

Abstract
Alagille syndrome is an inherited multisystemic disorder. We herein report an atypical case of a Japanese adult patient with Alagille syndrome. He had been diagnosed with Alagille syndrome as an infant based on a liver biopsy. At 27 years of age, he needed to start hemodialysis therapy, but an arteriovenous fistula was not created because his peripheral blood vessels were too narrow. He also had a recurrent brain infarction due to cerebral vascular stenosis. Alagille syndrome is generally recognized as a pediatric hepatic disease, but general physicians should be aware of its potential existence with renal involvement and vascular abnormalities.
Keywords: adult, Alagille syndrome, brain infarction, renal involvement, vascular abnormality
Introduction
Alagille syndrome is a rare autosomal-dominant multisystemic disorder that is classically defined by the presence of intrahepatic bile duct paucity and three or more of the following five clinical features: cholestasis associated with bile duct paucity on a liver biopsy, facial abnormalities (e.g. prominent forehead, hypertelorism, pointed chin, straight nose), cardiac abnormalities, dominantly pulmonary artery stenosis, skeletal abnormalities (mainly butterfly vertebrae), and ocular abnormalities (1). The JAG1 gene mutation and NOTCH2 gene mutation are major causes of Alagille syndrome. The JAG1 gene encodes jagged1 ligands, which are the main molecules for the Notch signaling pathway, and the NOTCH2 gene encodes Notch2 receptors, which are one of the four Notch transmembrane receptors. In addition to the above symptoms, renal abnormalities and vascular anomalies are also important manifestations of Alagille syndrome and are major causes of later mortality (2, 3).
The presence of an atypical type of Alagille syndrome that shows at least one of the major five symptoms with mutations of JAG1 or NOTCH2 has been reported. In general, although Alagille syndrome is thought to be a pediatric disease with chronic cholestasis, there are adult patients who show predominantly renal features without other manifestations (4, 5), and the prevalence of renal involvement is reported to be relatively high at 39% (2). Although vascular anomalies-especially pulmonary artery stenosis and intracranial bleeding-are major manifestations of Alagille syndrome, failure to achieve vascular access for dialysis therapy due to peripheral vascular anomalies and/or brain infarction due to intracranial vascular stenosis is rarely observed.
We herein report the case of an adult patient with Alagille syndrome who showed mainly renal failure, peripheral vascular stenosis, and recurrent brain infarction without liver manifestations.
Case Report
A 27-year-old Japanese man was referred to our hospital for the initiation of dialysis therapy. At 1 month after birth, he had shown neonatal jaundice and been diagnosed with Alagille syndrome based on his family history and the results of a liver biopsy. His father, older sister, and younger sister also had Alagille syndrome. His father had undergone dialysis therapy, and his younger sister had shown renal dysfunction. His older sister had died at three years of age due to biliary atresia. Although the patient's jaundice improved with age, he also had renal dysfunction from a young age, which gradually worsened. His serum creatinine level was around 3.0 mg/dL until 22 years of age, and a urinalysis had shown urine protein (1 to 2+) and occult blood (-). He stopped follow-up based on his own judgment at 22 years old. At 27 years old, he suffered a gout attack and consulted a neighborhood clinic, where he was diagnosed with gout and end-stage kidney disease and referred to our hospital to start dialysis therapy.
On admission, the patient exhibited no cardiac or skeletal manifestations (Fig. 1). His blood pressure was 162/76 mmHg, and his pulse rate were 109 beats/min. His body temperature was 37.2°C. He had uremic symptoms, including nausea, fatigue, and appetite loss, but no pretibial edema. Laboratory data on admission showed a serum creatinine level of 8.79 mg/dL and blood urea nitrogen of 69.9 mg/dL; hemoglobin and serum albumin were 9.8 g/dL and 4.2 g/dL, respectively. The C-reactive protein level was 0.68 mg/dL. The white blood cell count, platelets, alanine aminotransferase, aspartate aminotransferase, total bilirubin, alkaline phosphatase, gamma-glutamyl transpeptidase, lactate dehydrogenase, and blood glucose values were all within normal limits. The patient had hyperuricemia (11.8 mg/dL) and hyperphosphatemia (5.8 mg/dL). A urinalysis showed only proteinuria (1.3 g/gCre). The patient showed no morphological kidney abnormalities on imaging except for atrophic kidney.
