

Abstract
Racemization is considered to be an intrinsic stereochemical feature of free radical chemistry as can be seen in traditional radical halogenation reactions of optically active tertiary C–H bonds. If the facile process of radical racemization could be effectively combined with an ensuing step of bond formation in an enantioselective fashion, then it would give rise to deracemizative functionalization of racemic tertiary C–H bonds for stereoselective construction of chiral molecules bearing quaternary stereocenters. As a demonstration of this unique potential in radical chemistry, we herein report that metalloradical catalysis can be successfully applied to devise Co(II)-based catalytic system for enantioconvergent radical amination of racemic tertiary C(sp3)–H bonds. The key to the success of the radical process is the development of Co(II)-based metalloradical catalyst with fitting steric, electronic, and chiral environments of the D2-symmetric chiral amidoporphyrin as the supporting ligand. The existence of optimal reaction temperature is recognized as an important factor in the realization of the enantioconvergent radical process. Supported by an optimized chiral ligand, the Co(II)-based metalloradical system can effectively catalyze the enantioconvergent 1,6-amination of racemic tertiary C(sp3)–H bonds at the optimal temperature, affording chiral α-tertiary amines in excellent yields with high enantiocontrol of the newly created quaternary stereocenters. Systematic studies, including experiments utilizing optically active deuterium-labeled C–H substrates as a model system, shed light on the underlying mechanistic details of this new catalytic process for enantioconvergent radical C–H amination. The remarkable power to create quaternary stereocenters bearing multiple functionalities from ubiquitous C–H bonds, as showcased with stereoselective construction of bicyclic N-heterocycles, opens the door for future synthetic applications of this new radical technology.
Graphical Abstract
INTRODUCTION
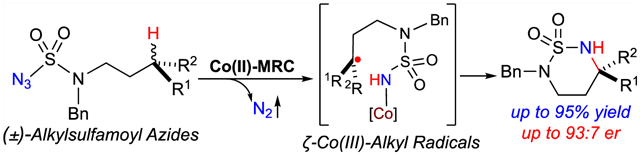
Homolytic radical chemistry has been increasingly explored for the development of new synthetic tools to construct organic molecules in light of its rich reactivities and practical attributes.1 Despite significant advancements, control of enantioselectivity remains a major challenge for many radical reactions, calling for innovative new solutions and fundamental mechanistic studies. Among recent approaches,2 metalloradical catalysis (MRC) represents a conceptually distinct strategy for achieving stereoselective radical reactions by exploiting metal-centered radicals as a new class of catalysts that are capable of activating substrates homolytically while translocating the spin density to generate metal-bonded organic radicals for subsequent radical processes under the catalyst control.3–5 To this end, Co(II) complexes of porphyrins, as stable 15emetalloradicals, have recently been demonstrated to be effective catalysts for the homolytic activation of organic azides to form the fundamentally new α-Co(III)-aminyl radicals.6 Supported by D2-symmetric chiral amidoporphyrin (D2-Por*) ligands, the resulting Co-stabilized N-centered radicals, which are situated inside a pocket-like chiral environment, can undergo common radical reactions, such as radical addition, H-atom abstraction, and subsequent radical substitution, but in a controlled and selective manner, creating new catalytic systems for asymmetric radical transformations.7 Our recent efforts to control the enantioselectivity of radical heterocyclization uncovered two basic modes of asymmetric induction in intramolecular radical C–H amination of organic azides via Co(II)-MRC (Scheme 1).7d,e,8 The two modes differ in the enantiodetermining step of the stepwise radical process: H-atom abstraction in Mode A (Scheme 1A) and radical substitution in Mode B (Scheme 1B). Although they were both derived from studying the amination reactions of secondary C–H bonds, we were fascinated by the exciting possibility of enantioconvergent amination of racemic tertiary C–H bonds via Mode B (Scheme 1C). As illustrated for 1,6-C–H amination of sulfamoyl azides, efficient H-atom abstraction of tertiary C–H bonds by the initially formed α-Co(III)-aminyl radicals I serves as a prerequisite. However, the central challenge for achieving such unparalleled enantioconvergent C–H amination lies in the contrasting ligand environments required by the two subsequent radical steps. While capacious space is needed for the two chiral faces in the resulting ζ-Co(III)-alkyl radicals (Re)-II and (Si)-II to undergo facile racemization, confined space will benefit asymmetric induction in the last step of radical substitution to ensure high enantioconvergence (Scheme 1C). Considering the tunability of the modular D2-Por* chiral platform, we envisioned the prospect of identifying a suitable ligand with fitting steric, electronic, and chiral environments that could accommodate the seemly opposite demands. If this feat could be achieved, then it would lead to a new radical process that would be able to deracemize racemic tertiary C(sp3)–H bonds in hydrocarbons for the production of valuable α-tertiary amines as a single enantiomer (see Figure S1 in Supporting Information, SI).9
Scheme 1.

Modes of Asymmetric Induction and Enantioconvergence in Radical C–H Amination via Co(II)-MRC
Catalytic intramolecular C(sp3)–H amination represents an attractive approach for the construction of chiral N-heterocycles through direct functionalization of ubiquitous C–H bonds in organic molecules.10 Despite considerable efforts, enantioselective variants of intramolecular C–H amination remain underdeveloped.11,12 Until now, there has been limited success of catalytic systems that are capable of converting both enantiomers of tertiary C–H substrates into a single enantiomer of α-tertiary amine derivatives.13 This is perhaps no surprise considering that existing catalytic systems for C–H amination typically proceed via a concerted C–H insertion pathway involving metallonitrene intermediates, which lacks a mechanism for racemization like the radical processes. We herein report a new catalytic radical process via Co(II)-based metalloradical catalysis that utilizes the facile nature of radical racemization and enables, for the first time, asymmetric intramolecular amination of tertiary C–H bonds. We show that the Co(II)-based metalloradical system can catalyze enantioconvergent 1,6-C(sp3)–H amination of various sulfamoyl azides containing racemic tertiary C–H bonds, affording six-membered chiral cyclic sulfamides in excellent yields with high enantiocontrol of the newly generated quaternary stereocenters. Additionally, we present mechanistic studies that offer insights into the underlying radical mechanism and reveal the origin of enantioconvergence in the Co(II)-catalyzed amination of racemic tertiary C–H bonds. To showcase the synthetic applications of this new methodology, we also describe the stereoselective construction of several fused-bicyclic N-heterocycles.
RESULTS AND DISCUSSION
Catalyst Development.
