.8. Cytokine analysis
Blood was collected at 0 h, 6 h and day 14 after the implantation of the pump. Serum was obtained and analyzed for the levels of IL-6 by ELISA (eBioscience) according to the manufacturer’s instructions.
2.9. Quantitative PCR (qPCR) analysis
For analysis of OC-related genes, FACS-sorted cells from BM or SPL were cultured in 24-well plates (105 cells/well) in osteoclastogenic medium for 48 h. For validation of RNA-seq, FACS-sorted cells from SPL were directly used for RNA extraction. Total RNA was extracted using a miRNeasy Mini kit (QIAGEN), and reversed transcribed to cDNA with a SuperScript™ III First-Strand Synthesis System (Invitrogen). qPCR was done with TB Green™ Advantage® qPCR Premix (Clontech) on a Roche Real-Time PCR System. Relative expression of OC-related genes was normalized to β-actin gene. Primers used are listed in Supplementary Table 1 [39–41].
2.10. RNA-seq and data analysis
Total RNAs were isolated from SPL CD11b+c-fms+Ly6Chi population of control and Pg-infected mice, and were sent to the GENEWIZ company (South Plainfield, NJ) for sequencing and bioinformatics analysis. Heatmap was generated from significantly regulated genes using R (v3.1.1). For downstream statistical analysis, differentially expressed genes (DEGs) were used, with an average mRNA expressions above 50 and a fold change greater than 1.5 [42]. Gene Set Enrichment Analysis (GSEA) was performed according to the instructions from the Broad Institute.
2.11. Statistical analysis
All results are expressed as mean ± SD. Statistical significance was determined by a two-tailed Student’s t-test or ANOVA analysis using GraphPad Prism 8 (San Diego, CA). A P value less than 0.05 was considered significant.
3. RESULTS
3.1. Osteoclastogenic activity of BM and peripheral cells is enhanced following Pg infection
In this study, we established a chronic infection in mice, using a subcutaneous osmotic pump system that continually released Pg for 14 days [35]. The in vitro osteoclastogenic potential of BM, SPL and PB cells following Pg infection was determined by the induction of TRAP+ multinuclear cell (MNC) formation in the presence of RANKL and M-CSF. Our results showed that RANKL induced a significantly higher number of OC formation in BM cell cultures from Pg-infected mice, compared with non-infected control mice (Fig. 1A,B). Similarly, OC formation from SPL and PB cells were also significantly increased following Pg infection (Fig. 1A,C and D). These results demonstrate that the osteoclastogenic potential of BM and peripheral cells is enhanced following chronic subcutaneous infection of mice with Pg.
FIGURE 1. Osteoclastogenic potential of BM, SPL and PB cells is enhanced following Pg infection.

(A) Representative TRAP staining of BM, SPL and PB cells of control (Ctrl) and Pg-infected mice after 5 days (BM), 6 days (SPL) and 7 days (PB) in the presence of M-CSF and RANKL. Scale bar, 200 μm. (B) Numbers of TRAP+ multinucleated cells (MNC) from BM cell cultures (n=6). (C) Numbers of TRAP+ MNC from SPL cell cultures (n=8). (D) Numbers of TRAP+ MNC from PB cell cultures (n=6). Data are expressed as mean ± SD. ***P < 0.001.
3.2. CD11b+c-fms+Ly6Chi population accumulates in BM and periphery following Pg infection
An enhancement in the osteoclastogenic activity of BM and peripheral cells following Pg infection could result from an increased OCP pool in BM and periphery, or from an augmented ability of OCP to differentiate into OC. To determine if there is an increase in the OCP pool in BM and periphery following Pg infection, we analyzed the percentage of potential OCP populations in BM, SPL and PB by flow cytometry with antibodies to CD11b, c-fms and Ly6C. Mice infected with Pg showed a significant increase in the frequencies of CD11b+, c-fms+, and CD11b+c-fms+ cells in BM compared with control mice, while the frequency of the CD11b−c-fms+ population was significantly decreased within BM after Pg infection (Fig. 2A). Further characterization of the percentage distribution of Ly6C− and Ly6C+ populations within the CD11b+c-fms+ population showed that the frequency of CD11b +c-fms + Ly6Chi population in BM was significantly elevated after Pg infection, while there was no significant difference in the frequency of CD11b+c-fms+Ly6C− population. (Fig. 2A). Moreover, the frequency of CD11b+c-fms+Ly6Chi population was over 20 fold higher than that of CD11b+c-fms+Ly6C− population in BM after Pg infection. No significant difference was observed in the total numbers of BM cells between Pg-infected mice and non-infected controls (Fig. 2B).
FIGURE 2. CD11b+c-fms+Ly6Chi population accumulate in BM, SPL and PB following Pg infection.
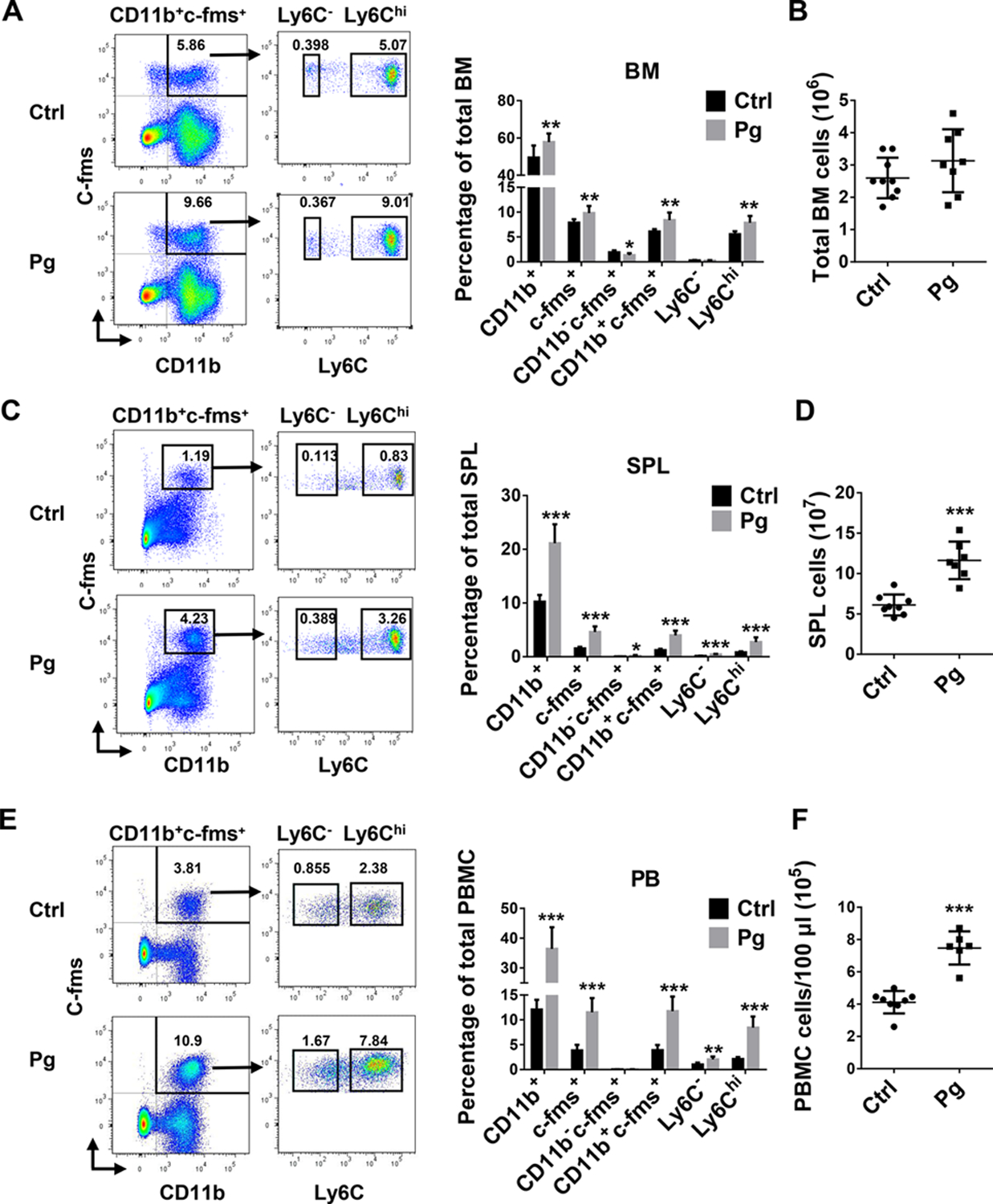
