

Abstract
Background & Aims:
Countries endemic for parasitic infestations have a lower incidence of Crohn’s disease (CD) than non-endemic countries, and there have been anecdotal reports of the beneficial effects of helminths in CD patients. Tuft cells in the small intestine sense and direct the immune response against eukaryotic parasites. We investigated the activities of tuft cells in patients with CD and mouse models of intestinal inflammation.
Methods:
We used microscopy to quantify tuft cells in intestinal specimens from patients with ileal CD (n = 19), healthy individuals (n = 14), and TNFΔARE/+ mice, which develop Crohn’s-like ileitis. We performed single-cell RNA-sequencing, mass spectrometry, and microbiome profiling of intestinal tissues from wildtype and ATOH1-knockout mice, which have expansion of tuft cells, to study interactions between microbes and tuft cell populations. We assessed microbe dependence on these populations using microbiome depletion, organoids, and microbe transplant experiments. We used multiplex imaging and cytokine assays to assess alterations in inflammatory response following expansion of tuft cells with succinate administration in TNFΔARE/+ and anti-CD3E CD mouse models.
Results:
Inflamed ileal tissues from patients and mice had reduced numbers of tuft cells, compared with healthy individuals or wild-type mice. Expansion of tuft cells was associated with increased expression of genes that regulate the tricarboxylic acid cycle, which resulted from microbe production of the succinate. Experiments in which we manipulated the intestinal microbiota of mice revealed the existence of an ATOH1-independent population of tuft cells that was sensitive to metabolites produced by microbes. Administration of succinate to mice expanded tuft cells and reduced intestinal inflammation in TNFΔARE/+ mice and anti-CD3E- treated mice, increased GATA3+ cells and type 2 cytokines (IL22, IL25, IL13), and decreased RORGT+ cells and type 17 cytokines (IL23) in a tuft cell-dependent manner.
Conclusions:
We found that tuft cell expansion reduced chronic intestinal inflammation in mice. Strategies to support tuft cells might be developed for treatment of CD.
Keywords: IBD, heterogeneity, epithelium, metabolism
Graphical Abstract
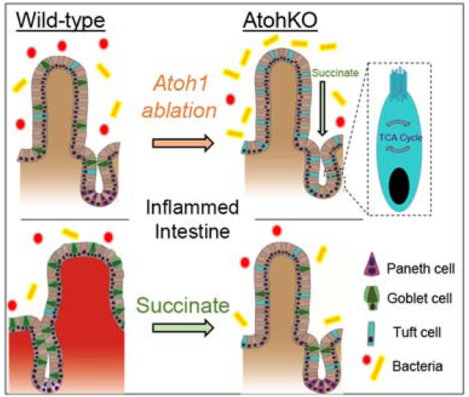
Introduction
Global incidences of communicable disease are inversely correlated with rates of Inflammatory Bowel Disease (IBD)1. Part of the “hygiene hypothesis,” this phenomenon is thought to result from improved hygiene practices associated with decreased tolerance to environmental antigens, such as those from the commensal microbiome2. This paradoxical effect has led to emerging interest in the use of helminths, or parasitic worms, for the treatment of IBD3. Enhanced anti-parasite immune responses characterized by type 2 cytokines, such as IL-25 and IL-13, have been shown to suppress T-helper (Th)1 and Th17 activities4,5. However, clinical trial data from IBD patients have been inconclusive, with many discontinued due to lack of efficacy3. Moreover, helminth therapy has its drawbacks given that prolonged infection causes complications3. Precision therapies using intermediary products may circumvent the majority of these issues.
In acute mouse models of eukaryotic infection, small intestinal tuft cells were found to sense the parasites and orchestrate the anti-parasite response6–8. Through the release of IL-25, tuft cells promote their own specification through a positive feedback loop mediated by type 2 immune cells, and tuft cell expansion is critical for parasite clearance. While these studies considered tuft cells as a single population, recent work has revealed substantial heterogeneity within the tuft cell lineage9, which led us to hypothesize that a distinct subpopulation of tuft cells is responsive to changes in the intestinal luminal environment. We aim to investigate heterogeneity of tuft cell function with regards to their origins of specification, and the role of these cell populations in intestinal inflammation. Initial studies identified a secretory cell route of intestinal tuft cell specification, along with barrier-promoting goblet and Paneth cells, under the regulation of the master secretory transcription factor ATOH110. More recent studies have demonstrated that small intestinal tuft cells may have an alternative lineage specification route independent of the ATOH1-controlled secretory lineage11,12. Here, we demonstrate that ATOH1-independent tuft cells expand upon luminal microbiome perturbation through a metabolic communication network, and this mechanism can be leveraged to suppress inflammation and restore epithelial architecture in small intestinal CD.
Methods
All human and animal studies were approved by the Vanderbilt IRB and IACUC, respectively, in accordance with NIH guidelines.
Results
Reduced tuft cell numbers are correlated with localized inflammation in the ileum
Eukaryotic parasites largely colonize the small intestine and up to 60% of CD patients have terminal ileal involvement. Thus, we assessed the correlation between intestinal tuft cell numbers and local tissue inflammation in ileal specimens from CD patients (n = 14), compared to normal ileal specimens (n = 11) from patients without a CD diagnosis (Supplementary Figure 1A). Because the canonical mouse tuft cell gene DCLK1 is not expressed in human TRPM5+ tuft cells (Supplementary Figure 1B), we used a previously published strategy for tuft cell identification in both human and mouse using pEGFR(Y1068) and COX2 co-expression11,13. Double-positive cells in the normal human ileal epithelium were distinct from single-positive pEGFR or COX2 cells in the lamina propria, and they possessed prominent pEGFR-positive apical “tufts” indicative of genuine tuft cells (Figure 1A). Compared to health controls, CD specimens had characteristic, heterogeneous inflammation, with areas of severe villus blunting, increased MUC2+ granules, decreased LYZ+ Paneth cells with diffuse LYZ granule staining, and increased LYZ+ lamina propria immune cells (Supplementary Figure 1C–F), consistent with previous reports14 In parallel, pEGFR+ COX2+ tuft cells were significantly reduced in CD ileal specimens (Figure 1B–C). In regions with less disease involvement and more organized tissue architecture in CD specimens, tuft cells could still be detected (Figure 1B - right). We speculate that suppression of tuft cell specification may contribute to the loss of inflammation control in CD and thus is associated with disease development and/or progression.
Figure 1. Tuft cell number is decreased in inflamed tissue with ileal inflammatory disease.
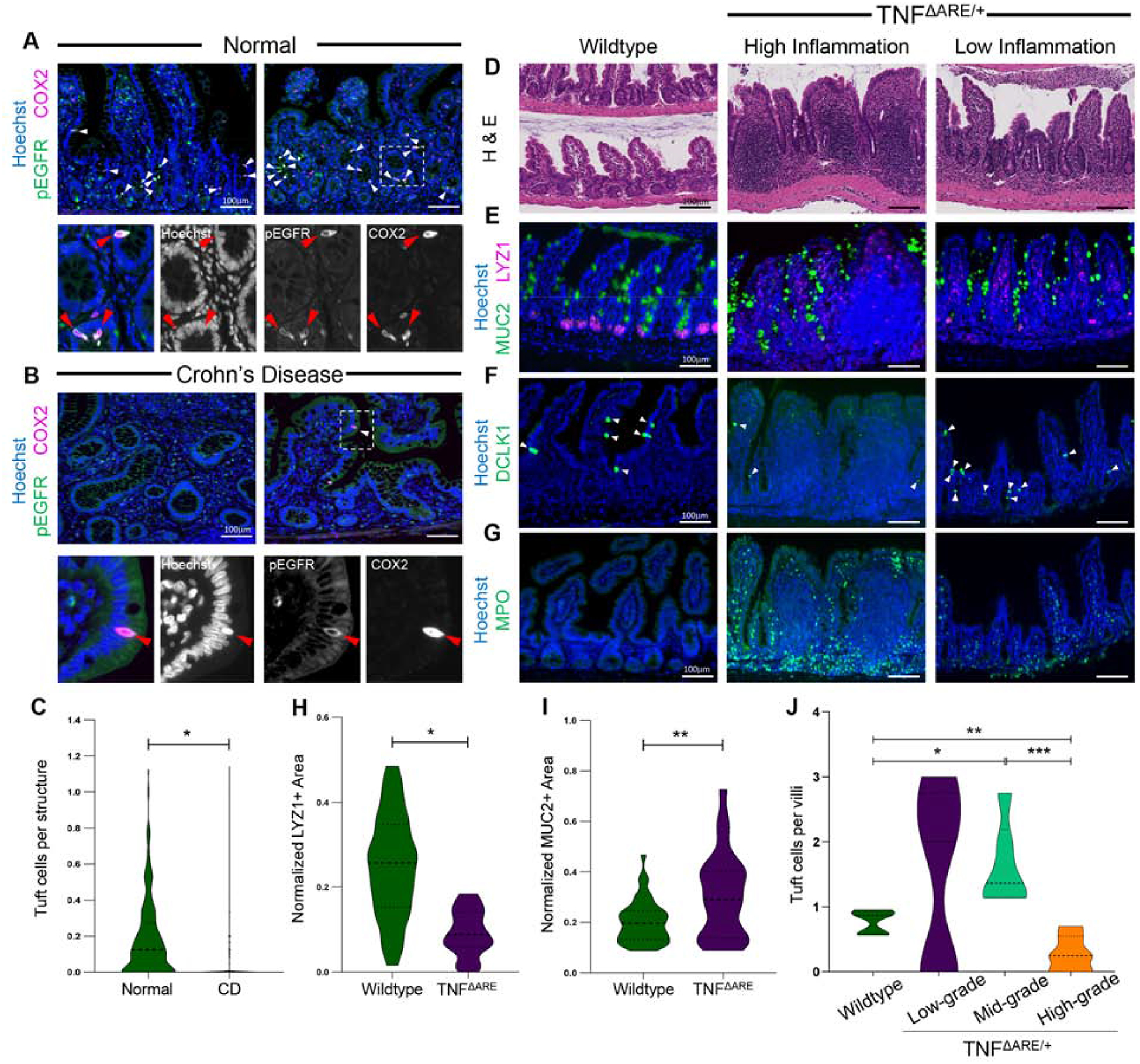
IF of tuft cell staining in human ilea of (A) healthy and (B) CD patients. Arrows denote co-expression. (C) IF quantification for tuft cells per crypt/villus. SEM for n = 11 normal and n = 14 CD patients. H&E (D) and (E-G) IF of cell markers of inflamed and uninflamed distal ilea from mice. (H-I) IF quantification for marker area normalized to Hoechst area per crypt. SEM for n = 4 mice for LYZ and 3 for MUC2. (J) IF quantification of DCLK1+ tuft cells in wildtype (n = 146 villi) and TNFΔARE/+ villi, by low- (n = 11), mid- (n = 125), and high-grade (n = 62) inflammation. p-value * < 0.05, ** < 0.01, *** < 0.001.