At the onset of this study, we selected N-benzyl sulfamoyl azide (±)-1a bearing racemic tertiary C–H bonds substituted with ester and phenyl groups as a testing substrate for the proposed enantioconvergent amination via Co(II)-MRC (Scheme 2). After initial optimization of conditions, the catalytic intramolecular C–H amination of azide (±)-1a was conducted at 50 °C in benzene with the use of different [Co(D2-Por*)] complexes as chiral metalloradical catalysts. As detailed below (Scheme 3B), the reaction temperature was recognized as an important factor in achieving the enantioconvergent process. To our delight, the first-generation catalyst [Co(P1)] (P1 = 3,5-DitBu-ChenPhyrin)14 could effectively catalyze 1,6-amination of the racemic tertiary C–H bond of (±)-1a, affording six-membered cyclic sulfamide 2a in high yield (93%) at a significant level of enantioselectivity (81:19 er) with (S)-2a as the major enantiomer. These preliminary results serve as a proof of concept for the enantioconvergent amination of racemic C–H bonds. In the hopes of improving the enantioconvergence of the Co(II)-catalyzed process, the effect of D2-Por* chiral ligands was systematically evaluated (see Figure S2 in the SI). Switching the catalyst to [Co(P2)] (P2 = 2,6-DiMeO-ChenPhyrin),14 which bears the same chiral amide units but more sterically hindered achiral meso-aryl groups, led to no change in the enantioselectivity and sense of asymmetric induction, but substantial decrease in product yield (22%). When the second-generation catalyst [Co(P3)] (P3 = 3,5-DitBu-QingPhyrin)15 was used, a considerable improvement in enantioselectivity (from 81:19 er to 86:14 er) was observed while the high product yield (94%) was restored. Interestingly, the major enantiomer switched from (S)-2a to (R)-2a. Further enhancement in asymmetric induction was achieved with the replacement of the catalyst by [Co(P4)] (P4 = 2,6-DiMeOQ-ingPhyrin), which maintained the high yield for 2a production with (R)-2a as the major enantiomer. Under optimized conditions, the enantioconvergent 1,6-amination of the racemic tertiary C–H bond of (±)-1a could be effectively catalyzed by [Co(P4)] to deliver the cyclic α,γ-diamino acid derivative 2a in 93% yield with 93:7 er. The absolute configuration of the newly created quaternary stereocenter in the major (R)-2a was established by X-ray crystallography.
Scheme 2. Ligand Effect on Co(II)-Catalyzed Enantioconvergent Amination of Racemic Tertiary C–H Bondsa,b.

aCarried out in benzene at 50 °C for 24 h using 2 mol % [Co(Por*)] on 0.10 mmol scale under N2 in the presence of 4 Å MS; [azide 1a] = 0.1 M; Isolated yields. b[R] Absolute configuration determined by X-ray crystallography.
Scheme 3.

Time Course and Temperature Dependence of Co(II)-Based Enantioconvergent Tertiary C–H Amination
Substrate Scope.
Under the optimized conditions, the [Co(P4)]-based system proved to be generally effective for the enantioconvergent 1,6-amination of racemic tertiary C(sp3)–H bonds in sulfamoyl azides (±)-1 bearing different substituents (Table 1). In addition to the reaction of sulfamoyl azide (±)-1a with ethyl ester, the Co(II)-based enantioconvergent amination could be effectively applied to tertiary C–H bonds in sulfamoyl azides having various ester functionalities such as methyl ((±)-1b), isopropyl ((±)-1c), and isobutyl ((±)-1d) esters, affording the cyclic α,γ-diamino esters (+)-2a–2d in equally high yields with similar enantioselectivities (Table 1; entries 1–4). Similarly, tertiary C–H bonds in sulfamoyl azides bearing different aryl substituents with varied electronic properties, including those with 4-methyl ((±)-1e), 4-methoxy ((±)-1f), 3-methoxy ((±)-1g), 4-bromo ((±)-1h), 4-chloro ((±)-1i), 4-fluoro ((±)-1j), 4-nitro ((±)-1k), and 4-trifluoromethyl ((±)-1l) phenyl groups, could all be enantioconvergently aminated to form the corresponding six-membered cyclic sulfamides (+)-2e–(−)-2l bearing quaternary stereocenters in high yields with high enantioselectivities (Table 1; entries 5–12). Besides the substrates with ester functionalities, the Co(II)-catalyzed enantioconvergent amination system could also be applied to tertiary C–H substrates with ketone functionality as exemplified by the enantioselective transformation of sulfamoyl azide (±)-1m to cyclic α,γ-diamino ketone (+)-2m in high yield (Table 1; entry 13). Likewise, the catalytic system by [Co(P4)] could tolerate substrates containing formyl functionality as demonstrated with the chemoselective amination of C–H bonds in sulfamoyl azides (±)-1n and (±)-1o, affording the desired chiral cyclic α,γ-diamino aldehydes (+)-2n and (+)-2o in high yields with high enantioconvergence (Table 1; entries 14 and 15). Additionally, allylic tertiary C–H bonds could also be enantioconvergently aminated by [Co(P4)] as illustrated by the effective reaction of cyclopentene-derived azide (±)-1p, leading to the stereoselective construction of spirobicyclic sulfamide (−)-2p in high yield without affecting the endocyclic C=C double bond (Table 1; entry 16). It is worthy to appreciate the peculiar ability of catalyst [Co(P4)] to discriminate the two endocyclic chiral faces that differ by only one π-bond for enantioconvergent formation of (−)-2p, albeit with the relatively lower enantioselectivity. It should be noted that the catalytic system by [Co(P4)] was found to be sensitive to both the steric hindrance and the nature of the substituents around the tertiary C–H bond. For example, sulfamoyl azides bearing ortho-substituted arenes and esters, including even those derived from 1-naphthalen and 3-indole, were shown to be ineffective substrates for the catalytic transformation. When the aryl/ester substituents in the sulfamoyl azides were replaced by either alkyl/ester or aryl/alkyl groups, they afforded the corresponding C–H amination products in similarly high yield but with much lower enantioselectivities. It is evident that new Co(II)-based catalytic systems supported by different D2-symmetric chiral amidoporphyrins need to be developed in order to effectively catalyze enantioconvergent 1,6-amination of racemic tertiary C(sp3)–H bonds in sulfamoyl azides bearing substituents beyond the combination of aryl and ester groups.
Table 1.
[Co(P4)]-Catalyzed Enantioconvergent Amination of Racemic Tertiary C–H Bonds in Different Sulfamoyl Azidesa,b

|
Carried out in benzene at 50 °C for 24 h using 2 mol % [Co(P4)] on 0.10 mmol scale under N2 in the presence of 4 Å MS; [azide 1] = 0.1 M.
Isolated yields.
Time Course and Temperature Dependence.
