

Abstract
The tight junction (TJ) and barrier function of colonic epithelium is highly sensitive to ionizing radiation. We evaluated the effect of lysophosphatidic acid (LPA) and its analog, Radioprotein-1, on γ-radiation-induced colonic epithelial barrier dysfunction using Caco-2 and m-ICC12 cell monolayers in vitro and mice in vivo. Mice were subjected to either total body irradiation (TBI) or partial body irradiation (PBI-BM5). Intestinal barrier function was assessed by analyzing immunofluorescence localization of TJ proteins, mucosal inulin permeability, and plasma lipopolysaccharide (LPS) levels. Oxidative stress was analyzed by measuring protein-thiol oxidation and antioxidant mRNA. In Caco-2 and m-ICC12 cell monolayers, LPA attenuated radiation-induced redistribution of TJ proteins, which was blocked by a Rho-kinase inhibitor. In mice, TBI and PBI-BM5 disrupted colonic epithelial tight junction and adherens junction, increased mucosal permeability, and elevated plasma LPS; TJ disruption by TBI was more severe in Lpar2−/− mice compared to wild type mice. RP1, administered before or after irradiation, alleviated TBI and PBI-BM5-induced TJ disruption, barrier dysfunction, and endotoxemia accompanied by protein thiol oxidation and downregulation of antioxidant gene expression, cofilin activation, and remodeling of the actin cytoskeleton. These data demonstrate that LPAR2 receptor activation prevents and mitigates γ-irradiation-induced colonic mucosal barrier dysfunction and endotoxemia.
Keywords: Intestine, tight junction, lysophosphatidic acid, endotoxemia, irradiation
Introduction
Radiological accidents or malicious use of a nuclear device are ever-present global concerns. Thus, the development of radiation countermeasures that mitigate acute radiation syndrome (ARS) is a priority for every nation to protect first responders, the military, and civilians at large. At this time, no FDA-approved radio-mitigator exists. The gastrointestinal (GI) tract has been shown to be a highly vulnerable target of irradiation. Radiation-induced GI dysfunction is commonly referred to as GI-ARS. GI-ARS is characterized by rapid-onset diarrhea during the early stage of radiation injury and endotoxemia, bacteremia, and septicemia in the later stages. Endotoxemia and bacteremia are significant factors contributing to multiple organ dysfunction in ARS. Colonic microbiota is the primary source of endotoxins, which highlights the critical importance of colonic mucosal barrier dysfunction in the pathogenesis of ARS.
The epithelial tight junction and mucosal barrier function prevent diffusion of toxins and pathogens from the lumen into intestinal tissue (1–3). The tight junction, a multiprotein complex that seals the epithelium’s intercellular space, is tightly regulated by cell signaling networks (2, 4–9). Occludin, one of the transmembrane protein components of the tight junction, interacts with a scaffold protein, ZO-1, which is an essential interaction for the assembly and maintenance of tight junction. The adherens junction, located beneath the tight junction, is known to regulate the integrity of tight junction indirectly (10). The tight junction and adherens junction integrated into the underlying actin cytoskeleton define the apical junctional complex (AJC) of the epithelium. Our recent study indicated that the tight junction and adherens junction of the mouse colon are highly sensitive to radiation (11). These junctions are disrupted as early as two hours post-irradiation, with sustained damage lasting for at least five days. These findings emphasize the importance of early disruption of colonic epithelial tight junction and barrier dysfunction resulting in endotoxemia, bacteremia, and septicemia in GI-ARS, which we propose to designate as “Colonic Radiation Sub-syndrome or CRS”. Understanding the cellular and molecular mechanisms of CRS is crucially essential for the development of effective radio-mitigators and treatment strategies.