We then assessed tuft cells in the TNFΔARE/+ mouse model, which develops Crohn’s-like ileitis due to TNF-α overexpression15. In the terminal ilea of four-month-old animals, we observed distorted crypt structure and blunted villi, decreased number of LYZ1+ Paneth cells, increased MUC2+ granule staining, and increased myeloperoxidase (MPO)+ neutrophils infiltration into the lamina propria (Figure 1D–I; Supplementary Figure 2A–B), similar to human CD. To address regional heterogeneity of inflammation, we performed spatially-resolved analysis by quantifying DCLK1+ tuft cells in wildtype and TNFΔARE/+ villi, stratifying the latter by low-, mid-, and high-grade inflammation (Supplementary Figure 2C). Compared to wildtype, we observed increased tuft cells in low- and mid-grade inflamed villi, while high-grade villi in TNFΔARE/+ animals were almost completely devoid of tuft cells (Figure 1J). We confirmed this regional heterogeneity by semi-automated image analysis based on infiltrated MPO+ neutrophils number as a surrogate for region-specific inflammation (Supplementary Figure 2D–F). We conclude that tuft cell frequency is inversely correlated with severity of disease, raising the possibility that increasing tuft cell specification may reduce inflammation.
ATOH1-independent tuft cells are an inducible cell population responsive to the commensal microbiota
To potentially leverage tuft cells as a strategy to alleviate intestinal inflammation, we sought to understand how they are specified. Previous studies reported heterogeneous tuft cell populations9,11,12. Our previously published Atoh1 knockout (AtohKO) mouse model (Lrig1CreERT2/+; Atoh1fl/fl) revealed that, while colonic tuft cells are ATOH1-dependent, AtohKO triggered significant small intestinal tuft cell expansion (Supplementary Figure 3A), which is in contrast to their ATOH1-dependence observed in prior studies10. Thus, we focus on ATOH1-independent tuft cells as flexible population that can be induced to expand, presenting a viable target that can be manipulated for the treatment of ileal CD. Consistent with the canonical intestinal differentiation hierarchy16, AtohKO intestines possessed normal crypt-villus architecture but lacked MUC2+ goblet cells and LYZ1+ Paneth cells (Supplementary Figure 3B–C). We observed increased MPO+ neutrophils in the villi of AtohKO animals accompanied by decreased weight gain (Supplementary Figure 3D–E), suggesting increased mucosal interaction with the microbiota due to the loss of barrier-forming secretory cell types. Despite increased neutrophils, overt inflammation, characterized by massive immune cell infiltration and villus blunting, was not observed. These observations led us to investigate whether ATOH1-independent tuft cell specification is driven by extrinsic cues originating from the luminal microbiota.
To investigate the nature of luminal perturbation that results in ATOH1-independent tuft cell expansion in the intestine, we first determined that our mouse colony is devoid of large parasites known to trigger tuft cell expansion7,8 (Supplementary Figure 3F). To assess the necessity of the commensal microbiota in driving tuft cell expansion, we used a previously published antibiotic cocktail to deplete a broad range of gram-positive and gram-negative bacteria17. Microbiome depletion with loss of Atoh1 did not affect Paneth cells, but surprisingly suppressed ATOH1-independent tuft cell specification in a dose-dependent manner (Figure 2A–B; Supplementary Figure 3G). Antibiotic administration reduced MPO+ staining but not weight loss, suggesting the former but not the latter was due to microbiome regulation by barrier-forming cells (Supplementary Figure 3E, H). However, in wildtype mice that possess both ATOH1-dependent and -independent tuft cell populations, microbiome depletion with high dose antibiotics and germ-free housing did not suppress tuft cell specification (Figure 2C–D; Supplementary Figure 3G). We reason that ATOH1-independent tuft cells are sensitive to luminal changes, while coexisting ATOH1-dependent tuft cells are largely insensitive to these changes.
Figure 2. ATOH1-independent tuft cell expansion is microbiome-dependent.
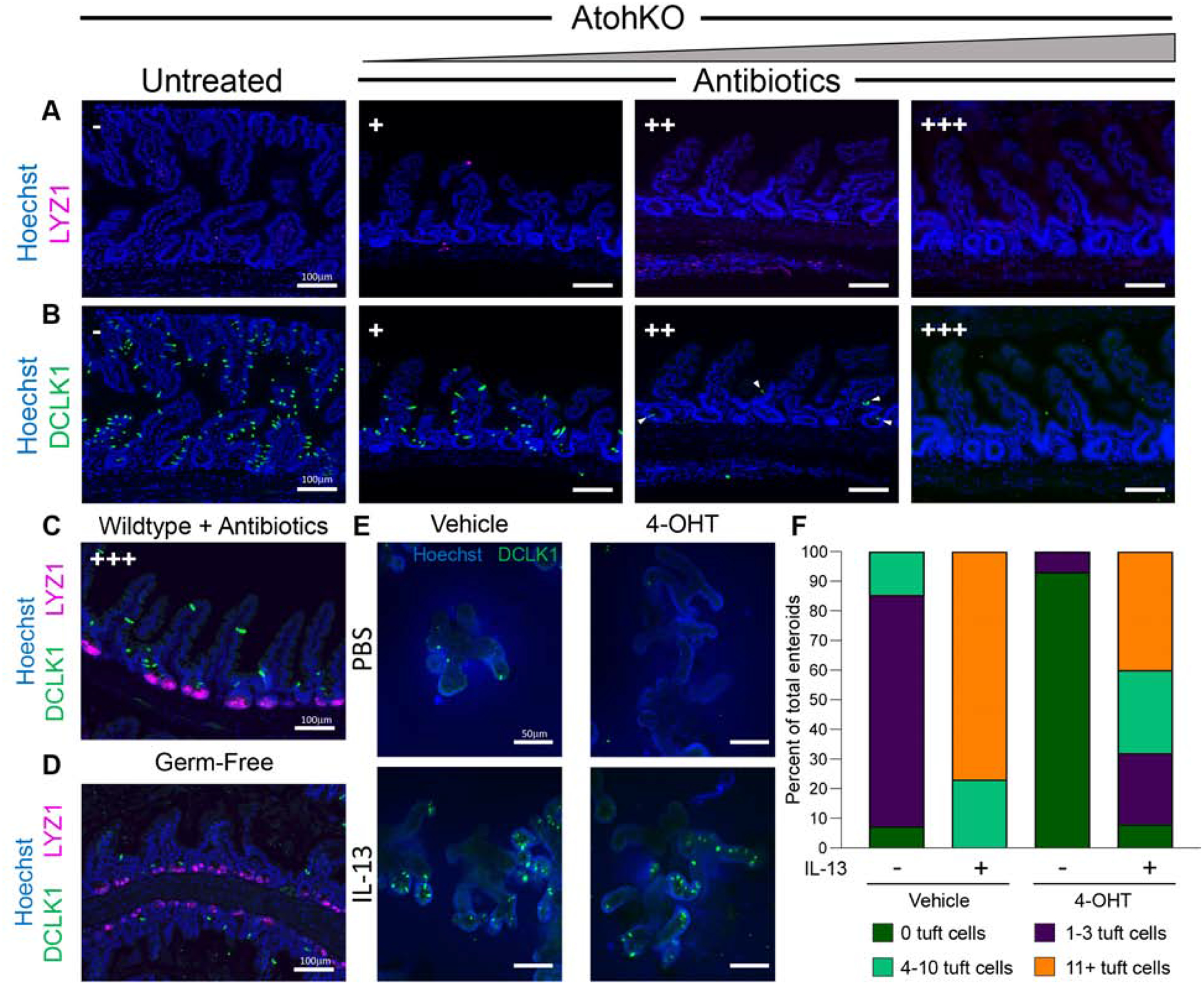
IF of cell markers in (A-B) AtohKO ilea under increasing doses of antibiotics (-, +, ++, +++), and (C-D) wildtype ilea under antibiotics and germ-free conditions. (E) IF of tuft cells in Lrig1CreERT2/+;Atoh1fl/fl small intestinal enteroids with or without 4-OHT to abate Atoh1, and with or without exogenous IL-13. (F) Fraction of enteroids with 0, 1–3, 4–10, 11+ tuft cells under different conditions.
