

Abstract
Borrelia burgdorferi (B. burgdorferi), the causative agent of Lyme disease, is transmitted by the bite of an infected tick. Once inoculated into the host dermis, it disseminates to various organs including distant skin sites, the heart, the joint and the nervous system. Most humans will develop an early skin manifestation called erythema migrans at the tick bite site. This can be followed by symptoms such as carditis, neuritis, meningitis or arthritis if not treated. A specific mouse strain, C3H/HeN develops arthritis with B. burgdorferi infection while another strain, C57BL/6 develops minimal to no arthritis. Neither strain of mice shows any skin signs of rash or inflammation. Factors that determine the presence of skin inflammation and the joint arthritis susceptibility in the host are only partially characterized. We show here that murine fibroblast-like synoviocytes (FLS) display trained immunity, a program in some cells that results in increased inflammatory responses if the cell has previously come in contact with a stimulus, and that trained immunity in FLS tested ex vivo correlates with Lyme arthritis susceptibility. Conversely, skin fibroblasts do not exhibit trained immunity which correlates with the absence of skin symptoms in these mice. Moreover, we demonstrate that the trained phenotype in FLS is affected by the cell environment which depends on the host genetic background. Future studies expanding this initial report of the role of trained immunity on symptoms of B. burgdorferi infection may provide insight into the pathogenesis of disease in murine models.
Keywords: Borrelia burgdorferi, Fibroblast-like synoviocyte, Trained immunity, Tolerance, Arthritis
Introduction
Infection with Borrelia burgdorferi (B. burgdorferi2), the causative agent of Lyme disease, results in a multi-system infection in both humans and in murine models of disease. In both humans and animals, the bacteria move from the skin, where they are deposited by a tick vector and then spread to involve sites such as the heart, the nervous system and the joints (1). Manifestations of the disease are thought to be due to the immune response to the organism and not to toxins or tissue destroying enzymes produced by the organism itself. While humans develop a characteristic rash called erythema migrans at the site of inoculation, mice do not typically develop any skin manifestations. For both humans and inbred strains of mice, joint arthritis is typically the latest developing and longest lasting of the manifestations and can be characterized by spontaneous remission of inflammation with recurrence in the same or a different joint (2).
B. burgdorferi differs from most infectious agents because the organism causes longstanding infection without killing its host. In mice, infection is lifelong unless treated with antibiotics (3). In humans, the infection can last several years in the absence of treatment (4), with rare cases of organisms found in a skin manifestation called acrodermatitis chronica atrophicans (ACA3) up to 10 years later (5, 6). The longevity of the organism in the host makes it likely that immune cells will have repeated contacts with B. burgdorferi or its antigens. It is currently not well understood how repeated exposure of cells to B. burgdorferi antigens could affect subsequent cellular responses. However, repeated responses to other stimuli have been shown to result in either innate immune tolerance or trained immunity (7).
Both responses describe the long-term reprogramming of innate immune cells induced by an endogenous or exogeneous ligand. Memory is established, thereby allowing cells to have an increased (trained immunity) or decreased (tolerance) inflammatory responses following subsequent exposure to an antigen compared to the initial exposure. Trained immunity has been shown to result in upregulation of inflammatory cytokines in in other bacterial infections. Prior exposure to muramyl dipeptide induces inflammatory cytokines and results in protection against Streptococcus pneumoniae and Toxoplasma gondii; prior exposure to flagellin protects against subsequent S. pneumoniae infection(8, 9). It has been also observed in vector-borne pathogen infections such as malaria and leishmaniasis where pre-infection with plasmodium or pretreatment with β-Glucan respectively increase the inflammatory response and confer some protection during subsequent parasite infections (10, 11). In these examples, the increased responses conferred by trained immunity occurs through nonspecific mechanisms involving epigenetic reprogramming of innate immune cells. The converse of trained immunity, innate immune tolerance, protects the host from tissue damage induced by excessive inflammation. It has been well studied using LPS, where repeated exposures confer protection against septic shock through reduction of cytokine induction (12, 13). Tolerance is also involved in the regulation of the immune response toward the microbiome, as defects in this mechanism have been associated with auto-inflammatory diseases such as the inflammatory bowel disease in the gut (14). A balance in regulation of trained immunity and innate immune tolerance allows the host to control the infection while simultaneously avoiding overly exuberant responses that could lead to host damage.
Synovial fibroblasts, also called fibroblast-like synoviocytes (FLS4) are a major cell type present in the joint. They are involved in the inflammation observed in B. burgdorferi infected mice as well as in human arthritis (14–16). FLS develop a persistent inflammatory phenotype through trained immunity following repeated exposures to inflammatory cytokines (17). In this study, we examine the role of FLS in the pathogenesis of Lyme arthritis. We found that FLS exhibit a phenotype that is consistent with trained immunity and that correlates with arthritis development in C3H/HeN mice. This contrasts with murine skin fibroblasts that show tolerance upon re-exposure to B. burgdorferi. This suggests that the continued presence of B. burgdorferi in the host induces trained immunity in murine FLS resulting in a highly inflammatory phenotype. Trained immunity may then play a role in explaining why arthritis is a prominent feature of infection with B. burgdorferi in mice (3).
Material and methods
Bacteria and Mice
An infectious isolate of B. burgdorferi, N40, was used. Four- to 8-wk-old female C3H/HeN, C57BL/6 and TLR2 ko mice were purchased from the National Cancer Institutes and Charles River Laboratories and co-housed at our facility until use. MyD88 ko mice were kindly provided to us by Dr. Pilar Alcaide (Tufts University, Boston, USA). For infection, mice were inoculated with one subcutaneous injection of 105 bacteria per mouse.
Cells isolation
Joint cells.
The joint cell isolation protocol was performed as previously described (15). Briefly, joints were extracted from euthanized mice and skin was removed. Tissues were cut in 2–3 mm pieces and digested with liberase™ (Roche), a cocktail of highly purified collagenase I, collagenase II and thermolysin. Whole cells were collected and seeded on a 24-well plate and used right away for incubation studies.
FLS.
The FLS isolation protocol was performed as previously described (18). Briefly, mice were euthanized, and hip synovium were extracted. Tissues were digested with type IV collagenase and seeded on a flask containing DMEM + 10% FBS and antibiotics. Cells were passaged once they reached 80% confluency and split 1:3. Cells were used for further studies between passage three and five.
Fibroblasts.
The fibroblasts isolation protocol was performed as previously described (19). Briefly, ears and tails were collected from euthanized mice. Tissues were cut in small pieces and treated with an enzymatic cocktail containing collagenase D (Roche) and pronase (Roche). Detached cells were collected and culture in flask containing DMEM + 10% FBS and antibiotics. Cells were passaged once they reached 80% confluency and split 1:2. Cells were used for further studies between passage three and four.
Ex vivo/In vitro assays
Ex vivo.
FLS, fibroblasts or joint cells from naïve or mice infected 4–8 weeks B. burgdorferi were collected and used immediately (mixed joint tissue) or incubated in vitro beyond passage three (FLS and fibroblasts). Cells were seeded on a 12-well (FLS and fibroblasts) or 24-well (Joint cells) plate and fresh B. burgdorferi were added at a MOI of 10 for six (FLS and fibroblasts) or 24 hours (joint cells). Then supernatants (FLS, fibroblasts and joint cells) and cells in TRIzol (FLS and fibroblasts) were collected for further analysis.
In vitro.
FLS or fibroblasts from naïve mice were collected and incubated in vitro beyond passage three. Cells were seeded on a 12-well plate and fresh B. burgdorferi were added at a MOI of 10 for 24 hours, washed with PBS, and incubated a second time with fresh fresh B. burgdorferi at a MOI of 10 for six hours. Then supernatants and cells in trizol were collected for further analysis.
ELISA
To measure IL-6, MIP1a, CXCL1 and TNF-a secreted by FLS and fibroblasts, enzyme-linked immunosorbent assays (ELISAs) were performed on cell supernatants as per the manufacturer’s instructions (R&D).