(A) Representative flow cytometry plots and percentage of myeloid cell population in BM of Ctrl and Pg-infected mice. (B) Total cell numbers in BM of Ctrl and Pg-infected mice (n=9 for Ctrl group and n=8 for Pg-infected group in A and B). (C) Representative flow cytometry plots and percentage of myeloid cell populations in SPL of Ctrl and Pg-infected mice. (D) Total cell numbers in SPL of Ctrl and Pg-infected mice (n=8 for Ctrl group and n=7 for Pg-infected group in C and D). (E) Representative flow cytometry plots and percentage of myeloid cell populations in PB of Ctrl and Pg-infected mice. (F) Total cell numbers in PB of Ctrl and Pg-infected mice (n=8 for Ctrl group and n=6 for Pg-infected group in E and F). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
In SPL, Pg infection led to a significant increase in the frequencies of CD11b+, c-fms+, CD11b+c-fms+ populations, as well as CD11b−c-fms+ population (Fig. 2C). However, the CD11b−c-fms+ population only represented less than 0.5% of the total SPL cell population, and were approximately 20-fold fewer than CD11b+c-fms+ population in SPL after Pg infection. Further analysis of the Ly6C expression on CD11b+cfms+ population showed that both CD11b+c-fms+Ly6Chi and CD11b+c-fms+Ly6C− populations were significantly elevated within Pg-infected mice. However, the frequency of CD11b+c-fms+Ly6Chi population was significantly higher than that of CD11b+c-fms+Ly6C− population in SPL after Pg infection (Fig. 2C). Furthermore, the number of total SPL cells increased significantly following Pg infection (Fig. 2D). Similar results were detected within PB cells following Pg infection (Fig. 2E,F). These results indicate that CD11b+c-fms+Ly6Chi population is the major monocytic population that increases in the BM and periphery following Pg infection.
3.3. Osteoclastogenic ability of CD11b+c-fms+Ly6Chi population is enhanced after Pg infection
Next, we assessed the ability of CD11b+c-fms+Ly6Chi population to differentiate into OC. CD11b+c-fms+Ly6Chi and CD11b+c-fms+Ly6C− populations were sorted from BM and SPL, then RANKL-induced OC formation was evaluated. We found that CD11b+c-fms+Ly6Chi population is able to generate significantly more OC than CD11b+c-fms+Ly6C− population (Fig. 3A,C). Moreover, Pg infection significantly promoted OC formation from CD11b+c-fms+Ly6Chi population. To examine the function of OC generated from CD11b+c-fms+Ly6Chi population, RANKL-induced F-actin ring formation and bone resorption activity were determined. F-actin ring formation was observed in CD11b+c-fms+Ly6Chi population within BM and SPL of control and Pg-infected mice (Fig. 3B,D). Notably, increased F-actin ring+ cells and bigger F-actin ring were observed in CD11b+c-fms+Ly6Chi population in Pg-infected mice. WGA staining of bone slices cultured with CD11b+c-fms+Ly6Chi population further confirmed an enhanced bone resorption activity of OC derived from CD11b+c-fms+Ly6Chi population of Pg-infected mice, relative to those of control mice (Fig. 3C,F). This demonstrates that the frequency of CD11b+c-fms+Ly6Chi population increased in BM and periphery, as well as their osteoclastogenic ability was enhanced after Pg infection.
FIGURE 3. Osteoclastogenic potential of CD11b+c-fms+Ly6Chi population in BM and SPL is enhanced following Pg infection.
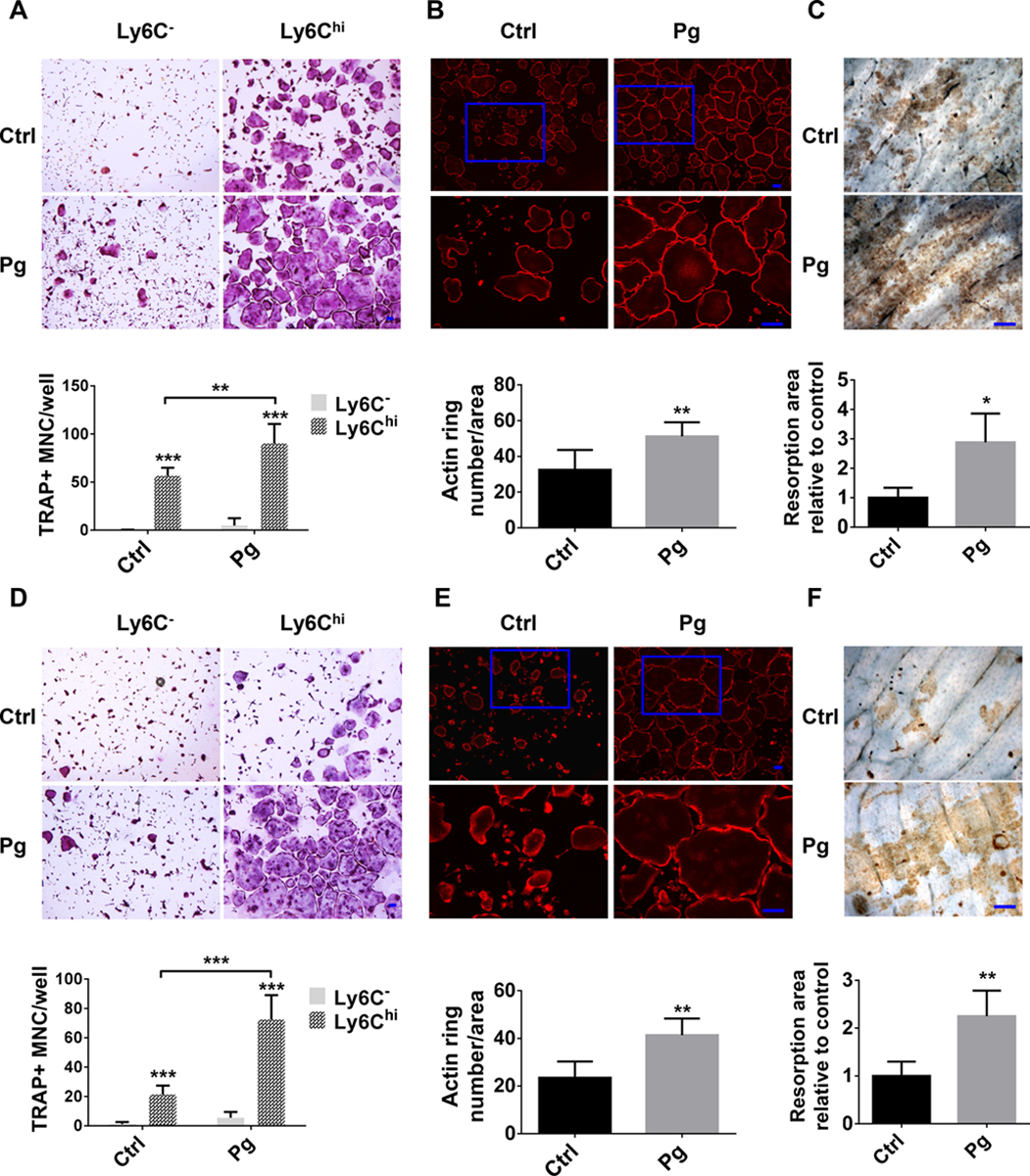
(A) TRAP+ MNC in the cultures of BM CD11b+ c-fms+Ly6C− and CD11b+c-fms+Ly6Chi populations in Ctrl and Pg-infected mice (n=6). (B) F-actin ring formation in the cultures of BM CD11b+c-fms+Ly6Chi populations of Ctrl and Pg-infected mice (n=6). (C) Bone resorption area by WGA staining in the cultures of BM CD11b+c-fms+Ly6Chi in Ctrl and Pg-infected mice (n=4). (D) TRAP+ MNC in the cultures of SPL CD11b+c-fms+Ly6C− and CD11b+c-fms+Ly6Chi populations of Ctrl and Pg-infected mice (n=6). (E) F-actin ring formation in the cultures of SPL CD11b+c-fms+Ly6Chi of Ctrl and Pg-infected mice (n=6). (F) Bone resorption area by WGA staining in the cultures of SPL CD11b+c-fms+Ly6Chi of Ctrl and Pg-infected mice (n=4). Data are expressed as mean ± SD. Scale bar, 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
3.4. CD11b+c-fms+Ly6Chi population is able to differentiate into OC in vivo
Next, we tracked the ability of CD11b+c-fms+Ly6Chi population to differentiate into mature OC in vivo. CD11b+c-fms+Ly6Chi population was sorted from the BM of Pg-infected mT/mG;c-fms-Cre mice as donor cells. Subsequently, sorted cells were injected intravenously into recipient C57BL/6 WT mice, in which calvarial inflammatory osteolysis had been initiated a day earlier. Six days after adoptive cell transfer, we were able to observe donor-derived GFP+/c-fms+ cells which are also TRAP+ and multinucleated, in the calvaria of recipient mice (Fig. 4). However, in the control recipient mice receiving PBS intravenously, only host-derived TRAP+ OC without GFP signal were detected (Fig. 4). This demonstrates that CD11b+c-fms+Ly6Chi population is capable of differentiating into OC in vivo.