To verify the heterogeneity of tuft cells in a controlled manner, we turned to enteroids18, which are cultured in an environment devoid of microbiota. The sterility of the culturing conditions did not inhibit the specification of tuft cells, consistent with the independence of tuft cell specification and the microbiota observed when ATOH1 is present in vivo. However, tuft cell specification in sterile enteroids was suppressed when Atoh1 loss was induced via Cre recombination (Figure 2E–F), demonstrating that tuft cell specification observed in the control condition was ATOH1-dependent. As expected, loss of Atoh1 led to suppression of Paneth and goblet cell specification (Supplementary Figure 3I–J). To confirm that ATOH1-independent tuft cells respond to luminal perturbations, we perused the literature and identified that the type 2 cytokine interleukin-13 (IL-13) drives tuft cell specification in response to luminal eukaryotic colonization7,8. In wildtype culture, IL-13 stimulated tuft cell specification, beyond the 0–3 number typically observed at baseline (Figure 2E–F). While AtohKO enteroids initially lacked tuft cells, IL-13 administration induced tuft cell specification and expansion in an ATOH1-independent manner, at levels similar to IL-13-treated wildtype enteroids (Figure 2E–F; Supplementary Figure 3K). These results support the existence of ATOH1-independent and - dependent tuft cell populations, with the former being highly malleable to luminal cues.
Trajectory analysis of single-cell RNA-sequencing data supports an ATOH1-independent tuft cell specification pathway
To investigate tuft cell heterogeneity and associated pathways, we generated scRNA-seq data from wildtype control, TNFΔARE/+, antibiotic-treated wild type (ATOH1-dependent tuft cells only), and AtohKO (ATOH1-independent tuft cells only) ileal epithelium, all across multiple biological replicates (Supplementary Figure 4A–D). t-SNE and clustering analyses performed on each of the combined datasets demonstrated that all clusters were represented in all cell replicates (Supplementary Figure 4E–L, 5). Stem and progenitor cells, enterocytes, tuft cells, goblet cells, Paneth cells, and enteroendocrine cells (or the lack thereof in AtohKO) were present in the correct proportions in all conditions (Figure 3A–D; Supplementary Figure 4I–L). The exception here was the TNFΔARE/+ datasets due to the admixing of regionally heterogeneous inflamed and uninflamed areas, as well as protein products, such as secretory MUC2+ granules, not necessarily reflecting cell types. For tuft cells, their numbers were significantly reduced in the antibiotic-treated wildtype ileum, consistent with the persistence of few ATOH1-dependent tuft cells when ATOH1-independent tuft cells were eliminated (Figure 3C, E). In contrast, AtohKO tuft cells were significantly expanded (Figures 3D–E); these results were largely consistent with cell type distributions obtained by microscopy analysis.
Figure 3. Trajectory analysis of scRNA-seq data supports alternative origins for ATOH1-dependent and -independent tuft cells.
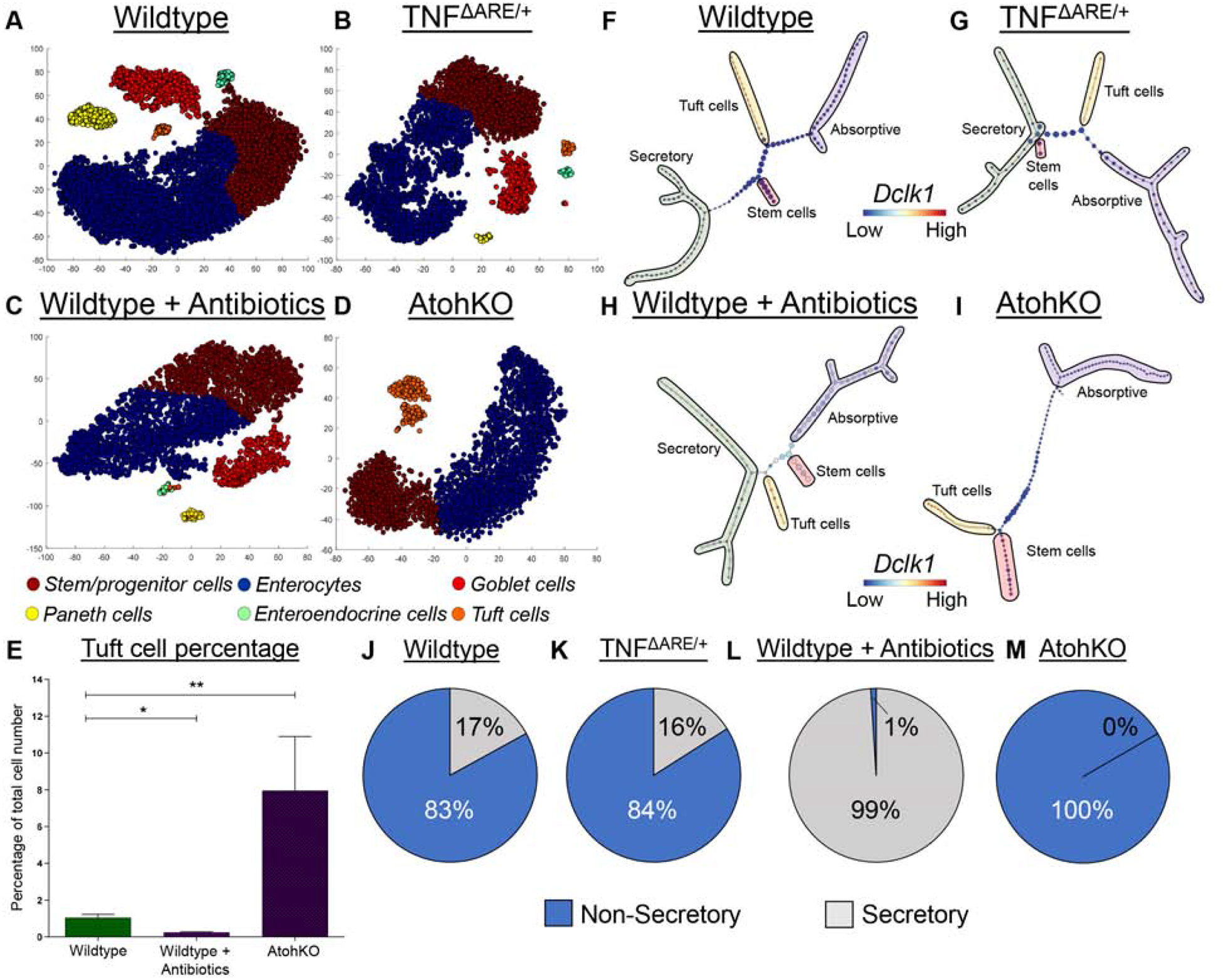
(A-D) t-SNE analysis of scRNA-seq data generated from different murine models annotated with cell type cluster. n = 6 wildtype, 3 TNFΔARE/+, 2 antibiotic-treated wildtype, 3 AtohKO. (E) Tuft cell percentage quantification from scRNA-seq. SEM for replicates plotted. (F-I) Top scoring p-Creode topologies of scRNA-seq data with lineage annotation. Node size represents cell state density. (J-M) Quantification of n = 100 p-Creode topology maps for either non-secretory or secretory tuft cell placement. p-value * < 0.05, ** < 0.01.
We next analyzed tuft cell specification pathways using the p-Creode algorithm to produce lineage trajectory representations of scRNA-seq data (Figure 3F–I; Supplementary Figure 6–7)11. The wildtype p-Creode map originated from stem cells and bifurcated into the absorptive and secretory lineages, with goblet and Paneth cells originating from a common secretory progenitor (Figure 3F). In contrast, the tuft cell lineage mainly shared a specification trajectory with absorptive cells, rather than secretory cells. In order to evaluate the robustness of this result, we generated bootstrapped p-Creode graphs by resampling the dataset and quantified tuft cell placement into different lineages. Tuft cell placement was non-secretory in 83% of wildtype trajectories and secretory in 17% (Figure 3J). Focused analysis of the tuft cell population in the wildtype ileum revealed two subclasses with divergent metabolism-related gene expression programs (Supplementary Figure 8A–B). These results are consistent with previous work documenting multiple tuft cell populations9, which may account for the varying tuft cell lineage placement in p-Creode trajectories (Figure 3J). We repeated p-Creode analysis to include rare enteroendocrine cells, as well as using another dataset by Haber et al.9, and observed similar tuft cell placement results (Supplementary Figure 9–10). p-Creode analysis of the TNFΔARE/+ dataset generated similar results, and illustrated that, even under inflammatory conditions, tuft cells can share a trajectory with absorptive cells (Figures 3G, K). These results show that tuft cell lineage branching from a non-secretory route was a robust and consistent feature of small intestinal cell differentiation across multiple datasets. Given the frequency of tuft cell placement outside the ATOH1-dependent secretory lineage, we surmised that ATOH1-independent tuft cells exist and, in a microbiome-replete intestinal environment, account for the majority of small intestinal tuft cells.
p-Creode analysis of the antibiotic-treated wildtype dataset, which we hypothesized to contain only ATOH1-dependent tuft cells, revealed that these tuft cells share a trajectory almost exclusively with secretory cells, with 99% placement with the secretory linage (Figures 3H, L). Differential gene expression analysis of antibiotic-treated wild type tuft cells revealed increased expression of genes associated with secretory cells, including Sox9, Muc2, and alpha-defensin Defa22 (Supplementary Figure 8C–E)19,20. In addition, t-SNE analysis showed that these tuft cells are clustered with enteroendocrine cells, although their memberships do not overlap (Figure 3C), consistent with prior work demonstrating shared differentiation of enteroendocrine cells and a tuft cell subset from common Prox1+ progenitors21. Similarly, ATOH1-dependent colonic tuft cells shared similar topological relationships with enteroendocrine cells (Supplementary Figure 8F–G).
