

Abstract
Interleukin-13 plays a critical role in mediating many biological processes responsible for allergic inflammation. Mast cells express Il13 mRNA and produce IL-13 protein in response to antigenic stimulation. Enhancers are essential in promoting gene transcription and are thought to activate transcription by delivering essential accessory co-factors to the promoter to potentiate gene transcription. However, enhancers mediating Il13 have not been identified. Furthermore, which Il13 enhancers detect signals triggered by antigenic stimulation have not yet been defined. In this study, we identified potential mouse Il13 enhancers using histone modification monomethylation at lysine residue 4 on histone 3 (H3K4me1) ChIP-seq and acetylation at lysine residue 27 on histone 3 (H3K27ac) ChIP-seq. We used Omni-ATAC-seq to determine which accessible regions within the potential Il13 enhancers that responded to IgE receptor crosslinking. We also demonstrated that the transcription factor (TF) cluster consisting of the NFATC2, STAT5, GATA2, AP1, and RUNX1 binding sites at the proximal Il13 enhancer, the TF cluster consisting of the EGR2-binding site at the distal Il13 E+6.5 enhancer, are critical in sensing the signals triggered by antigenic stimulation. Those enhancers, which are responsive to antigenic stimulation and constitutively active, cooperate to generate greater transcriptional outputs. Our study reveals a novel mechanism underlying how antigenic stimulation induces robust Il13 mRNA expression in mouse mast cells.
Introduction
The mouse interleukin-13 (Il13) gene contains 4 exons and 3 introns and is located at chromosome 11 (1). IL-13 is a pleiotropic cytokine produced by CD4+ Th2 T cells, CD8+ TC2 cells, natural killer (NK) T cells, basophils, and mast cells (2–7). It plays a critical role in the biological processes responsible for allergic inflammation (8). IL-13 induces goblet cell differentiation in the mucosal linings of the airway and the gut (9, 10), nitric oxide synthase in airway epithelium (11), fibroblast to myoblast transition (12, 13), and smooth muscle proliferation of airways (14). It also induces IgE class switching in B cells (15), leading to the production of antigen-specific IgE antibodies, which, together with their cognate antigens, activate basophils and mast cells.
Mast cells reside in the mucosal surface and connective tissues and express the high-affinity IgE receptor (FcεRI). They become activated when IgE and its cognate antigen bind and crosslink the FcεRI receptor (16, 17). Activated mast cells rapidly release stored inflammatory mediators, including histamine, heparin, and proteases. Mast cell activation also leads to de novo gene transcription of additional inflammatory mediators. Mast cells upregulate Il13 gene expression and produce IL-13 protein in response to IgE receptor crosslinking (18) and also express the IL-33 receptor (ST) (19, 20). Stimulation of this receptor by IL-33, a cytokine produced by damaged epithelial cells, upregulates Il13 gene expression, and production of the IL-13 protein (21–23).
Although several groups have investigated Il13 gene regulation in T cells, enhancers that promote the Il13 gene transcription in mast cells have not been studied systematically. In T cells, it has been reported that transcription factor (TF) GATA-3 is essential for the Il13 gene transcription (24–27). GATA3 regulates Il13 gene transcription by increasing chromatin accessibility at the Il13 promoter to TF NFAT1 (28, 29). c-Myb promotes Il13 gene transcription by interacting with GATA-3 at the GATA response element (30). In mast cells, NFAT and GATA2 cooperate to regulate Il13 gene transcription (31). Masuda’s group described the interaction between GATA proteins and activator protein-AP1 at the promoter of Il13 is necessary for Il13 gene transcription in mast cells (32). Early growth response factor-1 (EGR1) is required for mast cells to transcribe Il13 gene in response to IgE and antigen stimulation (33, 34). However, it is unknown which enhancers at the Il13 gene respond to IgE and antigen stimulation.
TFs typically promote gene transcription by binding to clusters of TF-binding sites at enhancer regions. Binding of TFs to their corresponding binding sites at the enhancers leads to the recruitment of histone-modifying enzymes. The recruited histone methyltransferase MLL3/4 adds monomethylation at lysine residue 4 on histone 3 (H3K4me1), while the recruited histone acetyltransferase P300 acetylates lysine residue 27 on histone 3 (H3K27ac). Epigenomic studies demonstrate that H3K4me1 marks genes poised to be transcribed, whereas H3K27ac identifies genes that are actively transcribed (35–38). A newly developed technique, called Assay for Transposase-Accessible Chromatin (ATAC-seq), effectively identifies clusters of transcription factor binding sites at enhancers genome-wide (39, 40).
In this study, we used H3K4me1 and H3K27ac ChIP-seq to identify potential Il13 enhancers, and Omni-ATAC-seq to identify clusters of TF binding sites at the potential Il13 enhancers that responded to IgE receptor crosslinking. We constructed a series of Il13 enhancer reporter genes to functionally verify clusters of TF binding sites that responded to IgE receptor crosslinking. We demonstrated that the TF cluster consisting of the NFATC2, STAT5, GATA2, AP1, and RUNX1 binding sites at the proximal Il13 enhancer, the TF cluster consisting of the EGR2-binding sites at the distal Il13 E+6.5 enhancer, are critical in sensing the signals triggered by antigenic stimulation. The IgE responsive and non-responsive enhancers cooperate to increase Il13 gene transcription.
Materials and Methods
Animals and Cell culture
CFTL-15, a murine IL-3-dependent mast cell line (41), was provided by Dr. Melissa Brown (Northwestern University, Chicago, IL). CFTL-15 cells were grown in complete RPMI1640 medium supplemented with 10 % FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 20 ng/mL IL-3 in 5% CO2 at 37℃. C57BL/6J (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Bone marrow-derived mast cells (BMMCs) were cultured from bone marrow cells of C57BL/6 mice in Iscove’s DMEM (10016CV, Corning TM cellgro TM, Manassas, VA) plus 10% FBS, 2 mM beta-mercaptoethanol 100 units/mL penicillin, 100 μg/mL streptomycin, and 20 ng/mL IL-3 for four weeks. Over 99% of BMMCs were mast cells, as determined by FACS analysis (FcεRI+ c-Kit+). Handing animals was approved by the National Jewish Health Institutional Animal Care and Use Committee.
Chromatin immunoprecipitation ChIP
Chromatin immunoprecipitation and ChIP-seq libraries were prepared as described before (42). Briefly, 1×107 BMMCs were fixed by 1% formaldehyde. After sonication and pre-cleaning, the samples were incubated with either anti-H3K27ac antibody (ab4729, Abcam) or anti-H3K4me1 antibody (ab8895, Abcam) and then with protein A agarose/salmon sperm DNA slurry (Millipore, Cat# 16–157). The beads were washed and eluted according to protocols described before (42). The crosslinking of eluted immunocomplexes was reversed, and the recovered DNA was cleaned up using a QIAGEN QIAquick PCR purification kit (Qiagen, Valencia, CA). ChIP-seq data have been deposited in GEO (GSE145544).