PCR/qRT-PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) and transcribed into cDNA by using Superscript VILO cDNA synthesis kit (Thermo Fisher Scientific). PCR was performed using Phusion polymerase. The primers used are: !8s (F- CATGATTAAGAGGGACGGC, R- TTCAGCTTTGCAACCATACTC), icam (F- AGACACAAGCAAGAAGACCACA, R- TGACCAGTAGAGAAACCCTCG), vcam (F- GGAGACCTGTCACTGTCAACTG, R- TCCATTTCACCACTGTGTAACC), cd248 (F- GCCAGCAGATGTGTGTCAA, R- GTAGGTGCCAGCCATAGGAT), Prolyl4-hydroxylase (F- GATTGTGGAGTTCAGTGAGCC, R- TTCATCATAGGTCCTGTTGTCTG) and cd68 (F- GCTTCTGCTGTGGAAATGC, R- GGTAGGTTGATTGTCGTCTGC). DNA was loaded on an agarose gel and revealed using Chemidoc system. The qPCR was performed on the CFX96 instrument (Bio-Rad) with Power SYBR Green PCR Master Mix (Thermo Fisher Scientific). The primers used are: il-6 (F- GACTTCACAGAGGATACCAC, R- TATCCAGTTTGGTAGCATCC), mip1a (F- TTCTCTGTACCATGACACTCTGC, R- CGTGGAATCTTCCGGCTGTAG), cxcl1 (F- CTGGGATTCACCTCAAGAACATC, R- CAGGGTCAAGGCAAGCCTC) and tnf-a (F- ATGAGCACAGAAAGCATGATC, R- TACAGGCTTGTCACTCGAATT). Genes expression was normalized to the hprt gene expression and compared to untreated conditions using the 2^−ΔΔCt method.
Confocal microscopy
Cells were cultured in chamber slides until they reach 80% confluency. Cells were then fixed in 1% paraformaldehyde and permeabilized in 5% goat serum with 0.1% saponin. Cells were incubated with either FITC-anti-CD90.2 (1:100) or anti-CD248 (40 ug/mL), anti-fibronectin (1:100) or anti-vimentin (1/1000) followed by Alexa-fluor-647 secondary antibodies (1/1000). Slides were examined using a Nikon A1R Confocal microscope and the 63X oil objective. Images were merged using the Fiji (ImageJ) software.
Results
Joint cells from B. burgdorferi infected mice are hyperresponsive to repeat exposure to B. burgdorferi.
When infected with B. burgdorferi, C3H/HeN mice develop arthritis that peaks at around three weeks of infection and resolves after six to eight weeks with the development of antibodies and a decrease in the numbers of B. burgdorferi in tissue (3). To determine if prior exposure to B. burgdorferi affects cellular responses upon re-exposure, we extracted total joint cells from age matched C3H/HeN mice that were either uninfected or infected with B. burgdorferi for 4 weeks. These cells were then incubated ex vivo with B. burgdorferi for 24h hours before measuring pro-inflammatory cytokine expression (IL-6, TNFα, MIP1a and CXCL1). Cells from infected mice secreted a higher amount of all four cytokines as compared with cells from uninfected mice (Fig. 1A). This result suggests that prior exposure to B. burgdorferi alters the responsiveness of joint cells to subsequent exposures in such a way as to result in increased induction of inflammatory cytokines.
Fig. 1. Joint cells from B. burgdorferi infected C3H/HeN mice have a persistent inflammatory phenotype.
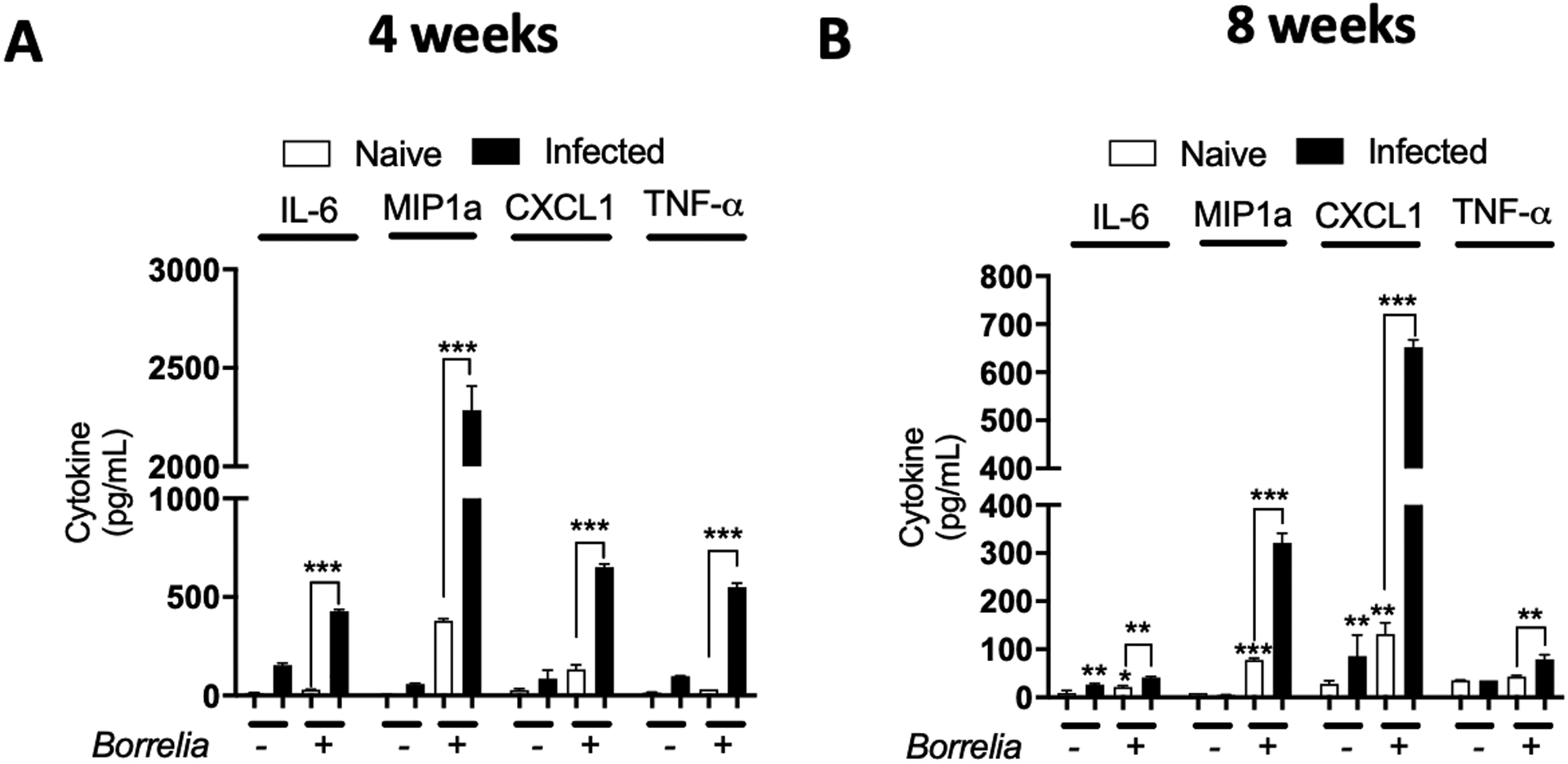
Joint from C3H/HeN mice infected or not with B. burgdorferi for four (A) or eight (B) weeks were extracted to collect whole cells and seeded on a plate. Fresh B. burgdorferi were then added to the cells at a MOI of 10 for 24 hours. Supernatants were collected and cytokines (IL-6, MIP1a, CXCL1 and TNF-a) expression was measured by ELISA. Bars represent the mean ± SEM of at least triplicates. Data are representative of two independent experiments conducted with three animals in each group. *P < 0.05; **P < 0.01; ***P < 0.001.