Finally, all bootstrapped p-Creode graphs generated from AtohKO data depicted tuft cells and absorptive cells as originating from a common progenitor (Figure 3I, M), again demonstrating their ATOH1-independence. Critically, these ATOH1-independent tuft cells expressed the canonical tuft cell gene signature derived from literature7 (Supplementary Figure 7D,8H), showing that they are not a damage-induced stem cell lineage; however, they had decreased expression of secretory genes compared to ATOH1-dependent tuft cells (Supplementary Figure 8C–E). These results further support the existence of ATOH1-dependent (secretory) and ATOH1-independent (non-secretory) tuft cell populations, where the ATOH1-independent population is sensitive to luminal cues and can be induced to expand. We sought to leverage the AtohKO condition in order to identify signals that drive ATOH1-independent tuft cell expansion.
Alterations in TCA metabolic pathways are associated with tuft cell expansion in a microbiome-dependent manner
To identify molecular pathways altered in the course of ATOH1-independent tuft cell expansion, we focused our analysis on dynamic alterations in gene expression along the tuft cell lineage between the wildtype and AtohKO condition, with the latter having significant tuft cell expansion. This approach circumvents technical batch effects, since the dynamics of gene expression along a trajectory is self-contained within individual analyses. Examples illustrating lineage-specific marker expression dynamics are shown in Supplementary Figure 11A.
We aimed to identify genes that switch their expression dynamics between the wildtype and AtohKO conditions in the tuft cell lineage. First, different types of gene dynamics were broadly classified into four categories, as well as a fifth category of unchanged or “flat” dynamics (Figure 4A). Group one genes, exemplified by Soux, trended upward along pseudotime of the stem-to-tuft cell trajectory and included genes known to be tuft cell markers (e.g., Ptgs1 and Sox9) (Supplementary Figure 11B)13. Group four genes, illustrated by Rps6, trended downwards and included stem cell markers downregulated during differentiation. Intermediate genes that trended upwards but returned to a lower baseline or those that initially trended downwards but returned to a higher baseline were categorized into groups two and three, respectively. When all expressed genes between the wildtype and AtohKO were visualized within these categories, a broad expansion of group two genes was observed upon loss of Atohl (Figure 4A). To identify gene expression changes accompanying tuft cell expansion in an unbiased manner, we extracted 1,755 genes that were positively enriched in the AtohKO group, namely, those that switched categories from a lower category in the wildtype data to a higher category in AtohKO (Supplementary Figure 11C). Over-representation analysis of positively enriched genes in the AtohKO epithelium identified pathways related to the tricarboxylic acid (TCA) cycle and oxidative phosphorylation based on KEGG, Wiki pathways, and Reactome databases (Figure 4B; Supplementary Figure 11D–E). Simplifying the analysis by re-classifying genes as having only “upward” or “downward” dynamics generated similar results enriching for metabolism-associated processes (Supplementary Figure 11F–H). We confirmed changes in TCA gene dynamics in the AtohKO tuft cell lineage by plotting expression trends fit to raw data from ten representative p-Creode maps, using tuft (Dclk1, Trpm5) and stem (Myc, Pcna) signature genes as controls with unaltered dynamics between conditions (Figure 4C–H; Supplementary Figure 11I). For instance, the TCA cycle enzyme gene Mdh2 trended down along the wildtype tuft cell specification trajectory, while its expression remained constant along the AtohKO trajectory (Figure 4D). Similarly, other TCA enzymes such as Idh3a, Sdhb, and Sdhd, and downstream ETC genes such as those coding for NADH dehydrogenases and ATP synthases all switched to more positive dynamic trends along the AtohKO tuft cell trajectory compared to wildtype (Figures 4C–H; Supplementary Figure 12). These results were confirmed by grouping cells within the tuft cell trajectory and performing standard differential expression analysis followed by gene set enrichment analysis (GSEA) between wild type and AtohKO cells, using a variety of databases such as MSigDB, KEGG, PANTHER, and Wikipathways (Figure 4I–M; Supplementary Figure 13A–F). Similar to the dynamic trend analysis, TCA cycle-related genes Idh3b, Mdh2, Sdha, Sdhb, Sdhc, Sdhd, Citrate synthase (Cs), Idh3a, and Mdh1 were significantly increased in AtohKO tuft cells compared to wildtype (Figure 4N–4S; Supplementary Figure 13G–L). To support the role of the microbiome in inducing these changes, we generated scRNA-seq data from AtohKO animals following antibiotic administration where microbiome depletion suppresses tuft cell expansion (Figure 2B). In the mid-dose condition (++), tuft cell expression of Idh3a, Idh3b, Idh3g, Sdha, and Sdhd was significantly reduced while others, such as Mdh1, Mdh2 and Sdhc, trended downwards (Supplementary Figure 13M–X). These results indicate that ATOH1-independent tuft cell expansion is accompanied by increases in TCA cycle gene expression, which is driven by extrinsic signals from the intestinal microbiome.
Figure 4. Analysis of AtohKO tuft cell gene expression identified upregulation in metabolic pathways.
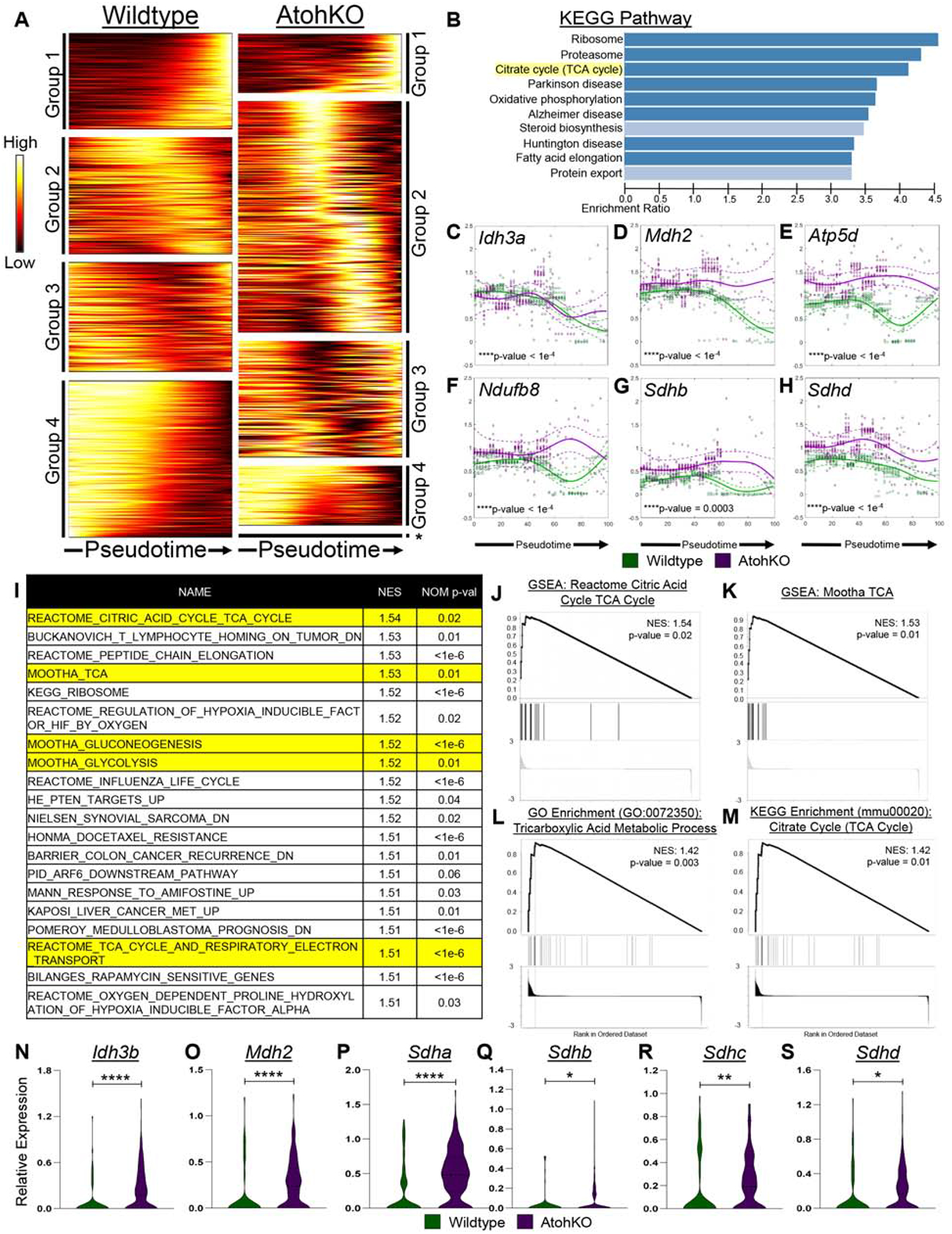
(A) Heatmap of gene expression trends grouped into different dynamic trends as denoted in Supplementary Figure 11. (B) KEGG enrichment of genes that class switched from lower order to higher order in AtohKO, ordered by NES. (C-H) Representative TCA cycle-related gene trends over the tuft cell lineage pseudotime. Solid lines represent mean expression trends and dashed lines represent confidence intervals fitted to raw data from 10 top-scoring p-Creode topologies. Datapoints are scaled expression data. (I) Top 20 GSEA gene sets enriched in AtohKO tuft cell transcriptomes, along with (J-M) positive enrichment plots with highest NES. (N-S) Scaled TCA cycle gene expression in tuft cells. p-value * < 0.05, ** < 0.01, **** < 0.0001.