Lysophosphatidic acid (LPA) is a growth factor-like lipid mediator that rescues cells from genotoxic stress, including radiation-induced apoptosis and cellular injury in vitro and in vivo administered post-irradiation (12). Exogenously delivered LPA is not suitable for use as a therapeutic due to its short half-life in plasma (~9 min). However, stabilized analogs of LPA such as Radioprotectin-1 (13, 14) have radioprotective (12, 15, 16) as well as radio-mitigative (16, 17) efficacy in animal models of GI-ARS. The structure, synthesis, and biological effects have been described before (13, 14, 18). In vitro receptor add-back studies and experiments conducted with Lpar2 knockout (Lpar2−/−) mice indicate that this receptor subtype is necessary and sufficient to protect cells from radiation-induced apoptotic cell death and tissue injury to the gut in mice (15, 19). Thus, improved non-lipid analogs specific to the LPA2 receptor, such as RP1, could be suitable for colonic ARS treatment without activation of the other LPA receptor subtypes.
In this study, we evaluated the protective and mitigating effects of RP1 on the radiation-induced disruption of intestinal epithelial AJC, an increase in mucosal permeability, and endotoxemia.
Materials and Methods
Cell culture
Caco-2bbe2 (ATCC, Rockville, MD) and m-ICCl2 (a kind gift from Dr. Alain Vandewalle, INSERM U773, Paris, France) cells (20) were grown under standard cell culture conditions as previously described (21). Experiments were conducted using cells grown in Transwell inserts of varying diameters (6.5 or 12 mm) for 3–4 or 15–17 days. Caco-2 cells are of human origin and develop tight junction and adherens junction when cultured on polycarbonate membrane in transwell inserts. It has been extensively used as an in vitro model of the intestinal epithelium. Unlike Caco-2 cells, the m-ICC12 cells are non-transformed cells of the intestinal crypts and have high expression of the LPAR2 receptors that have a high affinity for RP-1.
Epithelial barrier function
Transepithelial permeability to macromolecules was evaluated by measuring the unidirectional flux of FITC-inulin, as previously described (21).
Animals
All animal studies were performed under protocols approved by the University of Tennessee Health Science Center (UTHSC) Institutional Animal Care and Use Committee (IACUC). Four different studies were performed to answer specific questions. In the first study, 12–14 week-old adult wild type (WT) and Lpar2−/− mice were subjected to total body irradiation (TBI; 9.5 Gy). WT and Lpar2−/− mice were randomized into Sham (sham-treated), and IR (irradiated) groups. The integrity of colonic epithelial tight junction and adherens junction was examined at 2 hours after TBI. In the second study, mice were subjected to TBI with or without RP-1 administration (0.5 mg/kg, s.c., single dose 24 hours before irradiation, pretreatment group). Control mice were subjected to similar conditions without radiation (sham-treated). In the third study, mice were randomized into three groups: Sham (control), IR (vehicle administered 24 hours post-irradiation), and IR+RP-1 (RP1 was administered at 24 hours post-irradiation). All animals were sacrificed at 48 hours post-irradiation. The colonic epithelial tight junction, adherens junction, and endotoxemia were examined at 2 and 4 hours post-irradiation.
In the fourth study, mice were subjected to partial body irradiation, in which 5% of the bone marrow was shielded (PBI-BM5). The PBI-BM5 model is the standard model to study GI radiation injury and recovery. Radiation doses in excess of 6 Gy cause hematopoietic acute radiation syndrome, whereas radiation doses in excess of 10 Gy lead to intestinal damage (22–25). We chose the PBI-BM5 model where the bone marrow is shielded to the extent that it allows for the eventual recovery of the hematopoietic system by supplying growth and survival factors to the marrow cells, and even distant organs like the gut (26, 27). This part of the study is relevant to intestinal injury caused by radiation therapy. Our previous studies on LPA and its analogs, including the RP-1 class of compounds, have established that these agents protect the intestinal mucosal cells from oxidative damage, rescue cells from apoptotic cell death, and promote the recovery of GI and hematopoietic stem cells (28). These actions led to a significant decrease in lethality after doses as high as 16 Gy (12–15, 18, 29, 30). The experimental groups for this study consisted of Sham (control), IR (vehicle), and IR+RP-1 (0.5 mg/kg daily, s.c.). Vehicle or RP1 was administered at 0, 24, or 48 hours after irradiation. At 28, 52, or 76 hours post-irradiation, the integrity of colonic epithelial tight junction, adherens junction, and actin cytoskeleton were examined. At 28 hours post-irradiation, colons were collected and analyzed for oxidative stress and epithelial junctional integrity.