When we isolated and cultured total joint cells, we did not differentiate between the different cell types located in the joint. During steady state, approximately 20% of all cells in joints are leukocytes (CD45+ cells) (15). However, this proportion is higher in B. burgdorferi infected joints due to neutrophil and lymphocyte infiltration. As a result, the cell populations between naïve and infected mice might be comprised of significantly different cell types (20). To determine the influence of cell type on our results, we repeated the experiment using cells from uninfected mice or mice post-resolution of arthritis after B. burgdorferi infection (8 weeks after infection). At 8 weeks post-infection, the distribution of cell types in the joint is similar between naïve and infected mice (21). Despite this, cells from infected mice still secreted more cytokines following ex vivo incubation with B. burgdorferi than naïve cells (Fig. 1B). These data support the idea that joint resident cells develop an increased inflammatory phenotype upon re-exposure with B. burgdorferi, which fits with the concept of trained immunity. In addition, since the effect is maintained 8 weeks post infection in ex vivo experiments, it suggests this immune memory is fairly long lived.
Fibroblast-like synoviocytes, a joint resident cell-type, develop a trained immunity phenotype in B. burgdorferi-infected C3H/HeN mice while skin fibroblasts show a tolerance phenotype.
We next wanted to determine the specific cell type responsible for the increase in pro-inflammatory cytokine production following B. burgdorferi re-exposure in the joint. FLS have been shown to have a major role in inducing inflammation in the joint following B. burgdorferi infection. We thus purified FLS from the hip synovium of age matched B. burgdorferi infected C3H/HeN mice (8 weeks of infection) and uninfected controls (Fig. 2). Cells were cultured for at least three passages and checked for purity by qPCR and confocal microscopy (Fig. S1) before incubation ex vivo with B. burgdorferi for 24 hours. Unpassaged cells were positive for monocyte markers by qRT-PCR, but these were lost upon passaging of the cells. Staining of the cells using antibodies to fibroblast markers showed that 100% of passaged cells expressed these markers. Passaged FLS from infected mice showed a higher response against B. burgdorferi than cells from naïve mice as shown by a higher expression of IL-6, MIP1a, CXCL1 and TNFa at the protein (Fig. 2A) and mRNA level (Fig. S2A). Of note, this phenotype was maintained despite expanding the cells about 30-fold over two weeks. This shows that the phenotype is maintained despite prolonged culture in vitro and may be transmitted to newly divided cells.
Fig. 2. FLS from C3H/HeN mice infected with B. burgdorferi have a trained phenotype while FBs are tolerized.
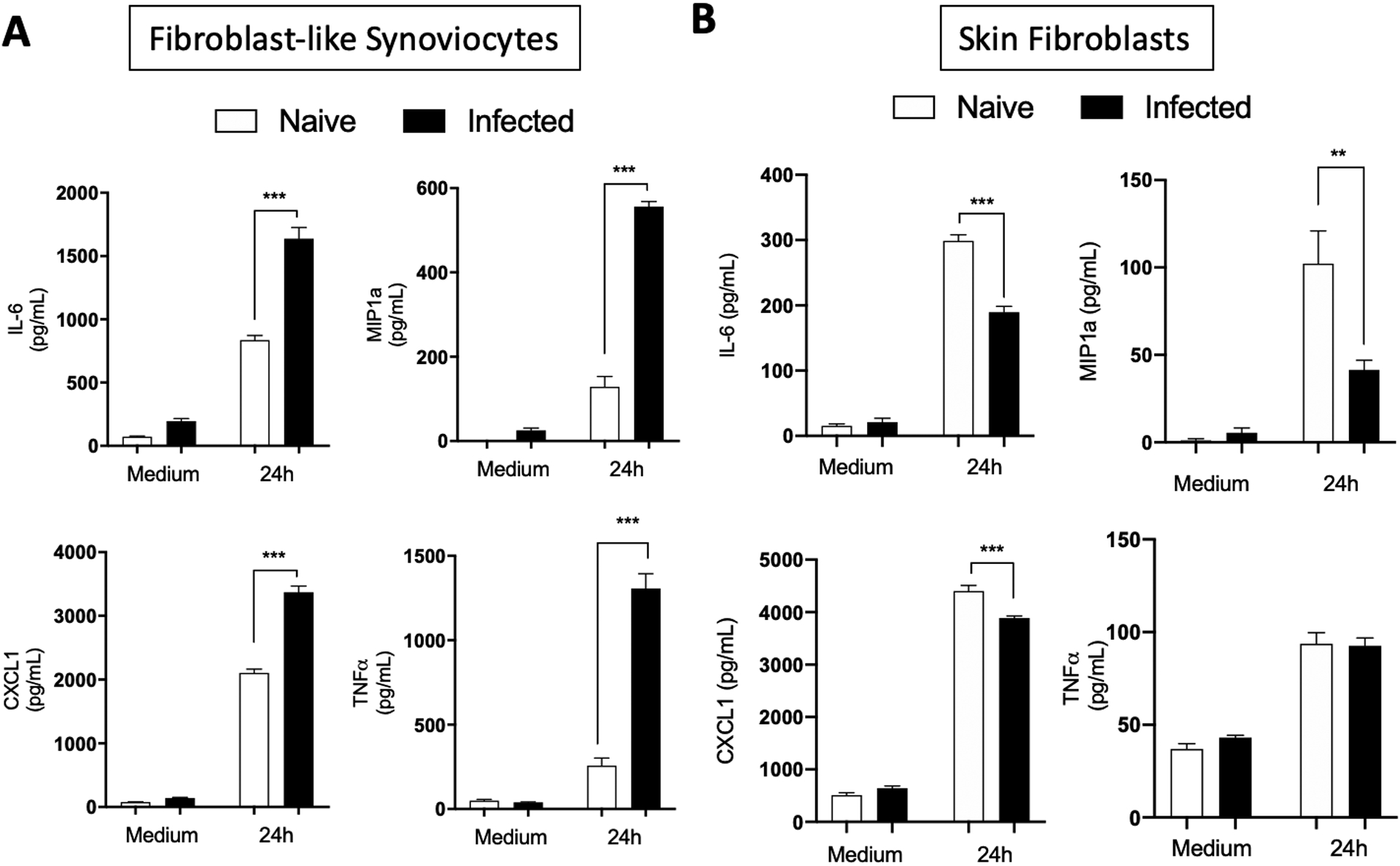
Hip synovium and ears/tails from naïve or eight weeks B. burgdorferi-infected C3H/HeN mice were collected and FLS (A) and FBs (B) were extracted. Cells were culture for three to five passages before being seeded on a plate. B. burgdorferi were then added at a MOI of 10 for 24 hours. Supernatants were collected and cytokines (IL-6, MIP1a, CXCL1 and TNF-a) expression was measured by ELISA. Bars represent the mean ± SEM of at least triplicates. Data are representative of at least two independent experiments conducted with three animals in each group. ***P < 0.001.
To determine the specificity of trained immunity in synovial fibroblasts, we tested the effect of repeat exposure to B. burgdorferi in skin fibroblasts. B. burgdorferi can be found in large numbers in the skin of infected mice with little or no inflammation. In contrast to FLS, skin fibroblasts extracted from C3H/HeN mice infected for 8-weeks had a lower inflammatory response compared to cells from naïve mice when incubated ex vivo with B. burgdorferi as seen by measuring IL-6, MIP1a, CXCL1 and TNFa at the protein level (Fig. 2B). The B. burgdorferi-induced tolerance was not observed at the mRNA level which showed similar levels in fibroblasts from infected and uninfected mice (Fig. S2B). This suggests that training against B. burgdorferi exposure and re-exposure is cell and tissue specific and likely linked to the composition of the inflammatory milieu in the joint tissue.