Non-parasite-derived sources of succinate in the luminal environment drive tuft cell expansion
Given the microbiome dependency and metabolic gene upregulation observed in ATOH1-independent tuft cell expansion, we sought to characterize the differences in (1) metabolites and (2) microbiota composition in the intestines of wildtype and AtohKO animals. O- benzylhydroxylamine (O-BHA) mass spectrometry analysis revealed that the relative concentration of succinate was significantly increased in the AtohKO cecal luminal contents but not in tissue, while levels of malate and butyrate were not altered (Figure 5A–C)22. Succinate is a metabolic intermediate in the TCA cycle and is converted by the enzyme succinate dehydrogenase into fumarate, which trended upwards in the AtohKO tissue, suggestive of host metabolic processing of luminal succinate (Figure 5D). Moreover, the disparity in succinate concentration between luminal contents and whole tissue was indicative of a commensal microbiota-derived, rather than a host-derived, origin for succinate (Figure 5A). Thus, we repeated the analysis in the AtohKO condition following microbiome depletion, which resulted in a significant decrease in luminal succinate, confirming that the commensal microbiota was primarily responsible for succinate increases in the AtohKO model (Figure 5E). We verified that exogenous succinate administration in wildtype animals induced tuft cell expansion, while major basic protein (MBP)-positive eosinophils and GATA3+ cells, components of the anti-parasite response, were increased in both succinate-treated wildtype and AtohKO intestinal tissues (Supplementary Figure 14A–C)23–26. To confirm that succinate is the intermediary downstream of the microbiome, we evaluated antibiotic-treated AtohKO animals, which are devoid of tuft cells, for the ability of succinate alone to rescue tuft cell expansion in a depleted microbiome environment. Succinate administration restored tuft cell expansion to microbiome replete levels in the small intestine of these animals (Supplementary Figure 14D), demonstrating that this TCA cycle metabolite alone is sufficient to drive specification of ATOH1-independent tuft cells.
Figure 5. Succinate production in the AtohKO small intestine drives ATOH1-independent tuft cell expansion.
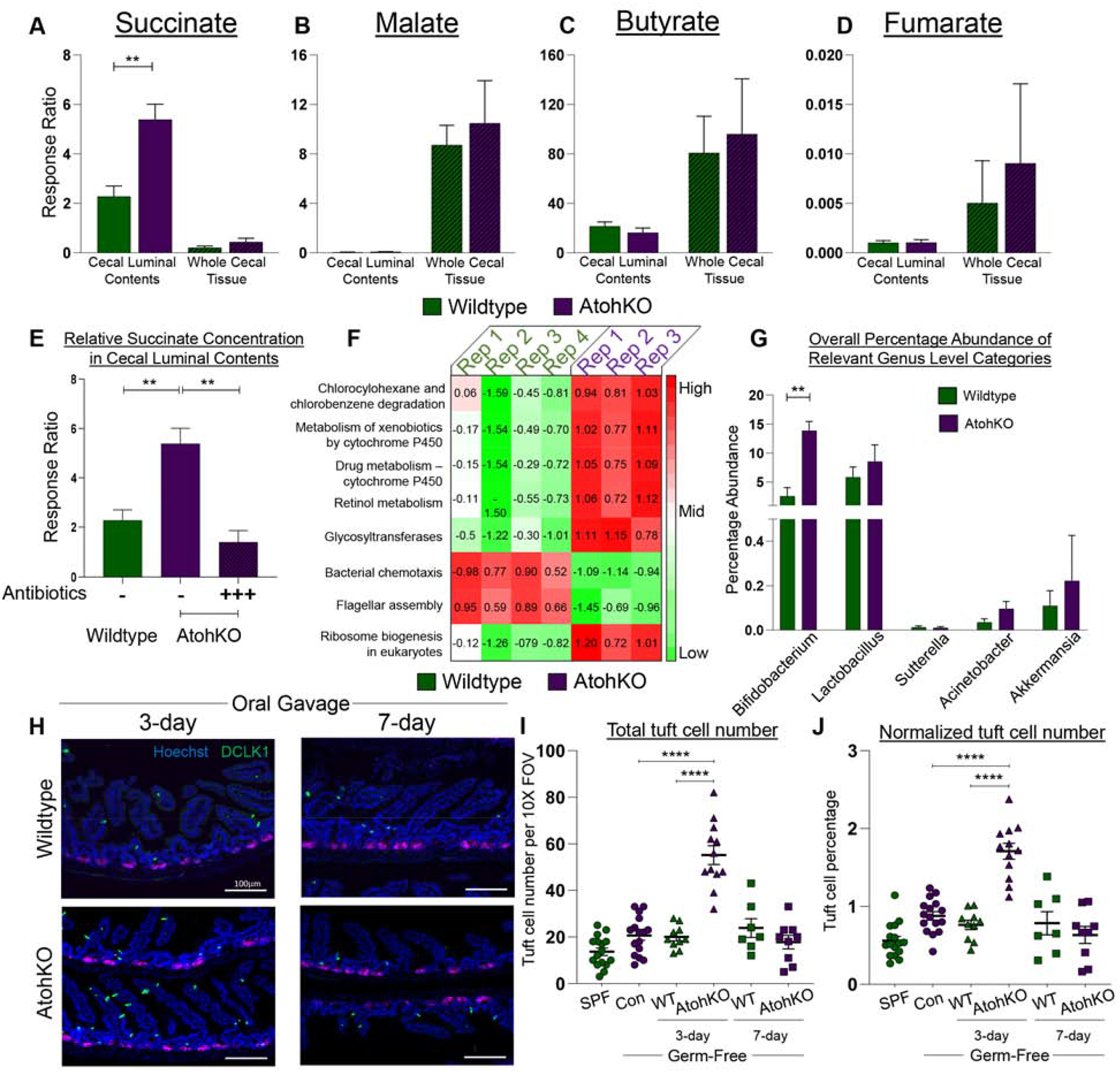
(A-E) Mass spectrometric measurements of metabolites from the cecal lumen and tissue. SEM for n = 5 wildtype, 3 AtohKO, and 3 antibiotic-treated AtohKO animals. (F) Heatmap of z-score normalized PICRUSt category scores between wild type and AtohKO. Categories p-values are all < 0.05. (G) Relative abundance of genus contributing to “Chlorocyclohexane and chlorobenzene degradation” category. SEM for n = 4 wildtype and 3 AtohKO. (H) IF for cell markers of GF animals gavaged with wildtype or AtohKO contents, at 3-days or 7-days. (I) Raw and (J) normalized (to nucleus count) tuft cell number in untreated and oral-gavaged animals. Datapoints represent FOVs and SEM across multiple biological replicates calculated. p-value ** < 0.01, **** < 0.0001.
To probe the response of tuft cells in response to succinate, we performed scRNA-seq in the ilea of succinate-treated wildtype mice. Consistent with IF imaging, quantification of scRNA-seq data showed induced tuft cell expansion by exogenous succinate administration (Supplementary Figure 15A–C). Consistent with TCA cycle gene induction in other conditions of tuft cell expansion, expression of Idh3g, Mdh2, Sdhb, Sdhc, Ogdh, and Rpl18a were significantly increased in succinate-treated tuft cells, while Cs and Idh3b trended upwards (Supplementary Figure 15D–L). This analysis provided an opportunity to investigate lineage-specific induction of these genes. Expression Idh3g, Sdhb, Sdhc, Rpl18a, and Idh3b was similarly increased in cells of the absorptive lineage by exogenous succinate administration, again confirming that these cells may share similar biology (Supplementary Figure 15D–O). In contrast, secretory lineage-specific expression of most of these genes were unchanged or even decreased. Differentiation trajectory and tuft cell lineage placement were similar to untreated wildtype (Supplementary Figure 15P–Q). These results are consistent with the ability of ATOH1-independent tuft cells outside of the secretory lineage to respond to luminal succinate.
We also queried the effect of succinate administration in the colon, where tuft cells were shown to be ATOH1-dependent. Wildtype colon was not responsive to succinate in tuft cell expansion nor downstream MBP+ type 2 cell infiltration (Supplementary Figure 16A–B). Colonic tuft cells do not express the succinate receptor (Sucnr1) (Supplementary Figure 16C)7,25. In antibiotic-treated AtohKO animals, where ATOH1-dependent colonic tuft cells are absent, succinate administration did not restore tuft cell specification (Supplementary Figure S16D). Succinate administration to wildtype mice did not trigger TCA cycle genes in colonic tuft cells (Supplementary Figure 16E–P). These results demonstrate that commensal-derived metabolic signals, specifically succinate, are necessary and sufficient to induce ATOH1-independent tuft cell expansion in the small intestine, but do not affect ATOH1-dependent tuft cells in the colon.