Omni-ATAC-seq
The Omni-ATAC-seq was performed according to the published method (40). Briefly, 50,000 BMMCs were either untreated or treated with DNP-IgE (1µg/mL, Sigma St Louis MO) for 12 hours followed by crosslinking with 10 µg/mL DNP-BSA (YAMASA, Tokyo, Japan) for 2 hours. The cells were spun down and washed with cold PBS once. Then the cells were resuspended in 50 μl cold ATAC-RSB-lysis buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 0.1% NP-40, 0.1% Tween-20 and 0.01% Digitonin) and incubated for 3 minutes. The lysis buffer was immediately washed out with 1 mL ATAC-RSB buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, and 0.1% Tween-20). The cell pellets were resuspended in 50 μl transposition mix (25 μl 2X TD buffer, 2.5 μl transposase (Illumina, FC-121–1030), 16.5 μl PBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20, 5 μl H2O) and incubated at 37 °C for 30 minutes. The reaction was stopped by adding 2.5 μL of 0.5M EDTA pH 8, and transposed DNA was purified using the Qiagen MiniElute PCR purification kit (Qiagen). Purified DNA was amplified using the following condition: 72℃ for 5 min, 98 ℃ for 30 s, and 7 cycles: 98 ℃ for 10 s, 63 ℃ for 30 s, 72 ℃ for 1 min. The amplified libraries were cleaned up, size-selected, and the quality and quantity of libraries were assessed on an Agilent Technologies 2100 Bioanalyzer. The pair-ended sequencing of DNA libraries was performed on an Illumina NovaSEQ6000 platform. ATAC-seq data has been submitted to GEO (GSE145542).
Il13 promoter and enhancer constructions
The Il13 minimal promoter (MP, −75 to −1 in relative to the transcription start site (TSS) of Il13 gene) and P150 (−150 to −1) were cloned into pGL3-basic luciferase vector (Promega, Madison, WI) in BglII and HindIII cloning sites. The E-1.0 (−3208 to −1038 in the relative to TSS of Il13 gene), E+0.2 (+238 to +1475), E+2.5 (+2533 to +4891), E+6.3 (+6534 to +8513), and E+8.9 (+8844 to +11372) enhancers were cloned into the DNA upstream of the Il13 minimal promoter-luciferase vector through the NheI and BglII restriction sites. The combination of the Il13 enhancers was generated using overlapping PCR as described (43) and cloned into the Il13 minimal promoter-luciferase vector through the NheI and BglII restriction sites. Mutations in transcription factor binding sites were generated by using overlapping PCR as described (43) or using mutated oligos. The primers used for the amplification of enhancer fragments and mutated oligos are listed in Table S1. All mutations were verified by sequencing. All polymerases, and restriction and modification enzymes were obtained from New England Biolabs (Beverly, MA, USA).
Transcription factor binding motif analysis
The putative transcription factor binding clusters were analyzed by using R/Bioconductor package TFBSTools (44). Transcription factor binding motif matrixes from the JASPAR 2016 database (http://jaspar.genereg.net/) were used in the analysis.
Mast cell transfection, activation and enhancer reporter assay
CFTL-15 cells (1.25 ×106) were electroporated with 10 μg luciferase plasmid and 0.5 μg Renilla vectors at 450 V and 500 μF using a Bio-Rad Gene Pulser. The transfected CFTL-15 mast cells were stimulated with DNP-IgE (1µg /mL) for 12 hours, followed by crosslinking with 10 µg/mL DNP-BSA for 6 hours (IgECL). Luciferase activities in the activated and control mast cells were measured with Infinite M1000® microplate reader (Tecan Systems, Inc., San Jose, CA) using Dual-Luciferase reporter assay system (E1960, Promega). The transcription activity was measured as the ratio of luciferase and Renilla activity.
Statistical analysis
All the error bars in this report represent mean ± standard deviation (mean ± SD). Statistical differences between the two samples were analyzed using the two-tailed student’s t-test.
Results
Identification of putative Il13 enhancers that respond to antigenic stimulation
We defined potential enhancers as noncoding DNA regions that are associated with both H3K4me1 and H3K27ac modifications. To identify potential Il13 regulatory regions, we analyzed H3K4me1 ChIP-seq and H3K27ac ChIP-seq peaks at the Il13 gene using the datasets generated and published by our laboratory (Il13 enhancers were not analyzed in the paper, GSE97253). We showed that there were six potential enhancer regions within 14.6 kb of the mouse Il13 gene, which covers the intergenic regions between the Il4 and Rad50 genes (Fig. 1A). We named these potential enhancers proximal enhancer (PE), E+8.9, E+6.5, E+2.5, E+0.2, and E-1.0 based on the distances of the putative enhancers to the transcription start site (TSS) of the Il13 gene (Fig. 1A). Enhancers contain clusters of TF binding sites that can bind to TFs constitutively or bind to TFs activated by external stimulations. Binding of TFs to enhancers creates chromatin accessible regions within the enhancers (45). The chromatin accessible regions surrounding the TF bound sites can be identified by Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq). To assess which of the chromatin accessible regions can respond to IgE receptor crosslinking, we performed Omni-ATAC-seq, an improved version of ATAC-seq (40). We treated or did not treat bone marrow derived mast cells (BMMCs) with IgE receptor crosslinking for two hours. We observed that the chromatin accessible regions within the PE, E+8.9, E+6.5, E+2.5, E+0.2, and E-1.0 showed more accessibility in response to IgE receptor crosslinking (Fig. 1B). Sequencing data has been deposited to GEO (GSE145542). These results suggest that TFs are activated by IgE receptor crosslinking bound to the chromatin accessible regions at the PE, E+8.9, E+6.5, E+2.5, E+0.2, and E-1.0 enhancers either directly or indirectly through the transcriptional looping mechanism (45).
Figure 1.