FLS from arthritis-resistant C57BL/6 mice do not develop a trained phenotype upon repeated B. burgdorferi exposure in vivo
We next decided to test for the phenotype using C57BL/6 mice, a mouse strain that shows resistance to B. burgdorferi-mediated arthritis. In contrast to C3H/HeN mice, FLS from infected C57BL/6 mice showed no differences between cells from uninfected or 8 weeks infected mice in levels of cytokines by ELISA (Fig. 3A). By measurements of mRNA transcripts, gene expression was even lower in infected cells than in naïve cells (Fig. 3B). These data shows that the FLS trained phenotype is specific to the C3H/HeN mice and suggests that it may participate in arthritis susceptibility observed in this strain.
Fig. 3. FLS from arthritis resistant C57BL/6 mice do not get trained during B. burgdorferi infection.
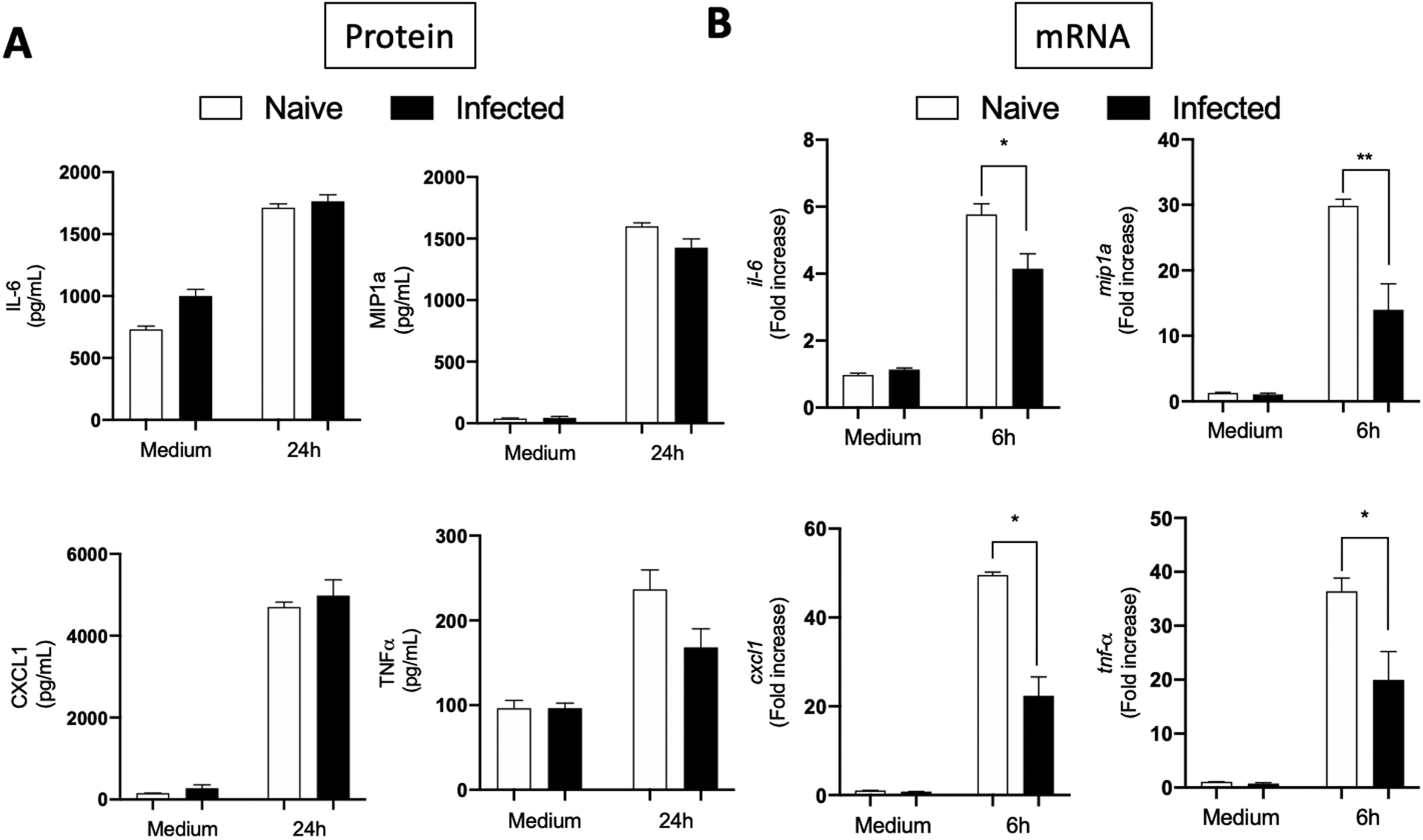
Hip synovium from naïve or eight weeks B. burgdorferi-infected C57BL/6 mice were collected and FLS were extracted. Cells were culture for three to five passages before being seeded on a plate. B. burgdorferi were then added at a MOI of 10 for six or 24 hours. Supernatants and cells were collected and cytokines (IL-6, MIP1a, CXCL1 and TNF-a) expression was measured by ELISA (A) and qPCR (B). Bars represent the mean ± SEM of at least triplicates. Data are representative of two independent experiments conducted with three animals in each group. *P < 0.05; **P < 0.01.
Stimulation and re-stimulation of FLS in vitro reproduces trained immunity from ex vivo cells.
To better characterize the trained immunity observed in FLS, we studied the responses of cultured FLS to stimulation and re-stimulation. FLS from naïve C3H/HeN or C57BL/6 mice were collected, incubated with or without B. burgdorferi for 24h, washed and stimulated a second time with B. burgdorferi for 24h. Similar to the ex vivo model, FLS from C3H/HeN mice preincubated with B. burgdorferi showed increased induction of the inflammatory cytokines IL-6, MIP1a and CXCL1 by ELISA (Fig. 4A) and MIP1a and CXCL1 by qRT-PCR (Fig. S3A). Of note, expression and production of one cytokine, TNFa did not match ex vivo results. In vitro, TNFa showed decreased rather than increased induction upon re-exposure by both qRT-PCR and ELISA.
Fig. 4. FLS from C3H/HeN and C57BL/6 share the same phenotype in vitro.