FIGURE 4. CD11b+c-fms+Ly6Chi population is able to differentiate into OC in vivo.
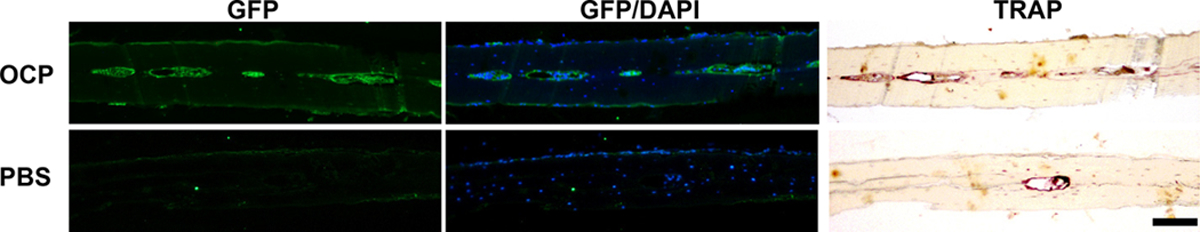
Pg was injected into the calvaria of C57BL/6 WT mice on day 0. Mice were then injected intravenously with PBS or with CD11b+c-fms+Ly6Chi population of Pg-infected mT/mG;c-fms-Cre mice on day 1 and on day 4. Calvarial sections were prepared on day 7 and stained for GFP, TRAP and DAPI. Images are representative of three independent experiments with similar results. Scale bar, 100 μm.
3.5. CD11b+c-fms+Ly6Chi population in Pg-infected mice have increased sensitivity to RANKL-induced OC gene expression
RANKL stimulates osteoclastogenesis through inducing the expression of numerous genes, such as Ctsk (Cathepsin K), MMP9 (Matrix metallopeptidase 9), OC-associated receptor (Oscar), OC stimulatory transmembrane protein (OCstamp) and Atp6v0d2 [43]. To address the molecular basis of the increased osteoclastogenic activity of CD11b+c-fms+Ly6Chi OCP in Pg-infected mice, we examined the expression of Ctsk, MMP9, Oscar, OCstamp and Atp6v0d2 mRNA in these cells from BM and SPL of the control and Pg-infected mice. Comparable levels of OC gene expression were seen between BM and SPL CD11b+c-fms+Ly6Chi population obtained in control and Pg-infected mice (data not shown). Significantly higher levels of OC gene expression were observed in the BM and SPL cells from Pg-infected mice relative to control mice following 48 h RANKL stimulation (Fig. 5A,B). These results indicate that the sensitivity of CD11b+c-fms+Ly6Chi OCP to RANKL-induced OC gene expression is increased after Pg infection.
FIGURE 5. CD11b+c-fms+Ly6Chi population of Pg-infected mice have increased sensitivity to RANKL-induced OC gene expression.
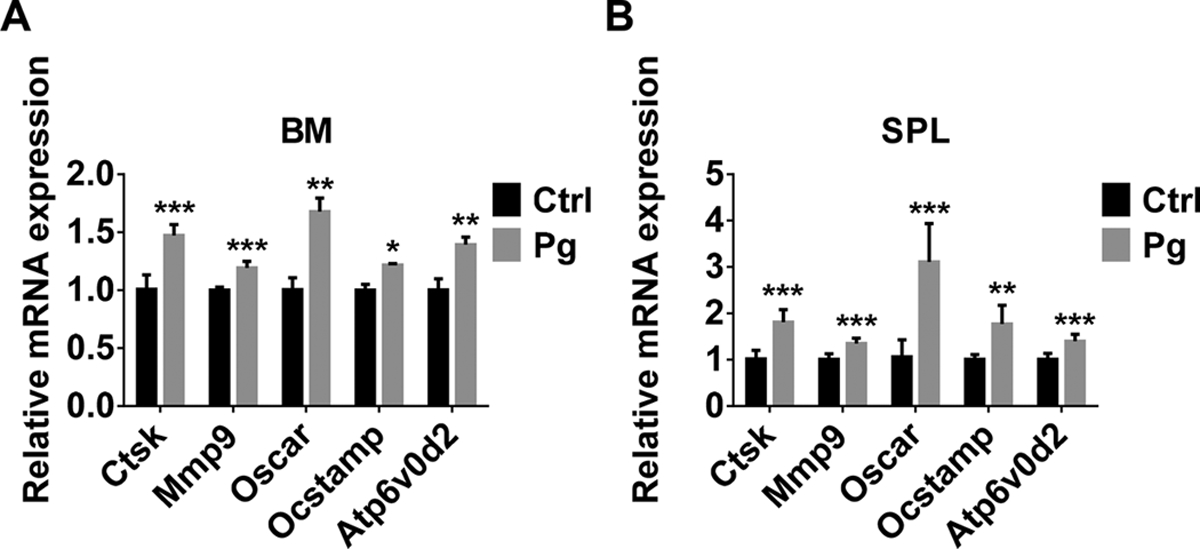
(A) mRNA levels of OC genes in BM CD11b+c-fms+Ly6Chi population of Ctrl and Pg-infected mice treated with RANKL for 48 h (n=6). (B) mRNA levels of OC genes in SPL CD11b+c-fms+Ly6Chi population of Ctrl and Pg-infected mice treated with RANKL for 48 h (n=6). Data are expressed as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
3.6. Pg infection induces a unique gene expression profile in CD11b+c-fms+Ly6Chi OCP
To further understand the possible mechanisms underlying the increased osteoclastogenic potential of OCP following Pg infection, RNA-Seq analysis was done with CD11b+c-fms+Ly6Chi population sorted from SPL of control and Pg-infected mice. Lots of gene expression in CD11b+c-fms+Ly6Chi population was affected by Pg infection, with a total of 1076 genes being upregulated and a total of 963 genes being downregulated (Fig. 6A). Importantly, gene expression of Irf8, Mafb, NR4A1 and ApoE, which have been shown to negatively regulate OC differentiation [40, 44], were significantly downregulated in CD11b+c-fms+Ly6Chi population following Pg infection (Fig. 6B). Moreover, gene expressions of S100A8 and S100A9, which are calcium-binding proteins known to promote OC differentiation [45, 46], were significantly increased in CD11b+c-fms+Ly6Chi population in Pg-infected mice (Fig. 6B). We also observed a significant upregulation of Foxm1 gene in CD11b+c-fms+Ly6Chi population of Pg-infected mice, which was recently reported as a critical regulator of the osteoclastogenic potential of OCP in the arthritic joints of mice, and contributes to arthritis-induced bone destruction [41]. The differential regulation of these genes in OCP following Pg infection was confirmed by qPCR (Fig. 6C).