Irradiation
Cells were irradiated with 2, 5, or 10 Gy γ-irradiation from a 137Cs source (using a J.L. Shepherd & Assoc. Mark I, Model 25, San Fernando, CA, USA) at a dose rate of 440 cGy/min. After irradiation, the culture medium was replaced with fresh complete culture medium. For TBI, mice were subjected to 9.5 Gy radiation (at a dose rate of ~76 cGy/min. For the PBI model, mice were anesthetized with 87 mg/kg ketamine and 13 mg/kg xylazine (i.p.) and placed in a plexiglass restrainer so that legs were shielded below the knee. The shielding protected tibiae, fibulae, and the paws from radiation that contained ~5% of the bone marrow. Mice placed into the isodose field of the irradiator were irradiated in groups of eight. In this model, 15.7 Gy delivered at a dose rate of 147 cGy/min yielded empirical ~LD85/10 mortality. Radiation field mapping and calibration by ion chamber dosimetry were done by the manufacturer. Also, routine validation and quality control measurements of exposure rates and exposure rate mapping in the chamber at positions of interest were conducted by a certified health physicist using a calibrated RadCal 0.6 cc therapy grade ion chamber/electrometer system. High-dose thermoluminescent dosimeters were used in most irradiations to validate the actual dose delivered to the mice (calibrated by MD Anderson Cancer Center Radiation Dosimetry Services). The isodose field was validated using Gafchromic film for high-dose dosimetry (10–50 Gy, Ashland Inc., Covington, KY). At the end of the experiment, gut permeability was measured as described below.
Gut permeability in vivo
Mucosal barrier dysfunction in vivo was evaluated by measuring gut permeability to FITC-inulin (6 kDa). On the last day of the experiment, mice were injected with FITC-inulin (6 kDa MW; 50 mg/ml solution; 2 μl/g body weight) via the tail vein. At one hour after injection, blood samples were collected by cardiac puncture under isoflurane anesthesia. Plasma was isolated using a heparin sulfate anticoagulant. Luminal contents from the colon and ileum were flushed with 0.9% saline. Fluorescence in plasma and luminal flushing was measured using a fluorescence plate reader. Fluorescence values in luminal flushing were normalized to fluorescence values in corresponding plasma samples and calculated as the percent of the amount injected. Several types of molecules, such as inulin, mannitol, fluorescein, and dextran have been used in the past as probes of intestinal epithelial paracellular permeability. Most of these probes provide similar results. We chose inulin as the paracellular permeability indicator as it has been historically used as the probe for extracellular fluid and renal function tests.
Immunofluorescence microscopy
Caco-2 and m-ICC12 cell monolayers were permeabilized with 0.2% Triton X-100, blocked, and stained for different junctional proteins as described for a prior study (31). Fluorescence was examined by using a Zeiss LSM 510/710 laser scanning confocal microscope. Cryo-sections of the colon (10 μm thickness) were fixed in acetone and methanol mixture (1:1) at 20°C for two minutes and rehydrated in 14 mM phosphate-buffered saline (PBS). Sections were permeabilized with 0.5% Triton X-100 in PBS for 15 minutes and blocked in 4% non-fat milk in TBST (20 mM Tris, pH 7.2 and 150 mM NaCl). Tissue sections were first incubated for one hour with primary antibodies (mouse monoclonal anti-occludin and rabbit polyclonal anti-ZO-1 antibodies, or mouse monoclonal E-cadherin and rabbit polyclonal anti-β-catenin antibodies). This was followed by incubation for one hour with the secondary antibodies (AlexaFluor-488-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG antibodies from Molecular Probes, Eugene, OR) containing Hoechst 33342 dye. In all cases, images from x-y (1 μm) sections were collected using LSM Pascal or Zen software (White Plains, NY, USA). Images from optical sections were stacked using ImageJ software (NIH, Bethesda, MD, USA) and processed with Adobe Photoshop (Adobe Systems, San Jose, CA, USA). We first selected the optimal conditions of laser strength, gain, and contrast for the intestinal sections from the Sham group of mice. All other images within the experiment were collected using the same optimal conditions. Images were processed in ImageJ and Adobe Photoshop soft wares using identical conditions for all groups of images so that quantitative comparisons were not compromised.