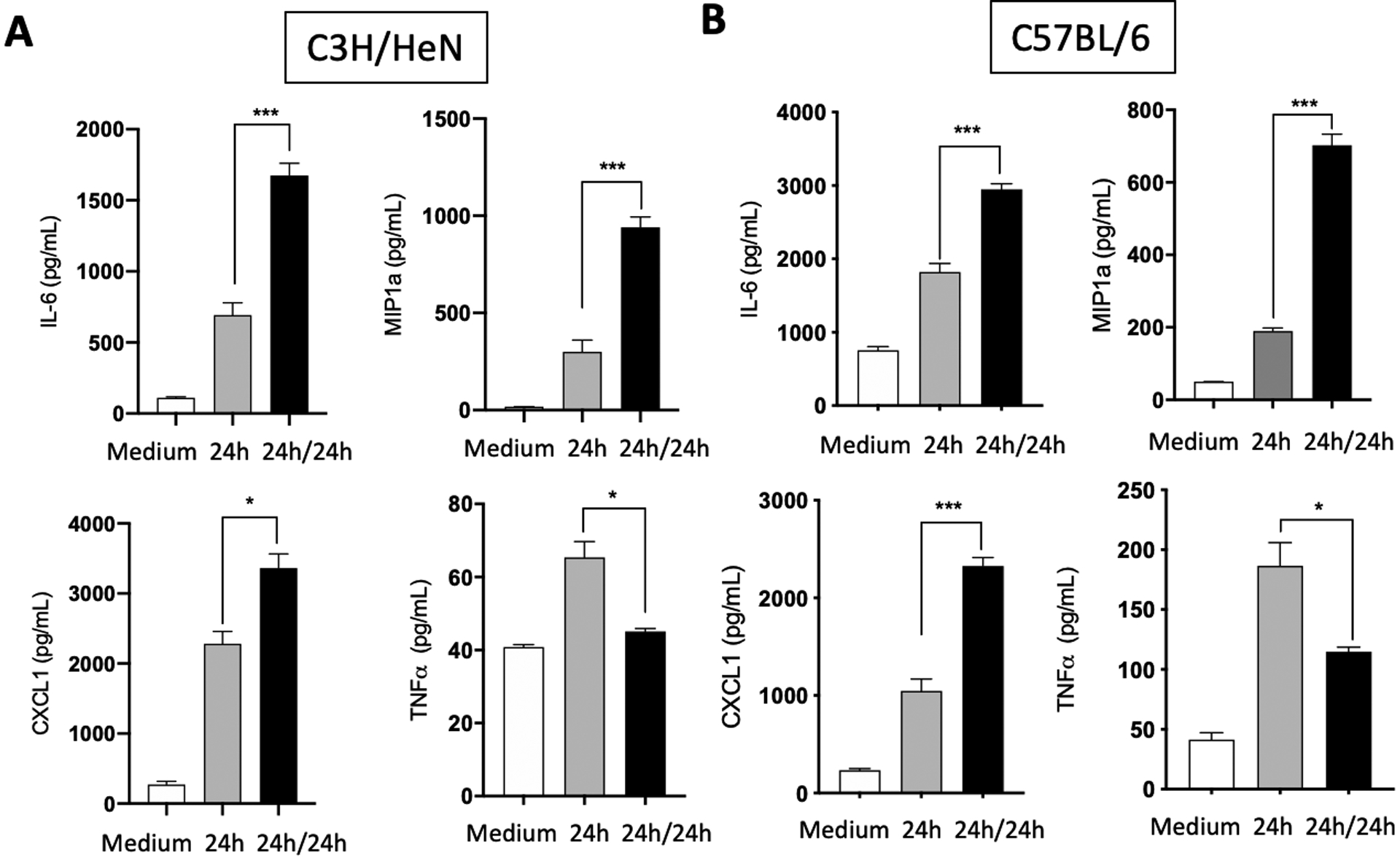
Hip synovium from naïve C3H/HeN (A) or C57BL/6 (B) mice were collected and FLS were extracted. Cells were culture for three to five passages before being seeded on a plate. B. burgdorferi were then added at a MOI of 10 for 24 hours (24h). Cells were washed with PBS and fresh B. burgdorferi were added to the cells at a MOI of 10 for another 24 hours (24h/24h). Supernatants were collected and cytokines (IL-6, MIP1a, CXCL1 and TNF-a) expression was measured by ELISA. Bars represent the mean ± SEM of at least triplicates. Data are representative of at least three independent experiments. *P < 0.05; ***P < 0.001.
For FLS from C57BL/6 mice, in vitro experiments showed similar increases in cytokine release as the C3H/HeN cells with repeat exposures, with IL6, MIP1a and CXCL1 being increased and TNFa being decreased both at the protein (Fig. 4B) and mRNA level (Fig. S3B). This contrasts with the ex vivo results seen in C57Bl/6 mice and suggests that the difference between the strains of mice is not in the intrinsic ability of their FLS cells to develop trained immune responses and is likely due to the presence of other inflammatory mediators or local cell to cell communication that alters cellular behavior.
The FLS trained phenotype does not require TLR2 nor MyD88 to be induced
Innate immunity and signaling through the toll-like receptors (TLR5) plays an important role in the inflammatory response to B. burgdorferi (22). TLR signaling has also been shown to induce epigenetic changes that alter cellular responsiveness to different stimuli (23). We explored if TLR2 which recognizes B. burgdorferi lipoproteins or the TLR adapter molecule, MyD88, are required for increased cytokine release upon repeat exposure of FLS to B. burgdorferi (24, 25). FLS from TLR2 and MyD88 ko mice were collected and tested by B. burgdorferi exposure and re-exposure. Interestingly, TLR2 knockout cells, while having an expected lower overall inflammatory response compared to WT, still exhibited increased cytokine release upon B. burgdorferi re-exposure for IL-6, MIP1a and CXCL1 (Fig. 5A). As expected, MyD88 knockout FLS showed greatly decreased cytokine induction upon exposure to B. burgdorferi overall, but still showed increased cytokine expression upon re-exposure (Fig. 5B). Together, these data suggest that TLR2 or MyD88 signaling is not required for induction of FLS trained immunity and that the low amounts of inflammatory mediators being produced in the deficient cells was still sufficient for the induction of immune memory.
Fig. 5. The B. burgdorferi-induced training in FLS does not require the TLR2-MyD88 pathway.
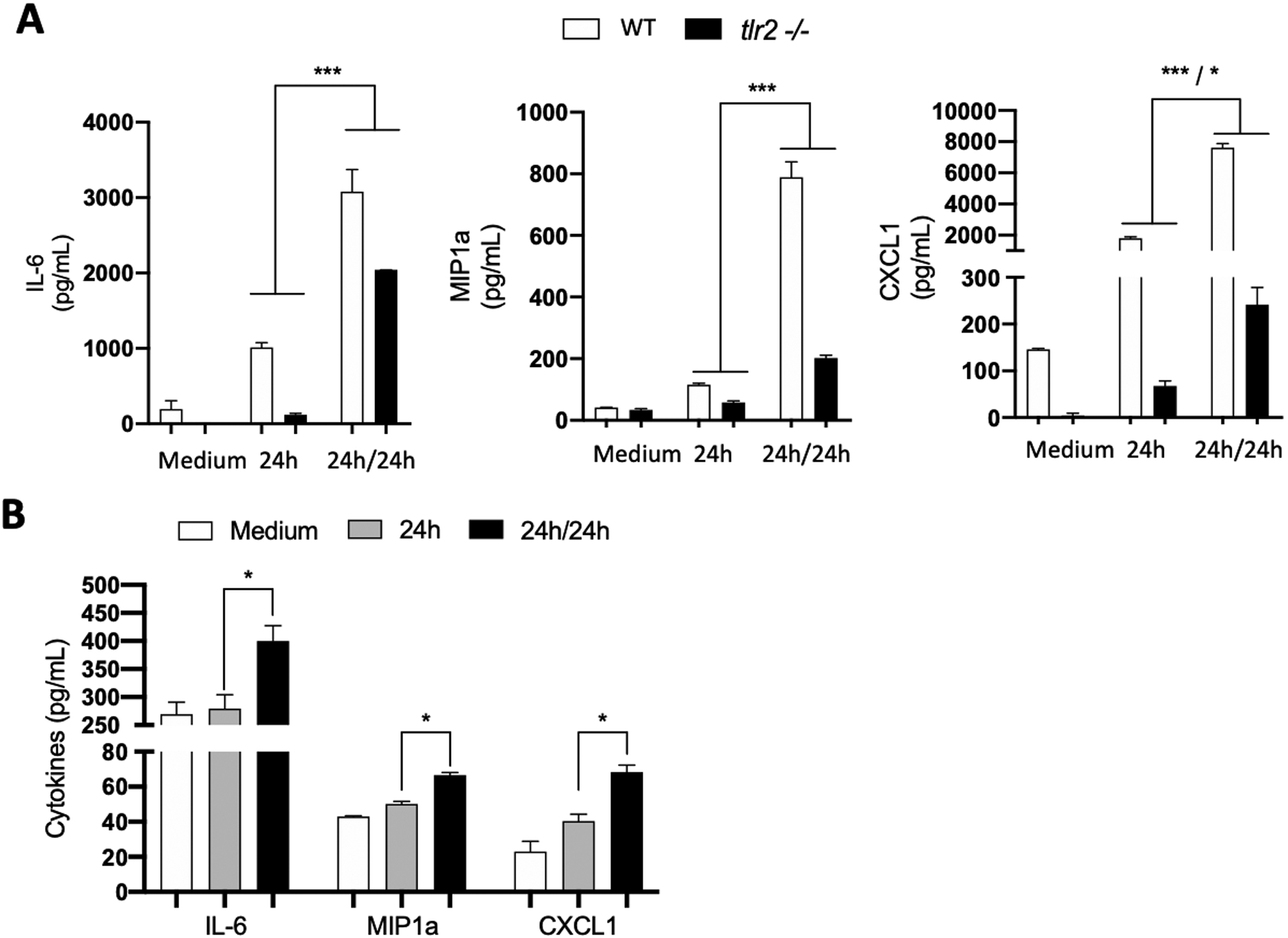
Hip synovium from WT C57BL/6 mice, tlr2 ko mice (A) or myd88 ko (B) mice were collected and FLS were extracted. Cells were culture for three to five passages before being seeded on a plate. B. burgdorferi were then added at a MOI of 10 for 24 hours (24h). Cells were washed with PBS and fresh B. burgdorferi were added to the cells at a MOI of 10 for another 24 hours (24h/24h). Supernatants were collected and cytokines (IL-6, MIP1a, CXCL1 and TNF-a) expression was measured by ELISA. Bars represent the mean ± SEM of at least triplicates. Data are representative of two independent experiments. *P < 0.05; ***P < 0.001.
