

Abstract
Immune dysregulation during sepsis is mediated by an imbalance of T cell costimulatory and coinhibitory signaling. CD28 is downregulated during sepsis and is significantly altered on memory vs. naïve T cells. Thus, in order to study the role of CD28 during sepsis in a more physiologically relevant context, we developed a “memory mouse” model in which animals are subjected to pathogen infections to generate immunologic memory, followed by sepsis induction via CLP. Using this system, we show that agonistic anti-CD28 treatment resulted in worsened survival in naïve septic animals but conferred a significant survival advantage in immunologically experienced septic animals. Mechanistically, this differential response was driven by the ability of CD28 agonism to elicit IL-10 production from Treg uniquely in memory but not naïve mice. Moreover, elevated IL-10 released by activated Tregs in memory mice inhibited sepsis-induced T cell apoptosis via the anti-apoptotic protein Bcl-xL. Together, these data demonstrate that immunologic experience is an important parameter that affects sepsis pathophysiology and can fundamentally change the outcome of modulating the CD28 pathway during sepsis. This study suggests that testing therapeutic strategies in immunologically experienced hosts may be one way to increase the physiologic relevance of rodent models in sepsis research.
Keywords: sepsis, αCD28 Ab, regulatory T cells, apoptosis, IL-10
INTRODUCTION
Sepsis, a dysregulated host response to infection, is a leading cause of organ dysfunction and a huge health burden estimated to kill 7.3 million people worldwide every year (1, 2). Despite increasing understanding of the pathophysiology of sepsis, there are no approved effective therapeutics available for sepsis once antibiotics and fluid resuscitation fail. Emerging evidence suggests that immune dysregulation during sepsis is mediated by an altered balance of T cell costimulatory and coinhibitory signaling (3). CD28, a critical T cell costimulatory receptor, binds B7 molecules to provide a costimulatory signal to sustain T lymphocyte activation and proliferation (4). It has been reported that loss of CD28 expression is a hallmark of decline in T cell function (5). In addition to its role in T cell priming, CD28 signaling regulates T cell adhesion, cytoskeletal reorganization, and is essential for inducing the trafficking of effector T cells to target tissues (6–8). Several studies have shown that CD28 expression on T cells is downregulated in the setting of sepsis, and decreases more in elderly septic patients compared with their middle-aged septic patients (9, 10). However, the mechanistic impact of the observed CD28 downregulation in the context of sepsis was not known: it could result in reduced ability of T cells to receive costimulatory signals, or it could be indicative of an activated T cell phenotype (11).
Similarly, studies to address the role of CD28, and the impact of its therapeutic manipulation during sepsis, have also yielded conflicting results. One study showed that CD28 is protective for intestinal immune responses during sepsis in that antibody-mediated CD28 stimulation resulted in upregulated T cell migration to gut independently of T cell receptor and gut-specific homing receptor expression (12). On the other hand, blockade of costimulatory signals, including CD40 and CD80/86, have also been shown to reduce mortality in polymicrobial sepsis (13). A separate study demonstrated that a CD28 mimetic peptide (AB103) that attenuates CD28 signaling and T-helper type 1 cytokine responses could attenuate inflammatory cytokine responses and bacterial burden and improve survival in a murine model of necrotizing soft-tissue infection (14, 15). Moreover, a clinical trial revealed that treatment with the anti-CD28 Ab TGN1412 led to rapid and severe cytokine storm in healthy humans (16). Taken together, these studies suggest that therapeutic targeting of CD28 pathway could be an effective strategy to prevent sepsis-induced immune dysfunction.
It is well known that CD28 expression is significantly altered on memory T cells in humans and mice. The specific pathogen free (SPF) laboratory mice possess much fewer memory T cells than adult human or feral mice. Recent studies demonstrated that laboratory mice contained fewer effector memory (TEM) and mucosally distributed memory T cells compared to pet store or feral mice with diverse microbial exposure. These antigen-experienced mice exhibited increased immune responses and altered resistance to infection (17). Another study demonstrated that wild mice possessed an increased number of Foxp3+ regulatory T (Treg) relative to laboratory mice (18). Thus, to better recapitulate immune features of adult patients, we developed a novel “memory mouse” model, in which mice receive sequential pathogen exposure and possess 30–50% memory T cells, similar to the frequency observed in adult humans (19). Here, we utilized this more physiologically relevant “memory mouse” model to interrogate the role of CD28 costimulatory signaling on T cells during sepsis.
We demonstrate that agonism of the CD28 pathway strikingly improved survival in septic memory mice. The apoptosis of memory T cells was inhibited in memory septic mice treated with CD28 agonist. Of note, Treg cells activated by CD28 agonist released more anti-inflammatory IL-10 in memory septic mice relative to naïve septic mice. Mechanically, increased IL-10 alleviated the apoptosis of memory T cells mediated by the mitochondrial pathway, contributing to a survival benefit in memory mice with sepsis.
MATERIALS AND METHODS
Memory mice model
Six week old male and female C57BL/6J (B6) mice were purchased from the Jackson Laboratory. Listeria monocytogenes (LM) was grown in brain heart infusion broth containing streptomycin 16h to log phase growth. Bacteria counts were calculated by Bio-photometer. The indicated groups of mice were injected intraperitoneally with 1×104 CFU of LM. Then 30 days later, the same mice were injected with a single dose of 2×105 PFU of the Armstrong strain of lymphocytic choriomeningitis virus (LCMV). Titers of the virus were determined by plaque assay on Vero cells. These generated memory mice were assessed on d0, 10, 25, 40, 55 post-LM infection in the blood by flow cytometry. Both of these acute infections are cleared on d59 after Listeria infection. Age-and gender-matched B6 laboratory mice, called “naïve mice”, served as controls. All mice were maintained in the BSL-2 facility of Emory University (Atlanta, GA). All mice were used in accordance with the Institutional Animal Care and Use Committee guidelines of Emory University (protocol number 2003238–082518).
Cecal ligation and puncture
Cecal ligation and puncture (CLP) are performed at d60 post-LM infection. Under isoflurane anesthesia, a midline incision was performed and the cecum was externalized. The cecum was ligated with a 4–0 silk suture and punctured through and through with a 25-gauge needle. All animals received buprenorphine (0.1mg/kg) preoperatively for pain relief. Immediately postoperatively, animals received 1mL of sterile saline for fluid resuscitation as well as antibiotics (ceftriaxone 50 mg/kg and metronidazole 35mg/kg) every 12h for 2 days. 100ug anti-CD28 antibody (BioXcell, clone:37.51) or Isotype antibody (BioXcell, Syrian hamster IgG) was injected intraperitoneally immediately after CLP and at d2, 4, 6 post CLP. Where indicated, 250ug anti-IL-10 antibody (BioXcell, clone: JES5–2A5) or isotype antibody (rat IgG1, clone: HRPN) were intraperitoneally administrated at the same time with αCD28Ab antibody. Mice were euthanized with CO2 asphyxiation at the indicated time points.
Flow cytometry
Mice were sacrificed and spleens were harvested at 24h after CLP. Splenocytes were stained with anti-CD3-Alexa 700 (BD), anti-CD4-PB (Biolegend, clone RM4–5), anti-CD8-PO (Invitrogen, clone MCD0830), anti-CD44-PerCP (Biolegend, clone IM7), anti-CD62L-PE (BD), anti-CD28-PE-Cy7 (Biolegend, clone E18) and anti-CD25-APC-Cy7 (BD). For detection of cell apoptosis, splenocytes were stained with a FITC Annexin V apoptosis detection kit with 7-AAD (Biolegend). Anti-Bcl-xL (54H6) and Bcl-2 (Biolegend, clone BCL/10C4) were used to detect engagement of the mitochondrial pathway of apoptosis while anti-CD95 (Biolegend, clone DX2) and anti-TNFR Type Ⅰ (Biolegend, clone 55R-286) were stained to detect expression of death receptors on T cells. Cells were intracellularly stained with anti-Ki-67 (Biolegend, clone 16A8) to assay for cell proliferation. Tregs were identified via intracellular staining for Foxp3-FITC (Ebioscience, clone FJK-16S) using the Foxp3 staining kit (Ebioscience). B cells were stained with anti-CD19-FITC (Biolegend). NK cells were stained with anti-NK1.1-PE (Ebioscience). Dendritic cells were stained with anti-CD11c-PE-Cy7 (BD). Neutrophils were stained with anti-Gr-1-Alexa 700 (Biolegend) and anti-CD11b-PerCP (Biolegend). Accucheck Counting Beads (Thermo Fisher Scientific) were added after staining to calculate the absolute number of cells per spleen.
Intracellular cytokine staining
2×106 splenocytes from each sample were plated in a 96-well plate. Cells were incubated in culture medium (R10) consisting of RPMI 1640 containing 10% FBS (Mediatech, Herdon, VA), 2mM L-glutamine, 0.01M HEPES buffer, 100mg/ml gentamicin (Mediatech), and 5×10−5M 2-mercaptoethanol (Sigma-Aldrich, St.Louis, MO). Cells were stimulated with 50ng/ml PMA and 1ug/ml ionomycin in the presence of Golgiplug (BD Pharmingen) for 5 hours at 37°C. Cells were surface-stained for anti-ICOS (C398.4A), anti-CD69 (H1.2F3), anti-CD127 (A7R34), anti-GITR (YGITR 765), and then permeabilized using Foxp3 kit and stained with anti-IL-10 (JES5–16E3), anti-Foxp3, anti-CTLA-4 (UC10–489), anti-Helios (22F6) (All Abs from Biolegend). Samples were analyzed on an LSRII flow cytometer (BD) and data were analyzed using FlowJo software (9.9.6 FlowJo, LLC).