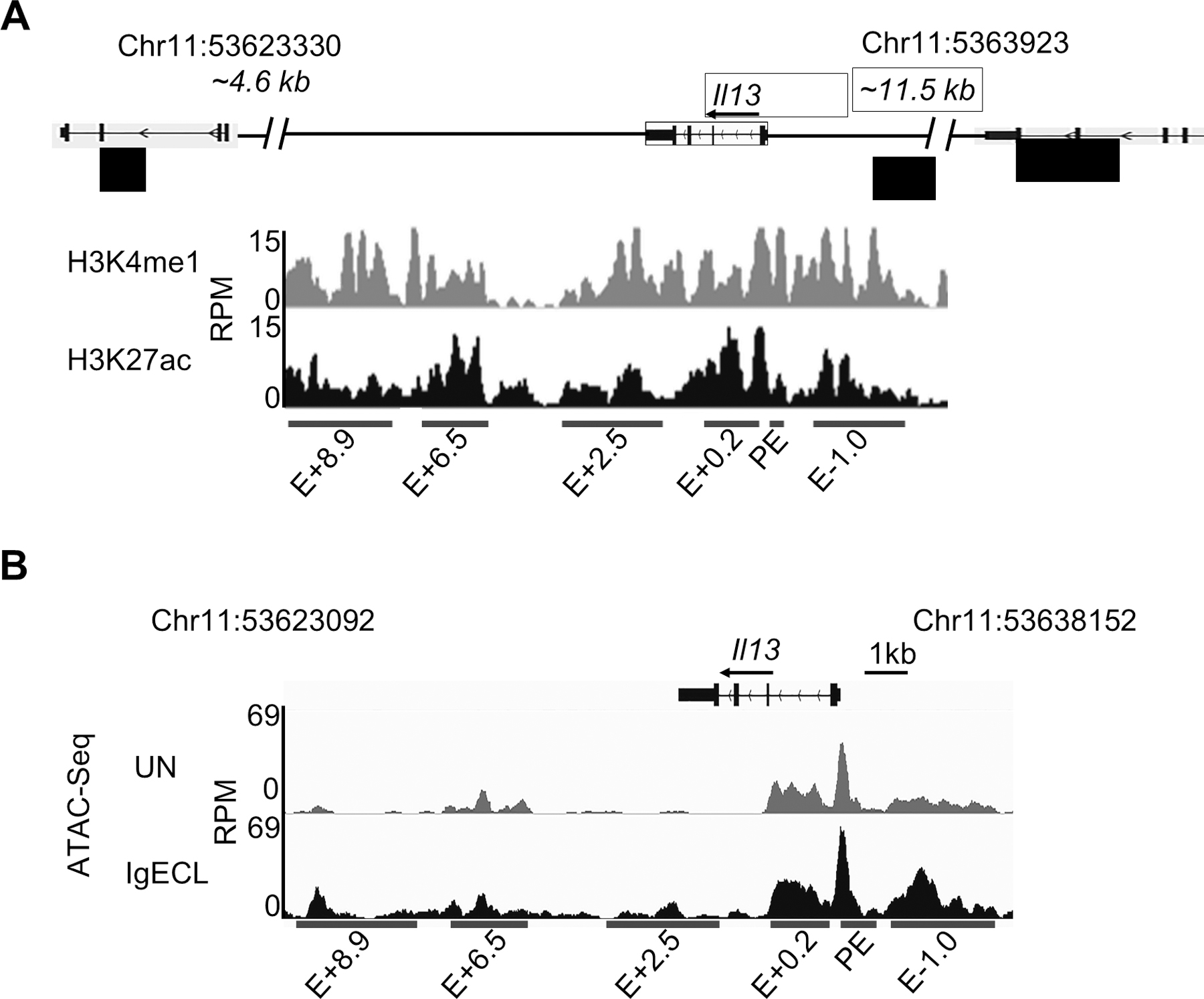
Identification of putative Il13 enhancers. (A) H3K27ac and H3K4me1 ChIP-Seq tracks from chromosome position 53623092 to 53630153 (7,061 bp), which is located in the DNA regions between the Il13 and Il4 gene (4.6 kb to the TSS of the Il4 gene) and between the Il13 gene and Rad50 gene (11.5 kb to TSS of the Il13 gene) in resting BMMCs. Red bars indicate putative Il13 enhancers and promoter positions which showed significant H3K4me1 and H3K27ac modifications. E: enhancer; +0.2, +2.5, +6.3, +8.9 indicates the distance (kilo base pair) from the beginning of the downstream Il13 enhancer to the TSS of the Il13 gene, and −1.0 indicates the distance from the beginning of the upstream Il13 enhancer to the TSS of the Il13 gene. RPM: reads per million. (B) Omni-ATAC-seq tracks at the Il13 gene obtained from BMMCs that were not stimulated (UN) or stimulated with IgE receptor crosslinking (IgECL). Data represent two biological samples.
Next, we used the luciferase reporter gene assay to determine which chromatin accessible regions at the six enhancers can respond to IgE receptor crosslinking directly. We constructed a series of enhancer reporter genes in which PE, E+8.9, E+6.5, E+2.5, E+0.2, and E-1.0 were linked to the Il13 minimal promoter that contained only essential elements for RNA POL II binding (−75 to +1 relative to the transcription start site) (Fig. 2A). Mast cells cell-line CFTL-15 is a non-transformed, IL-3-dependent mast cell line (41). These cells are mature mast cells that respond to IgE receptor crosslinking with much higher cytokine gene transcription compared to transformed, immature, mast cell lines, such as the PT18 and MC/9 mouse mast cell lines. We transfected CFTL-15 mast cells with the series of constructs and treated or did not treat the transfected cells with IgE receptor crosslinking for 6 hours. We found that the PE had the highest induced enhancer activity in response to IgE receptor crosslinking (2.5-fold higher compared to the unstimulated group, Fig 2B). The E+6.5 enhancers also showed a moderate induced-enhancer activity after IgE receptor crosslinking (40% increase compared the unstimulated group, Fig. 2B), while the E-1.0 and E+0.2 possessed constitutively active enhancer activity with or without IgE receptor crosslinking. The E+2.5 and E+8.9 enhancers did not show significant enhancer activity with or without IgE receptor crosslinking (Fig. 2B). The results demonstrate that the clusters of TF binding sites at the accessible regions of PE and E+6.5 enhancer may bind to TFs activated by IgE receptor crosslinking.
Figure 2.
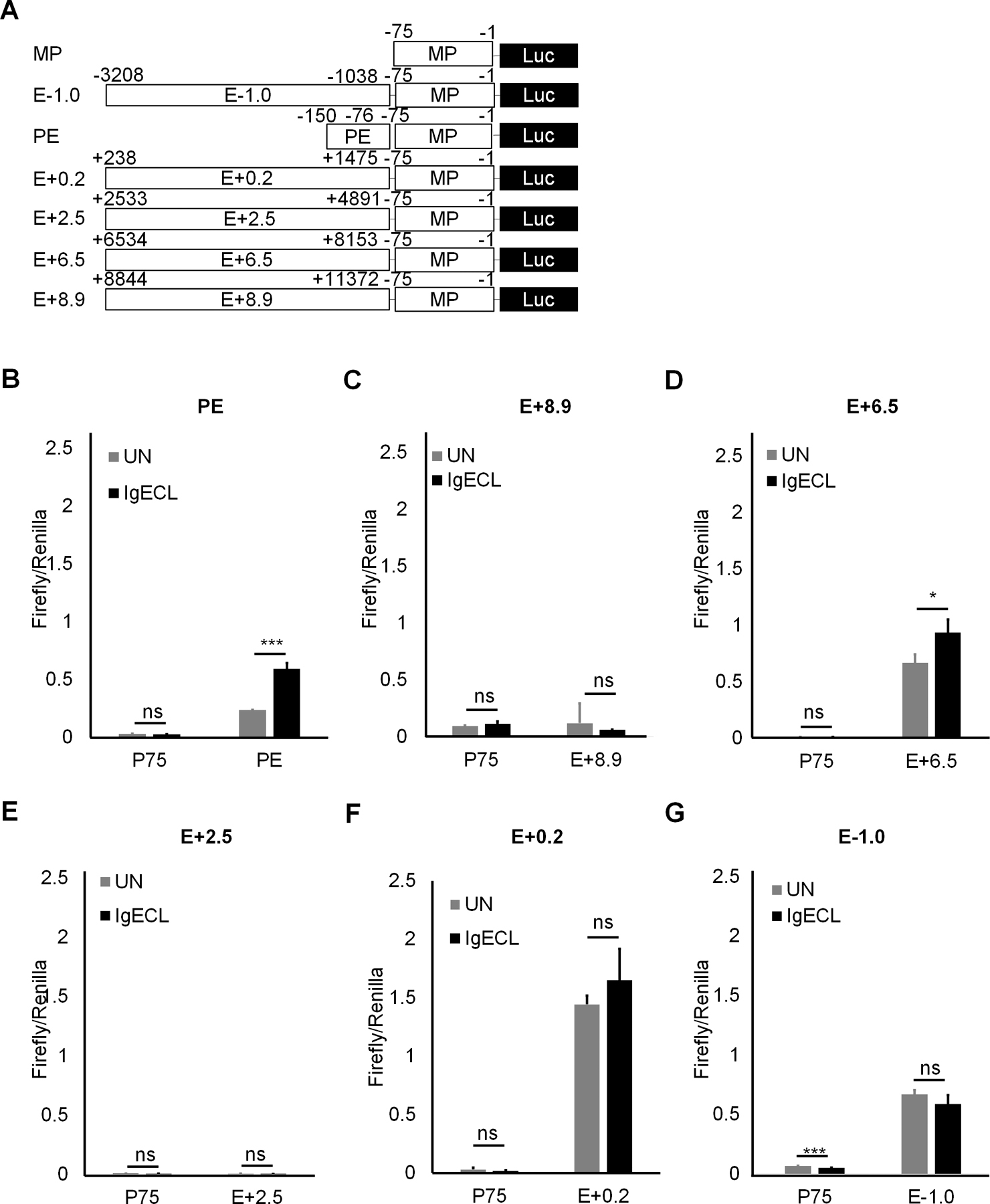