FIGURE 6. A unique gene expression profile in CD11b+c-fms+Ly6Chi population is induced by Pg infection.
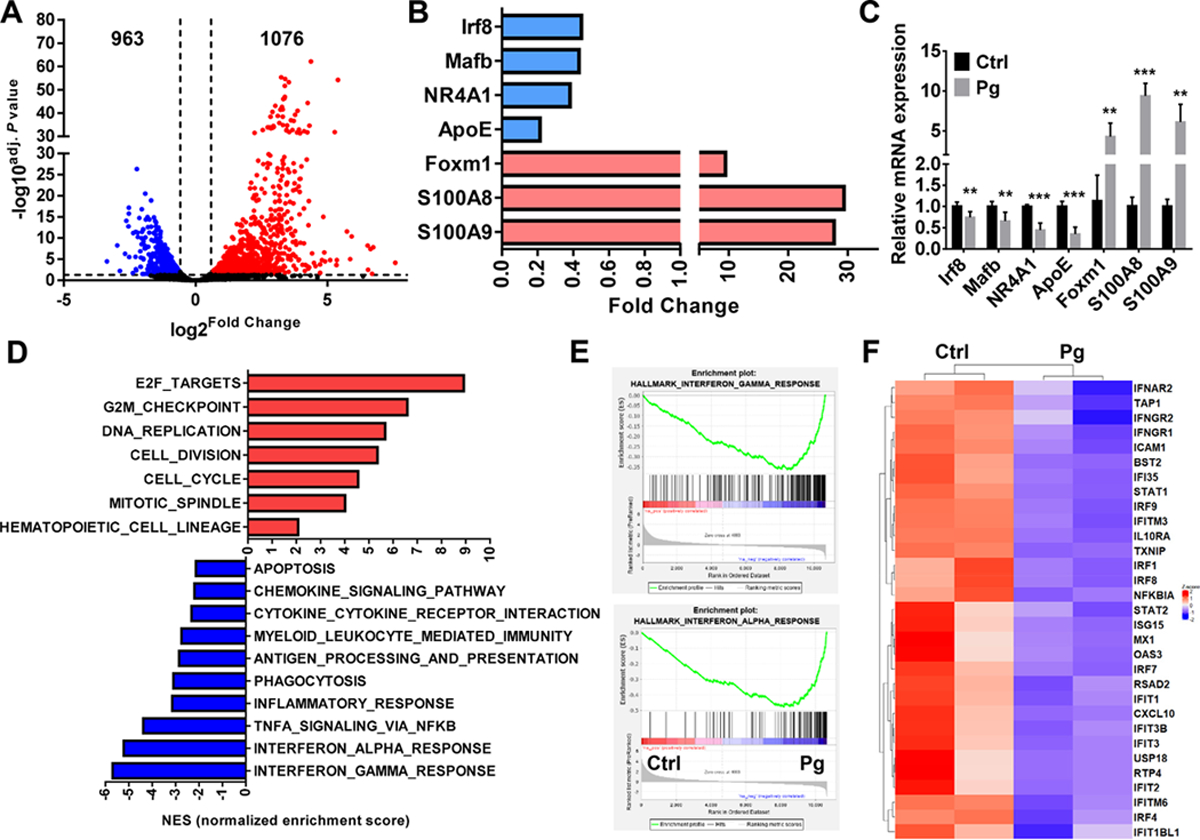
Cell sorting was followed by mRNA extraction and RNA-Seq (n=2). (A) Volcano plot showing upregulated (red) and downregulated genes (blue) in CD11b+c-fms+Ly6Chi population of Pg-infected mice relative to control ones. (B) Changed genes related with OC differentiation in CD11b+c-fms+Ly6Chi population of Pg-infected mice relative to control ones. (C) mRNA levels of indicated genes in CD11b+c-fms+Ly6Chi population of Ctrl and Pg-infected mice (n=6). (D) GSEA of upregulated and downregulated pathways in CD11b+c-fms+Ly6Chi population of Pg-infected mice relative to control ones ranked by normalized enrichment score (NES). (E) The GSEA enrichment plot of the genes related with IFN-α and IFN-r and response. (F) Heatmap of differentially expressed genes (DEGs) related with IFN-α and IFN-r pathway. Data are expressed as mean ± SD. **P < 0.01, ***P < 0.001.
Furthermore, GSEA showed that multiple signaling pathways related to cell proliferation were notably enhanced in CD11b+c-fms+Ly6Chi population from Pg-infected mice (Fig. 6D). Consistently, the apoptosis pathway was decreased in CD11b+c-fms+Ly6Chi population after Pg infection, which may explain the accumulation of this cell population in Pg-infected mice. GSEA also showed downregulation of pathways related to inflammation, phagocytosis and immunity, as well as antigen processing and presentation, in CD11b+c-fms+Ly6Chi population in Pg-infected mice. Noteworthy, the main downregulation involved interferon responses (IFN-γ and IFN-α) (Fig. 6D, E). Genes such as Ifit3, Irf1, Irf8, Ifna1, Ifngr1, Ifngr2 and Ifnar2 were dramatically downregulated in CD11b+c-fms+Ly6Chi population in Pg-infected mice compared with controls (Fig. 6F). Taken together, the data indicate that Pg infection promote the osteoclastogenic activity of BM and peripheral cells by priming CD11b+c-fms+Ly6Chi OCP toward OC lineage rather than inflammatory monocytes or macrophages.
3.7. IL-6 deficiency impairs Pg-induced accumulation of CD11b+c-fms+Ly6Chi OCP within BM and SPL
IL-6 is known to be induced by Pg, regulate bone homeostasis and inhibition of IL-6 is beneficial for the treatment of bone destruction in RA [47]. To determine if IL-6 participate in Pg-induced regulation of CD11b+c-fms+Ly6Chi OCP, we first analyzed the serum IL-6 response following Pg infection. Significantly increased serum IL-6 levels were observed at 6 h and 14 days after Pg infection in WT mice compared to control mice (Fig. 7A). We next investigated if Pg infection could induce the accumulation of CD11b+c-fms+Ly6Chi OCP of BM and SPL of IL-6 KO mice. Compared to IL-6 KO mice, the percentage of CD11b+c-fms+Ly6Chi population of BM (Fig. 7B) and SPL (Fig. 7D) of WT mice showed no significant difference. However, unlike WT mice, Pg infection of IL-6 KO mice was unable to induce a significant augmentation in the frequency of CD11b+c-fms+Ly6Chi population of BM and SPL. In addition, no significant difference in the number of total BM and SPL cells was observed in IL-6 KO mice following Pg infection (Fig. 7C,E). These results indicate that IL-6 participates in accumulation of CD11b+c-fms+Ly6Chi OCP in BM and SPL mediated by Pg.
Figure 7. IL-6 deficiency impairs Pg-mediated accumulation of CD11b+c-fms+Ly6Chi population in BM and SPL.