In vitro suppression assays
Splenic Treg cells were separately sorted from each memory mice treated with or without αCD28 Ab by magnetic cell sorting (MACS) with the CD4+ CD25+ Regulatory T cell isolation kit (MACS, Miltenyi Biotec.). Meanwhile, splenic naïve CD4+ T cells from naïve mice were negatively selected by MACS with the naïve CD4+ T cell isolation kit (MACS, Miltenyi Biotec.). In general, we obtained Treg cells of about 90% purity and naïve T cells of about 99% purity. Sorted Treg cells and naïve CD4+ T cells had >90% viability. As the responder cells, the sorted naïve CD4+ T cells were stained with CellTrace Violet (CTV, Thermo Fisher Scientific) and plated in a 96-well round-bottom plate at a density of 5×105 responder cells per well. The sorted Treg cells were added at a ratio of 1:4 and 1:8 with responder cells and 5ug/ml anti-CD3 (Clone: 145–2C11, BD) and incubated for 96 hours. The proliferation of responder cells (gated as CD3+ CD4+ Foxp3−) was quantified by flow cytometry based on the dilution of Cell Trace Violet (CTV). The % inhibition was calculated by the following formula: % inhibition = [1 − (% divided cells in experimental/the average of % divided cells of responder-only control)] × 100.
Measurement of cytokines in blood and supernatant
Blood was withdrawn via cardiac puncture after sacrifice. Samples were centrifuged at 4°C with 10,000rpm for 10 min to procure serum. Plasma cytokines concentration was determined using the Bio-Plex suspension array system and Bio-Plex Mouse Cytokine 8-Plex Panel according to the manufacturer’s instructions (Bio-Rad, Hercules, CA). The concentration of IL-10 in the coculture supernatant from Treg and naïve CD4+ T cells was also determined using Bio-plex system. Levels of IL-1β, IL-2, IL-6, IL-10, IL-13, MCP-1, TNF, and IFN-γ were reported in pg/mL. All samples were run in duplicate. Results were analyzed using Bio-Plex Manager™ 3.0 software.
Statistics
Data shown and described depict mean ± SEM. The one-way ANOVA and multiple comparison tests were used to compare the difference among the four groups. The Mann-Whitney U tests were used to compare continuous variables between two groups. All statistical analyses were conducted using GraphPad Prism 7.0 software (San Diego, CA). Two-tailed p-values <0.05 were considered statistically significant.
RESULTS
CD28 expression on both CD4+ and CD8+ T cells decreases during sepsis
To assess CD28 expression in the setting of sepsis, splenocytes from naive B6 mice that had undergone either CLP or sham surgery were analyzed by flow cytometry at 24h post-CLP (Fig 1A). CD28 MFI was significantly reduced on both CD4+ and CD8+ T cells in septic mice compared with sham animals (CD4: 555.0±14.9 vs. 323.6±16.7, p=0.002, Fig 1B; CD8: 321.2±9.1 vs. 228.0±11.6, p=0.002, Fig 1C). Multiple seminal studies have shown that CD28 expression is significantly altered on memory T cells compared to naïve counterparts (20), and that memory T cells have differential requirements for CD28-mediated costimulation relative to their naïve counterparts. Thus, in order to study CD28 biology during sepsis in a more physiologically relevant context, we generated immunologically-experienced “memory mice” via sequential infection of naïve B6 mice with Listeria monocytogenes followed by lymphocytic choriomeningitis virus (LCMV) as described in Methods. We chose to use Listeria and LCMV because they are 1) two very common pathogens that are frequently used in murine studies of immune responses, 2) one bacterial and one viral pathogen, and 3) well-documented to be completely cleared from the mouse by at most day 7 post-infection, which facilitated the study of the role of memory T cells, and not the impact of lingering concurrent latent or persistent infection, on sepsis pathogenesis. These antigen-experienced memory mice possessed a significantly increased frequency of memory (CD44hi) T cells in both the CD4+ and CD8+ T cells compartments by d59 after Listeria/LCMV infection. Memory mice possessed approximately 32% memory cells among CD4+ and 53% memory cells among CD8+ T cells in the blood on d59 post-infection, which is similar to the frequency of memory T cells found in adult humans (Supplemental Fig. 1A). In contrast, CD4+ and CD8+ T cell compartments of age-matched naïve mice were comprised of only 18% and 28% memory T cells, respectively (Supplemental Fig. 1B and 1C). We confirmed that CD28 was also significantly downregulated on CD44hiCD4+ and CD44hiCD8+ T cells in this memory mouse model (CD44hiCD4: 2217±92.2 vs. 1464±72.7, p=0.0079, Fig 1D and 1E; CD44hiCD8: 1254±53.5 vs. 1118±16.1, p=0.03, Fig 1D and 1F).
Figure 1. αCD28Ab improves survival in septic memory mice but not naïve septic mice.
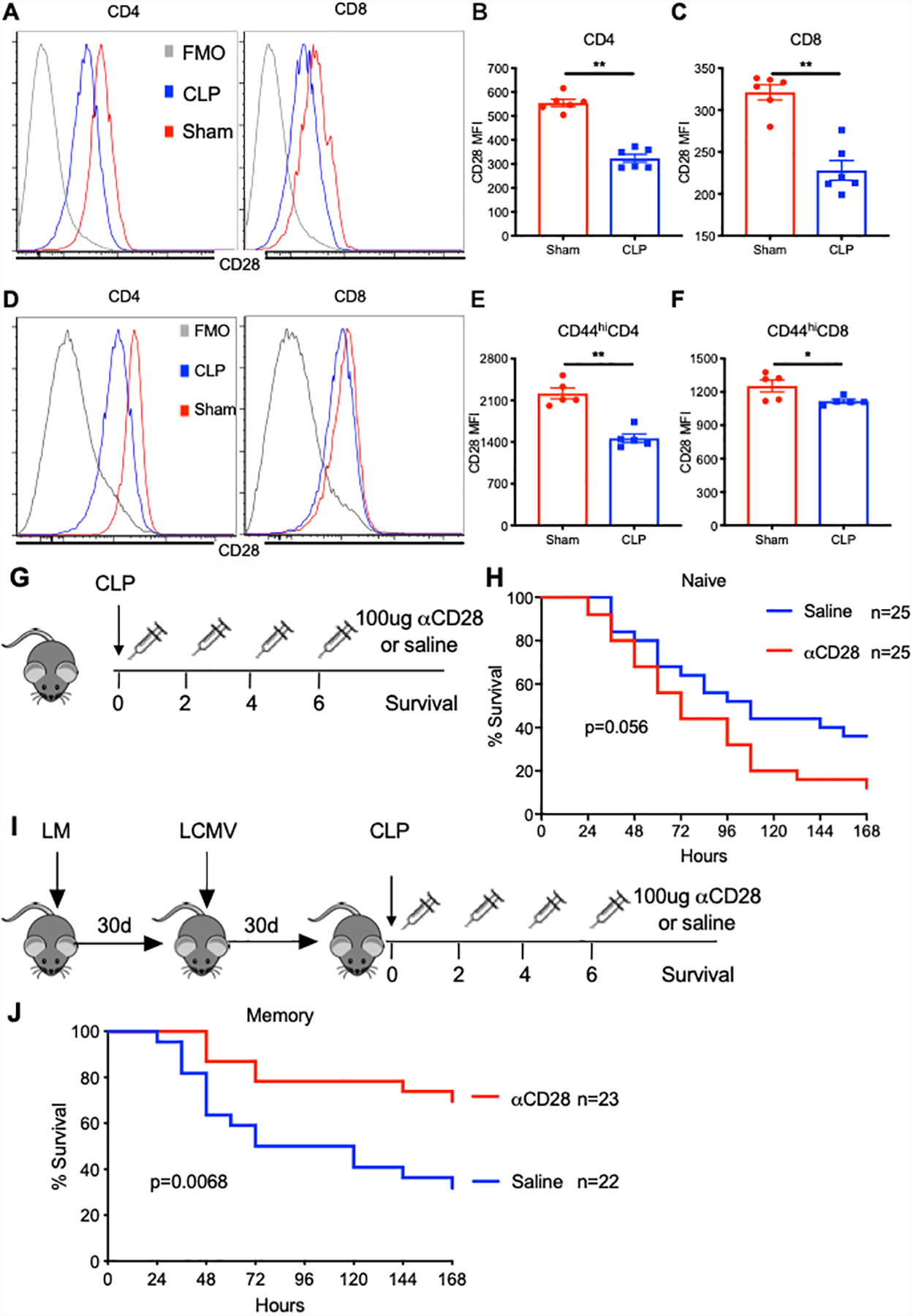
B6 naïve and memory mice received CLP or sham surgery and were sacrificed at 24h post-surgery. Spleens were harvested and CD28 expression on T cells was determined by flow cytometry. (A) Representative flow histogram of CD28 expression on bulk CD4+ and CD8+ T cells in naïve mice. (B and C) Summary data of MFI of CD28 on bulk CD4+ and CD8+ T cells at 24 hours after sham or CLP surgery. (D) Representative flow histogram of CD28 expression on CD44hiCD4+ and CD44hiCD8+ T cells in memory mice. (E and F) Summary data of MFI of CD28 on CD44hiCD4+ and CD44hiCD8+ T cells at 24 hours after sham or CLP surgery. Groups (n=5–6/group) were compared with the Mann-Whitney nonparametric test. **, p<0.01. *, p<0.05. All data expressed as mean ± SEM. FMO, Fluorescence Minus One. (G) Schematic of experiment setup for CLP sepsis study in naïve mice. (H) All naïve mice received CLP surgery and were randomly injected intraperitoneally with either αCD28 Ab (n=25) or saline (n=25) at specified time points. Mice were monitored for 7-day survival. (I) Schematic of experiment setup for CLP study in memory mice. (J) Memory mice underwent CLP and received αCD28Ab (n=23) or saline (n=22). Mice were monitored for 7-day survival. All data depicted a minimum of two independent experiments. The log-rank (Mantel-Cox) test was used to test for significance.