The PE and E+6.5 enhancer contain IgE-responsive elements. (A) Schematic diagrams showing enhancer reporter constructions. (B) Enhancer reporter analysis of PE enhancer transfected CFTL-15 cells that were not stimulated (UN) or stimulated with IgE receptor crosslinking (IgECL). (C) Enhancer reporter analysis of the E+8.9 enhancer. (D) Enhancer reporter analysis of the E+6.5 enhancer. (E) Enhancer reporter analysis of the E+2.5 enhancer. (F) Enhancer reporter analysis of the E+0.2 enhancer. (G) Enhancer reporter analysis of the E-0.1 enhancer. Mean ± SD were calculated from 3 transfectants in one experiment, representing two experiments with similar results. Statistical differences were analyzed by student’s t test. *: P<0.05, **: P<0.01, ***: P<0.001. MP, the minimal Il13 promoter.
Two inducible enhancers PE and E+6.5 cooperate with two constitutively active enhancers E-1.0 and E+0.2 to produce higher gene transcriptional outputs in response to IgE receptor crosslinking
Enhancers are thought to activate transcription by delivering necessary accessory co-factors to the promoter to potentiate gene transcription. Individual enhancers bound by TFs in the absence of IgE receptor crosslinking, and individual enhancers that bind to TFs activated by IgE receptor crosslinking, may loop to the promoter by the interaction of co-factors that bind to individual enhancers with the promoter. To determine whether the constitutively active enhancers and the IgE receptor crosslinking activated enhancers can act cooperatively and accumulatively, we constructed three enhancer reporter genes. In one, we linked all four enhancers that showed enhancer activity to the Il13 minimal promoter. The second construct contained two inducible enhancers, and the third had two constitutively active enhancers (Fig. 3A). We observed that the constructs with two inducible enhancers and two constitutively active enhancers possessed the highest levels of transcriptional outputs under both resting and activated conditions (51-fold change in the resting and 86-fold in the activated condition compared to the minimal promoter). The construct that contained two inducible enhancers responded to IgE receptor crosslinking with increased gene transcription (28-fold increase in the resting, 54-fold in the activated CFTL-15 cells, respectively). The construct that contained two constitutively active enhancers did not respond to IgE receptor crosslinking. We concluded from these results that two inducible enhancers and two constitutively active enhancers cooperate to produce higher gene transcriptional outputs in response to IgE receptor crosslinking.
Figure 3.
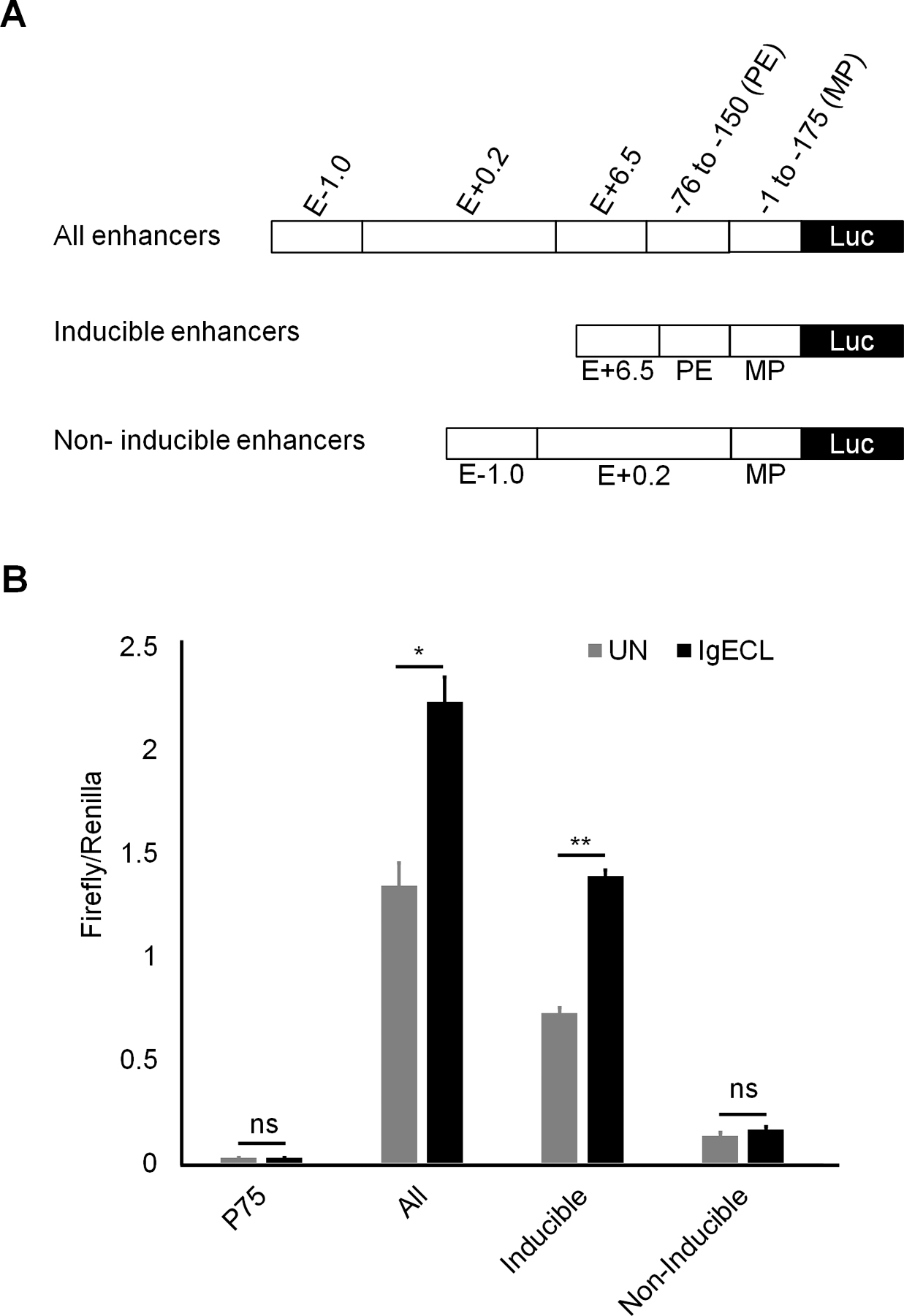
Two inducible enhancers PE and E+6.5 cooperate with two constitutively active enhancers E-1.0 and E+0.2 to generate higher gene transcriptional outputs. (A) Schematic diagrams showing enhancer reporter constructions. (B) Enhancer reporter analysis of the transfected CFTL-15 cells that were not stimulated (UN) stimulated with IgE receptor crosslinking (IgECL). Mean ± SD were calculated from 3 transfectants in one experiment, representing two experiments with similar results
