

Summary
The bleomycin mouse model is the extensively used model to study pulmonary fibrosis; however, the inflammatory cell kinetics and their compartmentalization is still incompletely understood. Here we assembled historical flow cytometry data, totaling 303 samples and 16 inflammatory-cell populations, and applied advanced data modeling and machine learning methods to conclusively detail these kinetics.
Three days post-bleomycin, the inflammatory profile was typified by acute innate inflammation, pronounced neutrophilia, especially of SiglecF+ neutrophils, and alveolar macrophage loss. Between 14 and 21 days, rapid responders were increasingly replaced by T and B cells and monocyte-derived alveolar macrophages. Multicolour imaging revealed the spatial-temporal cell distribution and the close association of T cells with deposited collagen.
Unbiased immunophenotyping and data modeling exposed the dynamic shifts in immune-cell composition over the course of bleomycin-triggered lung injury. These results and workflow provide a reference point for future investigations and can easily be applied in the analysis of other datasets.
Subject Areas: Immunology, Immune Response, Artificial Intelligence
Graphical Abstract
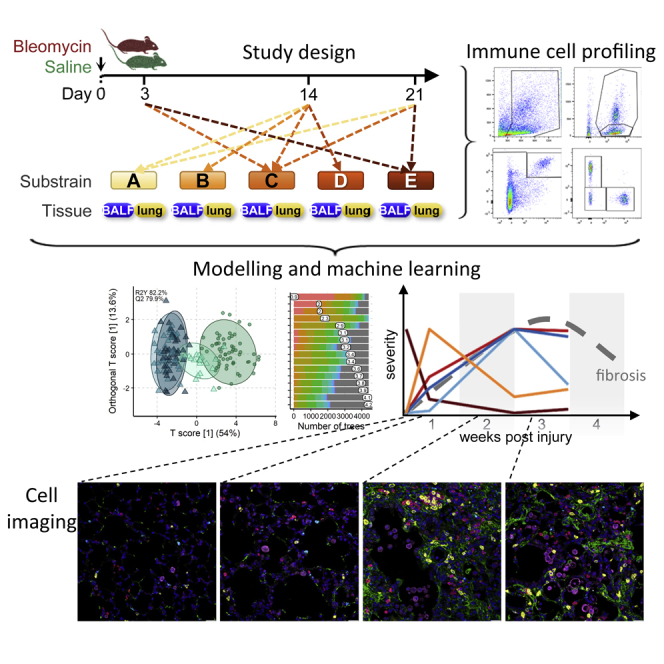
Highlights
-
•
The inflammatory cell landscape continually evolves after bleomycin exposure
-
•
Data modeling provides the most complete description of immune cell-trajectories
-
•
Chronic inflammation persists in the late-stage bleomycin-treated mice
-
•
Distinct inflammatory cells changes are observed in BALF and lung tissue
Immunology; Immune Response; Artificial Intelligence
Introduction
Animal models of human disease are an invaluable tool to decipher disease-relevant pathomechanisms, discover therapeutic targets, and drive translation into clinical practice. To date, the mouse bleomycin-induced lung injury model is the most frequently used animal model to investigate pulmonary fibrosis (Moore et al., 2013; Della Latta et al., 2015; Tashiro et al., 2017; Biasin et al., 2020). Similar to the human situation, in mice bleomycin exposure is characterized by epithelial damage, inflammatory cell infiltration, and expansion of fibroblasts and myofibroblasts as well as ECM deposition (Biasin et al., 2017, 2020; El Agha et al., 2017; Tashiro et al., 2017; Xie et al., 2018). Although, the bleomycin model does not completely recapitulate human idiopathic pulmonary fibrosis (IPF), it still remains the most common and important animal model to study this disease.
IPF is a severe, rapidly progressing interstitial lung disease with high mortality rates and short median survival of 1.5–4 years (Wuyts et al., 2013; Marshall et al., 2018). IPF is characterized by extensive lung tissue scarring, limited inflammation, and extracellular matrix remodeling (Meltzer and Noble, 2008). Current treatment options slow the loss of lung function but are unable to halt or reverse disease progression (Maher and Strek, 2019). Accordingly, there is an urgent unmet clinical need for novel therapies for IPF patients. To date, the etiology and pathogenesis of IPF is still insufficiently understood; however, the role of inflammation remains undeniable yet controversial. The older concept that IPF is an inflammatory-driven process has been gradually replaced by the theory of recurrent injury and aberrant repair (Selman et al., 2001; Selman and Pardo, 2002; Wuyts et al., 2013). However, multiple inflammatory cells have been implicated in disease pathogenesis, including macrophages (Misharin et al., 2017; Reyfman et al., 2019) and T cells (Todd et al., 2013), which are connected with poorer prognosis (Balestro et al., 2016).
In the bleomycin model, the early phase post bleomycin administration is characterized by acute lung injury and inflammation, which is observed to last between 1 and 7 days (Peng et al., 2013). This inflammatory phase is followed by active fibrosis, between 7 and 14 days, and late fibrosis, between 21 and 28 days (Izbicki et al., 2002; Peng et al., 2013; Della Latta et al., 2015; Tashiro et al., 2017). As most studies have only analyzed specific cell populations or time points, a comprehensive description of the inflammatory cell kinetics is still missing. For the detection and quantification of inflammatory cells, flow cytometry (FCM) is the method of choice. FCM is able to differentiate and quantify immune cell populations in unprecedented detail, not only from the circulation but also from disease relevant tissue (Misharin et al., 2017; Marsh et al., 2018; Tighe et al., 2019). In contrast to traditional immunofluorescent staining, which generally use 1–3 markers for cell identification, FCM applies multiple markers to simultaneously quantify numerous cell populations at a single cell resolution. Thus, FCM generates large quantities of complex data, where the analysis, visualization, and interpretation of which requires sophisticated analysis techniques, such as computational flow cytometry (Saeys et al., 2016; Marsh et al., 2018).
In order to conclusively detail the inflammatory cell kinetics in the bleomycin model, we here assembled historical FCM data from 15 different experiments and applied advanced data modeling, including univariate [Box 1], multivariate [Box 1], and machine learning [Box 2] methods. We show how the combination of advanced data modeling and in-depth immune profiling can detail the dramatic changes in the inflammatory landscape in this model and also serves as a reference point for future studies.