αCD28 Ab improves survival in septic memory mice but not naïve septic mice
Given the finding that CD28 expression was downregulated on CD4+ and CD8+ T cells during sepsis in both naïve and memory animals, we queried the impact of improving CD28 signaling using an agonistic anti-CD28 mAb on sepsis mortality in both memory mice and naïve mice. We first demonstrated that this antibody is indeed functioning as a CD28 agonist by analyzing both T cell cytokine production and proliferation in both naïve and memory septic mice, and results indicated that administration of the agonistic anti-CD28 increased cytokine secretion and proliferation in both naïve and memory septic mice relative to untreated controls (Supplemental Fig. S2). To assess the impact of agonistic anti-CD28 on sepsis mortality, both naïve and memory mice underwent CLP surgery and groups of mice received either αCD28 Ab or saline on d0, 2, 4, 6 post-CLP. Results demonstrated that αCD28 mAb failed to significantly impact 7-day survival in naïve mice with sepsis, although it trended toward worsened survival (p=0.056, Fig. 1G and 1H). In contrast, septic memory mice treated with αCD28 Ab showed a robust survival benefit relative to saline-treated mice. Sepsis survival was markedly improved from 30% to 70% in untreated vs. anti-CD28-treated memory mice (p=0.0068, Fig. 1I and 1J). To exclude the possibility of a non-specific Fc-mediated effect of the administered antibody, an anti-CD28 Ab isotype control antibody was administrated to both naïve and memory septic mice. There were no differences in 7-day survival between isotype control vs. saline-treated animals in either the naïve or memory groups (Supplemental Fig. S3).
Apoptosis of CD44hi memory CD4+ T cells is inhibited by αCD28 Ab in memory septic mice
To interrogate the cellular changes associated with decreased mortality in memory mice treated with αCD28 Ab, we assessed apoptosis within splenic T cell compartments in naïve vs. memory mice treated with αCD28 Ab vs. vehicle control at 24 hours following CLP using Annexin V and 7-AAD staining. Results indicated overall, memory septic mice possessed increased frequencies of apoptotic T cells relative to naïve septic mice in both the CD4+ and CD8+ T cell compartments regardless of treatment with αCD28 Ab. While there was no difference in the frequency of Annexin V+7-AAD+ apoptotic cells among CD44loCD4+ T cells in control vs aCD28-treated memory septic mice, memory septic mice treated with αCD28 Ab contained a lower frequency of apoptotic cells among CD44hiCD4+ T cells relative to untreated memory controls (8.0±1.1% vs.13.7±1.4%, p=0.014) (Fig. 2A–2C). In contrast, αCD28 Ab had no significant impact on the frequency of apoptotic cells among either CD44hi or CD44lo CD8+ T cells (Fig. 2D, 2E) in septic memory mice. In sum, these data suggest that memory mice generate more apoptotic T cells than naïve mice during sepsis, and that αCD28 Ab ameliorates the apoptosis of memory CD4+ T cells in memory mice with sepsis.
Figure 2. Apoptosis of CD44hi memory CD4+ T cells is rescued by αCD28 Ab in memory septic mice.
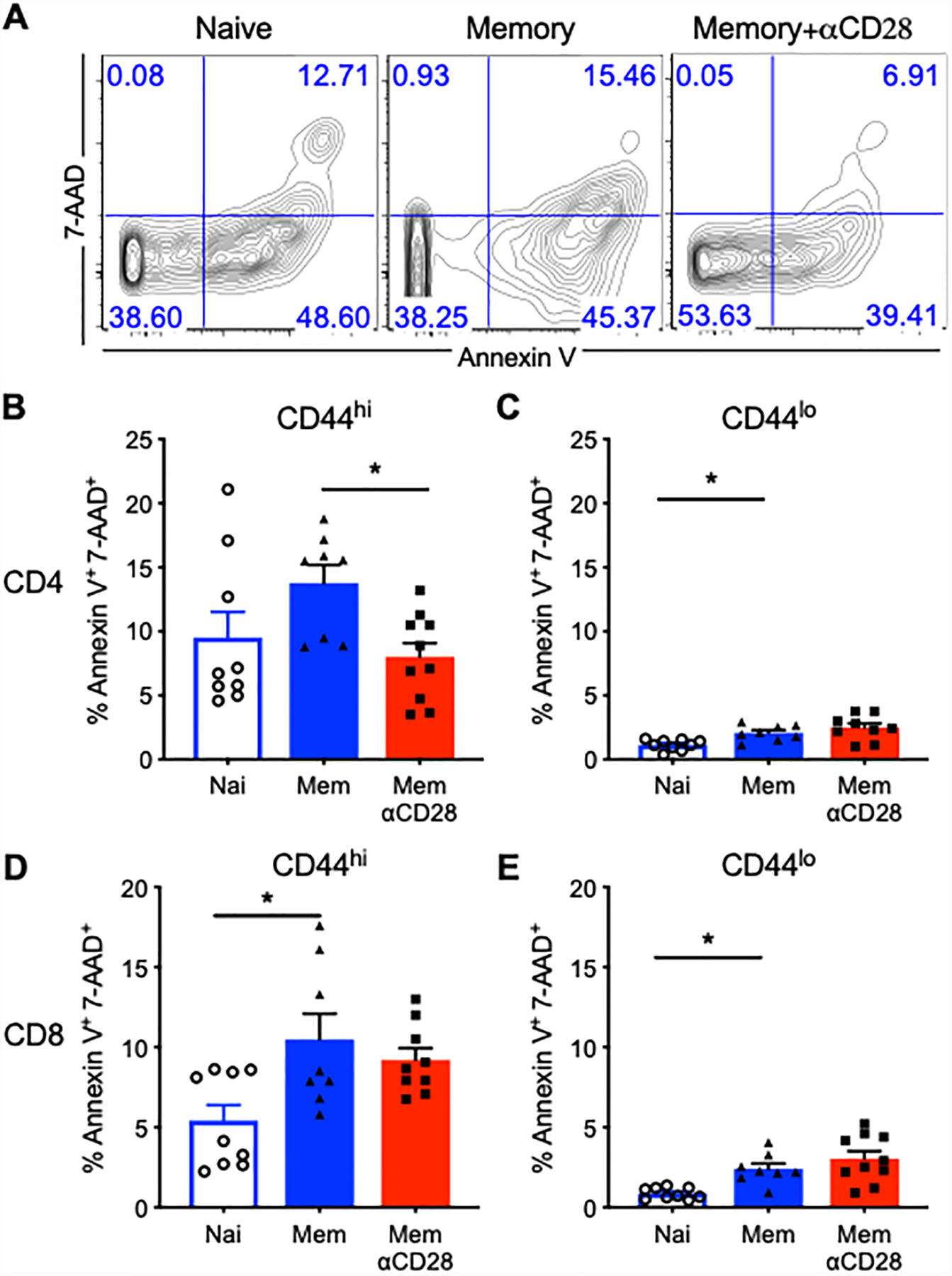
Memory mice and age-matched naïve controls received CLP, followed by injection of αCD28 Ab or saline. Mice were sacrificed and spleen was harvested at 24 hours after CLP. Splenocytes were stained with Annexin V and 7-AAD for T cell apoptosis by flow cytometry. (A) Representative flow cytograms for Annexin V+ and 7-AAD+ staining gated on CD44hiCD4+ T cells. (B and C) Summary data depicting frequency of apoptotic (AnnexinV+7-AAD+) CD44hiCD4+ and CD44loCD4+ T cells between untreated naïve mice and memory mice treated with/without αCD28Ab (n=8–9/group). (D and E) Summary data of the percentage of apoptotic memory and naïve CD8+ T cells among three groups (n=8–10/group). The data shown were compiled from two independent experiments. Groups were compared using one-way ANOVA analysis and Turkey multiple comparison tests. *, p<0.05.
Systemic IL-10 levels are increased in septic memory mice treated with αCD28 Ab
To determine the effect of αCD28Ab on the systemic cytokine environment during sepsis, we examined the levels of multiple circulating pro-inflammatory and anti-inflammatory cytokines at 24h post-CLP. αCD28Ab had no impact on the systemic IL-10 levels in naïve septic mice (Fig. 3A). In contrast, levels of serum IL-10 were significantly increased in memory septic mice treated with αCD28Ab relative to both untreated memory mice and to naïve septic mice treated with αCD28Ab. Further, there was a trend toward increased serum concentration of IL-6 in untreated memory mice, which was not observed in memory mice treated with αCD28Ab (Fig. 3B). No significant differences in other serum cytokines were observed, including TNF, IFN-γ, IL-1β, IL-2, IL-13 and MCP-1 in either memory or naïve mice treated with or without αCD28Ab (Fig. 3C to 3H).
Figure 3. Systemic IL-10 levels are increased in septic memory mice treated with αCD28Ab.