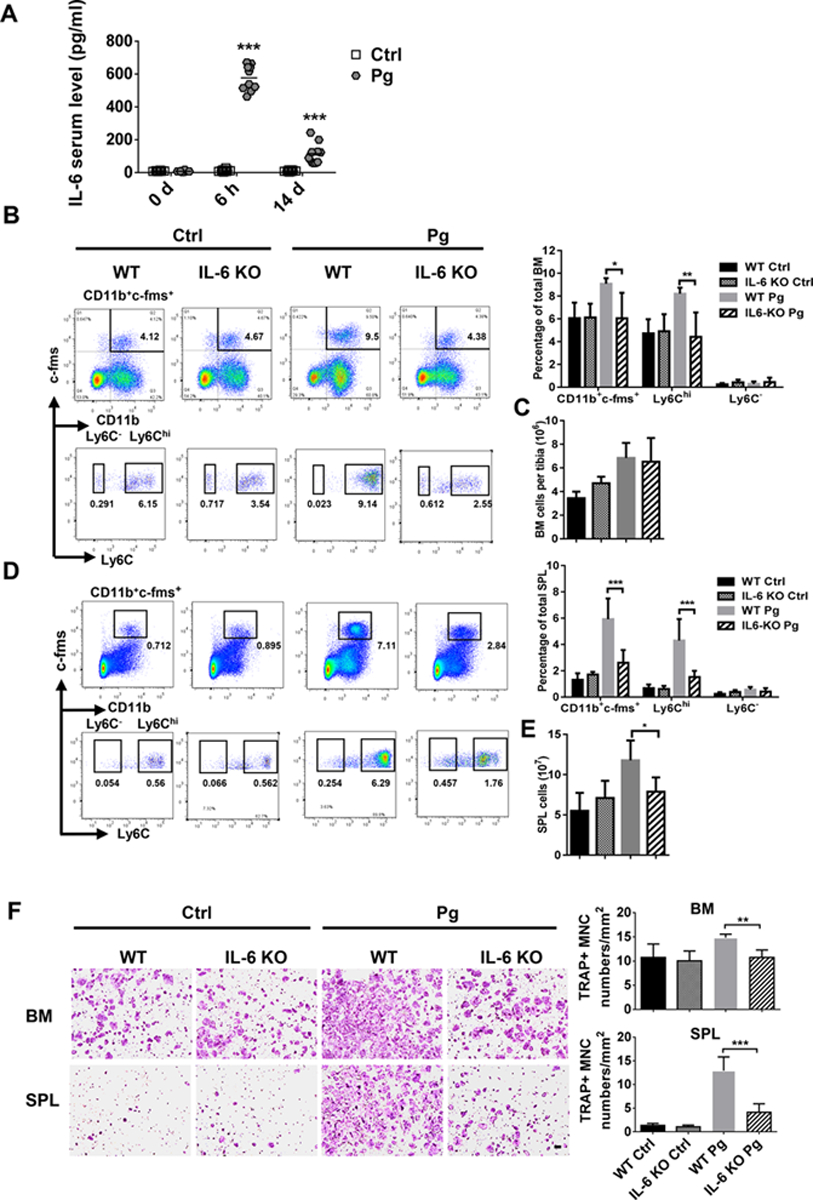
(A) Serum IL-6 level at 0 h, 6 h and day 14 after Pg infection (n=10). (B) Percentage of myeloid cells subpopulations in BM of Ctrl and Pg-infected WT and IL-6 KO mice. (C) Total cell numbers in the BM of Ctrl and Pg-infected groups of WT and IL-6 KO mice (n=4 for WT and IL-6 KO control mice and n=6 for WT and IL-6 KO Pg-infected mice in B and C). (D) Percentage of myeloid cells subpopulations in SPL of Ctrl and Pg-infected groups of WT and IL-6 KO mice. (E) Total cell numbers in SPL of Ctrl and Pg-infected groups of WT and IL-6 KO mice (n=8 for WT control mice, n=6 for IL-6 KO control mice, n=7 for WT Pg-infected control mice and n=6 for IL-6 KO Pg-infected mice in D and E). (F) Multinucleated TRAP+ OC formation in the cultures of BM and SPL cells from WT and IL-6 KO mice with or without Pg infection (n=4 for WT and IL-6 KO control mice and n=6 for WT and IL-6 KO Pg-infected mice of BM; and n=5 for WT and IL-6 KO control mice and n=6 for WT and IL-6 KO Pg-infected mice of SPL). Data are expressed as mean ± SD. Scale bar, 200 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
We further tested the osteoclastogenic ability of BM and SPL cells from IL-6 KO mice after Pg infection. Results showed that basal levels of OC formation were comparable between WT and IL-6 KO mice; however, Pg infection failed to enhance the osteoclastogenic ability of BM and SPL cells from IL-6 KO mice, as seen in WT mice (Fig. 7F). These results suggest that although IL-6 participates in Pg-induced accumulation of CD11b+c-fms+Ly6Chi OCP, endogenous IL-6 does not affect RANKL-induced OC differentiation of these cells.
4. DISCUSSION
Periodontitis is a chronic infectious disease induced by the key pathogen Pg, and characterized by irreversible bone loss. Elucidating the mechanisms leading to Pg-induced bone loss is of great importance for us understanding the pathogenesis of the disease and developing more effective treatments. In recent years, the focus of numerous investigations has been on the importance of OCP in autoimmune/autoinflammatory diseases of bone disorders; however, there is still limited knowledge on the role of OCP in bacterial infections. Furthermore, the identification of OCP in different tissues and in different disease conditions is a controversial issue. In this study, using a mouse model of chronic infection, we show that the Pg significantly enhances the osteoclastogenic activity of BM, SPL and PB cells, which is predominantly mediated by the increased frequency and osteoclastogenic activity of CD11b+c-fms+Ly6Chi population.
The current identification of cell surface markers of OCP are remarkably varied. Positive expression of c-fms is well established in OCP from the BM, SPL and PB under physiological and arthritic conditions [10, 11]. CD11b is another important marker used for the identification of OCP. While BM CD11b−/low cells in mice were reported as OCP under physiological condition and in an inflammatory arthritis model [9, 11], other studies identified positive expression of CD11b as an important marker of OCP in SPL and PB under physiological conditions and in the BM and PB of the hTNF-α–Tg arthritis model [4, 11, 13]. In the current study, we observed that both CD11b−c-fms+ and CD11b+c-fms+ populations were able to differentiate into OC in vitro in the presence of RANKL (data not shown). However, a significantly higher percentage of CD11b+c-fms+ population, as compared to CD11b−c-fms+ population, was found within the BM, SPL and PB. Furthermore, Pg chronic infection significantly increased the frequency of CD11b+c-fms+ population in BM, SPL and PB. Although controversial, Ly6C has been used as an important marker for the identification of OCP [9, 10, 41, 48]. In the present study, the frequency of CD11b+c-fms+Ly6Chi population increased significantly within the BM, SPL and PB after Pg infection. Furthermore, a stronger osteoclastogenic activity than that seen within CD11b+c-fms+Ly6C− cells was shown in vitro, and the ability of CD11b+c-fms+Ly6Chi population to differentiate into OC in vivo was confirmed. Along these lines, studies have demonstrated that CD11b+c-fms+Ly6Chi population is critical for bone erosion in arthritis [8, 13]. However, current research on OCP markers is somewhat perplexing. It’s well known that OC are derived from HSC and during this process, CD11b and c-fms expression changes from negative to positive [6, 49]. On the other hand, it has been reported that Ly6C− cells are converted from Ly6Chi cells [50]. Hence, it is reasonable to suggest that OCP express different markers under different circumstances and disease conditions. This might explain the various findings by different investigators [8–10, 13].
It has been shown that RANKL-induced osteoclastogenesis from OCP requires the activation of a subset of genes, such as Ctsk, Mmp9, Oscar, Ocstamp, and Atp6v0d2 [43]. Our results demonstrated a significant increase in these OC-related gene expression in CD11b+c-fms+Ly6Chi population of Pg-infected mice following RANKL stimulation. This observation supports the notion that OC derived from these cells exhibited an increase in size, bigger F-actin rings, and an increase in bone resorptive function. However, no significant difference was observed in the basal expression level of these OC genes in CD11b+c-fms+Ly6Chi population of control and Pg-infected mice in the absence of RANKL stimulation, suggesting that although CD11b+c-fms+Ly6Chi population of Pg-infected mice have enhanced osteoclastogenic potential, they are not set to the OC lineage. Therefore, these cells should still be able to differentiate into macrophages and dendritic cells under certain circumstances, as OCP are highly plastic [9, 11, 12].