Box 1. Glossary of Analysis Terms.
| Robustness | A measure for how easily outlier values distort results:
|
| Unbalanced | Describes unequal group sizes or missing values, methods assuming balanced groups will have misleading results |
| Positive skew | Asymmetric distribution of data with more small than large values, common in flow cytometry and many other biological measures |
| Data pre-processing | Pre-processing aims to normalize data distribution (i.e. make a bell-shape) by changing all values according to one or several defined mathematical equations
Cell count differences are not per se reflective of their biological importance; thus centering and scaling reduces the stark differences of cell numbers between the cell populations to allow comparisons of different cell populations. Are vital for multivariate statistical methods, otherwise results will be dominated by cells with highest counts or highest noise
|
| Data contaminations | Denotes all kinds of problematic values in the data, such as sample outliers, single value outliers, or missing values |
| Outlier | A value so different from the rest that it could be for example an analytical error |
| Univariate or multivariate | Univariate methods investigate each measured data on its own (e.g. analyzing only CD3+ T cells irrespective of the 15 other cell populations), whereas multivariate methods analyze multiple/all measured data at once (e.g. all 16 cell populations)
|
Box 2. Glossary of Multivariate Methods.
| Unsupervised or supervised (multivariate) | Unsupervised methods analyze measured data unbiased without any knowledge of biological factors (e.g. treatment, substrain), whereas supervised methods specifically use biological knowledge
|
| PCA | Unsupervised method projecting the measured data into new principal components with decreasing variability per component (PC 1 to PC n)
|
| UMAP | Unsupervised method projecting the measured data (each marker for each cell) into new UMAP components but optimizing to represent high-dimensional data structure as similar as possible in the low dimensions
|
| Machine learning | Algorithms that automatically learn from existing measured data (training data) with the aim to predict unknown data without specific human input
|
| OPLS-DA | Supervised machine learning method separating predictive variability into the T score (x axis) from non-predictive into the orthogonal T score (y axis) yielding a score plot similar to PCA
|
| Random forest | Supervised machine learning method classifying samples into their given biological group based on measured data using an ensemble of many decision trees
|
Results
Pre-Processing of Flow Cytometric Data Substantially Improves Statistical Analysis Performance
Intra-tracheal administration of bleomycin in mice results in a time-dependent development of fibrosis (Figures 1A and 1B). To comprehensively describe the inflammatory cell kinetics following bleomycin treatment, we assembled and conjointly analyzed historical FCM data from 15 independent experiments, which resulted in 159 bronchoalveolar lavage fluid (BALF) and 144 lung tissue samples (Table S1). Using standard gating strategies (Misharin et al., 2013; Biasin et al., 2017; Nagaraj et al., 2017; Gungl et al., 2018), a total of 16 cell populations covering the main myeloid and lymphoid cell types (Table 1) were identified (Figure 1C). The aggregation of historical experiments inherently led to an unbalanced [Box 1] experimental design (Table S1), which was handled by robust statistical methods [Box 1].
Figure 1.

Overview of Study Design, Pathological Changes, and Gating Strategy
(A) Historical flow cytometry data from the bleomycin mouse model were pooled and collectively analyzed. Samples were collected 3, 14, or 21 days afterbleomycin or saline administration from the compartments BALF (159 samples) and lung tissue (144 samples). Five different C57BL/6 substrains were included.
(B) Representative Masson's trichrome staining of lung sections, showing pathologic alterations in the bleomycin model. Zoomed images exemplify the increasing fibrosis accumulation from day 3–21 after bleomycin challenge; scale bar represents 1 mm and 100 μm, respectively. High-resolution versions of these images for use with the Virtual Microscope are available as eSlides: VM06176, VM06160, VM06162, VM06177.
(C) Representative flow cytometry gating strategy. The 16 cell populations taken for further analysis are highlighted in bold. Alveolar macrophages (AM), dendritic cells (DC), interstitial macrophages (IM), monocyte-derived AM (MoAM), monocyte-macrophages (MoMp), neutrophils (PMN); forward scatter (FSC), area (A), height (H), side scatter (SSC), and monolymph gate (ML).
See also Table S1 for overview of group distribution and Table S2 for antibody details.
Table 1.
Inflammatory Cell Identification and Corresponding Markers
| Panel | Cell Type | Abbreviation | Cell Population | SSC | Panel I |
Panel II |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD45 | SiglecF | CD11c | Gr-1 | CD64 | CD24 | MHC-II | CD11b | CD3 | CD4 | CD8 | CD19 | gdTCR | |||||
| Cell count | Cell count | CD45+ live cell | + | ||||||||||||||
| Myeloid | Macrophagesa | AMa | Alveolar macrophages | hi | + | + | + | + | +/− | ||||||||
| MoAMa | Monocyte-derived macrophages | + | Lo | + | + | – | + | +/− | |||||||||
| IMa | Interstitial macrophages | + | – | – | + | – | + | +/− | |||||||||
| DCs | DCb | CD11b+ dendritic cells | + | – | – | + | + | + | |||||||||
| Monocytesa | Gr1+ MoMpa | Inflammatory monocyte macrophages | + | + | + | – | + | ||||||||||
| Gr1- MoMpa | Constitutive monocyte macrophages | + | – | + | – | + | |||||||||||
| Granulocytesa | EOSa | Eosinophils | hi | + | + | – | + | ||||||||||
| PMNa | Mature neutrophils | hi | + | +/− | – | + | + | ||||||||||
| SiglecF+ PMNa | SiglecF+ neutrophils | hi | + | + | – | + | + | ||||||||||
| Immature PMNa | Immature neutrophils | hi | + | – | + | – | + | ||||||||||
| Lymphoid | B cellsb | CD19+ B cellsb | B cells | lo | + | – | + | ||||||||||
| T cells | CD3+ T cellsb | T cells | lo | + | + | – | |||||||||||
| CD4+ T cellsb | T helper cells | lo | + | + | + | – | – | ||||||||||
| CD8+ T cellsb | Cytotoxic T cells | lo | + | + | – | + | – | ||||||||||
| γδ T cellsb/a | γδ T cells | lo | + | + | + | – | – | + | |||||||||
Innate immune cells.
Adaptive immune cells.
In both tissues the distribution of all 16 analyzed cell populations was significantly non-normal with a positive skew [Box 1] (Figures 2A and S1 and Supplementary Data 1). To improve distribution, we trialed several common transformations: square root, reciprocal, Freeman Tukey, logit, LOG, LOGx+1, and 4RT. Only LOG, LOGx+1 and 4RT improved data distribution (pBH>0.05, Supplementary Data 1). As both LOG and LOGx+1 gave virtually equivalent results, but as LOGx+1 has additionally the advantage of not introducing missing values for zero value counts, consequent analysis was performed with only LOGx+1 and 4RT (Figures 2A and 2B).
Figure 2.

Data Transformation Improves Data distribution and Analytical Power
Analysis of cell count data (untransformed) or following transformation using LOGx+1 or 4RT (fourth root) using 159 BALF and 144 lung samples. Cell counts in BALF are 105 and in lung 104/mg tissue. Examples of data distribution of neutrophils (PMN) as one representative population in BALF and lung samples by (A) histograms show the frequency of PMN cell counts. Data were grouped into 30 equal intervals (binned cell counts).