To shed light on the working details of the enantioconvergent process, a time-course study for asymmetric amination of the racemic tertiary C–H bond in azide (±)-1a by [Co(P4)] was performed under the standard conditions. The yield and enantiomeric excess of the amination product (+)-2a, along with the remaining azide 1a and its enantiomeric excess, were continuously monitored over the course of the reaction (Scheme 3A). The enantiomeric excess of amination product (+)-2a reached to a maximum level at the beginning that remained unchanged throughout the entire reaction. Interestingly, the remaining azide 1a was found to undergo steady enantioenrichment during the course of the catalytic reaction, starting from racemic and becoming highly enriched by the end. This result indicates that the pocket-like chiral catalyst [Co(P4)] preferentially bound one of the two enantiomers of azide (±)-1a and then irreversibly activated it for generation of the corresponding α-Co(III)-aminyl radical intermediate I1a, which was in turn transformed to the specific (Re)- or (Si)-ζ-Co(III)-alkyl radical II1a by subsequent 1,6-H-atom abstraction (Scheme 1C). However, the stereoselective generation of intermediates (Re)-II1a or (Si)-II1a was clearly nonconsequential to the enantioselectivity of amination product 2a as it remained the same throughout the catalytic process. Together, it is evident that the two chiral faces in the resulting ζ-Co(III)-alkyl radicals (Re)-II and (Si)-II could undergo facile racemization, rendering the final radical substitution as the enantiodetermining step (Scheme 1C). During the course of the study, we uncovered an unusual dependence of enantioselectivity on reaction temperature, which further illustrates the combined importance of racemization and radical substitution in the enantioconvergent process. As shown in Scheme 3B, the C–H amination reaction of azide (±)-1a by [Co(P4)] was carried out at varied temperatures under the otherwise same standard conditions. When the reaction temperature was gradually elevated from room temperature, the enantioselectivity progressively increased until reaching a maximum ee of 86% at 50 °C (Scheme 3B). Further elevation of the reaction temperature up to 70 °C led to a decrease in enantioselectivity. Collectively, these results agree well with the proposed catalytic mechanism for enantioconvergent amination that demands a high enough temperature to accelerate the racemization between the radical intermediates (Re)-II and (Si)-II but a low enough temperature to favor asymmetric induction for subsequent radical substitution (Scheme 1C). Consequently, there exists an optimal reaction temperature at which a given catalytic system can achieve maximal enantioselectivity for enantioconvergent C–H amination.
Asymmetric Induction Mode.
To gain insight into the likely origin of asymmetric induction in the enantioconvergent process, isotopomeric sulfamoyl azides (S)-1q and (R)-1q were prepared in optically pure forms and employed as the substrates to study kinetic isotope effects (KIE) (Table 2). It should be noted that (S)-1q and (R)-1q are the best possible deuterated tertiary C–H models for the actual substrates in Table 1 to study KIE of asymmetric amination of racemic tertiary C–H bonds. Using the achiral catalyst [Co(P5)] (P5 = 3,5-DitBu-IbuPhyrin), the intramolecular kinetic isotope effects (KIE) for both reactions of azides (S)-1q and (R)-1q were determined to have the same value of 13.0 (Table 2; entries 1 and 2). Similarly, matching KIE values of 11.0 were obtained for both of the reactions when the analogous achiral catalyst [Co(P6)] (P6 = 2,6-DiMeO-IbuPhyrin) was used (Table 2; entries 3 and 4). These high values of primary KIE are in accordance with the proposed step of C–H bond cleavage through the 1,6-H-atom abstraction of tertiary C–H bonds by the initially formed α-Co(III)-aminyl radicals I (Scheme 1C). When a chiral catalyst is employed, deviation from the intrinsic KIE will be anticipated (higher in the chirality-matched case and lower in the chirality-mismatched case) if the H-atom abstraction is enantioselective. With the use of chiral catalyst [Co(P1)], the same KIE values of 12.0 were obtained for both (S)-1q and (R)-1q (Table 2; entries 5 and. 6), indicating a non-enantiodifferentiative H-atom abstraction by [Co(P1)]. However, switching to chiral catalyst [Co(P2)] raised the KIE to 27.0 for (S)-1q while lowering it to 10.0 for (R)-1q, suggesting preferential abstraction of pro-R over pro-S H-atom by [Co(P2)] (Table 2; entries 7 and 8). Enantiodifferentiative pro-R H-atom abstraction was also observed with chiral catalysts [Co(P3)] (Table 2; entries 9 and 10) and [Co(P4)] (Table 2; entries 11 and 12) but to a lesser extent. The measured KIE values were translated to give the ratios of the initially established prochiral faces (Re)-II1q and (Si)-II1q. These ratios were then used to generate calculated enantiomeric excesses (ee%cal) for the amination product 2q by assuming no racemization of the facial chirality in the subsequent radical substitution step. The observation of much lower enantiomeric excesses than predicted for the reactions catalyzed by achiral catalysts [Co(P5)] and [Co(P6)] indicates significant but incomplete racemization of the initial facial chirality via α-C–C bond rotation prior to radical substitution (Table 2; entries 1–4). For both chiral catalysts [Co(P1)] and [Co(P2)], in addition to much lower enantiomeric excesses than predicted, they catalyzed the production of (S)-2q as the major enantiomer with identical ee values for both reactions with (S)-1q and (R)-1q (Table 2; entries 5–8), revealing facile interconversion between (Re)-II1q and (Si)-II1q and confirming radical substitution as the major enantiodetermining step. Enantioconvergence was also exhibited by chiral catalyst [Co(P3)], which produced the opposite (R)-2q as the major enantiomer with higher averaged enantioselectivity of 32% ee (Table 2; entries 9 and 10). Further enhancement in enantioconvergence was achieved by chiral catalyst [Co(P4)], which afforded (R)-2q as the same major enantiomer with averaged enantioselectivity of 70% ee (Table 2; entries 11–13). Specifically, [Co(P4)] enabled stereoretentive amination of (S)-1q to produce (R)-2q with 94% ee while catalyzing the stereoinvertive amination of (R)-1q to produce (R)-2q with 46% ee. Together with the time-course study (Scheme 3A) and the existence of optimal temperature (Scheme 3B), these results from the deuterated model system suggest Mode B (Scheme 1) as a major pathway for the observed enantioconvergence. In addition to effective enantiocontrol of the radical substitution step, these data clearly signified the equal importance of facile interconversion between intermediates (Re)-II and (Si)-II to achieve highly enantioconvergent process for the amination of racemic tertiary C–H bonds (Scheme 1C).
Table 2.
Assessment of Asymmetric Induction Mode in Radical 1,6-C–H Amination through KIE Study of Isotopomersa

|
Carried out in benzene at 50 °C for 48 h using 2 mol % [Co(Por)] on 0.10 mmol scale under N2 in the presence of 4 Å MS; [azide 1q] = 0.1 M; yields in SI.
4 mol % [Co(Por)].
Ratio of H:D determined by 1H NMR spectroscopy.
Calculated based on the ratio of H:D.
ee of 2q calculated on the basis of stereoretentive RS.
ee of 2q determined by chiral HPLC analysis, which offered no separation of (R)-2qH from (R)-2qD and (S)-2qH from (S)-2qD.
[R] Absolute configuration determined by comparison of HPLC data with that of nondeuterated (R)-2q, the structure of which was confirmed by X-ray crystallography.
Detection, Trapping, and Probing of Radical Intermediates.