As the microbiome was necessary for in vivo tuft cell expansion, we used sequencing of the V4 region of the 16S rRNA gene to investigate altered microbiome distribution in the ileal luminal contents of co-housed wildtype and AtohKO littermates. Quality control analyses of sequencing data demonstrated that wildtype and AtohKO replicates clustered together due to biological variation rather than cage effects (Supplementary Figure 17A–F). Analysis of microbiome composition revealed a decrease in genus Barnesiella within AtohKO replicates compared to wildtype, while the relative abundance of Parasutterella and Bifidobacterium was increased (Supplementary Figure 17G–I). Bifidobacterium infantis, Bifidobacterium breve, and Bifidobacterium pseudolongum are components of the VSL3 probiotic that can induce remission in a subset of patients with active IBD27. We performed PICRUSt analysis on our 16S gene sequence data and identified eight functional categories that were positively enriched in the AtohKO microbiome, including “Chlorocyclohexane and chlorobenzene degradation” and “Retinol metabolism” (Figure 5F)28. A simplified diagram of the Chlorocyclohexane and chlorobenzene degradation pathway (ko00361) shows that this process is associated with succinate production (Supplementary Figure 17J). Further analysis revealed that Bifidobacterium, Lactobacillus, Sutterellla, Acinetobacter, and Akkermansia contributed to the positive enrichment of this pathway in the AtohKO microbiome; however, only Bifidobacterium was significantly increased compared to the wildtype microbiome (Figure 5G). Specifically, we observed that Bifidobacterium pseudolongum, a known producer of succinic acid29, was increased six-fold in the AtohKO microbiome (Supplementary Figure 17K).
To confirm the contribution of the microbiome to tuft cell expansion, we transferred the cecal microbiome from AtohKO or wildtype animals into germ-free (GF) wildtype animals for a short- (3-day) and long-term (7-day) period, with the caveat that the intact anti-microbial functions of Paneth and goblet cells may counteract effects of the inoculum. DCLK1+ tuft cells were significantly increased in both the duodenum and the ileum after three days of inoculation with AtohKO contents compared to controls (uncolonized GF or SPF), or GF mice colonized with wildtype contents, although the increase was not as pronounced as other tuft cell expansion conditions (Figure 5H–J; Supplementary Figure 18A–F). After seven days however, tuft cell numbers were not significantly different between wildtype- and AtohKO-colonized animals in either the duodenum or the ileum, consistent with previous results13. In contrast, colonic tuft cells did not increase from AtohKO content gavage under any conditions (Supplementary Figure 18G–L). 16S sequencing of the gavage inocula and intestinal lumen contents in post-gavage animals showed that Bifidobacterium pseudolongum trended upward in the AtohKO inoculum compared to the wildtype inoculum, and was enriched at three days, but not seven days, post-gavage in the small intestine of AtohKO-gavaged animals compared to wildtype-gavaged controls (Supplementary Figure 18M). These findings reveal the metabolic potential of certain commensal communities to drive ATOH1-independent tuft cell expansion.
Succinate administration ameliorates inflammation in the TNFΔARE/+ model
Given the hypothesis that expanding tuft cells can suppress inflammatory disease, we administered succinate in the drinking water of adult TNFΔARE/+ mice following disease onset to activate the microbiome-responsive ATOH1-independent population. Succinate administration in TNFΔARE/+ animals markedly improved intestinal tissue organization compared to age-matched, untreated TNFΔARE/+ controls, based on restored crypt-villus architecture, minimized villus distortion, inflammation, and injury scored by a pathologist (Figure 6A–C). Inflammation-associated immune cell subsets enhanced in the TNFΔARE/+ model15, including MPO+ neutrophils and FOXP3+ T-regulatory cells were significantly reduced in long-term succinate-treated animals, indicative of decreased infiltrative disease (Figure 6D–G).
Figure 6. Succinate administration enhances anti-parasitic immune response to counteract inflammation in TNFΔARE/+ animals.
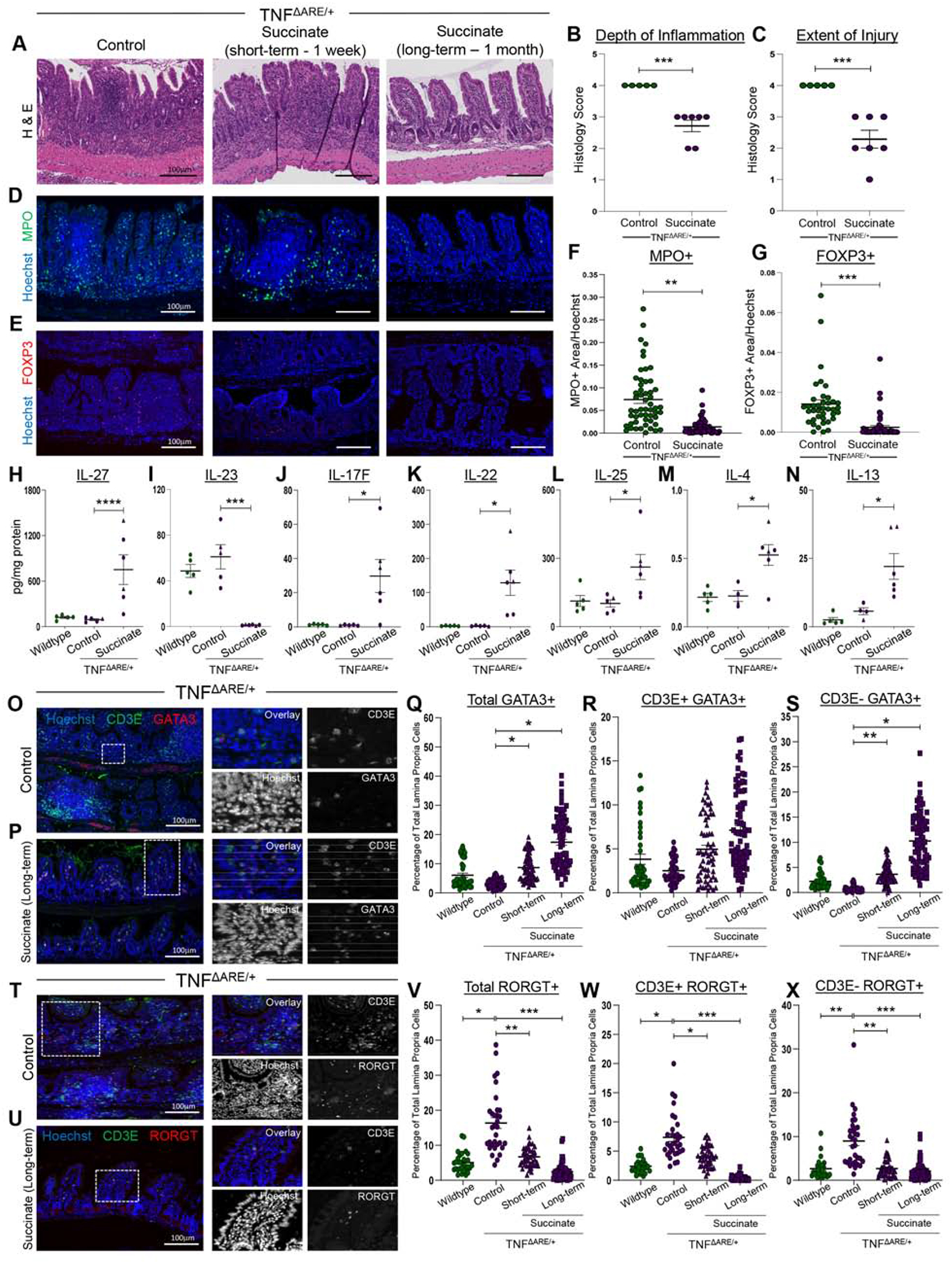
(A) H&E of TNFΔARE/+ ilea with succinate. (B-C) Histopathological scoring of control (n = 5) and long-term succinate-treated (n = 7) TNFΔARE/+ mice. SEM plotted. (D-E) IF of immune cell markers. (F-G) IF quantification of immune cell types normalized to nuclei area. Each dot represents a FOV and SEM across n = 4 animals. (H-N) Luminex cytokine measurements from ileal tissues. SEM across multiple biological replicates calculated (circles: males, triangles: females). (O-P) IF of type 2 immune markers. (Q-S) IF quantification of nuclear GATA3+ cells in the lamina propria. (T-U) IF of type 17 immune markers. (V-X) IF quantification of nuclear RORGT+ cells in the lamina propria. Datapoints represent FOVs and SEM from multiple biological replicates calculated. p-value * < 0.05, ** < 0.01, *** < 0.001. **** < 0.0001.
We used multiplex Luminex to assay cytokine levels of short-term succinate-treated animals to capture the initial changes to immune responses. Succinate-induced decrease in inflammation was not a result of decreased TNF- α levels in the TNFΔARE/+ model (Supplementary Figure 19A). Short-term succinate administration induced IL-27, which inhibits type 17 responses30, and suppressed IL-23 expression, which drives differentiation of Th17 cells31 (Figure 6H–I). Similar to past reports that showed IL-17+ IL-22+ cell increases in helminth treatment of IBD5, we observed significant increases in IL-17 and IL-22 in these animals, with the latter cytokine also shown to enhance mucosal regeneration and worm clearance (Figure 6J–K; Supplementary Figure 19B)32. Canonically, host response to eukaryotic infection is facilitated by type 2 cytokines; IL-25 is released by epithelial tuft cells while IL-13 and IL-4 are released by innate lymphoid type 2 cells (ILC2s)7. Short-term succinate administration in TNFΔARE/+ animals significantly increased levels of IL-25, IL-4, and IL-13, with other anti-parasite cytokines trending upward (Figures 6L–6N; Supplementary Figure 19C–E). Finally, IL-21, which is enhanced in patients with active CD, trended downwards with succinate administration (Supplementary Figure 19F)33. Cytokine analysis suggested that succinate-driven suppression of inflammation occurs due to a decrease in proinflammatory type 17 immunity and an activation of an anti-inflammatory profile analogous to an anti-parasite immune response.