(B) Violin plots, total represents combined saline and bleomycin samples.
(C) PCA scores plots [Box 2] with each point representing the combined inflammatory cell profile (16 populations) in one sample; plots are colored to highlight different experimental conditions. In B and C, dots represent single sample values.
(D) Heatmaps with hierarchical clustering of all 16 analyzed cell populations.
See also Figure S1 for data distribution and Figure S2 for macroPCA comparison.
Bleomycin Drives Strong Changes in the Inflammatory Profile
To identify global changes in the inflammatory cell profile, we first applied unsupervised [Box 2] principal component analysis (PCA [Box 2]). This method reduces dimensionality by creating new variables, which successively maximizes variance and thereby aids data interpretability. Without data transformation, the scores plot was dominated by single sample differences, which obscured any experimental effects (Figure 2C, left panel). After transformation, pronounced differences in the inflammatory profile were revealed (Figure 2C). Both LOGx+1 and 4RT substantially improved the performance of the hierarchical clustering, yielding clearer clustering and heatmap results (Figure 2D). The highest influence on the inflammatory landscape came from the tissue compartment (BALF or lung), causing samples to separate along the first principal component (PC1). The second highest difference was caused by bleomycin, separating samples in the BALF along the second principal component (PC2; Figure 2C, middle and right panels). Similarly, hierarchical clustering was first driven by the tissue compartment, followed by some weaker subclustering due to bleomycin treatment. The majority of cell populations increased after bleomycin exposure, whereas alveolar macrophages (AM) decreased (Figure 2D). We next utilized macroPCA, a robust PCA method able to handle and identify all possible types of data contaminations [Box 1], including strong single value or sample outliers [Box 1] (Hubert et al., 2019). MacroPCA results were in good agreement with PCA (Figure S2A), which confirmed that this dataset is free of severe outliers, allowing the use of a wide variety of statistical methods (Rousseeuw and Hubert, 2018).
As the strong compartment effect could mask weaker drivers that alter the inflammatory landscape, we analyzed BALF and lung samples separately (Figure 3). In the BALF, bleomycin exposure completely altered the inflammatory landscape, separating samples along PC1 (explaining 63.9% of the variation in the dataset). However, the bleomycin effect only accounted for 12.4% of the variation in the lung, separating on PC2 (Figure S2A). Again, macroPCA gave similar results in the analysis of the separate compartments (Figure S2B), reconfirming the absence of critical outliers. Analogous to the PCA findings, hierarchical clustering showed a strong clustering after bleomycin exposure in BALF, which was less clear in lung tissue samples. The influence of day post-treatment and substrain (individual C57BL/6 lines) on cell population changes was less distinct, with only some indication toward a possible sub-clustering due to these factors (Figure 3B).
Figure 3.

Bleomycin Induces Stronger Changes in the Inflammatory Profile in the BALF than the Lung
The contribution of different biological factors to the inflammatory cell profile as determined by (A) PCA score plots [Box 2] are colored to highlight different experimental conditions and (B) heatmaps with hierarchical clustering. To aid, interpretation heatmaps are split into two main clusters based on dendrogram distances. Colors and shapes represent tissue, treatment (Saline, Bleo), mouse substrain, and day post-treatment. Cell counts from 16 populations in 159 BALF and 144 lung samples were LOGx+1 transformed prior to clustering.
Modeling of Inflammatory Cell Kinetics with Univariate Statistical Analysis
In order to examine the potential influence of other experimental factors in depth and to simultaneously control for the unbalanced [Box 1] design arising from the use of historical data, we applied univariate [Box 1] linear mixed models with log10-transformation (LOGLME, Figure S3). As the multivariate [Box 1] analysis showed a strong bleomycin effect, the fixed factor [Box 3] Treatment {Saline, Bleo} was included in all models [Box 3]. Other fixed factors included Day {3,14,21} and Substrain {A,B,C,D,E}. The addition of each factor, either alone or together and with or without their interaction [Box 3] with Treatment, notably improved the fit [Box 3] of all simple models, increasing the goodness of fit and reducing Akaike information criterion (AIC; Figure S3). Thus, both the Day post bleomycin exposure and Substrain significantly influenced the cellular landscape.
Box 3. Glossary of Univariate Model Terms (LOGLME).
| Model | A mathematical equation describing the relationship of measured data to biological factor(s)
|
| Fitting | Finding the parameter values best describing the measured data, often assessed by the residuals |
| Residuals | Difference between fitted value and measured value (in linear models the distance from the measured data dot to the line) |
| Fixed factor | Also called between-subject effect, a biological factor that (possibly) affects the outcome, e.g. treatment or day after treatment |
| Interaction | The impact of one biological factor depends on the occurrence of another biological factor
|
| Random factor | Also called within-subject effect, a factor that (possibly) affects baseline level such as repeated measures from the same source or working in experimental batches
|
| Simple/mixed | Simple models contain only fixed factors factor; mixed models include random factors |
| Fitted value | The value suggested by the model from the fitted equation (measured value minus the fitted values is the residual), if the model is correct that would be the real value without measurement error |
| Predicted value | Similar to fitted values the predicted value is suggested by the model equation, but for formerly unknown or not measured points (e.g. CD3+ T cells day 21 in BALF in Figure S4B) |
| Overfitting | The model contains more parameters than the existing data allow to fit well and thus the model will fail to predict new data correctly
|
As each independent experiment could have similarities, the experimental ID was then included as a random factor (~1|Exp_ID). These mixed models significantly outperformed the aforementioned simple models. Finally, complex mixed models (combining the mixed models with the interactions of Treatment with Substrain or Day) notably outperformed all simple models (with or without interactions). The most complex mixed model [Treatment + Day + Substrain + Treatment:Substrain + Treatment:Day, ~1|Exp_ID] outperformed all other models, although more prominently in BALF than in lung (Figure S3A).
As complex models risk overfitting [Box 3], especially in light of the unbalanced design, we then investigated model simplification. We first tested whether it was possible to create one control group of all saline animals. In all mixed and complex models (i.e. with random factor Exp_ID) only 4 of the over 10,000 investigated pairwise comparisons of a saline subgroup with another saline subgroup had a pBH<0.01 in any of the 16 cell types. This means saline-treated animals were sufficiently similar to be combined into one control group. Consequently, Treatment and Day can be then merged into one fixed factor with four groups: saline (all days) and bleomycin after days 3, 14, and 21, which was termed SalineDay {Saline,3,14,21}, generating the simplified model [SalineDay + Substrain] and the simplified mixed model [SalineDay + Substrain ~ 1| Exp_ID]. The performance of the simplified mixed model was slightly lower than in the most complex mixed model, but well within the range of the other top performing mixed models (Figure S3B).