While radical intermediates associated with secondary C–H amination were previously established,7d,e,8 we were intrigued by whether the α-Co(III)-aminyl radical I and especially the resulting ζ-Co(III)-alkyl radicals II from 1,6-H-atom abstraction of tertiary C–H bonds could be detected, trapped, and probed. To directly detect the radical intermediates, the isotropic electron paramagnetic resonance (EPR) spectrum was recorded at room temperature for the reaction solution of [Co(P5)] with azide (±)-1i in benzene after heating at 50 °C for 5 min (Scheme 4A). The spectrum displays diagnostic signals at g-value of ~2.00 that are characteristic of α-Co(III)-aminyl radicals and related radical species.7d,e,8 Consistent with the proposed stepwise radical mechanism involving the key step of 1,6-H-atom abstraction, the observed broad signals could be fittingly simulated as a mixture of α-Co(III)-aminyl radical and ζ-Co(III)-alkyl radical on the basis of couplings by 59Co (I = 7/2) and 14N (I = 1): 95% of N-centered radical at α-position I[Co(P5)]/1i (g: 2.00258; A(Co): 4.2 MHz; A(N): 33.8 MHz) and 5% of C-centered radical at ζ-position II[Co(P5)]/1i (g: 2.00440; A(Co): 0 MHz; A(N): 0 MHz). In addition to the EPR detection, we made multiple attempts in trapping ζ-Co(III)-alkyl radical intermediates from Co(II)-catalyzed tertiary C–H amination of sulfamoyl azides by TEMPO. However, formation of TEMPO-trapped products could not be identified. This negative outcome of the trapping experiments was attributed to the larger steric hindrance of tertiary alkyl radicals. Accordingly, sulfamoyl azide (±)-1r that contains an allylic tertiary C–H bond was designed as a substrate with the aim of probing the intermediacy of the tertiary allylic radical by trapping its less sterically hindered resonance form as a primary allylic radical (Scheme 4B). When the amination of azide (±)-1r was catalyzed by [Co(P5)] in the presence of excess TEMPO (5.0 equiv), no formation of the corresponding six-membered cyclic sulfamide 2r was detected, indicating complete inhibition of 1,6-C–H amination by TEMPO. While the acyclic sulfamide 3r from direct TEMPO-trapping of the tertiary radical intermediate II[Co(P5)]/1r was expectedly absent from the reaction mixture, the TEMPO-containing acyclic sulfamide 4r bearing a trisubstituted (E)-olefin unit was isolated in 62% yield. Successful formation of 4r from the trapping of the primary allyl radical III[Co(P5)]/1r by TEMPO implies that the tertiary allylic radical intermediate II[Co(P5)]/1r was generated following 1,6-H-atom abstraction of the tertiary C–H bond in the initially formed α-Co(III)-alkyl radical I[Co(P5)]/1r. The high (E)-stereoselectivity observed for the formation of 4r is likely a steric outcome of the trisubstituted olefin inside the amidoporphyrin ligand environment. Furthermore, azides (±)-1s and (±)-1t bearing a cyclopropyl ring were synthesized as radical-clock substrates to probe the lifetime of the corresponding ζ-Co(III)-alkyl radical intermediates (Scheme 4C). Under the catalysis of [Co(P5)], the reaction of (±)-1s afforded acyclic sulfamide 3s in 37% yield in addition to forming cyclic sulfamide 2s in 37% yield while cyclic sulfamide 2t was obtained in 89% yield without formation of acyclic sulfamide 3t from the reaction of (±)-1t. In both reactions, the ring-opening product 4s and 4t were not observed. When the reaction of (±)-1s was further carried out in the presence of TEMPO, there was no evidence for formation of any TEMPO-trapped ring-opening product either.16 Together, these results indicated that the C–N bond formation via intramolecular radical substitution was faster than the ring-opening of the tertiary cyclopropylcarbinyl radicals within the pocket-like ligand environment of the catalyst.17
Scheme 4.

Detection, Trapping, and Probing of Radical Intermediates in Co(II)-Catalyzed Amination of Tertiary C–H Bonds
Synthetic Applications.
Considering its capacity for creating quaternary stereocenters bearing multiple functionalities, Co(II)-catalyzed enantioconvergent C–H amination could potentially be utilized for a variety of synthetic applications. For instance, the resulting six-membered cyclic sulfamides serve as convenient precursors for chiral α,γ-diamino acid derivatives upon desufonylation7d and are also important chiral heterocycles in their own right (see Figure S1 in the SI).18 To further showcase their synthetic utilities, the amino and ester groups at the quaternary stereocenter were demonstrated as useful functionalities for stereoselective construction of fused-bicyclic N-heterocycles as exemplified with the three applications of enantioenriched cyclic α,γ-diamino ester (R)-2a (Scheme 5). The amino group in (R)-2a could be efficiently alkylated with 2-azidoethylmethanesulfonate to produce azide 3a in excellent yield (97%). Selective hydrogenation of the azide group in 3a led to the formation of primary amine, which was converted in situ to cyclic sulfamide-fused piperazinone 4a through lactamization in the presence of base. The [4.4.0]-bicyclic N-heterocycle (R)-4a was not only constructed in excellent yields after two steps (95%) but also fully retained the original optical purity (Scheme 5A). The absolute configuration of structure 4a was determined by X-ray crystallography (Scheme 5A). Alternatively, the ester group in 2a could be selectively reduced with DIBAL-H to give aldehyde 2n in 87% yield. Subsequent N-alkylation with 2-azidoethylmethanesulfonate afforded compound 3c, which contains both formyl and azido functionalities, in 90% yield. It was discovered that the aliphatic azide unit in 3c could be effectively activated by metalloradical catalyst [Co(P5)] to undergo an unusual intramolecular amination of the C(sp3)–C(sp2) bond associated with the formyl group (Schmidt-type reaction in the absence of acids). The resulting cyclic sulfamide-fused imidazoline (R)-4b was isolated in 91% yield with full retention of the original enantiopurity and absolute configuration as determined by X-ray crystallography (Scheme 5B). The [Co(P5)]-catalyzed construction of [4.3.0]-bicyclic N-heterocycle (R)-4b from cyclic sulfamide (R)-2a can be conceived to proceed through an unprecedented radical cascade process involving four different types of radicals: the Co(II)-based metalloradical as well as N-, O-, and C-centered organic radicals (Scheme 5B). Upon metalloradical activation of the aliphatic azide in (R)-3c by [Co(P5)], the initially generated α-Co(III)-aminyl radical intermediate I3c would proceed 6-exo-trig radical addition to the nearby-located carbonyl group to form γ-Co(III)-alkoxyl radical intermediate II3c, which would then undergo radical β-scission to generate the more stable ε-Co(III)-alkyl radical intermediate III3c. Subsequent 5-exo-tet radical substitution would transform intermediate III3c into the final product (R)-4b while regenerating catalyst [Co(P5)]. Remarkably, both the absolute configuration and the optical purity of the quaternary stereocenter were preserved during the catalytic radical transformation by achiral catalyst [Co(P5)], which is likely attributed to the configurational stability of the rigid chiral face in radical intermediate III3c within the catalyst environment. The amino group in aldehyde 2n could also be alkylated with o-azido-m-fluorobenzyl bromide to afford compound 3d in excellent yield (96%). In contrast with the reaction of aliphatic azide in 3c, it was found that the aryl azide in 3d could be activated by metalloradical catalyst [Co(P1)] to carry out intramolecular decarbonylative amination of the closely positioned formyl group, delivering cyclic sulfamide-fused tetrahydroquinazoline (R)-4c in 72% yield (Scheme 5C). As with the formation of the fused 5-membered imidazoline in (R)-4b, the fused six-membered tetrahydroquinazoline in (R)-4c was constructed with near complete preservation of the original enantiopurity and retention of the absolute configuration as determined by X-ray crystallography. It is evident that a different radical cascade process occurred for the construction of [4.4.0]-bicyclic N-heterocycle (R)-4c (Scheme 5C). Presumably, the corresponding α-Co(III)-aminyl radical intermediate I3d resulting from metalloradical activation of the aryl azide in (R)-3d by [Co(P1)] would prefer 1,7-H-atom abstraction from the aldehydic C(sp2)–H bond over 7-exo-trig radical addition to the C=O bond, forming η-Co(III)-acyl radical intermediate II3d. Subsequent radical α-scission would convert the acyl radical II3d to the more stable ζ-Co(III)-alkyl radical intermediate III3d upon release of CO. The final 6-exotet radical substitution would transform the tertiary radical III3d to product (R)-4c stereospecifically without racemization of the configurationally stable chiral face while regenerating catalyst [Co(P1)]. Considering their unique three-dimensional structures with the rigid bicyclic framework that contains a bridgehead quaternary stereocenter bearing multiple functionalities, this new class of bicyclic sulfamide-fused N-heterocycles 4a–4c may find potential applications in drug research and development.19
Scheme 5.