Figure 1.
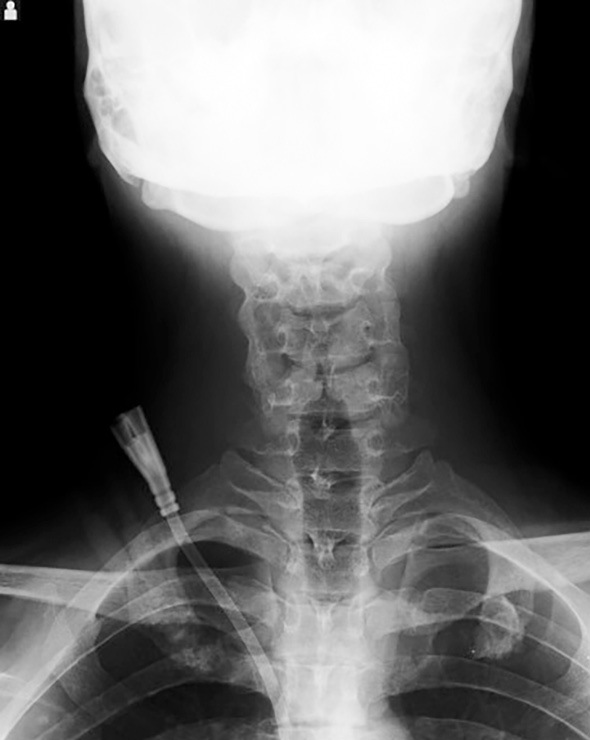
Cervical X-ray image of the patient, a 27-year-old man. Butterfly vertebrae were not observed.
Although he had already been diagnosed with Alagille syndrome as a child, we conducted a genetic analysis because we did not have his genetic information. An amplicon library of the target exons that contained the JAG1 and NOTCH2 was prepared, and next-generation sequencing was performed with an Ion Torrent Personal Genome Machine (Ion PGM) system (Life Technologies, Carlsbad, USA). A mutation analysis revealed a missense mutation c.2938T>C (p.C980R) in exon 24 of the JAG 1 gene; this mutation had been reported previously (6). The minor allele frequency of C980R in the Japanese Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp/index.html) and in the Exome Aggregation Consortium dataset (http://exac.broadinstitute.org/) had not been reported. These data indicated that C980R was a very rare variant. The effect of this missense mutation was predicted to be damaging by Sorting Intolerant From Tolerant (http://sift.jcvi.org/; J. Craig Venter Institute, Rockville, USA) and probably damaging by Polymorphism Phenotyping-2 (http://genetics.bwh.harvard.edu/pph2/; Harvard Medical School, Boston, USA). These findings led to the diagnosis of Alagille syndrome in our patient.
Since the patient selected hemodialysis therapy as renal replacement therapy, a preoperative check of the radial artery and cephalic vein was conducted. Although we could not detect the cephalic vein of the carpal region with the naked eye, it was observed as a narrow vein after avascularization by vascular ultrasound. Vascular calcification of the radial artery was not observed by vascular ultrasound, but the vascular diameter of the radial artery was small. An arteriovenous fistula (AVF) was surgically created but unfortunately failed due to radial arterial and cephalic vein stenosis, particularly cephalic vein stenosis. Instead of an AVF, the implantation of a brachioaxillary arteriovenous graft (AVG) was attempted. Although the blood flow of the AVG was observed after the operation, the patient showed left ptosis and oculomotor nerve paralysis the following day. He was diagnosed with a brain infarction of the ventral midbrain, left medial thalamus, and left cerebellum by brain magnetic resonance imaging (MRI) (Fig. 2) and started anticoagulant therapy. At that time, magnetic resonance angiography (MRA) revealed stenosis of both the vertebral artery and basilar artery in addition to a small aneurysm at the right internal carotid artery (Fig. 3).
Figure 2.