Preparation of the detergent-insoluble fraction
Actin-rich detergent-insoluble fraction was prepared as described previously (32). Mucosal scrapping from the colon and ileum were incubated on ice for 15 minutes with lysis buffer-CS (Tris buffer containing 1% Triton-X100, 2 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml bestatin, 10 μg/ml pepstatin-A, 10 μl/ml of protease inhibitor cocktail, 1 mM sodium vanadate and 1 mM PMSF). Briefly, mucosal lysates were centrifuged at 15,600 x g for 4 min at 4°C to sediment the high-density actin-rich detergent-insoluble fraction. The pellet was suspended in 100 μl of preheated lysis buffer-D (20 mM Tris buffer, pH 7.2, containing 10 μl/ml of protease inhibitor cocktail, 10 mM sodium fluoride, 1 mM sodium vanadate and 1 mM PMSF), sonicated to homogenize the actin cytoskeleton, and heated at 100°C for 10 min. Protein content was measured by the BCA method (Pierce Biotechnology, Rockford, IL). Triton-insoluble and soluble fractions were mixed with an equal volume of 2X concentrated Laemmli’s sample buffer, heated at 100°C for 5 min, and 25–40 μg protein samples were used for SDS-polyacrylamide gel electrophoresis followed by immunoblot analysis.
Immunoblot analysis
Total protein extracts or the Triton-insoluble fractions of colonic mucosa were separated by SDS-polyacrylamide gel electrophoresis (7%) and transferred to PVDF membranes as described for a previous study (32). Membranes were immunoblotted for different proteins using specific antibodies for different tight junction and adherens junction proteins with β-actin as the housekeeping protein in combination with HRP-conjugated anti-mouse IgG or anti-rabbit IgG secondary antibodies. Blots were developed using the ECL chemiluminescence reagent (Pierce, Rockford, IL) and quantitated by densitometry using ImageJ software. The density of each band was normalized to the density of the corresponding actin band.
Protein thiol assay
Protein thiols in colonic sections were monitored as described previously (31). Briefly, reduced protein thiols were evaluated by staining cryosections of the colon with BODIPY FL-N-(2-aminoethyl) maleimide (FLM) and confocal microscopy at excitation and emission wavelengths (490 nm and 534 nm, respectively). For oxidized protein thiols, the reduced protein thiol was first alkylated with N-ethylmaleimide followed by reduction of oxidized protein thiols with tris (2-carboxyethyl) phosphine prior to staining with FLM. Control staining was done after N-ethylmaleimide treatment. Fluorescence images were collected and quantitated using ImageJ software.
RNA extraction and RT-qPCR
RNA was isolated from the colon by using the TRIzol kit (Invitrogen, Carlsbad, CA, USA) and quantified using a NanoDrop photometer. Total RNA (1.5 μg) was used for the generation of cDNA using the ThermoScript RT-PCR kit for first-strand synthesis (Invitrogen). Quantitative PCR (qPCR) reactions were performed using cDNA mix (cDNA corresponding to 35 ng RNA) with 300 nmole primers in a final volume of 25 μl of 2× concentrated RT2 Real-Time SYBR Green/ROX master mix (Qiagen, Germantown, MD, USA) in an Applied Biosystems QuantStudio 6 Flex Real-Time PCR instrument (Norwalk, CT, USA). The cycle parameters were: 50 °C for 2 min, one denaturation step at 95 °C for 10 min, and 40 cycles of denaturation at 95 °C for 10 s, followed by annealing and elongation at 60 °C. The relative gene expression of each transcript was normalized to the GAPDH gene transcripts using the ΔΔCt method. Sequences of primers used for qPCR are provided in the Supplemental Information section (Table-1).