To compare the models in more detail we also directly compared the fitted values [Box 3] of the simplified mixed model with the most complex mixed model. The fitted values from both models strongly correlated (Pearson correlation R2 > 0.96, Figure S3B). This underlines the validity of model simplification and that no unexpected or systematic skew was introduced. As the simplified mixed model [SalineDay + Substrain ~ 1|Exp_ID] also gives more easily interpretable results and has a lower risk of overfitting [Box 3], it was chosen to examine the inflammatory cell kinetics underlying bleomycin mouse model.
This model was then applied to explore how individual substrains may influence the kinetics of different inflammatory cells. All mice included in this study are on the C57BL/6 background; however, they were obtained from different sources, e.g. commercial sources (C57BL/6J, substrain A) or are the wild-type littermates from in-house breedings (substrains B-E). Although some lines were inbred for up to 15 generations, all lines produced similar inflammatory responses in both lung compartments, which only differed in magnitude (Figure S4). This consistency allows to readout the compartmental kinetics of each cell population after bleomycin treatment for all substrains combined.
Inflammatory Cell Kinetics after Bleomycin-Induced Lung Injury Are Robust and Reproducible
Analysis of the inflammatory response in the BALF identified a non-resolving inflammatory response, with the total number of inflammatory cells continuing to increase over the investigated time course of 21 days. In the lung tissue, inflammation was characterized by an immediate increase at day 3, stagnating at day 14, and mostly resolved 21 days post-bleomycin exposure (Figure 4). This suggests that the inflammatory response is persistent, yet compartment dependent.
Figure 4.

Linear Mixed Models with log10-Transformation Reveal Complex Immune Cell Dynamics Occurring in the Lung Following Bleomycin-Induced Lung Injury
Plot of back transformed, fitted cell counts (line represents mean ± 95% confidence intervals) using the simplified mixed model [SalineDay + Substrain ~ 1|Exp_ID] of LOGx+1 transformed cell counts for BALF (counts∙105) and lung tissue (counts∙104/mg tissue). Animal numbers were in BALF in total n = 159 (Saline 60; 3d 23; 14d 39; 21d 37) and in lung in total n = 144 (Saline 56; 3d 23; 14d 32; 21d 33). See also Figure S3 for model comparisons and Figure S4 for modeling of all 16 cell populations separated by strain.
Early inflammatory changes were mostly dominated by the innate immune system, including both immature and mature neutrophils (immature and mature PMN), monocyte-derived alveolar macrophages (MoAM), and interstitial macrophages (IM). In contrast we observed a concomitant decrease in AM. Interestingly, the (inverted) trajectories of AM were comparable to the rise in MoAM, suggesting a functional replacement by the latter, and support observations in earlier studies (Misharin et al., 2017). Following the rapid increase in the first-line responders, PMN, their numbers later stagnated or gradually decreased and even returned to baseline levels in the lung tissue. We also identified a time-dependent increase in SiglecF+ PMN following bleomycin application. These cells have recently been described to be important for cancer progression and murine myocardial infarction (Engblom et al., 2017; Vafadarnejad et al., 2020). Similarly, eosinophils (EOS) and dendritic cells (DC) exhibited a bell-shape response curve. In contrast, monocyte populations (both Gr1+ and Gr1− MoMp) exhibited a slower, but consistent, stepwise temporal increase, which could be attributed to their contribution to both the innate and adaptive immunity and their role in tissue repair.
At later time points, inflammation was dominated by immune cells from adaptive immunity, with a clear preference to the alveolar compartment. In the BALF, CD3+ T lymphocytes (CD4+ and CD8+ T cells, respectively) had a steep, yet non-resolving, rise early in the inflammatory response. CD19+ B cells peaked at 14 days post-bleomycin challenge. Interestingly, at the latest investigated time point, 21 days, T cell numbers continued to rise, implicating their involvement at later stages in this model (Figure 4A).
Taken together, each inflammatory cell population shows dynamic and distinct inflammatory kinetics with some compartmental preference. With time, the involved immune cells shifted from the innate (e.g. PMN) to the adaptive immune system (e.g. T and B cells), with the BALF being more prominently affected than the lung tissue. However, after 21 days the inflammatory profile was still chronically altered in both compartments, suggesting ongoing inflammation.
Based on these results we went back to our FCM data and visualized the kinetics of the most dynamically altered populations using uniform manifold approximation and projection (UMAP)[Box 2]-derived plots (Figure 5A). UMAP, like PCA, is a dimensionality reduction technique that can utilize the entire flow cytometry dataset (i.e. the positivity or negativity and intensity of each marker for each cell) and reduces this information into a new two-dimensional space. As predicted in our modeling data and easily apparent in the UMAP plots, AM populations strongly decreased following bleomycin exposure, whereas PMN vastly increased after three days. Adaptive immune cells, such as CD4+ T cells, CD8+ T cells, and CD19+ B cells, expanded more at later time points and were virtually absent in saline-treated mice (Figure 5A). Visualization, using multicolour immunofluorescence, revealed the presence of CD11c+/SiglecF+ AM and Ly6G+ PMN in saline and day 3 treated mice, whereas CD4+ T cells and CD19+ B cells were more prominent at later time points (days 14 and 21) (Figure 5B). Interestingly, CD4+ T cells were commonly spatially localized to the collagen-rich fibrotic lung tissue (Figure 5B).
Figure 5.

Temporal and Spatial Localization of Inflammatory Cell Kinetics in BALF and Lung Tissue
(A) Uniform Manifold Approximation and Projection (UMAP) plots of concatenated CD45+ populations (min 3 independent samples with max 10′000 CD45+ cells per sample), cells were clustered according to their similarity in signal intensity of all parameters measured by flow cytometry and overlayed with manually gated populations in the BALF. For clarity axis labels are shown only on the first panel of the model.
(B) Spatial localization of alveolar macrophages (AM: CD11c+/SiglecF+), neutrophils (PMN: LY6G+), CD19+ B cells, and CD4+ T cells during the time course of bleomycin challenge. Nuclei are stained with DAPI (dark blue). Representative pictures of three independent mice at each time point. D3, D14, and D21 represent days 3, 14, and 21 post-bleomycin treatment, respectively; scale bar represents 20 μm. High-resolution versions of these images are available in Virtual Microscope as eSlides: VM06172, VM06173, VM06174, VM06175, respectively.
See also Table S3 for antibody details and Table S4 for instrument configuration.