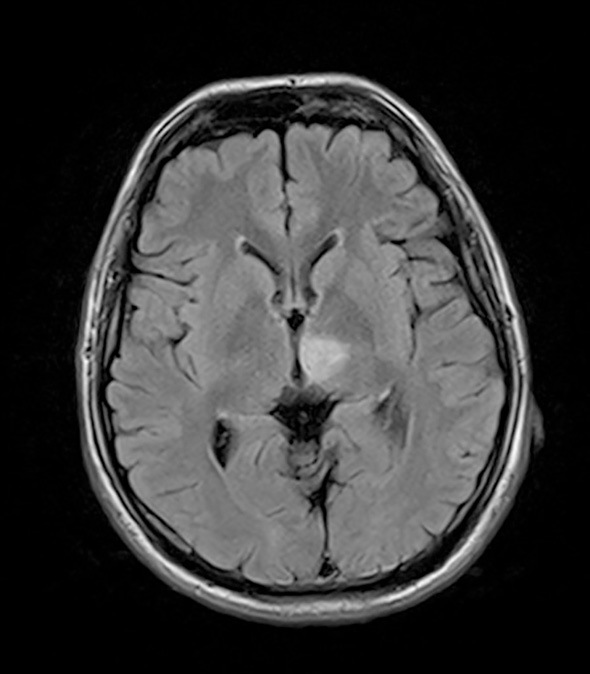
Brain MRI [fluid-attenuated inversion recovery (FLAIR) image]. A high-density area is observed in the ventral midbrain and left medial thalamus.
Figure 3.
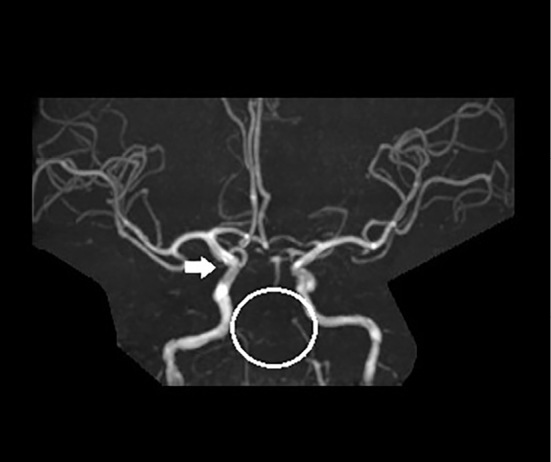
Brain MRA. A small aneurysm was observed at the branch of the right internal carotid artery and ophthalmic artery (white arrow). Vertebral artery stenosis and basilar artery stenosis were observed (white circle).
Two weeks after the generation of the AVG, the blood flow of the AVG had disappeared. Although percutaneous transluminal angioplasty (PTA) was conducted immediately for AVG occlusion, recanalization was not achieved. The day after PTA, brain infarction recurred. The symptoms were right hemiparesis and dysarthria, and we diagnosed him with a brain infarction of the left thalamus, cerebral peduncle, and cerebellar hemisphere. Eventually, a permanent hemodialysis catheter was inserted in the patient's internal jugular vein so that he could continue hemodialysis therapy. He is now undergoing hemodialysis therapy 3 times per week and rehabilitation to facilitate recovery from cerebral infarction sequelae.
Discussion
We described the case of an adult man with Alagille syndrome who showed mainly renal manifestations and recurrent brain infarction without liver manifestations. In infancy, the patient had exhibited one of the typical symptoms of Alagille syndrome (cholestasis due to bile duct paucity), but his main symptoms as an adult were renal failure and vascular manifestations. In light of these symptoms, he was categorized as having the atypical type of Alagille syndrome with a JAG1 mutation.
Renal involvement is one of the major features of Alagille syndrome. The reported prevalence of renal abnormalities in Alagille syndrome ranges from 40% to 70%, and the main categories of renal abnormalities are dysplasia, renal tubular acidosis, vesicoureteral reflux, obstruction, chronic renal failure, proteinuria, and hypertension (2, 7). Although the NOTCH2 mutation is thought to be related to the renal abnormalities (8), the genotype of our patient was a JAG1 mutation. We were unable to identify the reason for our patient's renal failure, as he had end-stage kidney disease on admission. He had no morphological abnormalities except for small kidney size and a long history of proteinuria, so renal parenchymal damage, such as glomerular mesangiolipidosis or focal segmental glomerulosclerosis, might have been a cause of his renal dysfunction (2, 7). Since there are reports of patients who were diagnosed with Alagille syndrome in adulthood based on only renal manifestations (4), nephrologists should consider the likelihood of Alagille syndrome when encountering a patient with renal failure and characteristic facial features of Alagille syndrome.