Plasma endotoxin assay
Plasma endotoxin concentrations were measured using Pierce LAL Chromogenic Endotoxin Quantitation Kit (Thermo Scientific, Cat# 88282) according to the manufacturer’s instructions.
Statistical Analysis
All data are expressed as Mean ± SEM. The differences among multiple groups were first analyzed by ANOVA (Prism 6.0, GraphPad, Inc. San Diego, CA, USA). When statistical significance was detected, Tukey’s t-test was used to determine the significance between multiple experimental groups and the corresponding control groups. Statistical significance was established at 95% confidence.
Materials
Hoechst 33342 dye and BODIPY FL-N-(2-aminoethyl) maleimide were purchased from Life Technologies (Grand Island, NY, USA). N-ethylmaleimide and tris (2-carboxyethyl) phosphine were from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were purchased from either Sigma-Aldrich or Thermo Fisher Scientific (Tustin, CA, USA). Anti-ZO-1, anti-occludin, anti-claudin-5 (CLDN-5), and anti-claudin-3 (CLDN-3) antibodies were purchased from Invitrogen (Carlsbad, CA, USA). Anti-E‐Cadherin and anti-β‐catenin antibodies were purchased from BD Biosciences (Billerica, MA, USA). Horseradish peroxidase-conjugated anti-mouse IgG, anti-rabbit IgG, and anti-β-actin antibodies were obtained from Sigma-Aldrich (St Louis, MO, USA). AlexaFlour‐488‐conjugated anti‐mouse IgG and Cy3‐conjugated anti‐rabbit IgG were purchased from Molecular Probes (Eugene, OR, USA). The anti-nrf2 antibody was purchased from Abcam (Cambridge, MA, USA). The anti-cofilinps3 antibody was purchased from Cell Signaling Technology (Danvers, MA, USA).
Results
LPA attenuates radiation-induced disruption of tight junctions and prevents barrier dysfunction in the intestinal epithelium
Caco-2 (Fig. 1, A–D) and m-ICC12 (Fig. 1, E) cell monolayers were incubated with LPA (10 μM) for 30 min before irradiation at varying doses, and tight junction integrity was examined by confocal microscopy for occludin and ZO-1. In control groups, Caco-2 cell monolayers, occludin, and ZO-1 were co-localized at the intercellular junctions, indicating the presence of intact tight junctions (Fig. 1A). Irradiation induced a dose-dependent redistribution of tight junction proteins, whereas pretreatment of the epithelium with LPA attenuated radiation-induced redistribution of tight junction proteins at both 5 Gy and 10 Gy doses. Densitometric analysis of ZO-1 fluorescence at the intercellular junctions indicated a significant reduction in junctional localization of ZO-1 post-irradiation. LPA-treated samples showed attenuation of radiation-induced ZO-1 redistribution (Fig. 1B). Tight junction protein complexes are anchored to the actin cytoskeleton. The disruption of the actin cytoskeleton is known to disrupt tight junction (33). Cofilin is an actin-severing protein that plays an essential role in the disruption of the actin cytoskeleton. Fluorescence staining for F-actin and cofilinpS3 (the inactive form) showed that radiation causes disruption of F-actin filaments and reduces levels of cofilinpS3. Pretreatment with LPA blocked radiation-induced reduction of inactive cofilin and disruption of F-actin filaments (Fig. 1C). Rho-GTPase is known to be involved in the activation of cofilin and the promotion of actin cytoskeletal organization (34, 35). The data showed that toxin-B, an inhibitor of Rho-GTPase, blocked LPA-mediated prevention of radiation effects on cofilin and F-actin organization (Fig. 1D). To confirm the effects of radiation on the tight junction, we examined the effects of radiation and LPA in I-mCC12 cell monolayers, a non-transformed mouse intestinal epithelial cell line with rich in LPA2 receptors (20). Data showed that radiation induces a dose-dependent redistribution of ZO-1 from the intercellular junctions, which was blocked by LPA treatment (Fig. 1E).
Figure 1: LPA attenuates radiation-induced disruption of tight junctions in the intestinal epithelial monolayers.