Combinatorial NFATC2, STAT5, GATA2, and AP1 binding sites at the PE enhancer are essential for the IgE receptor crosslinking-induced PE enhancer activity
To precisely determine which TF binding sites of the chromatin accessible regions are responsible for the IgE receptor crosslinking-induced PE enhancer activity, we performed TF binding motif analysis on the sequences found in the ATAC-seq peak in the PE enhancer (Fig. 4A). We found that consensus TFs NFATC2, STAT5, HOXA5, GATA2, AP1, and RUNX1 binding motifs at the region. To determine whether these TF binding motifs can promote the Il13 reporter gene transcription, we performed mutational analysis of the TF binding sites. To reduce the numbers of permutations, we first generated a mutant PE enhancer, in which all potential TF binding sites were mutated. We then generated PE with the mutations at the overlapping TF binding sites and selected double or triple mutations that failed to respond to IgE receptor crosslinking for single TF binding site mutational analysis (Fig. 4A). As expected, mutation at all TF binding sites abolished the IgE receptor crosslinking-induced PE enhancer activity (Fig. 4B). We found mutations at both NFATC2 and STAT5 binding sites, but not single NFATC2 or STAT5 binding sites, abolished the IgE receptor crosslinking-induced PE enhancer activity (Fig. 4B). Similarly, triple mutations at GATA2, AP1 and RUNX1 binding sites eliminated the IgE receptor crosslinking-induced PE enhancer activity (Fig. 4B). Individual TF binding site mutation revealed that GATA2, AP1 and RUNX1 binding sites were critical in mediating the IgE receptor crosslinking-induced PE enhancer activity (Fig. 4B). In contrast, mutation at the HOXA5 binding site did not affect the IgE receptor crosslinking-induced PE enhancer activity (Fig. 4B). These results demonstrate that combinatorial NFATC2, STAT5, GATA2, AP1 and RUNX1 binding sites at the PE enhancer are required for the IgE receptor crosslinking-induced PE enhancer activity.
Figure 4.
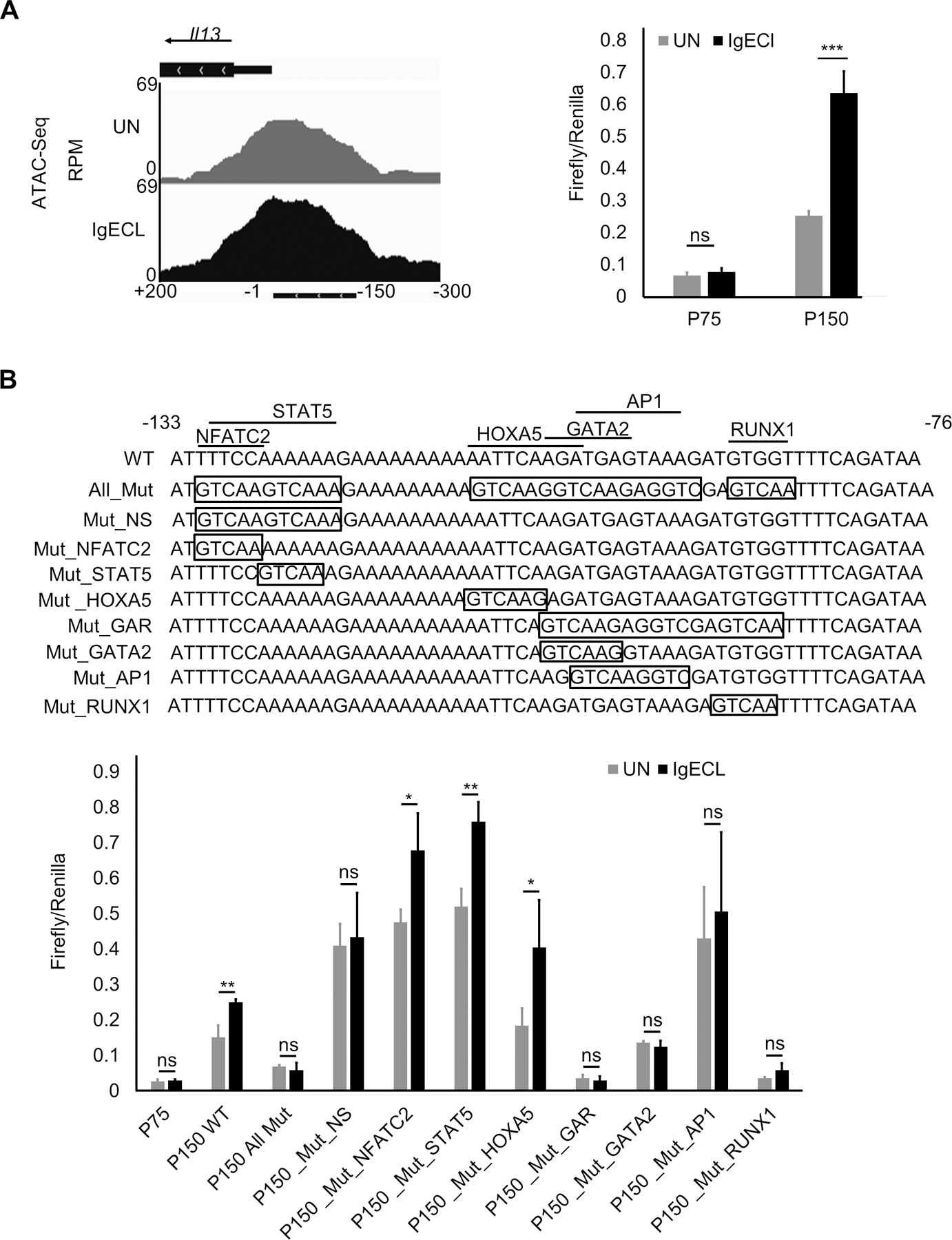
Combinatorial NFATC2, STAT5, GATA2, AP1 and RUNX1 binding sites at the PE enhancer are essential for the IgE receptor crosslinking-induced PE enhancer activity. (A) OmniATAC-seq tracks at the −300 to +200 region of the Il13 gene in BMMCs that were not stimulated (UN) or stimulated with IgE receptor crosslinking (IgECL). Lower panel represents enhancer reporter analysis of the P150 enhancer reporter transfected CFTL-15 cells that were not stimulated (UN) stimulated with IgE receptor crosslinking (IgECL). (B) Upper panel depicts schematic diagrams showing various mutations at the PE region. Mutated sequences are indicated inside the boxed. Lower panel, enhancer reporter analysis of transfected CFTL-15 cells that were not stimulated (UN) stimulated with IgE receptor crosslinking (IgECL).