The Inflammatory Cell Landscape Continually Evolves following Bleomycin Exposure
The combination of unsupervised multivariate methods and univariate modeling [Box 3] (LOGLME) identified the kinetics of each cell type with an early innate response followed later by adaptive immune response. However, the question how the entire landscape differs between different time points or which cell types define each stage is still open. In order to answer these questions, we applied three robust machine learning [Box 2] approaches.
Our first approach, OPLS-DA [Box 2] separates the dataset into predictive and non-predictive components. Predictive means the ability to discern between groups in the given classification factor, which was here SalineDay {Saline,3,14,21}. The OPLS-DA model quality was thoroughly investigated by cross-validation and permutations tests, which showed that in both compartments the models were highly significant (Q2>50%, p < 0.001). Similar to our PCA results (Figure 3), the inflammatory reaction was more pronounced in the BALF than in the lung, as apparent from a clearer group separation, higher percentages of variability in the predictive component, and higher predictive ability (Q2; Figure 6A). In BALF, the inflammatory landscape at 14 and 21 days post-bleomycin were very similar, but very different from the saline controls, whereas the landscape at 3 days bridged these two poles.
Figure 6.

Exploration of Inflammatory Cell Landscape Differences with Machine Learning in BALF and Lung Tissue
(A) Scores plot of OPLS-DA[Box 2] models per compartment for the factor SalineDay {Saline,3,14,21} with 95% confidence ellipses for each group. The predictive ability of the models Q2 was calculated by 7-fold cross validation, and 1,000 permutation tests reconfirmed model significance with p < 0.001.
(B) Conditional inference trees per compartment, showing cell types and cut-offs that define each group; saline, days 3, 14, and 21 post-bleomycin treatment (SalineDay). Model accuracy was evaluated with a stratified split into 65% trainings and 35% test set.
(C) Multidimensional scaling (MDS) [Box 2] plot (left panel) of the proximity matrix of random forest [Box 2] models grown with 5,000 trees. Model accuracy was evaluated with a stratified split into 65% trainings and 35% test set. The distribution of the minimal depth [Box 2] is shown for each cell type according to the number of trees; the mean of the minimal depth is shown (middle panel). The rank of the mean decrease in accuracy [Box 2] within each group is shown for each cell population (right panel). Animal numbers in all models from A-C were in BALF in total n = 159 (Saline 60; 3d 23; 14d 39; 21d 37) and in lung in total n = 144 (Saline 56; 3d 23; 14d 32; 21d 33). Models were based on LOGx+1-transformed cell counts for BALF (counts∙105) and lung tissue (counts∙104/mg tissue).
(D) Schematic, abstracted summary of the five lead cell types (highlighted in bold in panel C) and scaled 0 to 1 to highlight relative changes between cell types and compartments BALF and lung tissue.
We next investigated conditional inference trees and random forest [Box 2] models to infer which cell populations were the driving factors behind the group differences. Conditional inference trees in the BALF demonstrated that CD3+ T cells levels separated early (Saline, day 3) and later time points (days 14 and 21). Separating samples on low and high CD19+ B cells distinguishes between days 14 and 21, respectively. On the other hand, low levels of PMN strongly predicts saline-treated mice and the combination of low AM and SiglecF+ PMN aiding the separation between saline, days 3 and 14 (Figure 6B). In the lung compartment, both innate cells (MoAM, AM) and adaptive cells (CD4+ T cells and CD19+ B cells) were needed to define the different groups. Saline mice were defined by low levels of MoAM and high AM, whereas bleomycin treatment by high MoAM and CD4+ T cells. Similar to the BALF, day 21 was marked by high CD19+ levels, whereas D14 by was defined by lower B cell and MoAM levels (Figure 6B). A combination of low MoAM and low AM defined day 3.
Random forest models were then used to compare the ability of all cell populations to drive group separation. In agreement with previous results, again group separation was clearer in BALF than in lung, as demonstrated by multidimensional scaling plots of the random forest proximity matrix and higher accuracy (Figure 6C). In BALF, especially the adaptive immune cells CD8+ and CD3+ T cells as well as the innate SiglecF+ PMN differed most, as became apparent from their low minimal depth. Between the different groups high CD8+, CD3+, and CD19+ levels were most predictive for late inflammation, whereas low SiglecF+ PMN levels were most predictive for the cellular landscape in saline samples. The random forest suggests some fine but distinct differences between the global inflammatory landscape 14 and 21 days after bleomycin exposure (Figure 6C). Although both are highly inflamed (OPLS-DA), higher levels of adaptive cells are rather predictive for day 21 (e.g. all T and B cells), whereas higher levels of some innate cells are more predictive for day 14 than day 21 (e.g. DC, IM, immature PMN, MoAM, EOS) or day 3 (PMN). In contrast, lung models were dominated by macrophage cell populations differing most between the inflammatory stages, foremost the depletion of AM. The random forest models underline that the inflammatory landscape differs notably between lung and BALF.
Discussion
In this study, we have combined computation FCM, advanced data modeling, and machine learning approaches to conclusively define the inflammatory cell kinetics following bleomycin treatment in mice. By combining the data from 15 independent experiments, we amassed very large sample numbers, which were far in excess of those normally found in animal experiments. The aggregation of historical samples inherently led to an unbalanced experimental design, which was handled by sophisticated, robust statistical methods. By using pre-processing techniques such as data transformation, we substantially improved analysis power, which crucially contributed to clearer data interpretation. Changes in the inflammatory profile were dissected using multivariate and univariate statistical methods, including linear mixed models with log10 transformation. Only by applying these techniques in unison were we able to create the most comprehensive picture of inflammatory cell trajectories to date and characterize the sustained inflammation in the bleomycin model of pulmonary fibrosis. Importantly, these techniques and workflow can be easily applied for analysis of other datasets.
FCM data are normally highly asymmetric i.e. it has many larger values but no values smaller than zero; this non-normal distribution prevents the use of more powerful analysis methods. To re-establish normality we trialed several transformations but ultimately settled on LOGx+1 as it normalized the data distribution, can be easier to interpret, and also slightly improved the scedasticity compared with 4RT. Our data modeling approach resulted in a very large sample size, which notably increased statistical power and outweighed the potential drawbacks of added confounding variation from experimental runs or the use of different substrains. Furthermore, when experimental covariance was accounted for as random factor in LOGLME models, the inflammatory profiles in the BALF and lung tissue of all saline-treated animals, irrespective of experiment, were sufficiently similar to be combined into one large control group. Secondly, the trajectories of inflammatory cell profiles were found to be consistent for all five substrains, although their magnitudes slightly differed, which is important for experimental reproducibility in light of using different knockout lines or mice sourced from different companies.