Our RNA-seq data revealed comparable expression of OC-specific genes, e.g., Ctsk and MMP9, in CD11b+c-fms+Ly6Chi population of Pg-infected and non-infected mice (data not shown). However, a unique gene profile was identified in CD11b+c-fms+Ly6Chi population after Pg infection. Genes reported as positive regulators of OC differentiation, such as S100A8, S100A9, and Foxm1 [41, 45, 46], were significantly upregulated in CD11b+c-fms+Ly6Chi population of Pg-infected mice, whereas genes known as negative regulators of OC differentiation, like Irf8, Mafb, NR4A1 and ApoE [40, 44, 51, 52], were significantly downregulated in these cells. This outcome may account for the increased osteoclastogenic potential of these cells following Pg infection. Furthermore, GSEA results showed that the IFN pathway was the most downregulated signaling pathway, and that numerous genes associated with the IFN pathway were also downregulated in CD11b+c-fms+Ly6Chi OCP obtained from Pg-infected mice. It has been reported previously that Pg can negatively regulate the IFN signaling pathway in gingival epithelial cells [53]. Moreover, the IFN signaling pathway was shown to be involved in the inhibition of osteoclastogenesis [54–56], and IFN-γR−/− mice showed an exacerbated OC activity in endotoxin-induced bone resorption [57]. Thus, the decreased response to the IFN signal, could also be responsible for the enhanced osteoclastogenic activity of CD11b+c-fms+Ly6Chi population following Pg infection. Our RNA-Seq data also demonstrated an upregulation in cell division and differentiation pathways, as well as a downregulation in inflammatory, cytokine and antigen presenting pathways in CD11b+c-fms+Ly6Chi population, following Pg infection. These results further suggest that Pg infection skews CD11b+c-fms+Ly6Chi population toward OC and not to macrophage and dendritic cell differentiation. Overall, the RNA-seq data provides insight into potential downstream signaling pathways implicated in the enhanced osteoclastogenic activity of CD11b+c-fms+Ly6Chi population following Pg infection. However, elucidation of these pathways will be an important task of future investigations.
IL-6 is a pleiotropic cytokine that is critical in the pathogenesis of various bone diseases, including RA, osteoporosis, and bone-metastatic cancers [58–60]. Considerable evidence has accumulated indicating that serum IL-6 levels can predict postmenopausal bone loss [61]. Studies have also shown that IL-6 concentrations in the synovial fluid correlate with the severity of joint damage and the number of erosions in RA patients [62]. Blockade of IL-6 has been shown to mediate protection against joint destruction and to suppress disease activity in RA patients [47]. In the present study, we found that Pg infection resulted in a significant increase in serum IL-6 levels in WT mice, and that IL-6 deficiency impairs Pg-induced accumulation of CD11b+c-fms+Ly6Chi OCP within the BM and SPL. Elevated IL-6 concentrations were noted in salivary and periodontal tissues of periodontitis patients [63, 64]. In addition, lower numbers of OC and decreased bone lesions were found in a periodontitis model using IL-6 deficient mice [65]. Furthermore, it has been shown that IL-6 can regulate RANKL-induced OC differentiation and function, although both positive and negative effects have been reported [66–68]. Our study further showed that BM and SPL cells obtained from WT and IL-6 KO mice have comparable osteoclastogenic potential in response to RANKL, indicating that endogenous IL-6 is not required for RANKL-induced OC differentiation. Therefore, it is possible that IL-6 regulates pathological bone loss by regulating the expansion of the OCP cell pool. Understanding the molecular mechanisms that regulate Pg-induced OCP expansion is most relevant to the development of potential therapeutic targets aimed at reducing the number of OCP cells and controlling Pg-induced bone destruction.
There is growing evidence indicating that periodontal infection increases the risk of systemic diseases including RA, osteoporosis, diabetes, preterm births, cardiovascular diseases, and Alzheimer’s disease [19, 21]. It is believed that periodontitis can induce a systemic inflammatory state through mechanisms that include dissemination of inflammatory mediators, periodontal bacteria and/or their products [19, 69]. Periodontitis-associated bacteremias have been demonstrated in animal and human studies [26, 28–30]. In addition, studies have shown that Pg in the bloodstream can translocate into various tissues such as coronary arteries, placenta, and brain [25–27, 70]. Our results that regulation of precursor populations in BM and periphery following systemic Pg infection may provide new evidence underlying the linkage between periodontitis and systemic diseases. Indeed, consistent with our finding that Pg infection increases the number and osteoclastogenic function of OCP in BM and periphery, it has been shown that patients with periodontitis have increased osteoclastogenic function in peripheral blood mononuclear phagocytes [71]. In addition, a recent study using TNFtg mice as the chronic peripheral inflammation model, showed that the peripheral inflammation could cause a region-specific myeloid response in the central nervous system[72]. It is possible that such an increase of OCP will also contribute to bone loss in sites other than the alveolar bone in case of the presence of osteoclastogenic and inflammatory mediators at the remote sites. On the other hand, a marked increase in OCP frequency in the circulation was observed in psoriatic arthritis patients [73]. It is also possible that these OCP may reach the inflamed periodontal tissues, hence promoting alveolar bone loss.
Overall, the current study demonstrated that in a mouse model infected with the periodontal pathogen Pg, BM and peripheral CD11b+c-fms+Ly6Chi monocytic cells constitute an OCP population characterized by increased cell numbers and osteoclastogenic function. Moreover, our findings indicate that IL-6 is important for regulating the accumulation of these OCP. We further showed that following Pg infection there is a distinct transcriptional profile of genes related to OC differentiation and inflammatory signaling pathways that are not seen in the absence of infection. This is most relevant as specific transcriptional signatures induced by Pg infection can be associated with the resulting increase in OCP and their distinct profiles may provide new insights for effective treatment of infection/inflammation-mediated bone destruction.
Supplementary Material
ACKNOWLEDGMENTS
We appreciate Greg Harber for his technical support. This research was supported by grants from the National Institute of Dental and Craniofacial Research (R01 DE026465 to P.Z. and T90 DE022736 to Y.Z., Z.L. and L.S.). The University of Alabama at Birmingham Comprehensive Flow Cytometry Core is supported by the National Institutes of Health grants P30AI27667 and P30AR048311.