Synthetic Applications of Catalytic Products for Stereoselective Construction of Fused-Bicyclic N-Heterocycles
CONCLUSIONS
In summary, we have unveiled a new radical approach that promises deracemizative functionalization of racemic tertiary C(sp3)–H bonds for the stereoselective synthesis of chiral molecules with enantioconvergent construction of quaternary stereogenic centers. Fundamental knowledge acquired from our ongoing development of the emerging metalloradical catalysis (MRC) has enabled us to harness the potential of homolytic radical chemistry for the validation of this attractive approach by sequentially conjoining H-atom abstraction, radical racemization, and radical substitution in a controlled and enantioselective fashion. As the first application of this potentially general approach, we have successfully developed a new catalytic radical process via Co(II)-based metalloradical catalysis (Co(II)-MRC) that allows for the enantioconvergent amination of racemic tertiary C–H substrates. As suggested by the mechanistic studies, development of D2-symmetric chiral amidoporphyrin ligand platform that creates a well-balanced environment for the Co(II)-based metalloradical catalyst is the key to the success of the catalytic process. This environment facilitates 1,6-H-atom abstraction of tertiary C–H bonds and subsequent racemization of the resulting prochiral face while simultaneously enhancing asymmetric induction of radical substitution for construction of the quaternary stereogenic center. Furthermore, the existence of optimal reaction temperature is recognized as a characteristic feature of such an enantioconvergent radical process. Under the support of the optimized ligand 2,6-DiMeO-QingPhyrin, the Co(II)-based enantioconvergent system can catalyze 1,6-amination of racemic tertiary C–H bonds in sulfamoyl azides under mild conditions, leading to the stereoselective construction of six-membered cyclic sulfamides bearing chiral α-tertiary amines at the newly generated quaternary stereocenters in excellent yields with high enantioselectivities. Considering the unparalleled power to create quaternary stereocenters bearing multiple functionalities from ubiquitous C–H bonds, this new enantioconvergent amination methodology should find broad applications in organic synthesis, as showcased with the stereoselective construction of bicyclic N-heterocycles. It is our hope that this study will stimulate further research efforts in exploring the untapped potential of homolytic radical chemistry in modern organic synthesis.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH (R01-GM132471) and in part by NSF (CHE-1900375). We thank James G. Zhang (Johns Hopkins University) for helpful discussion with valuable suggestions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c11103.
Experimental details and analytical data for all new compounds (PDF)
Analytical data (ZIP)
Analytical data (ZIP)
The authors declare no competing financial interest.
Contributor Information
Kai Lang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States;.
Chaoqun Li, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Isaac Kim, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
X. Peter Zhang, Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, United States.
REFERENCES
- (1).For select books, see:; (a) Chatgilialoglu C; Studer A Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons: New York, 2012. [Google Scholar]; (b) Curran DP; Porter NA; Giese B Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications; John Wiley & Sons: New York, 2008. [Google Scholar]; (c) Zard SZ Radical Reactions in Organic Synthesis; Oxford University Press: Oxford, 2003. [Google Scholar]; (d) Renaud P; Sibi MP Radicals in Organic Synthesis; Wiley-VCH: Weinheim, 2001. For recent reviews, see: [Google Scholar]; (e) Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]; (f) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Quiclet-Sire B; Zard SZ Fun with Radicals: Some New Perspectives for Organic Synthesis. Pure Appl. Chem 2011, 83, 519–551. [Google Scholar]; (h) Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (i) Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]; (j) Sibi MP; Manyem S; Zimmerman J Enantioselective radical processes. Chem. Rev 2003, 103, 3263–3295. [DOI] [PubMed] [Google Scholar]
- (2).For select examples on approaches to controlling radical reactivity and stereoselectivity, see:; (a) Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: the Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rono LJ; Yayla HG; Wang DY; Armstrong MF; Knowles RR Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc 2013, 135, 17735–17738. [DOI] [PubMed] [Google Scholar]; (c) Hashimoto T; Kawamata Y; Maruoka K An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat. Chem 2014, 6, 702–705. [DOI] [PubMed] [Google Scholar]; (d) Du J; Skubi KL; Schultz DM; Yoon TP A Dual-Catalysis Approach to Enantioselective [2 + 2] Photocycloadditions Using Visible Light. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huo HH; Shen XD; Wang CY; Zhang LL; Röse P; Chen LA; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]; (f) Hoyt JM; Schmidt VA; Tondreau AM; Chirik PJ Iron-Catalyzed Intermolecular [2 + 2] Cycloadditions of Unactivated Alkenes. Science 2015, 349, 960–693. [DOI] [PubMed] [Google Scholar]; (g) Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem., Int. Ed 2015, 54, 3872–3890. [DOI] [PubMed] [Google Scholar]; (h) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Funken N; Mühlhaus F; Gansäuer A General, Highly Selective Synthesis of 1,3- and 1,4-Difunctionalized Building Blocks by Regiodivergent Epoxide Opening. Angew. Chem., Int. Ed 2016, 55, 12030–12034. [DOI] [PubMed] [Google Scholar]; (k) Brill ZG; Grover HK; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science 2016, 352, 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kern N; Plesniak MP; McDouall JJW; Procter DJ Enantioselective Cyclizations