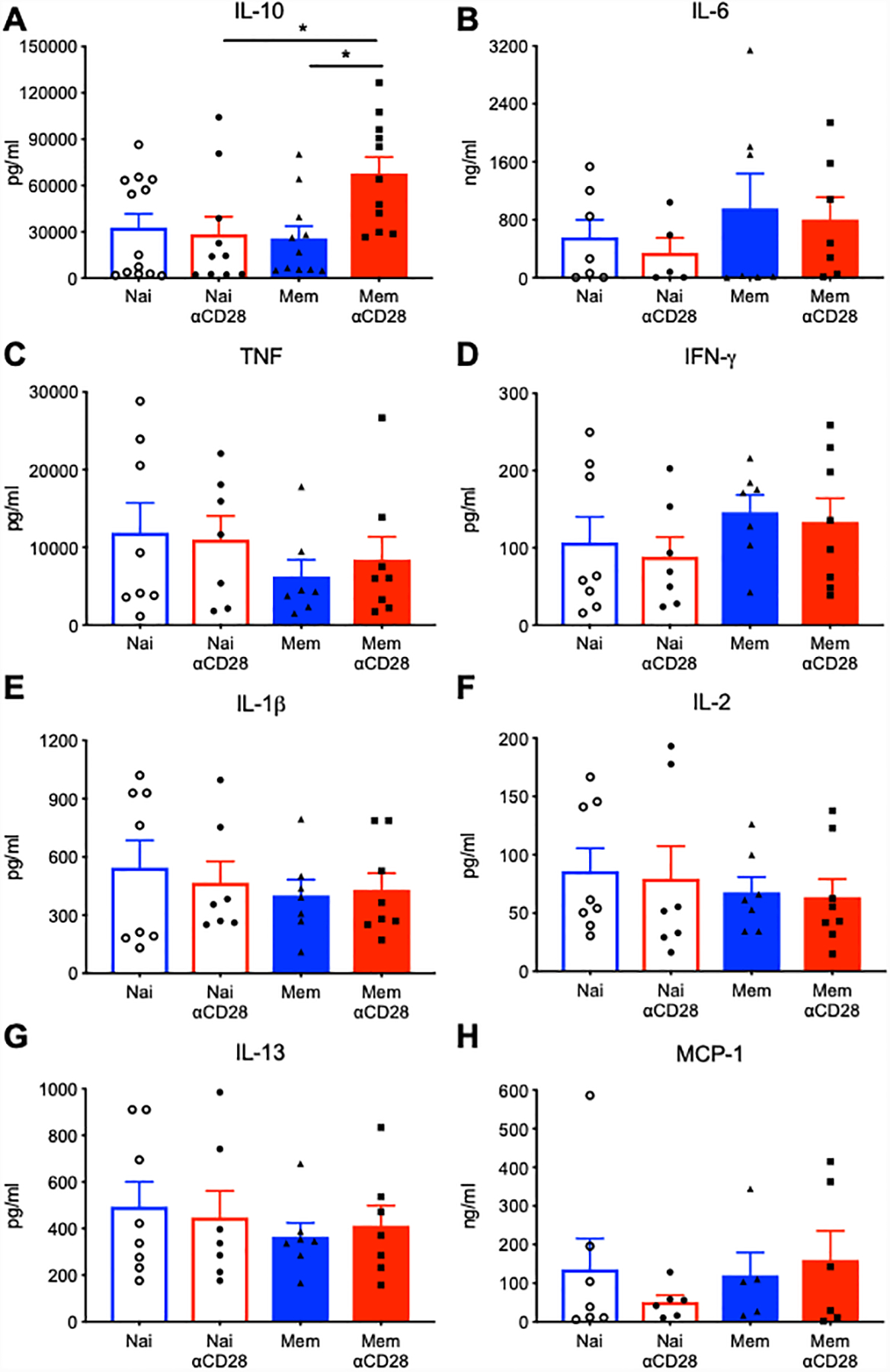
Blood samples were collected at 24h post-CLP from naïve mice treated with saline or αCD28Ab and memory mice treated with saline or αCD28Ab. Serum was analyzed using the Luminex Multiplex platform, examining the concentration of (A) IL-10, (B) IL-6, (C) TNF, (D) IFN-γ, (E) IL-1β, (F) IL-2, (G) IL-13, (H) MCP-1. Results were representative of three independent experiments. Error bars represent mean ± SEM. Groups (n=6–11/group) were compared using one-way ANOVA analysis and Turkey multiple comparison test. *, p<0.05.
Foxp3+ Treg cells in septic memory mice treated with αCD28 Ab exhibit increased IL-10 production
The finding that serum IL-10 levels were increased in memory septic mice treated with αCD28Ab prompted us to investigate the source of IL-10 production in αCD28Ab-treated memory mice. Naïve or memory mice received CLP in the presence or absence of the αCD28Ab and spleens were harvested at 24h and restimulated for 5h with PMA/ionomycin. Results indicated that memory septic mice possessed increased frequencies of IL-10-producing Foxp3+ Treg relative to naïve septic mice, even in untreated animals (Fig. 4B). This higher proportion of IL-10-producing Foxp3+ Treg was then further boosted by αCD28Ab treatment in memory septic mice: frequencies of IL-10 produced by regulatory T cells increased from 4.1% in untreated memory mice to 9.8% in αCD28Ab-treated memory mice (p<0.0001, Fig. 4B). Consistent with the above results on serum IL-10, there was no effect of αCD28Ab on IL-10 released by Foxp3+ regulatory T cells in naïve mice (Fig. 4A and 4B). Further, this boost was specific to IL-10 producing Foxp3+ Treg, as there was no difference in the absolute numbers of T cells in either naïve vs. memory septic mice in the presence or absence of αCD28Ab (data not shown). Additionally, there were no differences of IL-10 release by Foxp3− effector CD4+ T cells and CD8+ T cells among four groups (Fig. 4C and 4D).
Figure 4. Elevated IL-10 is secreted by Foxp3+ Treg cells in memory mice.

Memory and naïve mice treated with saline or αCD28Ab were sacrificed, and spleens were harvested 24 hours post-CLP. (A) Splenocytes were stimulated with PMA and ionomycin for 5 hours, followed by intracellular staining with anti-Foxp3 and anti-IL-10. (A) Representative flow plots and (B) summary data show IL-10 production by Foxp3+ Treg cells before and after stimulation among four groups (n=5–9/group). (C) The frequency of IL-10 secreted by Foxp3− effector CD4+ T cells in splenocytes from naïve and memory mice (n=5–10/group). (D) The frequency of IL-10 released by CD8+ T cells in splenocytes from naïve and memory mice (n=5–10/group). (E-H) Splenocytes were stimulated with LPS (10ug/ml) for 5 hours and then stained intracellularly with anti-IL-10 to evaluate IL-10 released by other immune cells, including B cells (E), dendritic cells (F), neutrophils (G) and natural killer cells (H). Data represented mean ± SEM. Groups (n=5–10/groups) were compared using one-way ANOVA analysis and Turkey multiple comparison test. *, p<0.05, ***, p<0.001 and ****, p<0.0001.
A large number of leukocytes, including dendritic cells (DC), natural killer (NK) cells, neutrophils, and B cells have been reported to produce IL-10 (21–23). Thus, we also assayed IL-10 production in these subsets. Data showed that B cells released little IL-10 following CLP, and αCD28Ab had no effect on the B cell-derived IL-10 in either memory or naïve mice (Fig. 4E). No differences in IL-10 production by NK cells or neutrophils in either memory or naïve mice were noted (Fig. 4F and 4G). DC produced less IL-10 in memory mice compared to DC in naïve mice (Fig. 4H). These data support the conclusion that the increased systemic IL-10 levels elicited by αCD28Ab in memory septic mice are the result of IL-10 production by Foxp3+ Treg.
αCD28 Ab accelerates proliferation and activation of Foxp3+ Treg in memory septic mice
Given the finding that agonistic αCD28Ab uniquely elicits IL-10 from Foxp3+ Treg in memory mice, we sought to determine the impact of CD28 agonism on the activation and differentiation status of Treg populations under these conditions. Memory mice and age-matched naïve mice underwent CLP, followed by injection of agonistic αCD28Ab or saline. Spleens were harvested at 24 hours post-CLP, and Treg proliferation and cell surface phenotype was analyzed by flow cytometry. Results indicated that there was no significant effect of αCD28Ab on the absolute numbers of Foxp3+ Treg cells in either memory or naïve groups (Fig. 5A). However, CD28 agonism restored the proliferation of Foxp3+ Treg cells (assessed by expression of intracellular Ki67), which was reduced in memory mice, to the levels observed in naïve mice (Fig. 5B). In contrast, proliferation of Foxp3+ Treg cells in naïve mice was not impacted by agonistic αCD28Ab (Fig. 5B).
Figure 5. αCD28 Ab accelerates proliferation and activation of Foxp3+ Treg in memory septic mice.
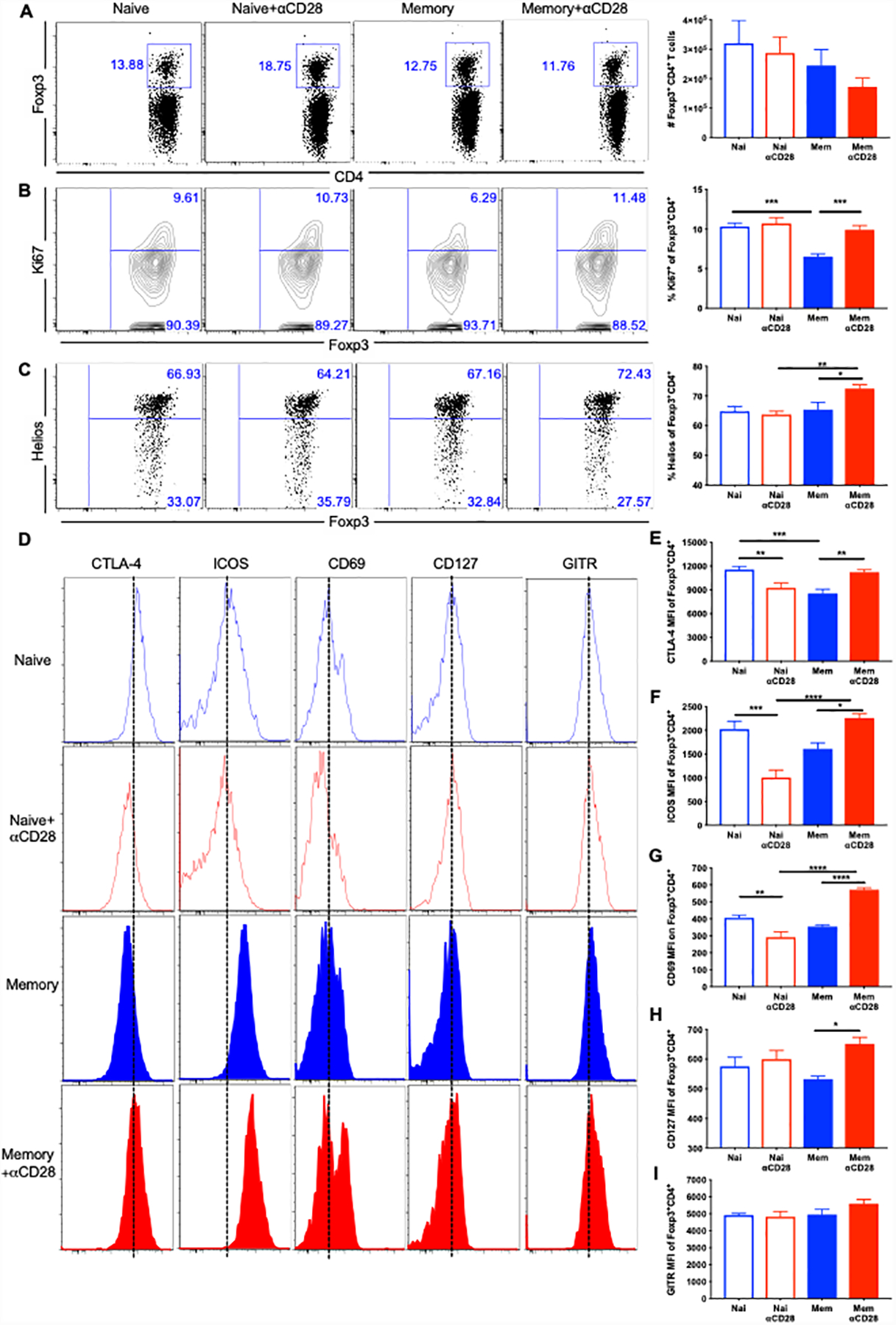
Memory mice and age-matched naïve controls underwent CLP, followed by injection of αCD28 Ab or saline. Mice were sacrificed and spleens were harvested at 24 hours post-CLP. (A) Absolute numbers of Foxp3+ Treg cells among four groups (n=8–9/group). (B) The proliferation of Treg cells was assessed by Ki67 staining. The percentages of Ki67+ Treg cells in the four groups (n=8–9/group) are shown. (C) Representative flow plots and summary data of the percentage of Helios+ in Tregs. (D-I) Flow histograms and summary data of CTLA-4 (E), ICOS (F), CD69(G), CD127 (H) and GITR (I) in memory vs naïve, memory vs memory+αCD28 and naïve vs naïve+αCD28 groups (n=4–9/group). The data were pooled from two independent experiments. Data represented as mean ± SEM and analyzed with one-way ANOVA analysis and Turkey multiple comparison test. *, p<0.05, **, p<0.01, ***, p<0.001 and****, p<0.0001.