The application of unsupervised and supervised as well as multivariate and univariate methods demonstrated that the majority of cell populations showed consistent trajectories in both compartments. However, the changes for most cells were more prominent in BALF than in lung tissue. This is in part due to fact that in healthy mice the vast majority of cells in the BALF are AM, whereas in the lung tissue even at baseline conditions, a highly heterogeneous pool of inflammatory cells exists, including macrophages, PMN, T, and B cells. The most informative results will be achieved by sampling both BALF and lung tissue. The investigation of lung tissue alone could lead to misinterpreting the duration or intensity of inflammation due to weaker changes, whereas analysis of the BALF alone could potentially miss cell populations that are not normally found within this compartment, e.g. interstitial macrophages or to a lesser extent DC. Therefore, deep inflammatory cell phenotyping requires the analysis of both compartments to give the full picture of the inflammatory status of the lung.
Our comprehensive analysis of multiple inflammatory cell population at several time points describes the kinetics not only during disease development but also when it is fully established. The initial inflammatory phase after bleomycin exposure was dominated by early responder cell types from the innate immune system of the myeloid lineage. Neutrophils constitute the first-line defense of the immune system and consequently show very acute kinetics, being rapidly recruited and also being the first cell type to resolve, visible as pronounced decreases from day 3 to day 14 after the challenge. In contrast, cells from adaptive immune system, such as B and T cells, increased much slower but continue to expand even at 21 days. The worth of subtyping cell populations is apparent by the inverse kinetics displayed within macrophages, which is only possible by using multicolour analysis. We could show that although the number of AM quickly decreased, MoAM increased. These contrary trajectories would explain the early observation that macrophages numbers were unchanged in this model (Izbicki et al., 2002), but the closer analysis of macrophage subtypes revealed strong dependent changes, as shown by (Misharin et al., 2013, 2017) and now reconfirmed by our results.
Increasingly macrophage heterogeneity has been suggested to play an important role in the pathogenesis of lung fibrosis and have implications for therapeutic strategies. MoAM undergo marked transcriptional changes during their differentiation in the injured lung tissue. These changes are not only associated with a continuous downregulation of genes typically expressed in monocytes and upregulation of genes expressed in AM but also with markedly elevated expression of proinflammatory and profibrotic genes related to M1 and M2 phenotype. This unique transcriptomic signature of MoAM provides an explanation on how bleomycin-induced lung fibrosis is attenuated following selective depletion of these cells (Misharin et al., 2017; McCubbrey et al., 2018; Joshi et al., 2020). Interestingly, the existence of common profibrotic pathways in MoAM harvested from mice during fibrosis development and profibrotic macrophages obtained from the lungs of IPF patients has been reported (Misharin et al., 2017; Aran et al., 2019). All these observations strongly suggest that selective targeting profibrotic macrophages, rather than the M1 or M2 phenotype, is more likely to be of benefit in such a complex disease as IPF. The potential contribution of MoAM to the resolution of lung fibrosis remains the subject of future studies, although very recent data supports this hypothesis (Cui et al., 2020). Hence, MoAM could represent a very plastic cell population with distinct functions in different phases of lung fibrogenesis.
Early and late fibrotic stages were characterized by increased numbers of T and B cells in the BALF, whereas numbers in the lung tissue remained relatively stable; this reflects earlier reports describing the presence of T cells in IPF lungs (Todd et al., 2013; Balestro et al., 2016). Here B cells are of particular interest, as abnormal B cell aggregates have been described in IPF lungs (Marchal-Sommé et al., 2006) and diverse circulating IgG autoantibodies were found in IPF plasma (Ogushi et al., 2001; Kurosu et al., 2008; Taillé et al., 2011). Furthermore, individual auto-immunoglobulins were linked to severity and/or poor prognosis of IPF (Ogushi et al., 2001; Kahloon et al., 2013), thus suggesting the causal role of certain autoantigens in IPF. Accordingly, transcriptome-profiling of lung tissue derived from pirfenidone-treated patients revealed downregulation of B-cell-related genes (Kwapiszewska et al., 2018). Future studies will, however, demonstrate whether these findings open an exciting new avenue for immunotherapy-based approaches in IPF.
Recommendations
This study explored fundamental aspects of the bleomycin animal model with good power owing to the high sample numbers so that constructive recommendations can be inferred.
-
(I)
In order to ascertain technical success of the experiment we strongly recommend to always include a negative control (saline) and a positive control (bleomycin, transgenic, or knock out otherwise untreated) group with each n ≥ 8. Foremost this serves to rate the strength of induced fibrosis and technical quality of the experiment. Statistical power gain is very high for every added sample in the single digit region. An n ≥ 8 leaves some safety margin to stay above the critical level of n = 5 to handle the occasional, unavoidable loss of samples due to premature death or technical problems.
-
(II)
For more sensitive and pronounced inflammatory readouts the BALF should be routinely sampled together with lung tissue and both samples subjected to analogous analysis.
-
(III)
For statistical analysis we strongly recommend to first investigate distribution and pre-processing (transformation, scaling, centering) paired with unsupervised multivariate methods.
-
(IV)
Specific and detailed analysis should be based on the main trends identified in unsupervised models. For detailed investigation of cell-specific differences univariate mixed models offer the highest flexibility and insights. Supervised multivariate methods are well capable to confirm wholistic trends and dissect main drivers of these trends. We strongly recommend to pre-process data before investigating first with unsupervised and second with supervised methods as well as relying on both multivariate and univariate methods, as they complement each other well in their type of generated insights.
Conclusions
The measurement of inflammatory cellular landscapes in the bleomycin-induced lung-injury mouse model with flow cytometry is very robust and suitable to quantify kinetic changes in multiple cell populations simultaneously. The results allowed to infer recommendations such as to add negative and positive control, apply data pre-processing, combine multivariate and univariate methods, and to routinely also investigate BALF. We also found that the unintended development of potential substrains does not per se hinder general reproducibility of results and that the approach to adapt bleomycin doses to the current experimental run is viable. This study underlines the relevance of combined analysis for more holistic insights into inflammatory profile changes. Cell populations show quite distinct trajectories in their kinetics. We also conclude that inflammatory cell-based response is active before, during, and after manifestation of fibrosis with a shift from the initial innate immune cell dominance toward the adaptive immunity, and importantly inflammatory cell accumulation is not resolved after three weeks.