To complement the cytokine analysis, we used imaging cytometry to examine cells involved in anti-parasite type 2 responses in spatially heterogeneous inflamed tissues. Directing our analysis to lamina propria immune cells (Supplementary Figure 19G)13, we focused on type 2 cells that are CD3+ (Th2) and CD3- (ILC2), first using a lineage panel to exclude B-cells, erythrocytes, and myeloid/dendritic cells (Supplementary Figure 19H–J), as well as non-nuclear GATA3 in the inflammation-activated submucosal stroma (Supplementary Figure 19K)26. While both inflamed ilea of untreated TNFΔARE/+ animals and uninflamed ilea of wild type animals had low numbers of CD3+/GATA3+ Th2 cells and CD3-/GATA3+ILC2s, both short- and long-term succinate treated animals had increased levels of these cell types, with ILC2s significantly increased (Figure 6O–6S; Supplementary Figure 19L–M). We applied a similar methodology to examine inflammatory type 17 cells through the transcription factor RORGT. RORGT+ cells, including both CD3+/RORGT+ Th17 cells and CD3-/RORGT+ ILC3s, which were rare in uninflamed tissues, were increased in inflamed TNFΔARE/+ animals, and were subsequently reduced and returned to baseline with short- and long-term succinate administration (Figure 6T–X; Supplementary Figure 19N–O). Thus, enhanced tuft cell specification and a commensurate increase in anti-parasite responses is implicated in the mechanism of succinate-mediated suppression of a type 17 inflammatory response.
Tuft cells are necessary for succinate-induced suppression of inflammation
Succinate administration to the TNFΔARE/+ model completely restored epithelial organization. LYZ1-expressing cells were restored to the base of the crypts and exhibited the typical Paneth cell morphology, while lamina propria LYZ1 expression was absent (Figure 7A). OLFM4+ stem cells were expanded beyond the +4 crypt position upon short-term succinate administration, consistent with ongoing restitution of inflammation-induced epithelial damage, and returned to their normal positions long-term (Supplementary Figure 19P–Q). As expected, DCLK1+ tuft cells were increased by succinate administration in TNFARE/+ animals (Figures 7B). To demonstrate universality, we evaluated another model of intestinal inflammation whereby the administration of an anti-CD3E antibody results in infiltrative disease of the small intestine (Figures 7C; Supplementary Figure S20)34. Succinate administration had protective effects in animals treated with the anti-CD3E agent as these animals had significantly less weight loss, tissue destruction based on histology, immune cell infiltration based on the reduced presence of MPO+ neutrophils, and restored tuft cell numbers (Figure 7C–E, H; Supplementary Figure S20A–B). To probe the necessity of tuft cells in succinate-induced inflammation suppression, we repeated these experiments in a Pou2f3-null mouse model, where intestinal and colonic tuft cells were absent and could not be induced by succinate (Supplementary Figure 20E–F)35. Succinate failed to rescue anti-CD3E-driven tissue destruction, weight loss, and neutrophil infiltration in Pou2f3-null animals (Figure 7C, F–H; Supplementary Figure 20C–D). These findings implicate a causal role of the succinate-tuft cell specification axis in modulating small intestinal inflammation.
Figure 7. Tuft cells are necessary for succinate-mediated inflammation suppression.
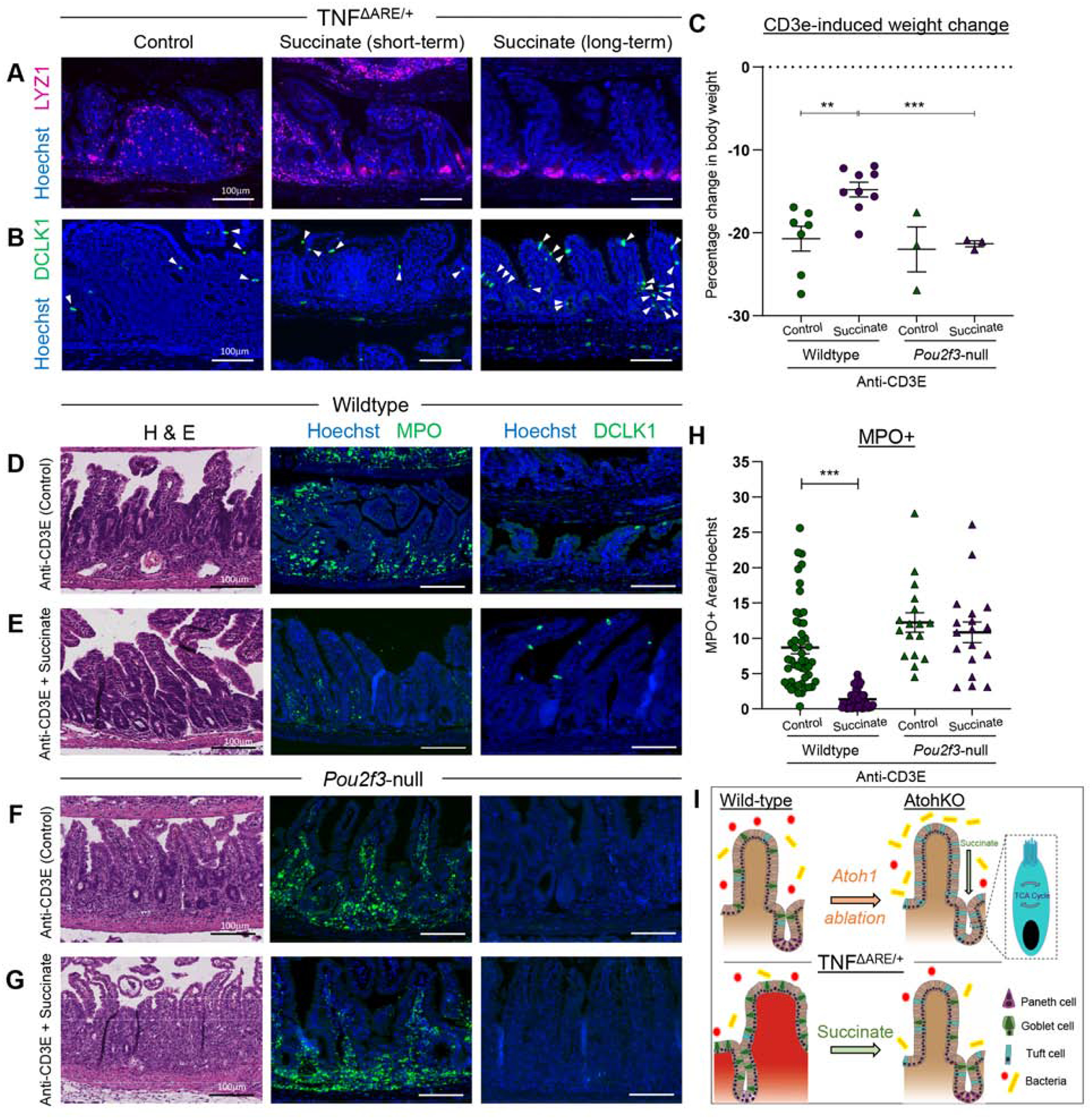
(AB) IF of cell markers of TNFΔARE/+ ilea with succinate. White arrows mark DCLK1+ tuft cells. (C) Percentage body weight change in succinate treated animals in the anti-CD3E model. SEM across multiple biological replicates calculated. (D-G) H&E and IF of cell markers in anti-CD3E-treated mice with or without tuft cells and succinate. (H) IF quantification of MPO+ cells normalized to nuclei area. Datapoints represent FOVs and SEM across multiple biological replicates calculated. (I) Summary diagram. p-value ** < 0.01, *** < 0.001.
Discussion
Chemosensory tuft cells have recently been identified to be necessary and sufficient to drive type 2 immune responses against eukaryotic colonization, possibly through parasite-derived succinate6,23–25. Despite being a global health concern, helminths have been proposed as a therapeutic option for CD3,5. Prevailing thought postulates that the anti-parasite immune response may counteract pro-inflammatory signaling driving CD3. We observed decreased numbers of tuft cells in inflamed ileal tissues from CD patients and mouse models, and thus, we hypothesized that tuft cells may be the conduit between parasite and host that can be leveraged for counteracting pro-inflammatory signals in the intestine.
We identified ATOH1-dependent and ATOH1-independent routes of specification that result in heterogeneous tuft cell populations (Figure 7I). Both ATOH1-dependent and - independent tuft cells exist in the small intestine, while only ATOH1-dependent tuft cells exist in the colon. Surprisingly, we found that ATOHI-independent tuft cells are a malleable cell population that can be expanded in the context of luminal perturbation, whereas the ATOH1-dependent population is invariant. Specifically, succinate derived from the commensal microbiome drives ATOHI-independent tuft cell gene expression and expansion; these may include several succinic acid producers of the Bifidobacterium genus known to maintain intestinal health28,30. Given the potential transmissibility of the tuft cell expansion phenotype by microbiome transfer, our results further revealed that the downstream metabolite succinate from a non-parasitic source is necessary and sufficient to drive ATOH1-independent tuft cell specification. Administration of succinate following disease onset in two mouse models suppressed inflammation and restored epithelial architecture, accompanied by enhanced anti-parasite, and reduced type 17 response. How succinate induces cytokine gene expression to initiate downstream responses remains an area of study, with possible hypotheses related to signaling pathway- or metabolite-regulation of the epithelial epigenome.