We next sought to characterize the effect of αCD28Ab on the activation and differentiation of splenic Treg cells in memory and naïve septic mice. Helios has been proposed as a marker to distinguish thymus-derived Treg cells from peripherally induced ones, which indicated more stable and functional Tregs (24). While αCD28Ab failed to impact Helios expression in Tregs isolated from naïve septic mice, αCD28Ab resulted in a significant increase in the frequency of Helios+ Tregs in memory septic mice compared with untreated memory controls (Fig. 5C).
Cytotoxic T lymphocyte antigen 4 (CTLA-4) has been reported to be critically required for the function of Treg cells in vivo (25).Our data demonstrated that CTLA-4 expression in Foxp3+ Tregs was upregulated in memory septic mice treated with αCD28Ab (Fig. 5D and 5E). Conversely, CTLA-4 was downregulated in naïve septic mice after treating with αCD28Ab (Fig. 5D and 5E). Inducible T cell co-stimulator (ICOS) is expressed by the majority of mouse Treg cells (26). αCD28Ab resulted in increased ICOS expression on Tregs in memory septic mice, whereas it dramatically reduced ICOS expression on Tregs in naïve septic mice (Fig. 5F). Following a similar pattern, treatment with αCD28Ab resulted in increased CD69 expression on Tregs in memory septic mice, but reduced CD69 expression in naïve septic mice (Fig. 5G). Furthermore, CD127 expression on Treg was increased by αCD28Ab in memory mice but not naïve mice (Fig. 5H). Finally, there was no effect of αCD28Ab on glucocorticoid-induced TNF-related receptor (GITR) expression on Treg isolated from either memory or naïve septic mice (Fig. 5I). These data demonstrate a differential effect of αCD28Ab on Foxp3+ Treg isolated from memory mice as compared to those isolated from naïve mice.
Based on the increased expression of CTLA-4, Helios and ICOS in αCD28Ab-treated memory mice, we queried whether Tregs isolated from αCD28 Ab-treated septic memory mice exhibited increased suppressive function relative to Tregs isolated from isotype control treated septic memory mice. To assess this, Tregs were sorted from spleens of αCD28Ab vs. isotype-treated septic memory mice, and naive CD4+ conventional T cells (Tconv) were used as responder cells (Figure 6A). As shown in Figure 6, results indicated that Tregs isolated from αCD28Ab-treated septic memory mice were significantly more effective at inhibiting Tconv proliferation than those isolated from isotype-treated animals at both ratios of 1:8 (40.6±3.6% vs. 26.3±5.7% inhibition, p=0.041, Figure 6B–6C). and 1:4 (59.5±1.6% vs. 45.7±4.4% inhibition, p=0.002, Figure 6B,D). Additionally, supernatants from co-cultures containing Treg isolated from αCD28Ab-treated memory mice contained significantly more IL-10 relative to those containing Treg isolated from isotype control treated mice (p=0.0087, Figure 6E). Collectively, these data show that treatment of septic memory mice with αCD28Ab results in increased suppressive capacity of Foxp3+ Tregs. Taken together, these data indicate that αCD28Ab inhibits the functionality and activation of Tregs in naïve animals following CLP, but in contrast enhances the proliferation, functionality, and activation of Tregs in memory mice during sepsis.
Figure 6. Treg cells from αCD28-treated memory mice exhibit increased suppressive capacity.
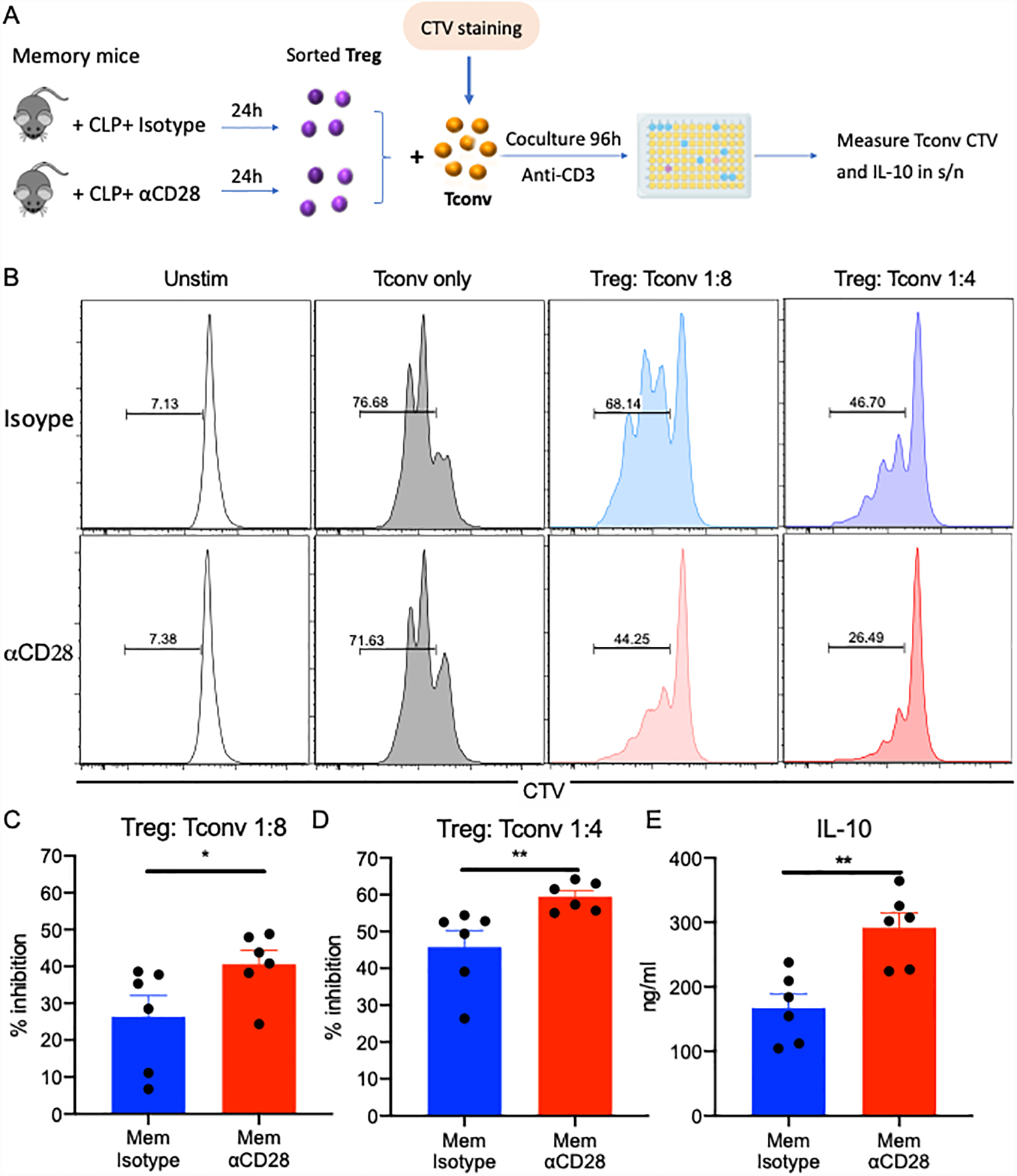
(A) Schematic of in vitro Treg suppression assay. (B) Representative flow histogram of CTV expression of Tconv cells after coculture with Treg cells for 96h. Responder T cells (Tconv) were gated on CD3+ CD4+ Foxp3− and their proliferation was analyzed based on the dilution of CTV dye. (C) Summary data of the suppressive capacity of Tregs (% inhibition) isolated from memory mice treated with isotype control Ab vs.αCD28 Ab when Treg: Tconv ratio was 1:8 (n=6/group). (D) Summary data of the suppressive capacity of Tregs (% inhibition) isolated from memory mice treated with isotype control Ab vs. αCD28 Ab when Treg: Tconv ratio was 1:4 (n=6/group). (E) IL-10 levels in the supernatant of coculture of Treg: Tconv (1:4) were compared between two memory groups. All data were collected from two independent experiments. Two groups were compared with the Mann-Whitney nonparametric test. **, p<0.01. *, p<0.05. Tconv, naïve conventional CD4+ T cells; CTV, Cell Trace Violet.