and Cyclization Cascades of Samarium Ketyl Radicals. Nat. Chem 2017, 9, 1198–1204. [DOI] [PubMed] [Google Scholar]; (m) Morrill C; Jensen C; Just-Baringo X; Grogan G; Turner NJ; Procter DJ Biocatalytic Conversion of Cyclic Ketones Bearing α-Quaternary Stereocenters into Lactones in an Enantioselective Radical Approach to Medium-Sized Carbocycles. Angew. Chem., Int. Ed 2018, 57, 3692–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Huang H-M; McDouall JJW; Procter DJ SmI2-Catalysed Cyclization Cascades by Radical Relay. Nat. Catal 2019, 2, 211–218. [Google Scholar]; (o) Li J; Zhang Z; Wu L; Zhang W; Chen P; Lin Z; Liu G Site-Specific Allylic C–H Bond Functionalization with a Copper-Bound N-Centred Radical. Nature 2019, 574, 516–521. [DOI] [PubMed] [Google Scholar]; (p) Shin NY; Ryss JM; Zhang X; Miller SJ; Knowles RR Light-Driven Deracemization Enabled by Excited-state Electron Transfer. Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Huo H; Gorsline BJ; Fu GC Catalyst-Controlled Doubly Enantioconvergent Coupling of Racemic Alkyl Nucleophiles and Electrophiles. Science 2020, 367, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Cheng Y-F; Liu J-R; Gu Q-S; Yu Z-L; Wang J; Li Z-L; Bian J-Q; Wen H-T; Wang X-J; Hong X; Liu X-Y Catalytic Enantioselective Desymmetrizing Functionalization of Alkyl Radicals via Cu(I)/CPA Cooperative Catalysis. Nat. Catal 2020, 3, 401–410. [Google Scholar]; (s) Wang Y; Carder HM; Wendlandt AE Synthesis of Rare Sugar Isomers through Site-Selective Epimerization. Nature 2020, 578, 403–408. [DOI] [PubMed] [Google Scholar]
- (3).For select reviews and highlights on Co(II)-based MRC, see:; (a) Huang H-M; Garduño-Castro MH; Morrill C; Procter DJ Catalytic Cascade Reactions by Radical Relay. Chem. Soc. Rev 2019, 48, 4626–4638. [DOI] [PubMed] [Google Scholar]; (b) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]; (c) Singh R; Mukherjee A Metalloporphyrin Catalyzed C–H Amination. ACS Catal. 2019, 9, 3604–3617. [Google Scholar]; (d) Xiong T; Zhang Q New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]; (e) Pellissier H; Clavier H Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev 2014, 114, 2775–2823. [DOI] [PubMed] [Google Scholar]; (f) Lu H; Zhang XP Catalytic C–H Functionalization by Metalloporphyrins: Recent Developments and Future Directions. Chem. Soc. Rev 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]; (g) Che CM; Lo VKY; Zhou CY; Huang JS Selective Functionalisation of Saturated C–H Bonds with Metalloporphyrin Catalysts. Chem. Soc. Rev 2011, 40, 1950–1975. [DOI] [PubMed] [Google Scholar]; (h) Driver TG Recent Advances in Transition Metal-Catalyzed N-Atom Transfer Reactions of Azides. Org. Biomol. Chem 2010, 8, 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fantauzzi S; Caselli A; Gallo E Nitrene Transfer Reactions Mediated by Metalloporphyrin Complexes. Dalton Trans. 2009, 5434–5443. [DOI] [PubMed] [Google Scholar]; (j) Doyle MP Exceptional Selectivity in Cyclopropanation Reactions Catalyzed by Chiral Cobalt(II)–Porphyrin Catalysts. Angew. Chem., Int. Ed 2009, 48, 850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For select examples of Ti(III)-based radical processes, see:; (a) Nugent WA; RajanBabu TV Titanium(III)-Induced Cyclization of Epoxy Olefins. J. Am. Chem. Soc 1988, 110, 8561–8562. [Google Scholar]; (b) RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (c) Gansäuer A; Rinker B; Pierobon M; Grimme S; Gerenkamp M; Mück-Lichtenfeld C A Radical Tandem Reaction with Homolytic Cleavage of a Ti–O Bond. Angew. Chem., Int. Ed 2003, 42, 3687–3690. [DOI] [PubMed] [Google Scholar]; (d) Gansäuer A; Fan C-A; Keller F; Keil J Titanocene-Catalyzed Regiodivergent Epoxide Openings. J. Am. Chem. Soc 2007, 129, 3484–3485. [DOI] [PubMed] [Google Scholar]; (e) Gansäuer A; Fleckhaus A; Lafont MA; Okkel A; Kotsis K; Anoop A; Neese F Catalysis via Homolytic Substitutions with C–O and Ti–O Bonds: Oxidative Additions and Reductive Eliminations in Single Electron Steps. J. Am. Chem. Soc 2009, 131, 16989–16999. [DOI] [PubMed] [Google Scholar]; (f) Gansäuer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers RA II Cationic Titanocene(III) Complexes for Catalysis in Single-Electron Steps. Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (g) Gansäuer A; Hildebrandt S; Vogelsang E; Flowers II RA Tuning the redox properties of the Titanocene(III)/(IV)-Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans. 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (h) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S Radical Redox-Relay Catalysis: Formal [3 + 2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (i) Yao C; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]; (j) Ye K-Y; McCallum T; Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]
- (5).For select examples of catalytic radical processes involving metalloradical intermediates, see:; (a) Smith DM; Pulling ME; Norton JR Tin-Free and Catalytic Radical Cyclizations. J. Am. Chem. Soc 2007, 129, 770–771. [DOI] [PubMed] [Google Scholar]; (b) Estes DP; Norton JR; Jockusch S; Sattler W Mechanisms by which Alkynes React with CpCr(CO)3H. Application to Radical Cyclization. J. Am. Chem. Soc 2012, 134, 15512–15518. [DOI] [PubMed] [Google Scholar]; (c) Li G; Han A; Pulling ME; Estes DP; Norton JR Evidence for Formation of a Co–H Bond from (H2O)2Co(dmgBF2)2 under H2: Application to Radical Cyclizations. J. Am. Chem. Soc 2012, 134, 14662–14665. [DOI] [PubMed] [Google Scholar]; (d) Kuo JL; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H• Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. For select examples of catalytic transformations via MRC, see: [DOI] [PubMed] [Google Scholar]; (e) Reddy AR; Hao F; Wu K; Zhou C-Y; Che CM Cobalt(II) Porphyrin-Catalyzed Intramolecular Cyclopropanation of N-Alkyl Indoles/Pyrroles with Alkylcarbene: Efficient Synthesis of Polycyclic N-Heterocycles. Angew. Chem., Int. Ed 2016, 55, 1810–1815. [DOI] [PubMed] [Google Scholar]; (f) Das BG; Chirila A; Tromp M; Reek JNH; de Bruin B CoIII-Carbene Radical Approach to Substituted 1H-Indenes. J. Am. Chem. Soc 2016, 138, 8968–8975. [DOI] [PubMed] [Google Scholar]; (g) Goswami M; de Bruin B; Dzik WI Difluorocarbene Transfer from a Cobalt Complex to an Electron-Deficient Alkene. Chem. Commun 2017, 53, 4382–4385. [DOI] [PubMed] [Google Scholar]; (h) Chirila A; Gopal Das B; Paul ND; de Bruin B Diastereoselective Radical-Type Cyclopropanation of Electron-Deficient Alkenes Mediated by the Highly Active Cobalt(II) Tetramethyltetraaza[14]annulene Catalyst. ChemCatChem 2017, 9, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Gu Z-Y; Liu Y; Wang F; Bao X; Wang S-Y; Ji S-J Cobalt(II)-Catalyzed Synthesis of Sulfonyl Guanidines via Nitrene Radical Coupling with Isonitriles: A Combined Experimental and Computational Study. ACS Catal. 