Limitations of the Study
Despite analyzing three independent time points, which cover the major stages of the bleomycin model, some time points are still missing. However, we consciously wanted to reuse existing experiments and avoid sacrifice of new animals. Future investigation would profit from an expansion, e.g. by inclusion of existing measurements from other groups, to cover also the progression from the initial inflammation toward active fibrosis phase by including analysis between days 3 and 14. Similarly, inflammatory profiling during fibrosis resolution, i.e. after 28 or 35 days, would deliver valuable insights on the involvement of specific subtypes during resolution. From a statistical point of view, the unbalanced study design with differing sample numbers in subgroups is unfavorable, which complicates analysis and loses some power. However, our use of robust methods such as LOGLME and machine learning methods (random forest) were able to overcome these limitations. Although over a dozen independent experimental runs were included, this is not a multicentric study. Quantitative comparison of results from other laboratories at other sites and other strains/substrains would allow to even better explore bleomycin model system robustness and reproducibility. In this study, manual gating was used to identify different cell populations, thereby including expert knowledge into the analysis, and gating specificity was confirmed by UMAP overlays. For some populations in the UMAP plots (e.g. AM), the populations were more spread than expected; this was most likely due to different marker intensity (in this case CD11c) between different experimental runs. The topic of auto-gating is rapidly developing and promises to considerably save hands-on time and foremost the potential to detect rare, otherwise undetected cell subpopulations. The focus of this study was to primarily determine the inflammatory cell kinetics; however, to further unravel the role of inflammation and potential therapeutic targets in fibrosis, a quantified link of cell subpopulations to fibrotic processes is warranted.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Leigh Marsh (leigh.marsh@lvr.lbg.ac.at)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All data needed to evaluate the conclusions in the paper are present in the paper; the supplementary data are available on Mendeley Data: https://doi.org/10.17632/7j485t986v.1.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are very grateful for the technical assistance from Kerstin Schweighofer, Sabine Halsegger, Thomas Fuchs, Nina Treitler, Eva Grasmann, Camilla Götz, and Betty Flasch, as well as Slaven Crnkovic, Mathias Hochgerner, Chandran Nagaraj and Horst Olschewski for the valuable discussions and advice. Diana Schnögl, F.V. and N.S. are members of the MolMed graduate program. Diana Schnögl is funded by FFG, project number 870904 awarded to Leigh Marsh. F.V. is funded by FFG, project number 874229 awarded to G.K.. V.B. is funded by Austrian Science Fund (FWF; project number, T1032-B34).
Author Contributions
Conceptualization, Data curation, Software, Validation, Methodology, N.B. and L.M.M.; Formal analysis and Visualization N.B., D.S., F.V., and L.M.M.; Investigation, Resources, N.B., V.B., D.S., F.V., K.J., B.M.N., N.S., and L.M.M.; Writing—original draft, N.B., V.B., G.K., M.W., L.M.M.; Writing—Review & Editing, all authors; Project administration, Supervision, and Funding acquisition, G.K. and L.M.M.
Declaration of Interests
The authors declare that they have no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101819.
Supplemental Information
References
- El Agha E., Kramann R., Schneider R.K., Li X., Seeger W., Humphreys B.D., Bellusci S. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 2017;21:166–177. doi: 10.1016/j.stem.2017.07.011. [DOI] [PubMed] [Google Scholar]
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestro E., Calabrese F., Turato G., Lunardi F., Bazzan E., Marulli G., Biondini D., Rossi E., Sanduzzi A., Rea F. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0154516. doi: 10.1371/journal.pone.0154516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg R.A., Hoefsloot H.C.J., Westerhuis J.A., Smilde A.K., van der Werf M.J. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006;7:142. doi: 10.1186/1471-2164-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasin V., Wygrecka M., Marsh L.M., Becker-Pauly C., Brcic L., Ghanim B., Klepetko W., Olschewski A., Kwapiszewska G. Meprin β contributes to collagen deposition in lung fibrosis. Sci. Rep. 2017;7:39969. doi: 10.1038/srep39969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasin V., Crnkovic S., Sahu-Osen A., Birnhuber A., El Agha E., Sinn K., Klepetko W., Olschewski A., Bellusci S., Marsh L.M., Kwapiszewska G. PDGFRα and αSMA mark two distinct mesenchymal cell populations involved in parenchymal and vascular remodeling in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2020;318:L684–L697. doi: 10.1152/ajplung.00128.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H., Jiang D., Banerjee S., Xie N., Kulkarni T., Liu R.-M., Duncan S.R., Liu G. Monocyte-derived alveolar macrophage apolipoprotein E participates in pulmonary fibrosis resolution. JCI Insight. 2020;5:e134539. doi: 10.1172/jci.insight.134539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom C., Pfirschke C., Zilionis R., Da Silva Martins J., Bos S.A., Courties G., Rickelt S., Severe N., Baryawno N., Faget J. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science. 2017;358:eaal5081. doi: 10.1126/science.aal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gungl A., Biasin V., Wilhelm J., Olschewski A., Kwapiszewska G., Marsh L.M. Fra2 overexpression in mice leads to non-allergic asthma development in an IL-13 dependent manner. Front. Immunol. 2018;9:2018. doi: 10.3389/fimmu.2018.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert M., Rousseeuw P.J., Van den Bossche W. MacroPCA: an all-in-one pca method allowing for missing values as well as cellwise and rowwise outliers. Technometrics. 2019;61:1–18. [Google Scholar]
- Izbicki G., Segel M.J., Christensen T.G., Conner M.W., Breuer R. Time course of bleomycin-induced lung fibrosis. Int. J. Exp. Pathol. 2002;83:111–119. doi: 10.1046/j.1365-2613.2002.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N., Watanabe S., Verma R., Jablonski R.P., Chen C.I., Cheresh P., Markov N.S., Reyfman P.A., McQuattie-Pimentel A.C., Sichizya L. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J. 2020;55:1900646. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahloon R.A., Xue J., Bhargava A., Csizmadia E., Otterbein L., Kass D.J., Bon J., Soejima M., Levesque M.C., Lindell K.O. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am. J. Respir. Crit. Care Med. 2013;187:768–775. doi: 10.1164/rccm.201203-0506OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene O.N. The log transformation is special. Stat. Med. 1995;14:811–819. doi: 10.1002/sim.4780140810. [DOI] [PubMed] [Google Scholar]