IL-10 neutralization reverses the protective effect of αCD28 Ab and aggravates inflammation in memory mice during sepsis
Given the findings of increased Treg activation and IL-10 production by Foxp3+ Treg in memory septic mice treated with αCD28 Ab, we next asked if the increased production of IL-10 mechanistically underlies the survival benefit conferred by anti-CD28 in memory septic mice. To do so, memory mice were subjected to CLP and were treated with (or without) αCD28Ab in the presence or absence of an anti-IL-10 neutralizing antibody (Fig. 7A). While anti-IL-10 also diminished sepsis survival in memory mice in the absence of αCD28Ab (data not shown), anti-IL-10 Ab eliminated the survival benefit observed in memory septic mice treated with αCD28 Ab (p=0.021, Fig. 7B).
Figure 7. IL-10 neutralization reverses the protective effect of αCD28 Ab and aggravates inflammation in memory mice during sepsis.

Naïve B6 mice were infected with Listeria monocytogenes and 30 days later infected with LCMV. The mice underwent CLP at day 60 and received αCD28 Ab and anti-IL-10 antibody (n=10) or isotype IgG (n=10) intraperitoneally at set time points. (A) Schematic of experiment design. (B) 7-day survival after CLP induction was analyzed using log-rank (Mantel-Cox) test. (C) Blood samples were collected at 24 hours post-CLP and systemic IL-6 concentration was measured in naïve and memory mice treated with αCD28 Ab and memory mice treated with αCD28 Ab and anti-IL-10 Ab (n=8/group). Results shown as mean ± SEM were pooled from two independent experiments. Statistical analysis was performed using one-way ANOVA analysis and Turkey multiple comparison test. *, p<0.05.
We also evaluated the serum concentration of the pro- and anti-inflammatory cytokines when IL-10 was blocked in memory septic mice. Results indicated that systemic IL-6 levels were significantly increased when IL-10 was neutralized in αCD28Ab-treated memory mice (p<0.05, Fig. 7C). Meanwhile, serum levels of TNF, IL-1β, IFN-γ, MCP-1 IL-2 and IL-13 were not different among three groups (data not shown). In sum, IL-10 blockade reversed the survival benefit of αCD28Ab in memory septic animals, and also exacerbated the IL-6 inflammatory response in memory septic mice.
IL-10 blockade induces downregulation of Bcl-xL and increased T cell apoptosis in memory mice treated with αCD28 Ab
Because these experiments suggested that survival benefit was reversed after blockade of IL-10 in memory mice with αCD28Ab, and results shown in Fig. 2 showed that αCD28 Ab ameliorated the apoptosis of memory T cells in memory septic mice, we next inquired whether the reduced T cell apoptosis observed in memory septic mice treated with αCD28Ab was dependent on IL-10. Memory and naïve mice were subjected to CLP, followed by treatment with either αCD28 Ab or αCD28Ab+anti-IL-10. Results indicated that total CD4+ T cells and CD8+ T cell populations contained significantly more apoptotic (Annexin V+7-AAD+) cells in memory mice treated with αCD28Ab+anti-IL-10 relative to those treated with αCD28Ab alone (Fig. 8A and 8B, CD4: 8.4±0.7% vs.5.8±0.5%, p=0.01; Fig. 8A and 8C, CD8: 7.0±1.4% vs.2.1±0.3%, p=0.0038).
Figure 8. IL-10 blockade induces downregulation of Bcl-xL and increased T cell apoptosis in memory mice treated with αCD28 Ab.
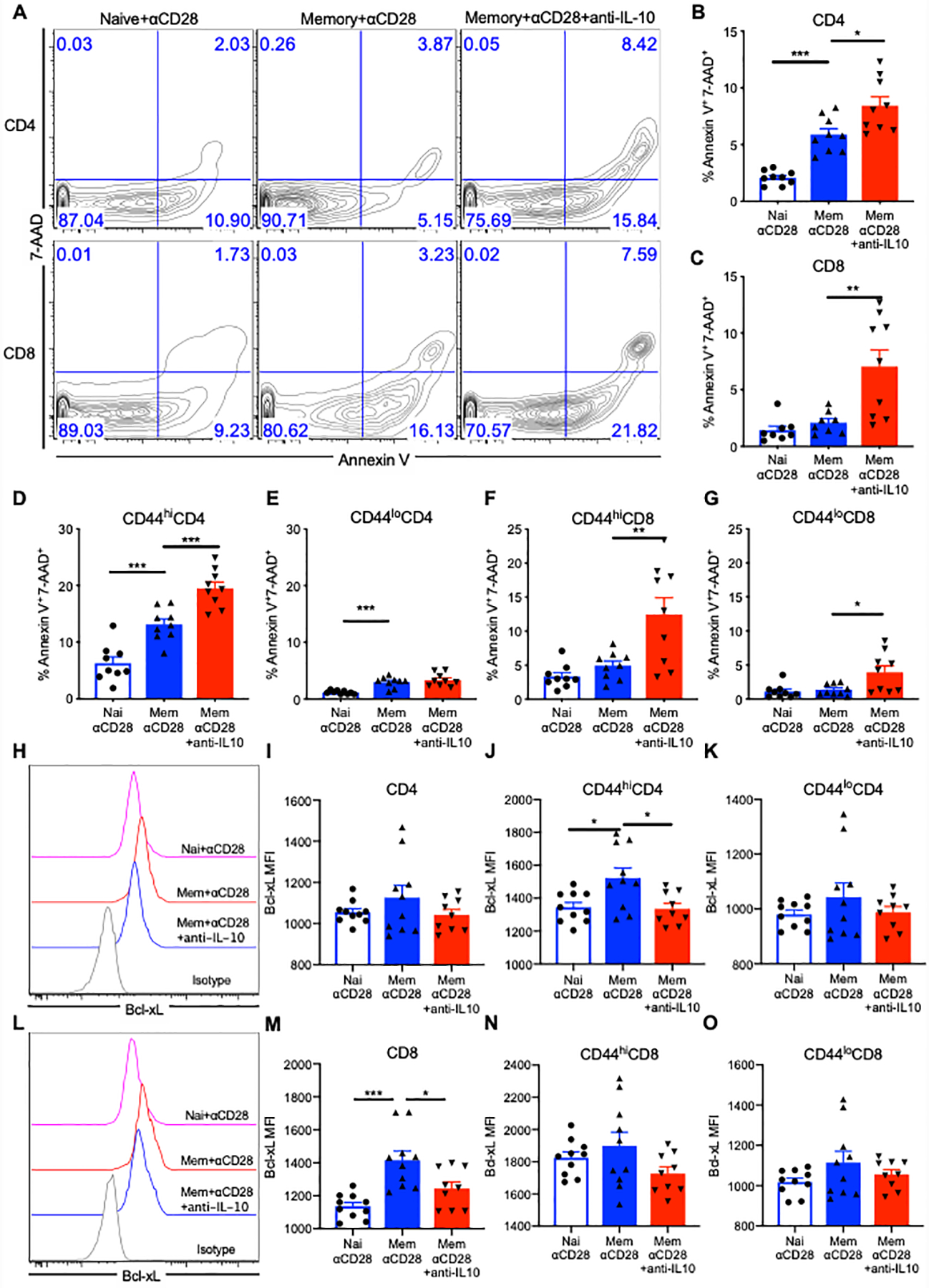
Memory and naïve mice received CLP and αCD28 Ab followed by either anti-IL-10 Ab or isotype IgG Ab. Splenocytes were stained for Annexin V and 7-AAD for apoptosis analysis of T cells and intracellularly stained with anti-Bcl-xL Ab among naïve mice with αCD28 Ab (n=8) vs. memory mice treated with αCD28 Ab (n=9) vs. memory mice treated with αCD28 Ab and anti-IL-10 Ab (n=9) at 24 hours after CLP. (A) Representative flow cytograms indicating the apoptosis of CD4+ and CD8+ T cells among three groups. (B) Summary of percentage of apoptotic CD4+ T cells. (C) Summary of percentage of apoptotic CD8+ T cells. (D and E) Data depicting the frequency of apoptotic memory (CD44hi) and naïve (CD44lo) CD4+ T cells. (F and G) Data depicting the frequency of apoptotic CD44hiCD8+ and CD44loCD8+ T cells. (H and I) Representative flow cytograms and summary data indicating Bcl-xL expression in CD4+ T cells among naïve + αCD28 vs memory + αCD28 vs memory + αCD28 + anti-IL-10 groups. (J and K) Summary of Bcl-xL MFI in CD44hiCD4+ and CD44loCD4+ T cells. (L and M) Representative flow cytograms and summary data indicating Bcl-xL expression in CD4+ T cells among three groups. (N and O) Summary of Bcl-xL MFI in CD8+, CD44hiCD8+ and CD44loCD8+ T cells. The results depict two independent experiments. Data represented mean ± SEM. Groups were compared with one-way ANOVA analysis and Turkey multiple comparison test. *, p<0.05, **, p<0.01 and ***, p<0.001.