2017, 7, 3893–3899. [Google Scholar]; (j) Liu J; Hu L; Wang L; Chen H; Deng L An Iron(II) Ylide Complex as a Masked Open-Shell Iron Alkylidene Species in Its Alkylidene-Transfer Reactions with Alkenes. J. Am. Chem. Soc 2017, 139, 3876–3888. [DOI] [PubMed] [Google Scholar]; (k) Roy S; Das SK; Chattopadhyay B Cobalt(II)-based Metalloradical Activation of 2-(Diazomethyl)pyridines for Radical Transannulation and Cyclopropanation. Angew. Chem., Int. Ed 2018, 57, 2238–2243. [DOI] [PubMed] [Google Scholar]; (l) Karns AS; Goswami M; de Bruin B Catalytic Synthesis of Indolines by Hydrogen Atom Transfer to Cobalt(III)-Carbene Radicals. Chem. - Eur. J 2018, 24, 5253–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Roy S; Khatua H; Das SK; Chattopadhyay B Iron(II)-Based Metalloradical Activation: Switch from Traditional Click Chemistry to Denitrogenative Annulation. Angew. Chem., Int. Ed 2019, 58, 11439–11443. [DOI] [PubMed] [Google Scholar]; (n) Das SK; Roy S; Khatua H; Chattopadhyay B Iron-Catalyzed Amination of Strong Aliphatic C(sp3)–H Bonds. J. Am. Chem. Soc 2020, 142, 16211–16217. [DOI] [PubMed] [Google Scholar]; (o) Zhang Z; Gevorgyan V Co-Catalyzed Transannulation of Pyridotriazoles with Isothiocyanates and Xanthate Esters. Org. Lett 2020, 22, 8500–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For detailed studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination, including EPR observation of α-Co(III)-aminyl radical intermediates (also known as Co(III)-nitrene radicals), see:; (a) Lyaskovskyy V; Olivos Suarez AI; Lu H; Jiang H; Zhang XP; de Bruin B Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)–Nitrene Intermediates. J. Am. Chem. Soc 2011, 133, 12264–12273. [DOI] [PubMed] [Google Scholar]; (b) Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović-Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc 2015, 137, 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related DFT studies on the radical mechanism of [Co(Por)]-catalyzed olefin aziridination and ligand hydrogen-bonding effect, see:; (c) Olivos Suarez AI; Jiang H; Zhang XP; de Bruin B The Radical Mechanism of Cobalt(II) Porphyrin-Catalyzed Olefin Aziridination and the Importance of Cooperative H-Bonding. Dalton Trans. 2011, 40, 5697–5705. [DOI] [PubMed] [Google Scholar]; (d) Hopmann KH; Ghosh A Mechanism of Cobalt-Porphyrin–Catalyzed Aziridination. ACS Catal. 2011, 1, 597–600. [Google Scholar]
- (7).(a) Subbarayan V; Ruppel JV; Zhu S; Perman JA; Zhang XP Highly Asymmetric Cobalt-Catalyzed Aziridination of Alkenes with Trichloroethoxysulfonyl Azide (TcesN3). Chem. Commun 2009, 4266–4268. [DOI] [PubMed] [Google Scholar]; (b) Jin L-M; Xu X; Lu H; Cui X; Wojtas L; Zhang XP Effective Synthesis of Chiral N-Fluoroaryl Aziridines through Enantioselective Aziridination of Alkenes with Fluoroaryl Azides. Angew. Chem., Int. Ed 2013, 52, 5309–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jiang H; Lang K; Lu H; Wojtas L; Zhang XP Asymmetric Radical Bicyclization of Allyl Azidoformates via Cobalt(II)-Based Metalloradical Catalysis. J. Am. Chem. Soc 2017, 139, 9164–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li C; Lang K; Lu H; Hu Y; Cui X; Wojtas L; Zhang XP Catalytic Radical Process for Enantioselective Amination of C(sp3)–H Bonds. Angew. Chem., Int. Ed 2018, 57, 16837–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hu Y; Lang K; Li C; Gill JB; Kim I; Lu H; Fields KB; Marshall M; Cheng Q; Cui X; Wojtas L; Zhang XP Enantioselective Radical Construction of 5-Membered Cyclic Sulfonamides by Metalloradical C–H Amination. J. Am. Chem. Soc 2019, 141, 18160–18169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereo-retentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Clayden J; Donnard M; Lefranc J; Tetlow DJ Quaternary Centres Bearing Nitrogen (α-Tertiary Amines) as Products of Molecular Rearrangements. Chem. Commun 2011, 47, 4624–4639. [DOI] [PubMed] [Google Scholar]; (b) Hager A; Vrielink N; Hager D; Lefranc J; Trauner D Synthetic Approaches towards Alkaloids Bearing α-Tertiary Amines. Nat. Prod. Rep 2016, 33, 491–522. [DOI] [PubMed] [Google Scholar]
- (10).For select reviews, see:; (a) Park Y; Kim Y; Chang S Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]; (b) Alderson JM; Corbin JR; Schomaker JM Tunable, Chemo- and Site-Selective Nitrene Transfer Reactions through the Rational Design of Silver(I) Catalysts. Acc. Chem. Res 2017, 50, 2147–2158. [DOI] [PubMed] [Google Scholar]; (c) Collet F; Lescot C; Dauban P Catalytic C–H Amination: the Stereo-selectivity Issue. Chem. Soc. Rev 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]