- Kurosu K., Takiguchi Y., Okada O., Yumoto N., Sakao S., Tada Y., Kasahara Y., Tanabe N., Tatsumi K., Weiden M. Identification of annexin 1 as a novel autoantigen in acute exacerbation of idiopathic pulmonary fibrosis. J. Immunol. 2008;181:756–767. doi: 10.4049/jimmunol.181.1.756. [DOI] [PubMed] [Google Scholar]
- Kwapiszewska G., Gungl A., Wilhelm J., Marsh L.M., Puthenparampil H.T., Sinn K., Didiasova M., Klepetko W., Kosanovic D., Schermuly R.T. Transcriptome profiling reveals the complexity of pirfenidone effects in idiopathic pulmonary fibrosis. Eur. Respir. J. 2018;52 doi: 10.1183/13993003.00564-2018. [DOI] [PubMed] [Google Scholar]
- Della Latta V., Cecchettini A., Del Ry S., Morales M.A. Bleomycin in the setting of lung fibrosis induction: from biological mechanisms to counteractions. Pharmacol. Res. 2015;97:122–130. doi: 10.1016/j.phrs.2015.04.012. [DOI] [PubMed] [Google Scholar]
- Maher T.M., Strek M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir. Res. 2019;20:205. doi: 10.1186/s12931-019-1161-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal-Sommé J., Uzunhan Y., Marchand-Adam S., Valeyre D., Soumelis V., Crestani B., Soler P. Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J. Immunol. 2006;176:5735–5739. doi: 10.4049/jimmunol.176.10.5735. [DOI] [PubMed] [Google Scholar]
- Marsh L.M., Jandl K., Grünig G., Foris V., Bashir M., Ghanim B., Klepetko W., Olschewski H., Olschewski A., Kwapiszewska G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018;51:1701214. doi: 10.1183/13993003.01214-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall D.C., Salciccioli J.D., Shea B.S., Akuthota P. Trends in mortality from idiopathic pulmonary fibrosis in the European Union: an observational study of the WHO mortality database from 2001–2013. Eur. Respir. J. 2018;51:1701603. doi: 10.1183/13993003.01603-2017. [DOI] [PubMed] [Google Scholar]
- McCubbrey A.L., Barthel L., Mohning M.P., Redente E.F., Mould K.J., Thomas S.M., Leach S.M., Danhorn T., Gibbings S.L., Jakubzick C.V. Deletion of c-FLIP from CD11b hi macrophages prevents development of bleomycin-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2018;58:66–78. doi: 10.1165/rcmb.2017-0154OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer E.B., Noble P.W. Idiopathic pulmonary fibrosis. Orphanet J. Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin A.V., Morales-Nebreda L., Mutlu G.M., Budinger G.R.S., Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013;49:503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin A.V., Morales-Nebreda L., Reyfman P.A., Cuda C.M., Walter J.M., McQuattie-Pimentel A.C., Chen C.-I., Anekalla K.R., Joshi N., Williams K.J.N. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017;214:2387–2404. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B., Lawson W.E., Oury T.D., Sisson T.H., Raghavendran K., Hogaboam C.M. Animal models of fibrotic lung disease. Am. J. Respir. Cell Mol. Biol. 2013;49:167–179. doi: 10.1165/rcmb.2013-0094TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj C., Haitchi H.M., Heinemann A., Howarth P.H., Olschewski A., Marsh L.M. Increased expression of p22phox mediates airway hyperresponsiveness in an experimental model of asthma. Antioxid. Redox Signal. 2017;27:1460–1472. doi: 10.1089/ars.2016.6863. [DOI] [PubMed] [Google Scholar]
- Ogushi F., Tani K., Endo T., Tada H., Kawano T., Asano T., Huang L., Ohmoto Y., Muraguchi M., Moriguchi H., Sone S. Autoantibodies to IL-1 alpha in sera from rapidly progressive idiopathic pulmonary fibrosis. J. Med. Invest. 2001;48:181–189. [PubMed] [Google Scholar]
- Peng R., Sridhar S., Tyagi G., Phillips J.E., Garrido R., Harris P., Burns L., Renteria L., Woods J., Chen L. Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease. PLoS One. 2013;8:e59348. doi: 10.1371/journal.pone.0059348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyfman P.A., Walter J.M., Joshi N., Anekalla K.R., McQuattie-Pimentel A.C., Chiu S., Fernandez R., Akbarpour M., Chen C.-I., Ren Z. Single-Cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019;199:1517–1536. doi: 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw P.J., Hubert M. Anomaly detection by robust statistics. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2018;8:e1236. [Google Scholar]
- Saeys Y., Van Gassen S., Lambrecht B.N. Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016;16:449–462. doi: 10.1038/nri.2016.56. [DOI] [PubMed] [Google Scholar]
- Selman M., Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir. Res. 2002;3:3. doi: 10.1186/rr175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman M., King T.E., Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Int. Med. 2001;134:136. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- Taillé C., Grootenboer-Mignot S., Boursier C., Michel L., Debray M.-P., Fagart J., Barrientos L., Mailleux A., Cigna N., Tubach F. Identification of periplakin as a new target for autoreactivity in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011;183:759–766. doi: 10.1164/rccm.201001-0076OC. [DOI] [PubMed] [Google Scholar]
- Tashiro J., Rubio G.A., Limper A.H., Williams K., Elliot S.J., Ninou I., Aidinis V., Tzouvelekis A., Glassberg M.K. Exploring animal models that resemble idiopathic pulmonary fibrosis. Front. Med. 2017;4 doi: 10.3389/fmed.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe R.M., Misharin A.V., Jakubzick C.V., Brinkman R., Curtis J.L., Duggan R., Freeman C.M., Herold S., Janssen W., Nakano H. Improving the quality and reproducibility of flow cytometry in the lung. Am. J. Respir. Cell Mol. Biol. 2019;61:150–161. doi: 10.1165/rcmb.2019-0191ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd N.W., Scheraga R.G., Galvin J.R., Iacono A.T., Britt E.J., Luzina I.G., Burke A.P., Atamas S.P. Lymphocyte aggregates persist and accumulate in the lungs of patients with idiopathic pulmonary fibrosis. J. Inflamm. Res. 2013;6:63–70. doi: 10.2147/JIR.S40673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafadarnejad E., Rizzo G., Krampert L., Arampatzi P., Arias-Loza A.-P., Nazzal Y., Rizakou A., Knochenhauer T., Bandi S.R., Nugroho V.A. Dynamics of cardiac neutrophil diversity in murine myocardial infarction. Circ. Res. 2020;127:e232–e249. doi: 10.1161/CIRCRESAHA.120.317200. [DOI] [PubMed] [Google Scholar]
- Wuyts W.A., Agostini C., Antoniou K.M., Bouros D., Chambers R.C., Cottin V., Egan J.J., Lambrecht B.N., Lories R., Parfrey H. The pathogenesis of pulmonary fibrosis: a moving target. Eur. Respir. J. 2013;41:1207–1218. doi: 10.1183/09031936.00073012. [DOI] [PubMed] [Google Scholar]
- Xie T., Wang Y., Deng N., Huang G., Taghavifar F., Geng Y., Liu N., Kulur V., Yao C., Chen P. Single-cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep. 2018;22:3625–3640. doi: 10.1016/j.celrep.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper; the supplementary data are available on Mendeley Data: https://doi.org/10.17632/7j485t986v.1.