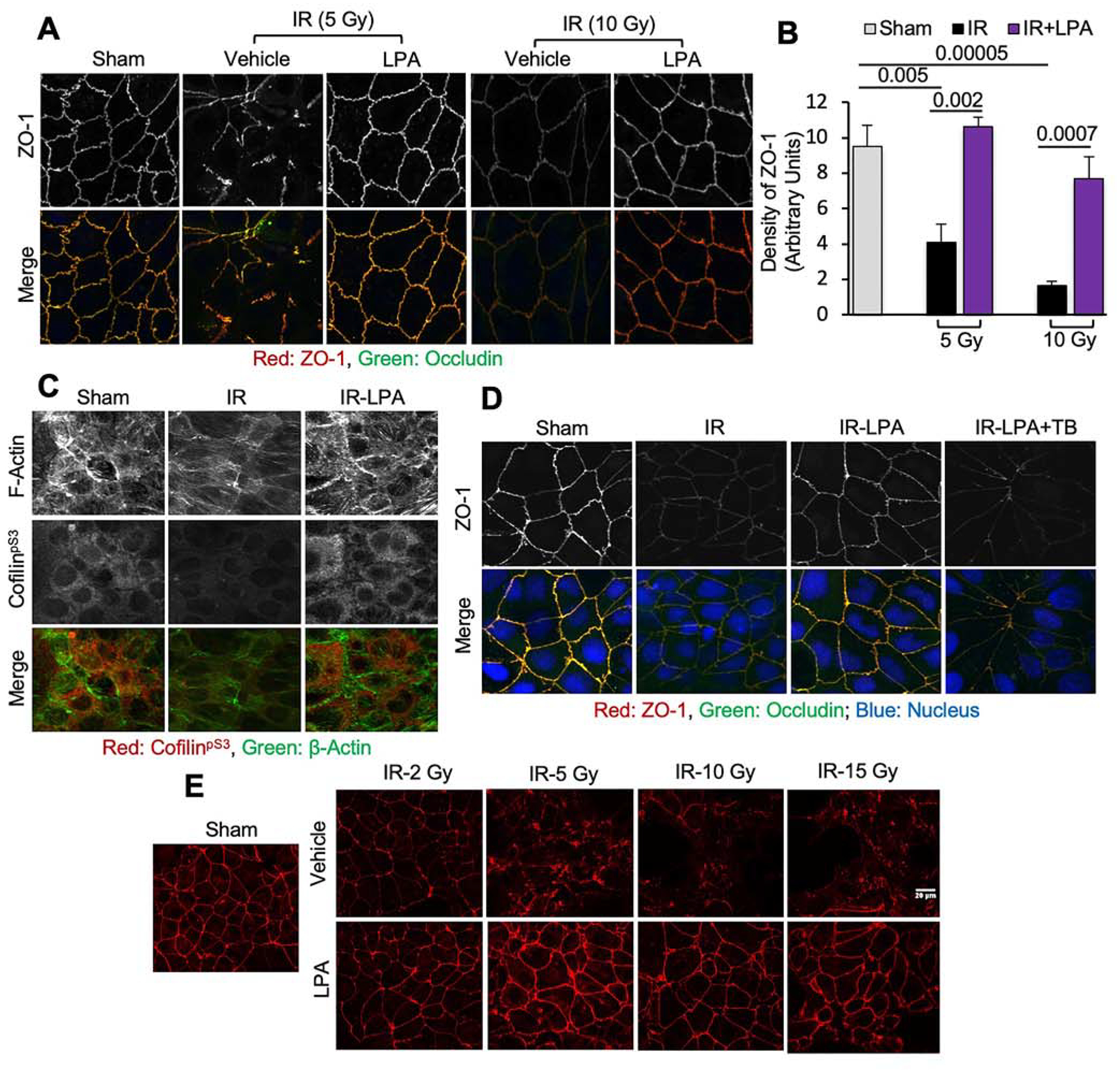
Caco-2 and m-ICC12 cell monolayers on Transwell inserts were irradiated (2–15 Gy) with or without LPA (10 μM) administered 30 min prior to irradiation. One hour after irradiation, tight junction integrity was examined. A & B: Caco-2 cell monolayers exposed to radiation with or without LPA were fixed and stained for occludin (green) and ZO-1 (red). Confocal images (A) and fluorescence density at the junctions (B) are presented. Values are Mean ± SEM (n = 4). The corresponding p-values (above bars) indicate significant differences between groups. NS indicates that the p-value is greater than 0.05. C: Caco-2 cell monolayers exposed to radiation (10 Gy) with or without LPA were stained for F-actin (green) and cofilinpS3 (red). D: Caco-2 cell monolayers were pretreated with toxin-B (TB) 30 min before LPA treatment. Monolayers were irradiated (10 Gy) 30 min after LPA treatment and stained for occludin (green), ZO-1 (red), and nucleus (blue). E: Confocal images for ZO-1 in m-ICC12 cell monolayers that were treated with LPA or vehicle before irradiation.