ABBREVIATIONS
- BM
bone marrow
- HSC
hematopoietic stem cells
- M-CSF
macrophage colony-stimulating factor
- OC
osteoclasts
- OCP
osteoclast precursors
- PB
peripheral blood
- Pg
Porphyromonas gingivalis
- RANKL
receptor activator of NF-κB ligand
- RA
rheumatoid arthritis
- SPL
spleen
- TRAP
tartrate-resistant acid phosphatase
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
REFERENCES
- 1.Cochran DL (2008) Inflammation and bone loss in periodontal disease. J Periodontol 79, 1569–76. [DOI] [PubMed] [Google Scholar]
- 2.Goldring SR and Gravallese EM (2000) Pathogenesis of bone erosions in rheumatoid arthritis. Current opinion in rheumatology 12, 195–9. [DOI] [PubMed] [Google Scholar]
- 3.Teitelbaum SL and Ross FP (2003) Genetic regulation of osteoclast development and function. Nature reviews. Genetics 4, 638–49. [DOI] [PubMed] [Google Scholar]
- 4.Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, Boyce BF, Xing L (2006) Tumor necrosis factor-alpha increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem 281, 11846–55. [DOI] [PubMed] [Google Scholar]
- 5.Kotani M, Kikuta J, Klauschen F, Chino T, Kobayashi Y, Yasuda H, Tamai K, Miyawaki A, Kanagawa O, Tomura M, Ishii M (2013) Systemic circulation and bone recruitment of osteoclast precursors tracked by using fluorescent imaging techniques. J Immunol 190, 605–12. [DOI] [PubMed] [Google Scholar]
- 6.Long CL and Humphrey MB (2012) Osteoimmunology: the expanding role of immunoreceptors in osteoclasts and bone remodeling. Bonekey Rep 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sucur A, Katavic V, Kelava T, Jajic Z, Kovacic N, Grcevic D (2014) Induction of osteoclast progenitors in inflammatory conditions: key to bone destruction in arthritis. Int Orthop 38, 1893–903. [DOI] [PubMed] [Google Scholar]
- 8.Seeling M, Hillenhoff U, David JP, Schett G, Tuckermann J, Lux A, Nimmerjahn F (2013) Inflammatory monocytes and Fcgamma receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc Natl Acad Sci U S A 110, 10729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charles JF, Hsu LY, Niemi EC, Weiss A, Aliprantis AO, Nakamura MC (2012) Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J Clin Invest 122, 4592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puchner A, Saferding V, Bonelli M, Mikami Y, Hofmann M, Brunner JS, Caldera M, Goncalves-Alves E, Binder NB, Fischer A, Simader E, Steiner CW, Leiss H, Hayer S, Niederreiter B, Karonitsch T, Koenders MI, Podesser BK, O’Shea JJ, Menche J, Smolen JS, Redlich K, Bluml S (2018) Non-classical monocytes as mediators of tissue destruction in arthritis. In Ann Rheum Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacome-Galarza CE, Lee SK, Lorenzo JA, Aguila HL (2013) Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J Bone Miner Res 28, 1203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao Y, Palomero J, Grabowska J, Wang L, de Rink I, van Helvert L, Borst J (2017) Macrophages and osteoclasts stem from a bipotent progenitor downstream of a macrophage/osteoclast/dendritic cell progenitor. Blood Adv 1, 1993–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ammari M, Presumey J, Ponsolles C, Roussignol G, Roubert C, Escriou V, Toupet K, Mausset-Bonnefont AL, Cren M, Robin M, Georgel P, Nehmar R, Taams L, Grun J, Grutzkau A, Haupl T, Pers YM, Jorgensen C, Duroux-Richard I, Courties G, Apparailly F (2018) Delivery of miR-146a to Ly6C(high) Monocytes Inhibits Pathogenic Bone Erosion in Inflammatory Arthritis. Theranostics 8, 5972–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gengenbacher M, Sebald HJ, Villiger PM, Hofstetter W, Seitz M (2008) Infliximab inhibits bone resorption by circulating osteoclast precursor cells in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis 67, 620–4. [DOI] [PubMed] [Google Scholar]
- 15.Perpetuo IP, Caetano-Lopes J, Rodrigues AM, Campanilho-Marques R, Ponte C, Canhao H, Ainola M, Fonseca JE (2017) Effect of Tumor Necrosis Factor Inhibitor Therapy on Osteoclasts Precursors in Rheumatoid Arthritis. Biomed Res Int 2017, 2690402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uster S, Coelho FM, Aeberli D, Stein JV, Hofstetter W, Engelhardt B, Seitz M (2018) TNFalpha blockade mediates bone protection in antigen-induced arthritis by reducing osteoclast precursor supply. Bone 107, 56–65. [DOI] [PubMed] [Google Scholar]
- 17.Cafiero C, Gigante M, Brunetti G, Simone S, Chaoul N, Oranger A, Ranieri E, Colucci S, Pertosa GB, Grano M, Gesualdo L (2018) Inflammation induces osteoclast differentiation from peripheral mononuclear cells in chronic kidney disease patients: crosstalk between the immune and bone systems. Nephrol Dial Transplant 33, 65–75. [DOI] [PubMed] [Google Scholar]
- 18.Hajishengallis G and Lamont RJ (2012) Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol 27, 409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajishengallis G (2015) Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 15, 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ (2012) Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res 91, 914–20. [DOI] [PubMed] [Google Scholar]
- 21.Sandal I, Karydis A, Luo J, Prislovsky A, Whittington KB, Rosloniec EF, Dong C, Novack DV, Mydel P, Zheng SG, Radic MZ, Brand DD (2016) Bone loss and aggravated autoimmune arthritis in HLA-DRbeta1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res Ther 18, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cutler CW, Kalmar JR, Genco CA (1995) Pathogenic strategies of the oral anaerobe, Porphyromonas gingivalis. Trends in microbiology 3, 45–51. [DOI] [PubMed] [Google Scholar]
- 23.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajishengallis G, Darveau RP, Curtis MA (2012) The keystone-pathogen hypothesis. Nat Rev Microbiol 10, 717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu SW, Huang CH, Huang HC, Lai YY, Lin YY (2006) Transvascular dissemination of Porphyromonas gingivalis from a sequestered site is dependent upon activation of the kallikrein/kinin pathway. J Periodontal Res 41, 200–7. [DOI] [PubMed] [Google Scholar]
- 26.Figuero E, Lindahl C, Marin MJ, Renvert S, Herrera D, Ohlsson O, Wetterling T, Sanz M (2014) Quantification of periodontal pathogens in vascular, blood, and subgingival samples from patients with peripheral arterial disease or abdominal aortic aneurysms. J Periodontol 85, 1182–93. [DOI] [PubMed] [Google Scholar]
- 27.Mougeot JC, Stevens CB, Paster BJ, Brennan MT, Lockhart PB, Mougeot FK (2017) Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. Journal of oral microbiology 9, 1281562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brodala N, Merricks EP, Bellinger DA, Damrongsri D, Offenbacher S, Beck J, Madianos P, Sotres D, Chang YL, Koch G, Nichols TC (2005) Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol 25, 1446–51. [DOI] [PubMed] [Google Scholar]
- 29.Horliana AC, Chambrone L, Foz AM, Artese HP, Rabelo Mde S, Pannuti CM, Romito GA (2014) Dissemination of periodontal pathogens in the bloodstream after periodontal procedures: a systematic review. PLoS One 9, e98271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ambrosio N, Marin MJ, Laguna E, Herrera D, Sanz M, Figuero E (2019) Detection and quantification of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans in bacteremia induced by interdental brushing in periodontally healthy and periodontitis patients. Arch Oral Biol 98, 213–219. [DOI] [PubMed] [Google Scholar]
- 31.Zhang P, Liu J, Xu Q, Harber G, Feng X, Michalek SM, Katz J (2011) TLR2-dependent modulation of osteoclastogenesis by Porphyromonas gingivalis through differential induction of NFATc1 and NF-kappaB. J Biol Chem 286, 24159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai X, Li Z, Zhao Y, Katz J, Michalek SM, Feng X, Li Y, Zhang P (2020) Enhanced dual function of osteoclast precursors following calvarial Porphyromonas gingivalis infection. J Periodontal Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Shi Z, Chen M, Li K, Li H, Wang S, Tang F, Xiong W, Xu H, Kesterson RA, Feng X (In preparation) A Csf1r-Cre knockin mouse model for studying the differentiation and function of cells of the monocyte/macrophage lineage. [Google Scholar]