Discussion
The manifestations of B. burgdorferi infection vary significantly between animal species and by tissue types. The skin is comprised of multiple cell types able to detect B. burgdorferi, including resident cells such as fibroblasts (26). Skin fibroblasts have been shown to express many pro-inflammatory cytokines and chemokines in response to B. burgdorferi in vitro (27). While in humans, most patients develop an early skin rash called erythema migrans, that is felt to be a result of the inflammatory response to the organism, mice do not develop a similar rash. Of note in humans with erythema migrans, the rash typically resolves spontaneously over the period of weeks although the bacteria remain present for much longer. Bacteria remain present in mouse skin almost indefinitely, without significant inflammation. This contrasts with the joint where C3H/HeN mice develop a severe arthritis in response to B. burgdorferi infection. The joint swelling is a late manifestation following B. burgdorferi infection in humans as well, although its severity is highly variable and can range from arthralgia to highly inflamed and proliferative synovial lesion. Murine Lyme arthritis is characterized by an infiltration of neutrophils and lymphocytes (20). Resident cells, including fibroblast-like synoviocytes (FLS), also participate to the inflammatory response in the joint in mice (28, 29). Human FLS have also been shown to express cytokines, chemokines and proteases in response to B. burgdorferi (30–32). Moreover, the bacteria in combination with IFNγ present in arthritic joints, have been shown to initiate the differentiation of human FLS into a highly inflammatory phenotype, notably through the upregulation of a large number of genes associated with antigen presentation (16).
Here, we showed that B. burgdorferi induces a highly pro-inflammatory trained phenotype in murine FLS that is persistent even after initial resolution of arthritis. In contrast, skin fibroblasts do not exhibit this type of pro-inflammatory response and even develop reduction of release of certain pro-inflammatory cytokines with repeat exposure that is typical of innate immune tolerance. Differential cell type responses to repeated stimulations has been demonstrated with TNFa. TNFa exposure of macrophages results in subsequent reductions in cytokine responses upon re-exposure (33), while exposure of FLS results in a sensitization that causes increased cytokine induction upon re-exposure (17). Fibroblasts from different origins have also been shown to respond differently to repeated exposures to LPS (34).
Beyond differences in cell type, it is possible that the pathogen may differentially express antigens that influence the development of trained immunity or tolerance to benefit its own survival. Antigens expressed by B. burgdorferi are different in different murine tissues (35) and this might result in the induction of different states of innate immune memory between the skin and the joint. Moreover, specific antigens might not be expressed at the same level between tissues, since B. burgdorferi gene expression depends on many elements of its external environment (including but not limited to temperature, pH, availability of micronutrients) (36–39) and these parameters are different between the skin and deeper tissues (40). Indeed, the antigen concentration is a critical parameter influencing innate immune memory (41). For example, low doses of LPS or Pam3CSK4, TLR4 and TLR2 ligands respectively, induce trained immunity while a higher dose induces tolerance in human monocytes (42). This could also potentially explain the difference seen in trained immunity responses of C57BL/6 FLS tested ex vivo vs in vitro as the levels of bacteria in C57BL/6 mice maybe lower than what is used to expose and re-expose in vitro.
In mice, specific genetic backgrounds have been linked to arthritis susceptibility. Notably, C3H/HeN mice develop significant and recurrent arthritis following B. burgdorferi infection while C57BL/6 show resistance to joint inflammation/swelling (3). Here we show that B. burgdorferi -induced trained immunity in FLS occurs ex vivo only in cells taken from infected, arthritis-susceptible mice, further suggesting it is a contributing factor promoting arthritis in these mice. Interestingly, the B. burgdorferi-induced training in FLS depends on the cell environment, which may be influenced at least partially by the host genetic background, rather than on the cell ability to be trained. Indeed, B. burgdorferi induce training in both C3H/HeN and C57BL/6 cells in vitro, but only C3H/HeN cells are trained in vivo. Additional factors present in C57BL/6 mice might prevent FLS from being trained in vivo. It may include high expression of the anti-inflammatory cytokine IL-10, which is higher in C57BL/6 mice than in C3H/HeN mice. IL-10 plays a role in the reduction of inflammation in the joint (43, 44). This anti-inflammatory cytokine induces a tolerant phenotype in macrophages (45–47). IL-10 present in the joint might then prevent FLS from developing trained immunity by downregulating inflammatory pathways and promoting tolerance. Alternatively, in the C3H/HeN background, the FLS training might be induced indirectly by B. burgdorferi through interferons. Repeated IFN-b exposure has been shown to induce trained immunity in embryonic fibroblasts through histone methylation (48). In vitro, human FLS become highly inflammatory in response to IFNg, a critical cytokine for synovial lesion development in human and murine hosts (16, 49). These events could train the cells upon re-exposure with the bacteria. In vivo, C3H/HeN mice highly express type-I IFN following B. burgdorferi infection and the interferon response has been linked to the increased inflammation observed in this genetic background as compared to C57BL/6 (15, 50, 51). Therefore, interferons might also be a contributing factor in the initiation of FLS training observed in C3H/HeN mice and leading to excessive inflammation.
Both the induction of innate immune tolerance in skin fibroblasts and trained immunity in joint FLS was long lasting and transmissible through several cell generations. This is consistent with prior studies that showed the durability of trained immunity in tendon fibroblasts from patients with chronic tendinopathy (52). This is thought to be mediated by priming effects from the initial exposure resulting in epigenetic changes that re-program the cells long term. It is unclear what types of changes are responsible for the phenotypes observed in B. burgdorferi induced training.
While the innate immune tolerance persistence observed here in skin fibroblasts correlates with the absence of skin symptoms in mice over time, the training persistence in FLS correlates with arthritis recurrence observed in the joint of C3H/HeN mice (3). It is tempting to speculate that the hyper-responsiveness to B. burgdorferi in the joint may help with clearance of the organism during early infection. B. burgdorferi have not been cultured from synovial fluid from human patients but have been cultured from skin up to a decade after the original infection in patients with ACA (53). Notably, the ability to survive in mouse skin is critical for B. burgdorferi to complete its lifecycle and be taken up by a feeding tick. Infection in the joint does not serve a known purpose for completion of the B. burgdorferi lifecycle in mice. In humans who have early Lyme disease and are not treated, there are reports of relapses of erythema migrans occurring after resolution of the initial rash (54). One possible explanation would be the waning of tolerance in the skin as the infection comes under control, but is not fully cleared by the adaptive immune system. This would, over time, leave naïve cells able to generate a new response to residual spirochetes. In patients with erythema migrans that are treated, they may contract a second episode of Lyme disease—most commonly marked by another erythema migrans lesion. This suggests that tolerance in the skin may be of limited duration in humans or that there is variability among patients in the tolerance response. Notably, repeat infection does not typically occur in the first year after the original infection (55). This is currently thought to be due to partial immunity from the original infection, but it is also possible that reinfection can occur during this period but symptoms are muted due to tolerance. As there is currently no good test for determining re-infection, reinfection is typically determined by clinical observation of the erythema migrans rash.