Our findings shed light on the heterogeneity of small intestinal tuft cells and their functional role in reducing intestinal inflammation through luminal succinate. Beyond succinate, our findings suggest that enhanced tuft cell specification could be a viable therapeutic strategy, where exploring the mechanisms linking tuft cells to anti-inflammatory signaling could lead to clinically-viable targets for CD therapy.
Supplementary Material
BACKGROUND AND CONTEXT
Inflammatory bowel disease incidence is inversely correlated with endemic parasite infestation. Clinical studies have suggested helminth therapy is beneficial in IBD treatment. Intestinal tuft cells modulate anti-parasite responses.
NEW FINDINGS
At homeostasis, heterogeneous tuft cell populations exist and tuft cell frequency is decreased under inflammatory conditions. Tuft cell expansion in models of intestinal inflammation resolved disease.
LIMITATIONS
Therapeutic tuft cell expansion was only conducted in mouse models. Further studies are necessary to elucidate mechanisms by which tuft cells resolve disease.
IMPACT
Tuft cells are an understudied sector of epithelial biology and could potentially provide new biomarkers and/or therapeutic targets for IBD treatment.
LAY SUMMARY
Tuft cell subpopulations in the intestinal tract respond differentially to external stimuli. Therapeutic expansion of tuft cells in models of small intestinal inflammation ameliorates disease.
Acknowledgements
K.S.L., H.Y.K., A.J.S., A.N.S., and M.C.M. are funded by R01DK103831. A.B. is funded by T32Al138932. K.S.L., M.A.R.S., Q.L., E.T.M., and R.J.C. are funded by P50CA236733. C.A.H. is funded by a training grant from T32HD007502 and a pre-doctoral F31GM120940. B.C. is funded by a training grant from T32LM012412. E.A.S. is funded by KL2TR002245. E.A.S. and M.K.W. are funded by P30DK058404. M.K.W. is funded by UM1CA183727. R.J.C. is funded by R35CA197570. J.A.G. is funded by K01DK106311. M.M.A. and K.T.W. are funded by I01BX001453, P01CA028842, R21AI142042, and Cure for IBD. Microbiome sequencing was funded by the Vanderbilt Microbiome Initiative. The authors would like to thank Cherie’ Scurrah and Paige Vega in the Lau lab for support in mouse experiments, Drs. Seth Bordenstein, Andrew Brooks, Nicholas Markham, Oliver McDonald, Izumi Kaji, James Goldenring, Eunyoung Choi, Joseph Roland, Damian Maseda, and Mariana Byndloss, the Vanderbilt Digestive Disease Research Center, and the Vanderbilt Epithelial Biology Center for helpful discussions centered around the microbiome, tuft cells, and metabolism. The authors would also like to thank various cores and centers at Vanderbilt for making this work possible, including the Vanderbilt Mass Spectrometry Research Center, Cooperative Human Tissue Network, VANTAGE Genomics Core, the Translational Pathology Shared Resource, and the Digital Histology Shared Resource. The authors would like to acknowledge the UNC Chapel Hill CGIBD Gnotobiotic Core and Dr. Jakob von Moltke for their assistance in the acquisition of mouse lines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
J.R.W. is the founder of Resphera Biosciences. All other authors declare no conflict of financial interests.
References
- 1.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012;142:46–54.e42. [DOI] [PubMed] [Google Scholar]
- 2.de Silva P, Korzenik J. The Changing Epidemiology of Inflammatory Bowel Disease: Identifying New High-risk Populations. Clin Gastroenterol Heptaol. 2015; 4:690–2. [DOI] [PubMed] [Google Scholar]
- 3.Summers RW, Elliott DE, Urban JF, et al. Trichuris suis therapy in Crohn’s disease. Gut 2005;54:87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su J, Chen T, Ji X-Y, et al. IL-25 Downregulates Th1/Th17 Immune Response in an IL-10–Dependent Manner in Inflammatory Bowel Disease. Inflamm Bowel Dis 2013;19:720–728. [DOI] [PubMed] [Google Scholar]
- 5.Broadhurst MJ, Leung JM, Kashyap V, et al. IL-22+ CD4+ T cells are associated with therapeutic trichuris trichiura infection in an ulcerative colitis patient. Sci Transl Med 2010;2:60ra88. [DOI] [PubMed] [Google Scholar]
- 6.Gerbe F, Sidot E, Smyth DJ, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016;529:226–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moltke J von, Ji M, Liang H-E, et al. Tuft cell derived IL25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016;259:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howitt MR, Lavoie S, Michaud M, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016;351:1329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haber AL, Biton M, Rogel N, et al. A single-cell survey of the small intestinal epithelium. Nat Publ Gr 2017;551:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerbe F, Es JH Van, Makrini L, et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J Cell Biol 2011;192:767–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herring CA, Banerjee A, Mckinley ET, et al. Unsupervised Trajectory Analysis of Single-Cell RNA-Seq and Imaging Data Reveals Alternative Tuft Cell Origins in the Gut. Cell Syst 2018;6:37–51.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gracz AD, Fordham MJ, Trotier DC, et al. Sox4 Promotes Atoh1-Independent Intestinal Secretory Differentiation Toward Tuft and Enteroendocrine Fates. Gastroenterology 2018;155:1508–1523.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKinley ET, Sui Y, Al-Kofahi Y, et al. Optimized multiplex immunofluorescence single-cell analysis reveals tuft cell heterogeneity. JCI insight 2017;2:e93487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wehkamp J, Wang G, Ku I, et al. The Paneth Cell alpha-Defensin Deficiency of Ileal Crohn’s Disease Is Linked to Wnt/Tcf-4. J Immunol 2007;179:3109–18. [DOI] [PubMed] [Google Scholar]
- 15.Kontoyiannis D, Pasparakis M, Pizarro TT, et al. Impaired On / Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements: Implications for Joint and Gut-Associated Immunopathologies. 1999;10:387–398. [DOI] [PubMed] [Google Scholar]
- 16.Shroyer NF, Helmrath MA, Wang VY– C, et al. Intestine-Specific Ablation of Mouse atonal homolog 1 (Math1) Reveals a Role in Cellular Homeostasis. Gastroenterology 2007;132:2478–2488. [DOI] [PubMed] [Google Scholar]
- 17.Meng D, Newburg DS, Young C, et al. Bacterial symbionts induce a FUT2-dependent fucosylated niche on colonic epithelium via ERK and JNK signaling. Am J Physiol Gastrointest Liver Physiol 2007;293:780–787. [DOI] [PubMed] [Google Scholar]
- 18.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459:262–265. [DOI] [PubMed] [Google Scholar]
- 19.Bastide P, Darido C, Pannequin J, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 2007;178:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farin HF, Karthaus WR, Kujala P, et al. Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell-derived IFN-γ. J Exp Med 2014;211:1393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan KS, Gevaert O, Zheng GXY, et al. Intestinal Enteroendocrine Lineage Cells Possess Homeostatic and Injury-Inducible Stem Cell Activity. Cell Stem Cell 2017;21:78–90.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan B, Lu Z, Dong S, et al. Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography–tandem mass spectrometry detection. Anal Biochem 2014;465:134–147. [DOI] [PubMed] [Google Scholar]
- 23.Schneider C, O’leary CE, Moltke J Von, et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 2018;174:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei W, Ren W, Ohmoto M, et al. Activation of intestinal tuft cell-expressed Sucnr1 triggers type 2 immunity in the mouse small intestine. PNAS 2018;115:5552–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nadjsombati MS, McGinty JW, Lyons-Cohen MR, et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 2018;49:33–41.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita M, Ukai-Tadenuma M, Miyamoto T, et al. Essential role of GATA3 for the maintenance of type 2 helper T (Th2) cytokine production and chromatin remodeling at the Th2 cytokine gene loci. J Biol Chem 2004;279:26983–90. [DOI] [PubMed] [Google Scholar]
- 27.Bibiloni R, Fedorak RN, Tannock GW, et al. VSL#3 Probiotic-Mixture Induces Remission in Patients with Active Ulcerative Colitis. Am J Gastroenterol 2005;100:1539–46. [DOI] [PubMed] [Google Scholar]
- 28.Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meulen R Van der, Adriany T, Verbrugghe K, et al. Kinetic analysis of bifidobacterial metabolism reveals a minor role for succinic acid in the regeneration of NAD+ through its growth-associated production. Appl Environ Microbiol 2006;72:5204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stumhofer JS, Laurence A, Wilson EH, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol 2006;7:937–945. [DOI] [PubMed] [Google Scholar]
- 31.Aggarwal S, Ghilardi N, Xie MH, et al. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 2003;278:1910–1914. [DOI] [PubMed] [Google Scholar]
- 32.Turner J-E, Stockinger B, Helmby H. IL-22 Mediates Goblet Cell Hyperplasia and Worm Expulsion in Intestinal Helminth Infection. 2013;10:9–e1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holm TL, Tornehave D, Søndergaard H, et al. Evaluating IL-21 as a potential therapeutic target in Crohn’s disease. Gastroenterol Res Pract 2018;2018:5962624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miura N, Yamamoto M, Fukutake M, et al. Anti-CD3 induces bi-phasic apoptosis in murine intestinal epithelial cells: possible involvement of the Fas/Fas ligand system in different T cell compartments. Int Immunol 2005;17:513–522. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita J, Ohmoto M, Yamaguchi T, et al. Skn-1a/Pou2f3 functions as a master regulator to generate Trpm5-expressing chemosensory cells in mice Ishimaru Y, ed. PLoS One 2017;12:e0189340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.