Mean ± SD were calculated from 3 transfectants in one experiment, representing two experiments with similar results. Mut, mutant; Mut_NS, NFATC2 and STAT5 double mutants; Mut_GAR, GATA2, AP1 and RUNX1 triple mutants.
The EGR2 binding site at the E+6.5 enhancer mediates the IgE receptor crosslinking-induced E+6.5 enhancer activity
To locate the IgE receptor crosslinking responsive TF binding sites within the E+6.5 enhancer, we first performed deletional analysis since the E+6.5 spanned over 2 kb long, making it challenging to conduct mutational analysis. We constructed a series of mutations by deleting 200 bp at a time (Fig. 5A, lower panel). We found that the deletion of a 200 bp DNA fragment from E+6.5_7370 to 7570, resulted in a loss of the IgE receptor-crosslinking-induced Il13 reporter gene transcription, indicating this 200 bp DNA region contains clusters of TF binding sites that drove Il13 reporter gene transcription in response to IgE receptor crosslinking. Indeed, this region was co-localized with an ATAC-seq peak (Fig. 5A), suggesting that this region is accessible to TFs. Further deletional analysis, in which we deleted DNA fragments 50 bp at a time, revealed that region between 7670 and 7720 was responsible for most of the Il13 reporter gene transcription in response to IgE receptor crosslinking (Fig 5 B). TF binding analysis showed three potential TFs: FOXA2, EGR2, and STRE binding sites in the 50 bp fragment. We mutated FOXA2, EGR2, and STRE binding sites and found that the triple mutations at the 50 bp fragment abolished the IgE receptor crosslinking-induced Il13 reporter gene transcription (Fig. 5C). Single mutation at the EGR2 binding site also dramatically reduced the IgE receptor crosslinking-induced Il13 reporter gene transcription (Fig. 5C). Conversely, neither the mutation at the FOX2 or STRE binding site significantly decreased Il13 reporter gene transcription in response to IgE receptor crosslinking (Fig. 5C). These results suggest that the EGR2 binding sites at the E+6.5 enhancer recruit TFs activated by IgE receptor crosslinking to promote Il13 gene transcription in response to antigenic stimulation.
Figure 5.
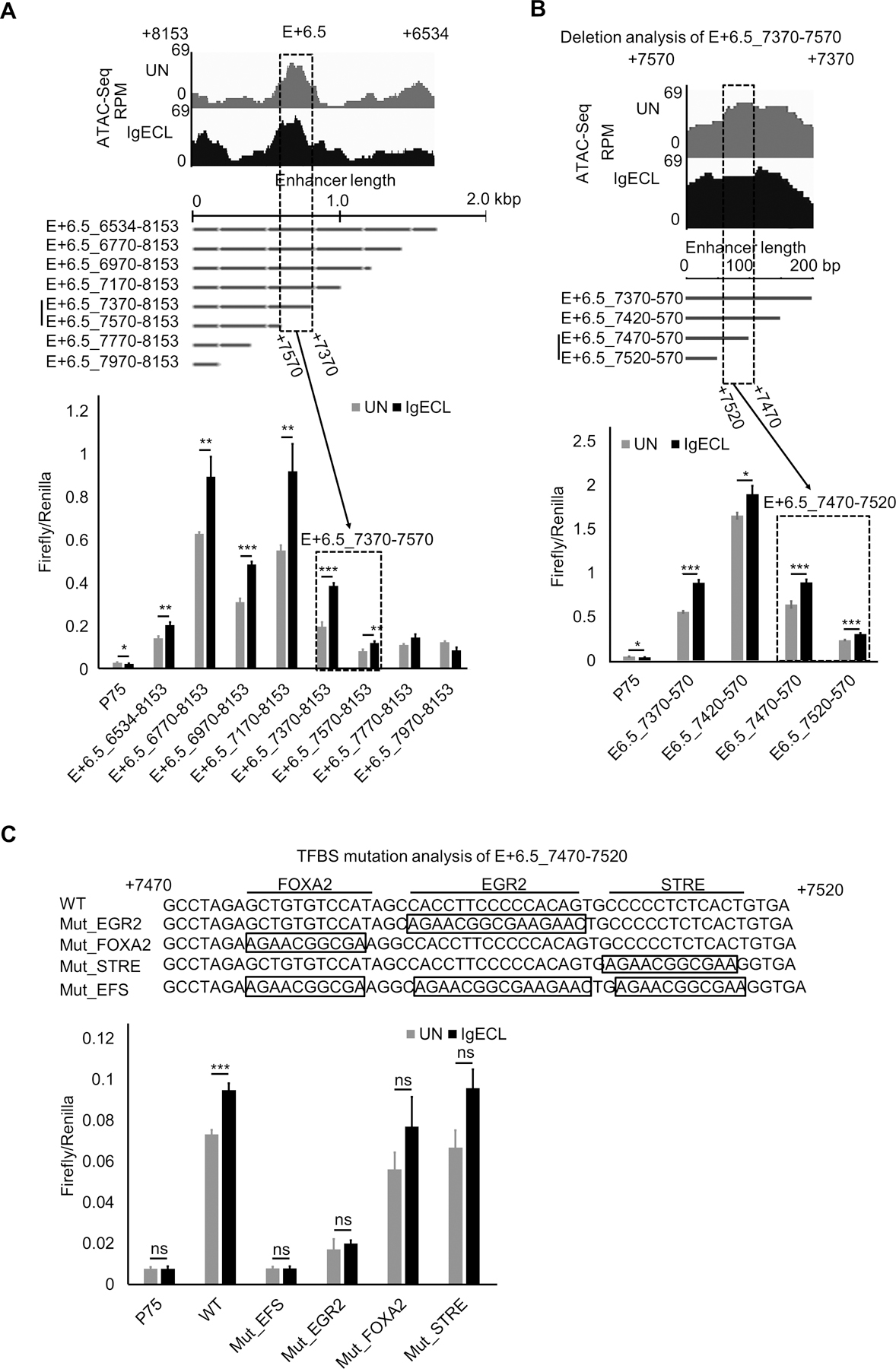
The EGR2 binding site at the E+6.5 enhancer mediates the IgE receptor crosslinking-induced E+6.5 enhancer activity. (A) Top panel shows Omni-ATAC-Seq peaks at the E+6.5 enhancers in resting (UN) and stimulated BMMC with IgE receptor crosslinking (IgECL). Middle panel is the schematic diagram of enhancer deletional analysis. The dotted box highlights the enhancer segments that lost its responsiveness to IgE receptor crosslinking. Lower panel, enhancer reporter analysis of the fragment from position +7370 to +7570 of the E+6.5 enhancer. (B) Enhancer reporter analysis of the fragment from position +7470 to +7520 of the E+6.5 enhancer. (C) Mutational analysis of the fragment from position +7470 to +7520 of the E+6.5 enhancer. Mut_EFS, EGR2, FOX2 and STRE triple mutants. Mean ± SD were calculated from 3 transfectants in one experiment, representing two experiments with similar results.