The role of trained immunity in human Lyme arthritis remains unknown. Antigens such as peptidoglycan have been found for prolonged periods in the joints of patients with Lyme arthritis and could serve as a persistent stimulus promoting inflammation in the setting of trained immunity (56). The tendency towards trained innate immunity in joints could provide an explanation for why post-treatment Lyme arthritis, which can persist for long periods after antibiotic therapy, improves with anti-inflammatory agents and for why similarly prolonged recurrence of inflammation does not occur at sites such as the skin that do not exhibit trained immunity. Studies in patients with rheumatoid arthritis have suggested a link between trained immunity and the induction of arthritis (57). Further studies will be needed to determine the presence and potential impact of trained immunity in humans with Lyme arthritis and whether blocking this phenotype in cells may reduce the duration of symptoms in patients who continue to have arthritis even after antibiotic therapy.
Supplementary Material
Key Points.
Cells show immunological learning to prior exposure to Borrelia burgdorferi.
Learned responses differ by cell type and include both training and tolerance.
Learned responses correlate with infectious symptoms observed in murine models.
Acknowledgement
We thank the members of the Hu lab, especially Urmila Powale and Dr. Tanja Petnicki-Ocwieja, for helpful conversations and critical evaluation of the data. The visual abstract was created with Biorender.
This work was supported by National Institutes of Health (NIH) (R21AI126757, R01AI15015 to LTH) and the Deborah and Charles Blackman-GLA Fellowship (to QB).
Footnotes
B. burgdorferi: Borrelia burgdorferi
ACA : Acrodermatitis chronica atrophicans
FLS: fibroblast-like synoviocytes
TLR: toll-like receptor
References
- 1.Cardenas-de la Garza JA, De la Cruz-Valadez E, Ocampo-Candiani J, and Welsh O. 2019. Clinical spectrum of Lyme disease. Eur. J. Clin. Microbiol. Infect. Dis 38: 201–208. [DOI] [PubMed] [Google Scholar]
- 2.Bockenstedt LK, and Wormser GP. 2014. Review: Unraveling Lyme Disease. Arthritis Rheumatol. 66: 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barthold SW, De Souza MS, Janotka JL, Smith AL, and Persing DH. 1993. Chronic lyme borreliosis in the laboratory mouse. Am. J. Pathol 143: 959–972. [PMC free article] [PubMed] [Google Scholar]
- 4.Steere AC, Schoen RT, and Taylor E. 1987. The clinical evolution of lyme arthritis. Ann. Intern. Med 107: 725–731. [DOI] [PubMed] [Google Scholar]
- 5.Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JWR, Li X, and Mead PS. 2016. Lyme borreliosis. Nat. Rev. Dis. Prim 2: 16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asbrink E, Brehmer-Andersson E, and Hovmark A. 1986. Acrodermatitis chronica atrophicans - A spirochetosis. Clinical and histopathological picture based on 32 patients; course and relationship to erythema chronicum migrans afzelius. Am. J. Dermatopathol 8: 209–219. [DOI] [PubMed] [Google Scholar]
- 7.Boraschi D, and Italiani P. 2018. Innate immune memory: Time for adopting a correct terminology. Front. Immunol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muñoz N, Van Maele L, Marqués JM, Rial A, Sirard JC, and Chabalgoity JA. 2010. Mucosal administration of flagellin protects mice from Streptococcus pneumoniae lung infection. Infect. Immun 78: 4226–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krahenbuhl J, Sharma S, Ferraresi R, and Remington J. 1981. Effects of Muramyl Dipeptide Treatment on Resistance to Infection with Toxoplasma gondii in Mice. Infect. Immun 31: 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.dos Santos JC, Barroso de Figueiredo AM, Teodoro Silva MV, Cirovic B, de Bree LCJ, Damen MSMA, Moorlag SJCFM, Gomes RS, Helsen MM, Oosting M, Keating ST, Schlitzer A, Netea MG, Ribeiro-Dias F, and Joosten LAB. 2019. β-Glucan-Induced Trained Immunity Protects against Leishmania braziliensis Infection: a Crucial Role for IL-32. Cell Rep. 28: 2659–2672.e6. [DOI] [PubMed] [Google Scholar]
- 11.Schrum JE, Crabtree JN, Dobbs KR, Kiritsy MC, Reed GW, Gazzinelli RT, Netea MG, Kazura JW, Dent AE, Fitzgerald KA, and Golenbock DT. 2018. Cutting Edge: Plasmodium falciparum Induces Trained Innate Immunity. J. Immunol 200: 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heagy MW 2002. Endotoxin tolerance: A review. Crit. Care Med 30. [PubMed] [Google Scholar]
- 13.Liu D, Cao S, Zhou Y, and Xiong Y. 2019. Recent advances in endotoxin tolerance. J. Cell. Biochem 120: 56–70. [DOI] [PubMed] [Google Scholar]
- 14.Ahluwalia B, Moraes L, Magnusson MK, and Öhman L. 2018. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand. J. Gastroenterol 53: 379–389. [DOI] [PubMed] [Google Scholar]
- 15.Lochhead RB, Sonderegger FL, Ma Y, Brewster JE, Cornwall D, Maylor-Hagen H, Miller JC, Zachary JF, Weis JH, and Weis JJ. 2012. Endothelial Cells and Fibroblasts Amplify the Arthritogenic Type I IFN Response in Murine Lyme Disease and Are Major Sources of Chemokines in Borrelia burgdorferi -Infected Joint Tissue. J. Immunol 189: 2488–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lochhead RB, Ordoñez D, Arvikar SL, Aversa JM, Oh LS, Heyworth B, Sadreyev R, Steere AC, and Strle K. 2019. Interferon-gamma production in Lyme arthritis synovial tissue promotes differentiation of fibroblast-like synoviocytes into immune effector cells. Cell. Microbiol 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowley T, O’Neil JD, Adams H, Thomas AM, Filer A, Buckley CD, and Clark AR. 2017. Priming in response to pro-inflammatory cytokines is a feature of adult synovial but not dermal fibroblasts. Arthritis Res. Ther 19: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, Ouyang Q, Hu Z, Huang Q, Wu J, Wang R, and Yang M. 2016. A protocol for the culture and isolation of Murine synovial fibroblasts. Biomed. Reports 5: 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan M, and Gasser S. 2016. Generating primary fibroblast cultures from mouse ear and tail tissues. J. Vis. Exp 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nardelli DT, Callister SM, and Schell RF. 2008. Lyme arthritis: Current concepts and a change in paradigm. Clin. Vaccine Immunol 15: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zlotnikov N, Javid A, Ahmed M, Eshghi A, Tang TT, Arya A, Bansal A, Matar F, Parikh M, Ebady R, Koh A, Gupta N, Song P, Zhang Y, Newbigging S, Wormser GP, Schwartz I, Inman R, Glogauer M, and Moriarty TJ. 2017. Infection with the Lyme disease pathogen suppresses innate immunity in mice with diet-induced obesity. Cell. Microbiol 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh SK, and Girschick HJ. 2006. Toll-like receptors in Borrelia burgdorferi-induced inflammation. Clin. Microbiol. Infect 12: 705–717. [DOI] [PubMed] [Google Scholar]
- 23.Perkins DJ, Patel MC, Blanco JCG, and Vogel SN. 2016. Epigenetic mechanisms governing innate inflammatory responses. J. Interf. Cytokine Res 36: 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, and Weis JJ. 2002. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol 168: 348–55. [DOI] [PubMed] [Google Scholar]
- 25.Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, Weis JH, and Weis JJ. 2004. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J. Immunol 173: 2003–10. [DOI] [PubMed] [Google Scholar]
- 26.Bernard Q, Grillon A, Lenormand C, Ehret-Sabatier L, and Boulanger N. 2020. Skin Interface, a Key Player for Borrelia Multiplication and Persistence in Lyme Borreliosis. Trends Parasitol. 36: 304–314. [DOI] [PubMed] [Google Scholar]