Vascular anomalies were another noteworthy finding of the present case, especially peripheral vascular abnormalities and intracranial vascular abnormalities. Vascular abnormalities have been documented as another main feature of Alagille syndrome. Although pulmonary artery stenosis is one of the five major clinical features, abnormalities of the aorta, renal arteries, celiac arteries, superior mesenteric arteries, and subclavian arteries are also often observed in Alagille syndrome (9). Peripheral vascular abnormalities are rare in Alagille patients. Our patient had extremity peripheral vascular stenosis, and vascular access for hemodialysis therapy could not be established. The condition of the peripheral arteries and veins is an important aspect of the creation of an AVF or AVG. Nephrologists should consider the condition of the peripheral vascular system when treating Alagille patients who show renal failure.
Severe potential complications of Alagille syndrome are intracranial bleeding related to Moyamoya disease and intracranial vessel aneurysms; noncardiac vascular complications also contribute substantially to the mortality of patients with Alagille syndrome (9). Although our patient had intracranial aneurysms, his major symptom was a brain infarction due to intracranial vascular stenosis. In our patient, the first brain infarction occurred after the generation of brachioaxillary AVG. Since we could not find any reports concerning the relationship between the generation of brachioaxillary AVG and brain infarction, we suspect that the generation of AVG was not of direct relevance to the brain infarction. However, as he lost a lot of blood during the operation, anemia might have influenced the onset of cerebral infarction.
His main features (vascular access failure for dialysis therapy and brain infarction) were due to noncardiac vascular abnormalities, i.e. extremity peripheral vascular stenosis and cerebral vascular stenosis. These atypical features of mainly vascular abnormalities without liver manifestations might have been due to the patient's JAG1 mutation, as the Notch signaling pathway plays an important role in vascular development. Kamath et al. proposed that vasculopathy might be the primary abnormality in Alagille syndrome, and they suggested that the clinical features of the syndrome might be explained by abnormal vascular development (9). Our patient showed peripheral-dominant vascular abnormalities. This specific abnormality might be attribute to the mutation JAG1 c.2938T>C (p.C980R). Thus far, the phenotype-genotype correlation has not been elucidated, as hotspot mutations have not been found in the JAG1 gene. In vitro studies have shown that cysteine-loss missense variants like those in our case reduce JAG1 signaling (10). Although the effects of this mutation are expected to be deleterious, we cannot fully explain the precise reason for our patient's atypical manifestations.
In conclusion, we encountered a rare case of Alagille syndrome in an adult man who mainly had peripheral vascular anomalies with renal failure. Nephrologists should consider the potential diagnosis of Alagille syndrome when they encounter patients with renal failure and vascular abnormality.
Informed consent was obtained from the patient for genetic testing and for his case to be reported.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
The author would like to thank Dr. Ryo Sumazaki, the Director of the Ibaraki Children's Hospital, for providing valuable advice, and Dr. Shogo Ito, Dr. Takao Togawa, and Professor Shinji Saitoh of the Department of Pediatrics and Neonatology, Nagoya City University Graduate School of Medical Science, for performing the mutation analysis.
References
- 1.Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet 9: 75-82, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamath BM, Podkameni G, Hunchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A 158A: 85-89, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation 109: 1354-1358, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Jacquet A, Guiochon-Mantel A, Noel LH, et al. Alagille syndrome in adult patients: it is never too late. Am J Kidney Dis 49: 705-709, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Harendza S, Hubner CA, Glaser C, et al. Renal failure and hypertension in Alagille syndrome with a novel JAG1 mutation. J Nephrol 18: 312-317, 2005. [PubMed] [Google Scholar]
- 6.Guegan K, Stals K, Day M, Turnpenny P, Ellard S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin Genet 82: 33-40, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Kamath BM, Spinner NB, Rosenblum ND. Renal involvement and the role of Notch signaling in Alagille syndrome. Nat Rev Nephrol 9: 409-418, 2013. [DOI] [PubMed] [Google Scholar]
- 8.McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet 79: 169-173, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation 109: 1354-1358, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Gilbert MA, Bauer R, Rajagopalan R, et al. Alagille syndrome mutation update: comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum Mutat 40: 2197-2220, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]