Discussion
Systematic investigation of how Il13 enhancers detect signals triggered by antigenic stimulation has not yet been explored in mast cells. In this study, we identified potential Il13 enhancers using histone mark H3K4me1 and H3K27ac ChIP-seq. We then used Omni-ATAC-seq to determine which accessible regions within the potential Il13 enhancers responded to IgE receptor crosslinking. We demonstrated that the TF cluster consisting of the NFATC2-, STAT5-, GATA2-, AP1- and RUNX1-binding sites in the Il13 proximal promoter, the EGR2-binding sites in the Il13E+6.5 enhancer, respond to IgE receptor crosslinking as measured by their ability to direct Il13 reporter gene transcription.
Our results support a model in that the clusters of TF binding sites at inducible PE and E+6.5 enhancers bind to activated TFs, and the binding recruits co-factors, such as mediators, that loop the distal E+6.5 to the Il13 minimal promoter and PE enhancer. The formation of the E+6.5 enhancer, PE enhancer, and Il13 promoter loop also bring together the constitutively active enhancers E-1.0 and E+0.2, which are located in the proximity of the Il13 promoter and PE enhancers, presumably through the interaction of TFs and co-factors. We propose that the clusters of NFATC2-, STAT5-, GATA2-, AP1-, and RUNX1-binding sites at the PE bind to NFATC2, STAT5, GATA2, AP1, and RUNX1 either activated by IgE and antigen-mediated crosslinking of IgE receptors on the surface of mast cells or by cytokines produced by activated mast cells (in the case of STAT5) and that the EGR2-binding site at the inducible E+6.5 enhancer bind to EGR2 activated by IgE receptor crosslinking to induce the formation of transcription complexes in response to antigenic stimulation. Indeed, our previous study provides evidence that GATA2 plays a critical role in the Il13 gene transcription in response to IgE receptor crosslinking (46). EGR2 was reported to be rapidly upregulated after IgE receptor crosslinking (47). It directly binds to the Ccl1 chemokine gene promoter and is essential for Ccl1, but not for Ccl3, and Ccl9 gene transcription and protein production (47). Thus, it is likely that EGR2 can bind to the EGR2-binding site at the E+6.5 enhancer to enhance the Il13 gene transcription in response to antigenic stimulation.
A previous study showed that the DNA sequence (ttcaagatgagtaaa) located at the proximal Il13 promoter from position −108 to −94 is a potential composite GATA2 and AP1 binding site (32). Single nucleotide mutation at the GATA2 and AP1-binding sites abolish luciferase reporter gene transcription in immature, transformed mast cell lines PT18 and MC9 mast cell lines that express low levels of Il13 mRNA in response to IgE receptor crosslinking (31). Our results confirm the importance of the GATA2 and AP-1 composite sites in the Il13 luciferase reporter gene transcription. Our results also demonstrate additional TFs NFATC2-, STAT5-, and RUNX1-binding sites in the PE enhancer that are important to mediating Il13 luciferase reporter gene transcription. Using mature, IL-3-dependent, non-transformed mast cells CFTL-15 in our study might be critical in revealing this new finding.
However, it is challenging to identify the specific TFs that bind to the identified clusters of transcription factor-binding sites in response to antigenic stimulation because many potential TFs can bind to the same consensus TF binding sites. Thus, our studies reveal that TF binding sites, rather than TFs, are critical in mediating Il13 gene transcription in response to antigenic stimulation. Future work that measures the physical binding of TFs to the particular TF binding sites and knocks out or knocks down individual TF genes in mast cells is needed to determine the exact TFs that bind to the consensus TF binding site in response to antigenic stimulation.
In summary, IL-13 plays a critical role in mediating allergic inflammation, the underlying cause of many allergic diseases. Our detailed analysis of how TF binding sites at chromatin accessible regions at the IgE responsive enhancers in the Il13 gene advances our understanding of mechanisms of allergic diseases.
Supplementary Material
Key points.
Inducible and constitutive enhancers cooperate to generate transcriptional outputs
Identification of TF binding site clusters critical in Il13 gene transcription
Acknowledgements
We thank lab members for thoughtful discussions. We are grateful to Ms. Marina Rahmani for technical assistance. We thank Ms. Kathleen Rauch for editor assistance.
Grant support:
This work was supported by National Institutes of Health Grant (RO1 AI107022 and R01AI135194 to H.H.)
Funds provided by Sun Yet-sen University (to J.L.)
Funds provided by the China Scholarship Council (201406270087 to B.L.)
Abbreviations
- BMMCs
Bone Marrow-derived Mast Cells
- IgE
Immunoglobulin E
- TF
Transcription Factor
- GATA2
GATA Binding Protein 2
- GATA3
GATA Binding Protein 3
- NFAT1
Nuclear Factor Of Activated T-Cells 1
- AP1
Activator Protein 1
- EGR1
Early Growth Response factor-1
- STAT5
Signal Transducer and Activator of Transcription 5
- RUNX1
Runt-related transcription factor 1
- FOXA2
Forkhead box protein A2
- NFATC2
Nuclear Factor Of Activated T Cells 2
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References:
- 1.McKenzie AN, Li X, Largaespada DA, Sato A, Kaneda A, Zurawski SM, Doyle EL, Milatovich A, Francke U, and Copeland NG 1993. Structural comparison and chromosomal localization of the human and mouse IL-13 genes. J Immunol 150: 5436–5444. [PubMed] [Google Scholar]
- 2.Brown KD, Zurawski SM, Mosmann TR, and Zurawski G 1989. A family of small inducible proteins secreted by leukocytes are members of a new superfamily that includes leukocyte and fibroblast-derived inflammatory agents, growth factors, and indicators of various activation processes. J Immunol 142: 679–687. [PubMed] [Google Scholar]
- 3.Cherwinski HM, Schumacher JH, Brown KD, and Mosmann TR 1987. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. The Journal of experimental medicine 166: 1229–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenzie AN, Culpepper JA, de Waal Malefyt R, Briere F, Punnonen J, Aversa G, Sato A, Dang W, Cocks BG, Menon S, and et al. 1993. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci U S A 90: 3735–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burd PR, Thompson WC, Max EE, and Mills FC 1995. Activated mast cells produce interleukin 13. The Journal of experimental medicine 181: 1373–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, DeKruyff RH, and Umetsu DT 2003. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nature medicine 9: 582–588. [DOI] [PubMed] [Google Scholar]
- 7.Ochensberger B, Daepp GC, Rihs S, and Dahinden CA 1996. Human blood basophils produce interleukin-13 in response to IgE-receptor-dependent and -independent activation. Blood 88: 3028–3037. [PubMed] [Google Scholar]
- 8.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, and Donaldson DD 1998. Interleukin-13: central mediator of allergic asthma. Science 282: 2258–2261. [DOI] [PubMed] [Google Scholar]
- 9.Kanoh S, Tanabe T, and Rubin BK 2011. IL-13-induced MUC5AC production and goblet cell differentiation is steroid resistant in human airway cells. Clin Exp Allergy 41: 1747–1756. [DOI] [PubMed] [Google Scholar]