Furthermore, IL-10 neutralization resulted in a significant increase in the frequency of apoptotic cells among CD44hiCD4+ memory T cells (Fig. 8D, 19.5±1.1% vs.13.1±0.9%, p=0.0007), but not among CD44loCD4+ naïve T cells (Fig. 8E). For CD8 T cells, both CD44hiCD8+ memory T cell (12.4±2.4% vs.4.9±0.6%, p=0.0047) and CD44loCD8+ naïve T cell populations (3.9±0.9% vs.1.3±0.2%, p=0.019) possessed higher frequencies of apoptotic cells in mice treated with αCD28Ab+anti-IL-10 group vs. αCD28Ab alone (Fig. 8F and 8G). Taken together, these data suggest that the increased IL-10 secreted by Foxp3+ Treg in αCD28Ab-treated memory septic animals is inhibiting apoptosis of memory CD4+ and CD8+ T cells in this model, in that apoptosis of memory CD4+ and CD8+ T cells is increased when IL-10 is neutralized.
Given the above findings, we next sought to identify the underlying mechanism of the increased T cell apoptosis when IL-10 was blocked. Two different pathways of T cell apoptosis were assessed: mitochondrial (Bcl-xL and Bcl-2) and death receptor-mediated (Fas and TNFR). Results indicated that there were no differences in Bcl-xL expression in total CD4+ T cells among any of the groups (Fig. 7H and 7I). Consistent with the findings presented above, the Bcl-xL expression in CD44hiCD4+ T cells was decreased in memory mice treated with αCD28Ab+IL-10 blockade vs. those treated with αCD28Ab alone (p=0.015, Fig. 7J). No differences in Bcl-xL expressed were observed among CD44loCD4+ cells in these groups (Fig. 7K). Total CD8+ T cells isolated from the αCD28Ab+anti-IL-10 memory group exhibited decreased Bcl-xL expression compared with CD8+ T cells isolated from αCD28Ab-treated memory mice (p=0.02, Fig. 7L and 7M). There were no differences of Bcl-xL expression in either CD44hiCD8+ or CD44loCD8+ T cell subsets among three groups (Fig. 7N and 7O).
In contrast to the observed effects of IL-10 blockade on Bcl-xL expression on CD44hiCD4+ memory T cells in memory mice treated with anti-CD28, blockade of IL-10 had no impact on Bcl-2 expression in either the CD4+ or CD8+ memory or naïve T cell compartments (Supplemental Fig. S4). Similarly, blockade of IL-10 did not alter Fas or TNFR1 expression on either CD4+ or CD8+ T cells or memory T cell subsets in septic memory mice (Supplemental Fig. S4). Overall, these data provide strong evidence that the administration of αCD28Ab may function to inhibit mitochondrial-mediated apoptosis by inducing an IL-10-dependent increase in Bcl-xL in CD44hi memory CD4+ T cells, which contributes to improved survival in memory mice during sepsis.
DISCUSSION
Despite decades of research using rodent models of sepsis, no effective therapies to treat sepsis-induced immune dysregulation have emerged from these studies, leading some to question the relevance of animal models for sepsis (27). Here, we hypothesized that one factor underlying the apparent lack of relevance to human sepsis pathophysiology might be the different degrees of immunologic experience between adult humans and SPF-housed laboratory animals. As such, we developed the antigen-experienced memory mouse model via sequential infection of mice with bacterial and viral pathogens to recapitulate the frequencies of memory T cells observed in the adult human immune system (17). In these animals, memory T cells constituted 32% and 53% of the CD4+ and CD8+ T cell compartments, respectively, in the blood. These frequencies are similar to those observed in adult humans. Using this system, we show that the response to CD28 immunomodulation during sepsis is fundamentally different in naïve vs. memory septic animals, in that αCD28Ab conferred a trend towards worsened survival in naïve septic animals but in sharp contrast resulted in significantly improved survival in immunologically experienced septic animals. Mechanistically, this differential response was driven by a unique ability of αCD28Ab to elicit IL-10 production from Treg in memory but not naïve mice. Together, these data demonstrate that relative immunologic experience is an important parameter that affects sepsis pathophysiology and can fundamentally change the outcome of therapeutic targeting of the CD28 pathway during sepsis. This study suggests that testing therapeutic strategies in immunologically experienced hosts may be one way to increase the physiologic relevance of rodent models in sepsis research.
Results presented here add further depth to our understanding of the role of CD28 during sepsis. Our findings that agonistic αCD28Ab resulted in a trend toward worsened mortality are consistent with several studies in mouse models of polymicrobial sepsis showing that hosts with CD28 deficiency (28–30), CD80/86 deficiency (13), or which received a CD28-blocking peptide mimetic (14) exhibited improved mortality. Importantly, however, our data show that CD28 agonism in immunologically experienced hosts had the opposite effect, mediated by potent release of IL-10 from “memory” Tregs. The extent to which these differential results are recapitulated in human physiology remains to be determined. However, it is known that mortality in human septic patients is associated with significantly reduced CD28 expression on splenic T cells (9, 10), and another study found that levels of soluble CD28, potentially indicative of shedding from surface of T cells, was higher in patients who died than who lived (13). Taken together with our results, these findings suggest that preserved CD28 signaling may be critical for the preservation of immune homeostasis in immunologically experienced septic hosts, be they mouse or human.
Over the last few years, several groups have begun investigating the effect of antigen or pathogen exposure prior to sepsis on the pathophysiology and outcome of sepsis, spurred by seminal studies showing that previous exposure to multiple microbial infections fundamentally improves the response to a subsequent infection, resulting in improved bacterial/viral clearance and increased effector T cell differentiation (17). Our previous report showed that despite the fact that memory mice exhibited increased T cell apoptosis compared with naïve hosts, there was no difference in survival between memory and naïve mice following sepsis (in the absence of any immune modulation (19). One study showed that mice treated with anti-CD3 activating Ab contained increased frequencies of memory T cells, and that these immunologically experienced mice exhibited improved survival of sepsis accompanied by increased bacterial clearance compared to naïve mice (31). Other groups have used pet store mice cohoused with SPF B6 mice to generate a “dirty” mice model to generate more memory T cells before sepsis (32, 33). Badovinac, Griffiths and colleagues showed that cohousing mice with pet-store derived “dirty mice” increased the sepsis-induced cytokine storm and mortality (33). We postulate that chronic, persistent viral infections might also be communicated in the “dirty” mice model and reactivate following sepsis, which themselves could contribute to immunopathology and outcome of sepsis independently of immunologic memory. Because the model used in the current study utilizes acute bacterial and viral infections are cleared well before CLP, the effects we observed can be attributed to the existence of immunologic memory and not latent or chronic infection.
Our results reveal a potent effect of αCD28Ab on Treg function in memory mice. It has previously been reported that CD28 controls both Treg proliferation and survival via the induction of several cell-intrinsic survival factors such as anti-apoptotic Bcl-xL (34). Here, we report a differential effect of αCD28Ab on Tregs in memory vs. naïve septic mice: αCD28Ab diminished the activation of Treg in naïve animals, but increased the proliferation, Helios expression, and activation of Treg in memory animals. The fact that increased Treg activity in αCD28Ab-treated memory septic mice is associated with increased sepsis survival is in line with previously published reports showing that Treg are beneficial during the early, hyper-inflammatory phase of sepsis (35, 36). Furthermore, these data suggest a fundamental difference in the response of memory vs. naïve Treg to modulation of CD28 signaling in the context of sepsis. This concept of “memory Treg” cells, which have responded to antigen and are capable of surviving for fairly long periods even in the apparent absence of antigen, has recently been reported in the literature. Memory Tregs can expand rapidly and mediate potent immune suppression upon secondary activation (37–39). A challenge in defining memory Treg cells has been a lack of specific phenotypic markers in human and mouse. Thus, further investigation is needed to dissect the role of memory Tregs during sepsis. However, the results presented here support the intriguing hypothesis that similar to the differential requirements for CD28 costimulation between naïve and memory Tconv (40, 41), CD28 signaling may likewise differentially impact naïve and memory Treg.
Further, our results revealed that Tregs activated by αCD28 Ab in memory septic mice released more IL-10, which inhibited mitochondrial-mediated apoptosis of T cells, leading to improved survival accompanied by decreased inflammatory responses in memory septic mice. Controversy exists concerning the role of IL-10 in sepsis: when IL-10 was used in models of endotoxemia, it improved survival (42), potentially via its anti-inflammatory properties, while in models of polymicrobial sepsis (CLP) worsened survival, potentially via immunosuppression (43). Similarly, studies of anti-IL-10 in CLP in previously naïve animals resulted in improved survival (43). In contrast, our data show that anti-IL-10 (both in the presence and absence of αCD28Ab) significantly reduced morality in memory septic mice. We speculate that the differential results could be the result of a heightened inflammatory response in memory mice vs. heightened immunosuppression in previously naïve mice during sepsis. Indeed, in human septic patients, high IL-10 in the circulation and a high ratio of IL-10 to TNF was associated with increased mortality (44). Whether this increase in IL-10 is a cause of human sepsis mortality or an effect of massive inflammation (that actually underlies mortality) remains an unanswered question.
In sum, the data presented here reveal the potent therapeutic effect of αCD28Ab on sepsis in a physiologically relevant immunologically experienced mouse model. Notably, the results indicate that activated Treg in memory mice are associated with decreased T cell apoptosis and increased survival during sepsis. The role of CD28 signaling on antigen-experienced Tregs warrants further investigation to explore its application to clinical sepsis in humans, in that targeting CD28 signaling on this subset could provide a potential immunomodulatory therapeutic strategy to improve sepsis survival in the clinic.
Supplementary Material
Key Points:
Agonism of CD28 improves sepsis mortality in memory but not naïve hosts
CD28 agonism results in enhanced production of IL-10 from Foxp3+Treg in memory mice
IL-10 is required for the survival benefit of CD28 agonism in septic memory mice
Acknowledgements
The authors would like to thank Dr. Jennifer Robertson (Emory Transplant Center) for flow cytometry assistance and Drs. Rafi Ahmed and Koichi Araki (Emory University) for providing LCMV.
This work was funded by R01s GM113228, GM104323, GM072808, AA027396, and AI129724 to MLF and CMC.