- 35.Su L, Xu Q, Zhang P, Michalek SM, Katz J (2017) Phenotype and Function of Myeloid-Derived Suppressor Cells Induced by Porphyromonas gingivalis Infection. Infect Immun 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD (2018) CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci 3, 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Shi Z, Jules J, Chen S, Kesterson RA, Zhao D, Zhang P, Feng X (2019) Specific RANK Cytoplasmic Motifs Drive Osteoclastogenesis. J Bone Miner Res 34, 1938–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Wang S, Zhang P, Said-Al-Naief N, Michalek SM, Feng X (2009) Molecular mechanism of the bifunctional role of lipopolysaccharide in osteoclastogenesis. J Biol Chem 284, 12512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cremers NAJ, van den Bosch MHJ, van Dalen S, Di Ceglie I, Ascone G, van de Loo F, Koenders M, van der Kraan P, Sloetjes A, Vogl T, Roth J, Geven EJW, Blom AB, van Lent P (2017) S100A8/A9 increases the mobilization of pro-inflammatory Ly6C(high) monocytes to the synovium during experimental osteoarthritis. Arthritis Res Ther 19, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scholtysek C, Ipseiz N, Bohm C, Krishnacoumar B, Stenzel M, Czerwinski T, Palumbo-Zerr K, Rothe T, Weidner D, Klej A, Stoll C, Distler J, Tuckermann J, Herrmann M, Fabry B, Goldmann WH, Schett G, Kronke G (2018) NR4A1 Regulates Motility of Osteoclast Precursors and Serves as Target for the Modulation of Systemic Bone Turnover. J Bone Miner Res 33, 2035–2047. [DOI] [PubMed] [Google Scholar]
- 41.Hasegawa T, Kikuta J, Sudo T, Matsuura Y, Matsui T, Simmons S, Ebina K, Hirao M, Okuzaki D, Yoshida Y, Hirao A, Kalinichenko VV, Yamaoka K, Takeuchi T, Ishii M (2019) Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nature immunology 20, 1631–1643. [DOI] [PubMed] [Google Scholar]
- 42.Yan Z, Yang W, Parkitny L, Gibson SA, Lee KS, Collins F, Deshane JS, Cheng W, Weinmann AS, Wei H, Qin H, Benveniste EN (2019) Deficiency of Socs3 leads to brain-targeted EAE via enhanced neutrophil activation and ROS production. JCI insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423, 337–342. [DOI] [PubMed] [Google Scholar]
- 44.Kim WS, Kim HJ, Lee ZH, Lee Y, Kim HH (2013) Apolipoprotein E inhibits osteoclast differentiation via regulation of c-Fos, NFATc1 and NF-kappaB. Exp Cell Res 319, 436–46. [DOI] [PubMed] [Google Scholar]
- 45.Grevers LC, de Vries TJ, Vogl T, Abdollahi-Roodsaz S, Sloetjes AW, Leenen PJ, Roth J, Everts V, van den Berg WB, van Lent PL (2011) S100A8 enhances osteoclastic bone resorption in vitro through activation of Toll-like receptor 4: implications for bone destruction in murine antigen-induced arthritis. Arthritis Rheum 63, 1365–75. [DOI] [PubMed] [Google Scholar]
- 46.Dapunt U, Giese T, Maurer S, Stegmaier S, Prior B, Hansch GM, Gaida MM (2015) Neutrophil-derived MRP-14 is up-regulated in infectious osteomyelitis and stimulates osteoclast generation. J Leukoc Biol 98, 575–82. [DOI] [PubMed] [Google Scholar]
- 47.Kang S, Tanaka T, Narazaki M, Kishimoto T (2019) Targeting Interleukin-6 Signaling in Clinic. Immunity 50, 1007–1023. [DOI] [PubMed] [Google Scholar]
- 48.Sprangers S, Schoenmaker T, Cao Y, Everts V, de Vries TJ (2016) Different Blood-Borne Human Osteoclast Precursors Respond in Distinct Ways to IL-17A. J Cell Physiol 231, 1249–60. [DOI] [PubMed] [Google Scholar]
- 49.Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, Miyata T, Anderson DM, Suda T (1999) Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med 190, 1741–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mildner A, Schonheit J, Giladi A, David E, Lara-Astiaso D, Lorenzo-Vivas E, Paul F, Chappell-Maor L, Priller J, Leutz A, Amit I, Jung S (2017) Genomic Characterization of Murine Monocytes Reveals C/EBPbeta Transcription Factor Dependence of Ly6C(−) Cells. Immunity 46, 849–862.e7. [DOI] [PubMed] [Google Scholar]
- 51.Kim K, Kim JH, Lee J, Jin HM, Kook H, Kim KK, Lee SY, Kim N (2007) MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood 109, 3253–9. [DOI] [PubMed] [Google Scholar]
- 52.Zhao B, Takami M, Yamada A, Wang X, Koga T, Hu X, Tamura T, Ozato K, Choi Y, Ivashkiv LB, Takayanagi H, Kamijo R (2009) Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med 15, 1066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jauregui CE, Wang Q, Wright CJ, Takeuchi H, Uriarte SM, Lamont RJ (2013) Suppression of T-cell chemokines by Porphyromonas gingivalis. Infect Immun 81, 2288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang M, Tian L, Luo G, Yu X (2018) Interferon-Gamma-Mediated Osteoimmunology. Front Immunol 9, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB (2008) TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nature immunology 9, 378–87. [DOI] [PubMed] [Google Scholar]
- 56.Zhao B and Ivashkiv LB (2011) Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Research & Therapy 13, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T (2000) T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408, 600–5. [DOI] [PubMed] [Google Scholar]
- 58.Tawara K, Oxford JT, Jorcyk CL (2011) Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer management and research 3, 177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hashizume M and Mihara M (2011) The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis 2011, 765624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boyce B, Broxmeyer H, Manolagas SC (1992) Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science (New York, N.Y.) 257, 88–91. [DOI] [PubMed] [Google Scholar]
- 61.Scheidt-Nave C, Bismar H, Leidig-Bruckner G, Woitge H, Seibel MJ, Ziegler R, Pfeilschifter J (2001) Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J Clin Endocrinol Metab 86, 2032–42. [DOI] [PubMed] [Google Scholar]
- 62.Holt I, Cooper RG, Hopkins SJ (1991) Relationships between local inflammation, interleukin-6 concentration and the acute phase protein response in arthritis patients. European journal of clinical investigation 21, 479–84. [DOI] [PubMed] [Google Scholar]
- 63.Batool H, Nadeem A, Kashif M, Shahzad F, Tahir R, Afzal N (2018) Salivary Levels of IL-6 and IL-17 Could Be an Indicator of Disease Severity in Patients with Calculus Associated Chronic Periodontitis. Biomed Res Int 2018, 8531961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ross JH, Hardy DC, Schuyler CA, Slate EH, Mize TW, Huang Y (2010) Expression of periodontal interleukin-6 protein is increased across patients with neither periodontal disease nor diabetes, patients with periodontal disease alone and patients with both diseases. J Periodontal Res 45, 688–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baker PJ, Dixon M, Evans RT, Dufour L, Johnson E, Roopenian DC (1999) CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun 67, 2804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Q, Zhou X, Huang D, Ji Y, Kang F (2017) IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell Physiol Biochem 41, 1360–1369. [DOI] [PubMed] [Google Scholar]
- 67.Axmann R, Bohm C, Kronke G, Zwerina J, Smolen J, Schett G (2009) Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum 60, 2747–56. [DOI] [PubMed] [Google Scholar]
- 68.Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S (2008) Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J Biol Chem 283, 11535–40. [DOI] [PubMed] [Google Scholar]
- 69.Cullinan MP and Seymour GJ (2013) Periodontal disease and systemic illness: will the evidence ever be enough? Periodontol 2000 62, 271–86. [DOI] [PubMed] [Google Scholar]
- 70.Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J (2019) Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Science advances 5, eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herrera BS, Bastos AS, Coimbra LS, Teixeira SA, Rossa C Jr., Van Dyke TE, Muscara MN, Spolidorio LC (2014) Peripheral blood mononuclear phagocytes from patients with chronic periodontitis are primed for osteoclast formation. J Periodontol 85, e72–81. [DOI] [PubMed] [Google Scholar]
- 72.Suss P, Hoffmann A, Rothe T, Ouyang Z, Baum W, Staszewski O, Schett G, Prinz M, Kronke G, Glass CK, Winkler J, Schlachetzki JCM (2020) Chronic Peripheral Inflammation Causes a Region-Specific Myeloid Response in the Central Nervous System. Cell Rep 30, 4082–4095 e6. [DOI] [PubMed] [Google Scholar]
- 73.Chiu YG, Shao T, Feng C, Mensah KA, Thullen M, Schwarz EM, Ritchlin CT (2010) CD16 (FcRgammaIII) as a potential marker of osteoclast precursors in psoriatic arthritis. Arthritis Res Ther 12, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.