- (11).For select examples of nonasymmetric intramolecular C–H amination, see:; (a) Huard K; Lebel H N-Tosyloxycarbamates as Reagents in Rhodium-Catalyzed C–H Amination Reactions. Chem. -Eur. J 2008, 14, 6222–6230. [DOI] [PubMed] [Google Scholar]; (b) Hennessy ET; Betley TA Complex N-Heterocycle Synthesis via Iron-Catalyzed, Direct C–H Bond Amination. Science 2013, 340, 591–595. [DOI] [PubMed] [Google Scholar]; (c) Alderson JM; Phelps AM; Scamp RJ; Dolan NS; Schomaker JM Ligand-Controlled, Tunable Silver-Catalyzed C–H Amination. J. Am. Chem. Soc 2014, 136, 16720–16723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hong SY; Park Y; Hwang Y; Kim YB; Baik M-H; Chang S Selective Formation of γ-Lactams via C–H Amidation Enabled by Tailored Iridium Catalysts. Science 2018, 359, 1016–1021. [DOI] [PubMed] [Google Scholar]; (e) Azek E; Khalifa M; Bartholoméüs J; Ernzerhof M; Lebel H Rhodium(II)-Catalyzed C–H Aminations using N-Mesyloxycarbamates: Reaction Pathway and By-Product Formation. Chem. Sci 2019, 10, 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Baek Y; Betley TA Catalytic C–H Amination Mediated by Dipyrrin Cobalt Imidos. J. Am. Chem. Soc 2019, 141, 7797–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]; For metal-free C(sp3)–H radical amination, see:; (g) Evoniuk CJ; Gomes G. d. P.; Hill SP; Fujita S; Hanson K; Alabugin IV Coupling N–H Deprotonation, C–H Activation, and Oxidation: Metal-Free C(sp3)–H Aminations with Unprotected Anilines. J. Am. Chem. Soc 2017, 139, 16210–16221. [DOI] [PubMed] [Google Scholar]; (h) Bafaluy D; Muñoz-Molina JM; Funes-Ardoiz I; Herold S; de Aguirre AJ; Zhang H; Maseras F; Belderrain TR; Pérez PJ; Muñiz K Copper-Catalyzed N–F Bond Activation for Uniform Intramolecular C–H Amination Yielding Pyrrolidines and Piperidines. Angew. Chem., Int. Ed 2019, 58, 8912–8916. [DOI] [PubMed] [Google Scholar]; (i) Ju M; Huang M; Vine LE; Dehghany M; Roberts JM; Schomaker JM Tunable Catalyst-Controlled Syntheses of β- and γ-Amino Alcohols Enabled by Silver-Catalysed Nitrene Transfer. Nat. Catal 2019, 2, 899–908. [Google Scholar]
- (12).For select examples of intramolecular asymmetric C–H amination, see:; (a) Liang J-L; Yuan S-X; Huang J-S; Yu W-Y; Che C-M Highly Diastereo- and Enantioselective Intramolecular Amidation of Saturated C–H Bonds Catalyzed by Ruthenium Porphyrins. Angew. Chem., Int. Ed 2002, 41, 3465–3468. [DOI] [PubMed] [Google Scholar]; (b) Milczek E; Boudet N; Blakey S Enantioselective C–H Amination Using Cationic Ruthenium(II)–Pybox Catalysts. Angew. Chem., Int. Ed 2008, 47, 6825–6828. [DOI] [PubMed] [Google Scholar]; (c) Wang H; Park Y; Bai Z; Chang S; He G; Chen G Iridium-Catalyzed Enantioselective C(sp3)–H Amidation Controlled by Attractive Noncovalent Interactions. J. Am. Chem. Soc 2019, 141, 7194–7201. [DOI] [PubMed] [Google Scholar]; (d) Zhou Z; Chen S; Qin J; Nie X; Zheng X; Harms K; Riedel R; Houk KN; Meggers E Catalytic Enantioselective Intramolecular C(sp3)–H Amination of 2-Azidoacetamides. Angew. Chem., Int. Ed 2019, 58, 1088–1093. [DOI] [PubMed] [Google Scholar]; (e) Xing Q; Chan C-M; Yeung Y-W; Yu W-Y Ruthenium(II)-Catalyzed Enantioselective γ-Lactams Formation by Intramolecular C–H Amidation of 1,4,2-Dioxazol-5-Ones. J. Am. Chem. Soc 2019, 141, 3849–3853. [DOI] [PubMed] [Google Scholar]; (f) Ju M; Zerull EE; Roberts JM; Huang M; Guzei IA; Schomaker JM Silver-Catalyzed Enantioselective Propargylic C-H Bond Amination through Rational Ligand Design. J. Am. Chem. Soc 2020, 142, 12930–12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For a recent example of enantioconvergent C–H amination via enzymatic catalysis, see:; (a) Yang Y; Cho I; Qi X; Liu P; Arnold FH An Enzymatic Platform for the Asymmetric Amination of Primary, Secondary and Tertiary C(sp3)–H Bonds. Nat. Chem 2019, 11, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent example of enantioconvergent C–H amination by Cu-based catalysts, see:; (b) Yang C-J; Zhang C; Gu Q-S; Fang J-H; Su X-L; Ye L; Sun Y; Tian Y; Li Z-L; Liu X-Y Cu-Catalysed Intramolecular Radical Enantioconvergent Tertiary β-C(sp3)–H Amination of Racemic Ketones. Nat. Catal 2020, 3, 539–546. [Google Scholar]
- (14).Chen Y; Fields KB; Zhang XP Bromoporphyrins as Versatile Synthons for Modular Construction of Chiral Porphyrins: Cobalt-Catalyzed Highly Enantioselective and Diastereoselective Cyclopropanation. J. Am. Chem. Soc 2004, 126, 14718–14719. [DOI] [PubMed] [Google Scholar]
- (15).Xu X; Lu H; Ruppel JV; Cui X; Lopez de Mesa S; Wojtas L; Zhang XP Highly Asymmetric Intramolecular Cyclopropanation of Acceptor-Substituted Diazoacetates by Co(II)-Based Metalloradical Catalysis: Iterative Approach for Development of New-Generation Catalysts. J. Am. Chem. Soc 2011, 133, 15292–15295. [DOI] [PubMed] [Google Scholar]
- (16).Wen X; Wang Y; Zhang XP Enantioselective Radical Process for Synthesis of Chiral Indolines by Metalloradical Alkylation of Diverse C(sp3)–H Bonds. Chem. Sci 2018, 9, 5082–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).For the rate constant of ring-opening of the parent cyclopropylmethyl radical, see:; Bowry VW; Lusztyk J; Ingold KU Calibration of a New Horologery of Fast Radical Clocks. Ring-Opening Rates for Ring- and Alpha-Alkyl-Substituted Cyclopropylcarbinyl Radicals and for the Bicyclo[2.1.0]pent-2-yl Radical. J. Am. Chem. Soc 1991, 113, 5687–5698. [Google Scholar]
- (18).(a) Corbett JW; Gearhart LA; Ko SS; Rodgers JD; Cordova BC; Klabe RM; Erickson-Viitanen SK Novel 2,2-Dioxide-4,4-Disubstituted-1,3-H-2,1,3-Benzothiadiazines as non-Nucleoside Reverse Transcriptase Inhibitors. Bioorg. Med. Chem. Lett 2000, 10, 193–195. [DOI] [PubMed] [Google Scholar]; (b) Shaw D; Best J; Dinnell K; Nadin A; Shearman M; Pattison C; Peachey J; Reilly M; Williams B; Wrigley J; Harrison T 3,4-Fused Cyclohexyl Sulfones as γ-Secretase Inhibitors. Bioorg. Med. Chem. Lett 2006, 16, 3073–3077. [DOI] [PubMed] [Google Scholar]
- (19).For bioactivity studies on related N-heterocyclic structures, see:; (a) Braghiroli D; Puia G; Cannazza G; Tait A; Parenti C; Losi G; Baraldi M Synthesis of 3,4-Dihydro-2H-1,2,4-benzo- thiadiazine 1,1-Dioxide Derivatives as Potential Allosteric Modulators of AMPA/Kainate Receptors. J. Med. Chem 2002, 45, 2355–2357. [DOI] [PubMed] [Google Scholar]; (b) Vadivelan S; Deeksha TN; Arun S; Machiraju PK; Gundla R; Sinha BN; Jagarlapudi SARP Virtual Screening Studies on HIV-1 Reverse Transcriptase Inhibitors to Design Potent Leads. Eur. J. Med. Chem 2011, 46, 851–859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.