Effect of LPAR2 deficiency on the radiation-induced disruption of tight junction and adherens junction in the mouse colon in vivo
The LPAR2 receptor plays a crucial role in many intestinal mucosal protective functions of LPA, including inhibition of cholera toxin-induced diarrhea (36), intestinal ion transport (37–39), and prevention of radiation-induced intestinal mucosal atrophy and stem cell ablation (28). In this study, we examined the effect of LPAR2 deficiency on the severity of radiation-induced disruption of colonic epithelial tight junctions in vivo at 2 hours after irradiation. Our previous study showed that radiation disrupts colonic epithelial tight junction as early as 2 hours post-irradiation and that the damage is sustained for at least 24 hours (11). In addition to disruption of tight junction, the apoptotic cell death may contribute to barrier dysfunction. However, radiation-induced apoptosis develops after 6–8 hours of radiation exposure (24, 25). Because the focus of this study was on tight junction disruption, we chose a 2-hour post-irradiation time interval for these analyses. Immunofluorescence confocal images show that redistribution of occludin and ZO-1 from the epithelial junctions was more severe in Lpar2−/− mice compared to that present in wildtype mouse colons (Fig. 2A). Similarly, radiation-induced redistribution of adherens junction proteins, E-cadherin, and β-catenin from the epithelial junctions was more severe in the colon of Lpar2−/− mice compared to that of wildtype mice (Fig. 2B).
Figure 2: Effect of LPA2 receptor deficiency on the radiation-induced disruption of tight junction and adherens junction.

Wild type (WT) and LPA2 receptor knockout (Lpar2−/−) mice were subjected to TBI (9.5 Gy). Two hours after irradiation, colonic sections were stained for tight junction and adherens junction proteins. A: Merged fluorescence images for occludin (green), ZO-1 (red), and nucleus (blue). B: Fluorescence images for E-Cadherin (green), β-Catenin (red), and nucleus (blue). Representative images from 4 mice per each irradiated group and 2 mice for control groups are presented.
RP1 blocks radiation-induced disruption of apical junctional complexes, barrier dysfunction, and endotoxemia
RP1, a stable non-lipid LPAR2-specific agonist (13), protects the intestinal mucosa from diarrhea and mucosal atrophy caused by various types of insults (14, 28, 39). In the current study, we evaluated the effect of RP1 on the radiation-induced disruption of intestinal epithelial tight junctions and adherens junctions, an increase in mucosal permeability, and endotoxemia. We analyzed barrier dysfunction at 2 and 4 hours post-irradiation to focus on barrier dysfunction that was due to tight junction disruption rather than apoptosis. Prophylactic administration of RP1 (0.5 mg/kg; s.c.) at 24 hours pre-irradiation blocked the radiation-induced loss of junctional distribution of occludin and ZO-1 when examined at 2 hours post-irradiation (Fig. 3A). Similarly, RP1 administration blocked radiation-induced redistribution of E-cadherin and β-catenin from the epithelial junctions (Fig. 3B). Densitometric analysis of ZO-1 (Fig. 3C) and E-cadherin (Fig. 3D) fluorescence at the junctions indicated that RP1 completely attenuated the radiation-induced redistribution of these proteins from the junctions. Immunoblot analysis (Fig. 3E) of total protein extracts from the colonic mucosa showed that irradiation significantly reduced the levels of occludin (Fig. 3F), claudin-3 (Fig. 3G), and β-catenin (Fig. 3H); this effect of radiation was blocked by RP1. Mucosal permeability in the colon and ileum in vivo was measured at 2 hours and 4 hours after irradiation. RP1 significantly reduced radiation-induced mucosal permeability in the colon (Fig. 3I). RP1 showed no significant effect on inulin permeability in the ileum at these time points (Fig. 3J). Prevention of radiation-induced colonic mucosal permeability by RP1 at 4 hours post-irradiation was associated with a significant (p = 0.04) reduction of the radiation-induced increase in plasma LPS levels (Fig. 3K).