Footnotes
Disclosures
The authors have declared no conflicts of interest.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, and Angus DC. 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daniels R 2011. Surviving the first hours in sepsis: getting the basics right (an intensivist’s perspective). J Antimicrob Chemother 66 Suppl 2: ii11–23. [DOI] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Monneret G, and Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poirier N, Blancho G, and Vanhove B. 2012. CD28-specific immunomodulating antibodies: what can be learned from experimental models? Am J Transplant 12: 1682–1690. [DOI] [PubMed] [Google Scholar]
- 5.Godlove Jason, C. WK, and Weng Nan-ping. 2007. Gene expression and generation of CD28-cd8 T cells mediated by IL-15. Exp Gerontol 42: 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salazar-Fontana LI, B. V, Samelson LE, Bierer BE. 2003. CD28 engagement promotes actin polymerization through the activation of the small Rho GTPase Cdc42 in human T cells. J Immunol 171: 2225–2232. [DOI] [PubMed] [Google Scholar]
- 7.Shimizu Y, v. SG, Ennis E, Newman W, Horgan KJ, Shaw S. 1992. Crosslinking of the T cell- specific accessory molecules CD7 and CD28 modulates T cell adhesion. The Journal of Experimental Medicine 175: 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turcovski-Corrales SM, F. R, Peltz G, Taub DD. 1995. CD28:B7 interactions promote T cell adhesion. Eur J Immunol 25: 3087–3093. [DOI] [PubMed] [Google Scholar]
- 9.Inoue S, Suzuki-Utsunomiya K, Okada Y, Taira T, Iida Y, Miura N, Tsuji T, Yamagiwa T, Morita S, Chiba T, Sato T, and Inokuchi S. 2013. Reduction of immunocompetent T cells followed by prolonged lymphopenia in severe sepsis in the elderly. Crit Care Med 41: 810–819. [DOI] [PubMed] [Google Scholar]
- 10.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, and Hotchkiss RS. 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306: 2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borken F, Markwart R, Requardt RP, Schubert K, Spacek M, Verner M, Ruckriem S, Scherag A, Oehmichen F, Brunkhorst FM, and Rubio I. 2017. Chronic Critical Illness from Sepsis Is Associated with an Enhanced TCR Response. J Immunol 198: 4781–4791. [DOI] [PubMed] [Google Scholar]
- 12.Mirenda V, Jarmin SJ, David R, Dyson J, Scott D, Gu Y, Lechler RI, Okkenhaug K, and Marelli-Berg FM. 2007. Physiologic and aberrant regulation of memory T-cell trafficking by the costimulatory molecule CD28. Blood 109: 2968–2977. [DOI] [PubMed] [Google Scholar]
- 13.Nolan A, Weiden M, Kelly A, Hoshino Y, Hoshino S, Mehta N, and Gold J. 2008. CD40 and CD80/86 Act Synergistically to Regulate Inflammation and Mortality in Polymicrobial Sepsis. Am J Respir Crit Care Med 177: 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramachandran G, Tulapurkar ME, Harris KM, Arad G, Shirvan A, Shemesh R, Detolla LJ, Benazzi C, Opal SM, Kaempfer R, and Cross AS. 2013. A peptide antagonist of CD28 signaling attenuates toxic shock and necrotizing soft-tissue infection induced by Streptococcus pyogenes. J Infect Dis 207: 1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramachandran G, Kaempfer R, Chung CS, Shirvan A, Chahin AB, Palardy JE, Parejo NA, Chen Y, Whitford M, Arad G, Hillman D, Shemesh R, Blackwelder W, Ayala A, Cross AS, and Opal SM. 2015. CD28 homodimer interface mimetic peptide acts as a preventive and therapeutic agent in models of severe bacterial sepsis and gram-negative bacterial peritonitis. J Infect Dis 211: 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, and Panoskaltsis N. 2006. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355: 1018–1028. [DOI] [PubMed] [Google Scholar]
- 17.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, and Masopust D. 2016. Recapitulating adult human immune traits in laboratory mice by normalizing environment. Nature 532: 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abolins S, King EC, Lazarou L, Weldon L, Hughes L, Drescher P, Raynes JG, Hafalla JCR, Viney ME, and Riley EM. 2017. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat Commun 8: 14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie J, Chen CW, Sun Y, Laurie SJ, Zhang W, Otani S, Martin GS, Coopersmith CM, and Ford ML. 2019. Increased attrition of memory T cells during sepsis requires 2B4. JCI insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Effros RB 1997. Loss of CD28 expression on T lymphocytes: A marker of replicative senescence. Dev Immunol 21: 471–478. [DOI] [PubMed] [Google Scholar]
- 21.O’Garra A, and Vieira P. 2007. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol 7: 425–428. [DOI] [PubMed] [Google Scholar]
- 22.Fillatreau S, Gray D, and Anderton SM. 2008. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol 8: 391–397. [DOI] [PubMed] [Google Scholar]
- 23.Kasten KR, Muenzer JT, and Caldwell CC. 2010. Neutrophils are significant producers of IL-10 during sepsis. Biochem Biophys Res Commun 393: 28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bin Dhuban K, d’Hennezel E, Nashi E, Bar-Or A, Rieder S, Shevach EM, Nagata S, and Piccirillo CA. 2015. Coexpression of TIGIT and FCRL3 identifies Helios+ human memory regulatory T cells. J Immunol 194: 3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wing K, Onishi Y, Martin PP, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S. 2008. CTLA-4 Control over Foxp3+ Regulatory T cell Function. Science 322: 271–275. [DOI] [PubMed] [Google Scholar]
- 26.Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, Kroczek RA, and Hutloff A. 2008. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol 180: 774–782. [DOI] [PubMed] [Google Scholar]
- 27.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, and Tompkins RG. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albertsmeier M, Prix NJ, Winter H, Bazhin A, Werner J, and Angele MK. 2017. Monocyte-Dependent Suppression of T-Cell Function in Postoperative Patients and Abdominal Sepsis. Shock 48: 651–656. [DOI] [PubMed] [Google Scholar]
- 29.Serbanescu MA, Ramonell KM, Hadley A, Margoles LM, Mittal R, Lyons JD, Liang Z, Coopersmith CM, Ford ML, and McConnell KW. 2016. Attrition of memory CD8 T cells during sepsis requires LFA-1. J Leukoc Biol 100: 1167–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singbartl K, Bockhorn SG, Zarbock A, Schmolke M, and Van Aken H. 2005. T cells modulate neutrophil-dependent acute renal failure during endotoxemia: critical role for CD28. J Am Soc Nephrol 16: 720–728. [DOI] [PubMed] [Google Scholar]
- 31.Taylor Matthew D. , C. S. D. M. N. A. a. 2018. Induction of nonsepcific memory T cells alters the immune response to cecal ligation and puncture (CLP). shock 49(S1): 132. [Google Scholar]
- 32.Guo Fei, N. Y, Wada Takeshi, Nguyen Jennifer, Cahill Laura, Lee Sue Chin, Norman Derek, Tigyi Gabor and Lederer James. 2018. Comparing the phenotype betwen naturally-colonized “dirty mice” and laboratory “clean” mice by a CYTOF-based systems immunology approach. SHOCK 149(S1): 128. [Google Scholar]
- 33.Griffith Thomas S., S. FV, Pierson Mark, Huggins Matthew A., Danahy Derek B., Badovinac Vladimir P. and Hamilton Sara E.. 2018. Physiology microbial exposure substantially influences de novo inflammatory responses in vivo. SHOCK 149(S1): 122. [Google Scholar]
- 34.Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, and Bluestone JA. 2003. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol 171: 3348–3352. [DOI] [PubMed] [Google Scholar]
- 35.Scumpia PO, Delano MJ, Kelly KM, O’Malley KA, Efron PA, McAuliffe PF, Brusko T, Ungaro R, Barker T, Wynn JL, Atkinson MA, Reeves WH, Clare Salzler MJ, and Moldawer LL. 2006. Increased Natural CD4+CD25+ Regulatory T Cells and Their Suppressor Activity Do Not Contribute to Mortality in Murine Polymicrobial Sepsis. The Journal of Immunology 177: 7943–7949. [DOI] [PubMed] [Google Scholar]
- 36.Okeke EB, Okwor I, Mou Z, Jia P, and Uzonna JE. 2013. CD4+CD25+ regulatory T cells attenuate lipopolysaccharide-induced systemic inflammatory responses and promotes survival in murine Escherichia coli infection. Shock 40: 65–73. [DOI] [PubMed] [Google Scholar]
- 37.Rosenblum MD, Way SS, and Abbas AK. 2015. Regulatory T cell memory. Nature Reviews Immunology 16: 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez AM, Zhu J, Huang X, and Yang Y. 2012. The development and function of memory regulatory T cells after acute viral infections. J Immunol 189: 2805–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brincks EL, Roberts AD, Cookenham T, Sell S, Kohlmeier JE, Blackman MA, and Woodland DL. 2013. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J Immunol 190: 3438–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bachmann MF, G. A, Linkert S, Cerundolo V, Lanzavecchia A, Kopf M, and Viola A. 1999. Developmental Regulation of Lck Targeting to the CD8 Coreceptor Controls Signaling in Naive and Memory T Cells. J Exp Med 163: 4125–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sprent J, and Surh CD. 2002. T cell memory. Annu Rev Immunol 20: 551–579. [DOI] [PubMed] [Google Scholar]
- 42.Wang R, Fang Q, Zhang L, Radvany L, Sharma A, Noben-Trauth N, Mills GB, and Shi Y. 1997. CD28 ligation prevents bacterial toxin-induced septic shock in mice by inducing IL-10 expression. J Immunol 158: 2856–2861. [PubMed] [Google Scholar]
- 43.Song GY, Chung CS, Schwacha MG, Jarrar D, Chaudry IH, and Ayala A. 1999. Splenic immune suppression in sepsis: A role for IL-10-induced changes in P38 MAPK signaling. J Surg Res 83: 36–43. [DOI] [PubMed] [Google Scholar]
- 44.van Dissel JT, van Langevelde P, Westendorp RG, Kwappenberg K, and Frolich M. 1998. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet 351: 950–953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.