- 27.Schramm F, Kern A, Barthel C, Nadaud S, Meyer N, Jaulhac B, and Boulanger N. 2012. Microarray analyses of inflammation response of human dermal fibroblasts to different strains of Borrelia burgdorferi sensu stricto. PLoS One 7: e40046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lochhead RB, Sonderegger FL, Ma Y, Brewster JE, Cornwall D, Maylor-Hagen H, Miller JC, Zachary JF, Weis JH, and Weis JJ. 2012. Endothelial Cells and Fibroblasts Amplify the Arthritogenic Type I IFN Response in Murine Lyme Disease and Are Major Sources of Chemokines in Borrelia burgdorferi-Infected Joint Tissue. J. Immunol 189: 2488–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasky CE, Pratt CL, Hilliard KA, Jones JL, and Brown CR. 2016. T cells exacerbate lyme borreliosis in TLR2-deficient mice. Front. Immunol 7: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang X, Izadi H, Coleman AS, Wang P, Ma Y, Fikrig E, Anguita J, and Pal U. 2008. Borrelia burgdorferi lipoprotein BmpA activates pro-inflammatory responses in human synovial cells through a protein moiety. Microbes Infect. 10: 1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh SK, Morbach H, Nanki T, Faber C, Baar V, and Girschick HJ. 2004. Differential expression of matrix metalloproteinases and cyclooxygenases in synovial cells exposed to Borrelia burgdorferi. Inflamm. Res 53: 689–696. [DOI] [PubMed] [Google Scholar]
- 32.Singh SK, Morbach H, Nanki T, and Girschick HJ. 2005. Differential expression of chemokines in synovial cells exposed to different Borrelia burgdorferi isolates. Clin. Exp. Rheumatol 23: 311–322. [PubMed] [Google Scholar]
- 33.Park SH, Park-Min KH, Chen J, Hu X, and Ivashkiv LB. 2011. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat. Immunol 12: 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein K, Frank-Bertoncelj M, Karouzakis E, Gay RE, Kolling C, Ciurea A, Bostanci N, Belibasakis GN, Lin L-L, Distler O, Gay S, and Ospelt C. 2017. The epigenetic architecture at gene promoters determines cell type-specific LPS tolerance. J. Autoimmun 83: 122–133. [DOI] [PubMed] [Google Scholar]
- 35.Hodzic E, Feng S, and Barthold SW. 2013. Assessment of transcriptional activity of Borrelia burgdorferi and host cytokine genes during early and late infection in a mouse model. Vector-Borne Zoonotic Dis. 13: 694–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samuels DS, and Samuels LRN. 2016. Gene regulation during the enzootic cycle of the lyme disease spirochete. For. Immunopathol. Dis. Therap 7: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll JA, Garon CF, and Schwan TG. 1999. Effects of environmental pH on membrane proteins in Borrelia burgdorferi. Infect. Immun 67: 3181–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyde JA, Trzeciakowski JP, and Skare JT. 2007. Borrelia burgdorferi alters its gene expression and antigenic profile in response to CO2 levels. J. Bacteriol 189: 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Troxell B, Ye M, Yang Y, Carrasco SE, Lou Y, and Frank Yang X. 2013. Manganese and zinc regulate virulence determinants in borrelia burgdorferi. Infect. Immun 81: 2743–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webb P 1992. Temperatures of skin, subcutaneous tissue, muscle and core in resting men in cold, comfortable and hot conditions. Eur. J. Appl. Physiol. Occup. Physiol 64: 471–476. [DOI] [PubMed] [Google Scholar]
- 41.Bauer M, Weis S, Netea MG, and Wetzker R. 2018. Remembering Pathogen Dose: Long-Term Adaptation in Innate Immunity. Trends Immunol. 39: 438–445. [DOI] [PubMed] [Google Scholar]
- 42.Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L, Gow NAR, Williams DL, Van Der Meer JWM, and Netea MG. 2014. Trained immunity or tolerance: Opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin. Vaccine Immunol 21: 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown JP, Zachary JF, Teuscher C, Weis JJ, and Wooten RM. 1999. Dual role of interleukin-10 in murine lyme disease: Regulation of arthritis severity and host defense. Infect. Immun 67: 5142–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sonderegger FL, Ma Y, Maylor-Hagan H, Brewster J, Huang X, Spangrude GJ, Zachary JF, Weis JH, and Weis JJ. 2012. Localized Production of IL-10 Suppresses Early Inflammatory Cell Infiltration and Subsequent Development of IFN-γ–Mediated Lyme Arthritis. J. Immunol 188: 1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung Y, Zhang N, and Wooten RM. 2013. Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, Mascanfroni ID, AlAdham Z, Lavoie S, Ibourk M, Nguyen DD, Samsom JN, Escher JC, Somech R, Weiss B, Beier R, Conklin LS, Ebens CL, Santos FGMS, Ferreira AR, Sherlock M, Bhan AK, Müller W, Mora JR, Quintana FJ, Klein C, Muise AM, Horwitz BH, and Snapper SB. 2014. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 40: 706–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mia S, Warnecke A, Zhang XM, Malmström V, and Harris RA. 2014. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-β yields a dominant immunosuppressive phenotype. Scand. J. Immunol 79: 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, Dey A, Wakabayashi Y, Sakaguchi K, Fujita T, Tamura T, Zhu J, and Ozato K. 2018. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl. Acad. Sci. U. S. A 115: E9162–E9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuo J, Warner TF, and Schell RF. 2017. Arthritis is inhibited in Borrelia-primed and infected interleukin-17A-deficient mice after administration of anti-gamma-interferon, anti-tumor necrosis factor alpha and anti-interleukin-6 antibodies. Pathog. Dis 75: 73. [DOI] [PubMed] [Google Scholar]
- 50.Paquette JK, Ma Y, Fisher C, Li J, Lee SB, Zachary JF, Kim YS, Teuscher C, and Weis JJ. 2017. Genetic Control of Lyme Arthritis by Borrelia burgdorferi Arthritis–Associated Locus 1 Is Dependent on Localized Differential Production of IFN-β and Requires Upregulation of Myostatin. J. Immunol 199: 3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller JC, Ma Y, Bian J, Sheehan KCF, Zachary JF, Weis JH, Schreiber RD, and Weis JJ. 2008. A Critical Role for Type I IFN in Arthritis Development following Borrelia burgdorferi Infection of Mice. J. Immunol 181: 8492–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dakin SG, Buckley CD, Al-Mossawi MH, Hedley R, Martinez FO, Wheway K, Watkins B, and Carr AJ. 2017. Persistent stromal fibroblast activation is present in chronic tendinopathy. Arthritis Res. Ther 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Åsbrink E, and Hovmark A. 1985. Successful Cultivation of Spirochetes from Skin Lesions of Patients With Erythema Chronicum Migrans Afzelius and Acrodermatitis Chronica Atrophicans. Acta Pathol. Microbiol. Scand. Ser. B Microbiol 93 B: 161–163. [DOI] [PubMed] [Google Scholar]
- 54.Steere AC, Bartenhagen NH, Craft JE, Hutchinson GJ, Newman JH, Rahn DW, Sigal LH, Spieler PN, Stenn KS, and Malawista SE. 1983. The early clinical manifestations of Lyme disease. Ann. Intern. Med 99: 76–82. [DOI] [PubMed] [Google Scholar]
- 55.Krause PJ, Foley DT, Burke GS, Christianson D, Closter L, and Spielman A. 2006. REINFECTION AND RELAPSE IN EARLY LYME DISEASE,. [PubMed]
- 56.Jutras BL, Lochhead RB, Kloos ZA, Biboy J, Strle K, Booth CJ, Govers SK, Gray J, Schumann P, Vollmer W, Bockenstedt LK, Steere AC, and Jacobs-Wagner C. 2019. Borrelia burgdorferi peptidoglycan is a persistent antigen in patients with Lyme arthritis. Proc. Natl. Acad. Sci. U. S. A 116: 13498–13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Włodarczyk M, Druszczyńska M, and Fol M. 2019. Trained innate immunity not always amicable. Int. J. Mol. Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.