- 10.Khan WI, Blennerhasset P, Ma C, Matthaei KI, and Collins SM 2001. Stat6 dependent goblet cell hyperplasia during intestinal nematode infection. Parasite immunology 23: 39–42. [DOI] [PubMed] [Google Scholar]
- 11.Suresh V, Mih JD, and George SC 2007. Measurement of IL-13-induced iNOS-derived gas phase nitric oxide in human bronchial epithelial cells. Am J Respir Cell Mol Biol 37: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito A, Okazaki H, Sugawara I, Yamamoto K, and Takizawa H 2003. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol 132: 168–176. [DOI] [PubMed] [Google Scholar]
- 13.Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, and Izuhara K 2006. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. The Journal of allergy and clinical immunology 118: 98–104. [DOI] [PubMed] [Google Scholar]
- 14.Moynihan BJ, Tolloczko B, El Bassam S, Ferraro P, Michoud MC, Martin JG, and Laberge S 2008. IFN-gamma, IL-4 and IL-13 modulate responsiveness of human airway smooth muscle cells to IL-13. Respir Res 9: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, de Waal Malefyt R, and de Vries JE 1993. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A 90: 3730–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galli SJ, Grimbaldeston M, and Tsai M 2008. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nature reviews. Immunology 8: 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galli SJ, and Tsai M 2012. IgE and mast cells in allergic disease. Nature medicine 18: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toru H, Pawankar R, Ra C, Yata J, and Nakahata T 1998. Human mast cells produce IL-13 by high-affinity IgE receptor cross-linking: enhanced IL-13 production by IL-4-primed human mast cells. The Journal of allergy and clinical immunology 102: 491–502. [DOI] [PubMed] [Google Scholar]
- 19.Chen CC, Grimbaldeston MA, Tsai M, Weissman IL, and Galli SJ 2005. Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci U S A 102: 11408–11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moritz DR, Rodewald HR, Gheyselinck J, and Klemenz R 1998. The IL-1 receptor-related T1 antigen is expressed on immature and mature mast cells and on fetal blood mast cell progenitors. J Immunol 161: 4866–4874. [PubMed] [Google Scholar]
- 21.Moussion C, Ortega N, and Girard JP 2008. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PloS one 3: e3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP, Stappenbeck TS, Patterson GA, Pierce RA, Brody SL, and Holtzman MJ 2013. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. The Journal of clinical investigation 123: 3967–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allakhverdi Z, Smith DE, Comeau MR, and Delespesse G 2007. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol 179: 2051–2054. [DOI] [PubMed] [Google Scholar]
- 24.Zheng W, and Flavell RA 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89: 587–596. [DOI] [PubMed] [Google Scholar]
- 25.Lavenu-Bombled C, Trainor CD, Makeh I, Romeo PH, and Max-Audit I 2002. Interleukin-13 gene expression is regulated by GATA-3 in T cells: role of a critical association of a GATA and two GATG motifs. J Biol Chem 277: 18313–18321. [DOI] [PubMed] [Google Scholar]
- 26.Takemoto N, Kamogawa Y, Jun Lee H, Kurata H, Arai KI, O’Garra A, Arai N, and Miyatake S 2000. Cutting edge: chromatin remodeling at the IL-4/IL-13 intergenic regulatory region for Th2-specific cytokine gene cluster. J Immunol 165: 6687–6691. [DOI] [PubMed] [Google Scholar]
- 27.Kishikawa H, Sun J, Choi A, Miaw SC, and Ho IC 2001. The cell type-specific expression of the murine IL-13 gene is regulated by GATA-3. J Immunol 167: 4414–4420. [DOI] [PubMed] [Google Scholar]
- 28.Yao X, Zha W, Song W, He H, Huang M, Jazrawi E, Lavender P, Barnes PJ, Adcock IM, and Durham AL 2012. Coordinated regulation of IL-4 and IL-13 expression in human T cells: 3C analysis for DNA looping. Biochem Biophys Res Commun 417: 996–1001. [DOI] [PubMed] [Google Scholar]
- 29.Yao X, Yang Y, He HY, Wang M, Yin KS, and Huang M 2010. GATA3 siRNA inhibits the binding of NFAT1 to interleukin-13 promoter in human T cells. Chin Med J (Engl) 123: 739–744. [PubMed] [Google Scholar]
- 30.Kozuka T, Sugita M, Shetzline S, Gewirtz AM, and Nakata Y 2011. c-Myb and GATA-3 cooperatively regulate IL-13 expression via conserved GATA-3 response element and recruit mixed lineage leukemia (MLL) for histone modification of the IL-13 locus. J Immunol 187: 5974–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monticelli S, Solymar DC, and Rao A 2004. Role of NFAT proteins in IL13 gene transcription in mast cells. J Biol Chem 279: 36210–36218. [DOI] [PubMed] [Google Scholar]
- 32.Masuda A, Yoshikai Y, Kume H, and Matsuguchi T 2004. The interaction between GATA proteins and activator protein-1 promotes the transcription of IL-13 in mast cells. J Immunol 173: 5564–5573. [DOI] [PubMed] [Google Scholar]
- 33.Li B, Power MR, and Lin TJ 2006. De novo synthesis of early growth response factor-1 is required for the full responsiveness of mast cells to produce TNF and IL-13 by IgE and antigen stimulation. Blood 107: 2814–2820. [DOI] [PubMed] [Google Scholar]
- 34.Li B, Berman J, Wu P, Liu F, Tang JT, and Lin TJ 2008. The early growth response factor-1 contributes to interleukin-13 production by mast cells in response to stem cell factor stimulation. J Immunotoxicol 5: 163–171. [DOI] [PubMed] [Google Scholar]
- 35.Calo E, and Wysocka J 2013. Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, and Jaenisch R 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, and Ren B 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- 38.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, Kathiria A, Cho SW, Mumbach MR, Carter AC, Kasowski M, Orloff LA, Risca VI, Kundaje A, Khavari PA, Montine TJ, Greenleaf WJ, and Chang HY 2017. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14: 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pierce JH, Di Fiore PP, Aaronson SA, Potter M, Pumphrey J, Scott A, and Ihle JN 1985. Neoplastic transformation of mast cells by Abelson-MuLV: abrogation of IL-3 dependence by a nonautocrine mechanism. Cell 41: 685–693. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Liu B, Harmacek L, Long Z, Liang J, Lukin K, Leach SM, O’Connor B, Gerber AN, Hagman J, Roers A, Finkelman FD, and Huang H 2018. The transcription factors GATA2 and microphthalmia-associated transcription factor regulate Hdc gene expression in mast cells and are required for IgE/mast cell-mediated anaphylaxis. J Allergy Clin Immunol 142: 1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bryksin AV, and Matsumura I 2010. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques 48: 463–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan G, and Lenhard B 2016. TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics (Oxford, England) 32: 1555–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spitz F, and Furlong EE 2012. Transcription factors: from enhancer binding to developmental control. Nature reviews. Genetics 13: 613–626. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Qi X, Liu B, and Huang H 2015. The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J Immunol 194: 4328–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Z, Macneil AJ, Junkins R, Li B, Berman JN, and Lin TJ 2013. Mast cell FcepsilonRI-induced early growth response 2 regulates CC chemokine ligand 1-dependent CD4+ T cell migration. J Immunol 190: 4500–4507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.