Figure 3: RP1 blocks TBI-induced disruption of tight junction and adherens junction, barrier dysfunction, and endotoxemia.

A-D: Wild type mice were injected with RP1 (0.5 mg/kg) or vehicle (Veh) 30 min prior to TBI (9.5 Gy) (IR and IR+RP1); the control group was sham-treated. At 2 hours post-irradiation, colonic sections were co-stained for occludin, ZO-1, and nucleus (A) or E-cadherin, β-catenin, and nucleus (B). Confocal images were captured, and fluorescence densities at the junctions were measured. Fluorescence densities for ZO-1 (C) and E-cadherin (D) are presented. Values are Mean ± SEM (n = 4; each value is an average of fluorescence values from 10 regions within the individual colonic section). E-H: At 4 hours post-irradiation, colonic mucosal extracts were immunoblotted (E), and the band densities for occludin (OCLN; F), claudin-3 (CLDN; G), and β-catenin (H). Values are Mean ± SEM (n = 4). I-K: At 2 and 4 hours post-irradiation, mucosal permeability in vivo in the colon (E) and ileum (F) and the plasma LPS levels (G) were measured. Values are Mean ± SEM (n = 4). In all graphs, the numbers above the bars are p-values for significant differences between groups indicated by the horizontal bars. “NS” indicates no significant difference between groups with the p-values greater than 0.05. The experiment was repeated with similar results.
RP1 mitigates radiation-induced disruption of apical junctional complexes, barrier dysfunction, and endotoxemia
This study was performed to determine whether RP1 administered 24 hours after irradiation is effective in reversing radiation damage. The United States Government is actively seeking radio-mitigators that are effective when administered 24 hours after irradiation (40). Therefore, in this study, we administered RP1 at 24 hours after irradiation (0.5 mg/kg; s.c.) and examined the barrier function at 24 hours after RP1 treatment to determine the reversibility of radiation damage. Post-irradiation treatment with RP1 reversed the radiation-induced loss of junctional occludin and ZO-1 when examined at 24 hours after RP1 treatment (Fig. 4A). Similarly, RP1 reversed radiation-induced redistribution of E-cadherin and β-catenin from the epithelial junctions (Fig. 4B). Radiation also induced redistribution of claudin-3, another transmembrane protein of tight junction in mouse colons (Fig. 4C). Post-irradiation treatment with RP1 reversed this effect of radiation on the colonic epithelial distribution of claudin-3. Densitometric analysis of ZO-1 (Fig. 4D), β-catenin (Fig. 4E), and claudin-3 (Fig. 4F) at the junctions confirmed that RP1 completely attenuated radiation-induced redistribution of these proteins from the junctions. Mucosal permeability in colon and ileum in vivo was measured at 24 and 48 hours RP1 treatment. RP1 significantly alleviated radiation-induced mucosal permeability in the colon (Fig. 4G) and ileum (4H). Restoration of radiation-induced loss of intestinal mucosal permeability by RP1 was associated with a significant (p = 0.015 at 24 hours & 0.0001 at 48 hours) reduction in radiation-induced elevation of plasma LPS (Fig. 4I).
Figure 4: RP1 mitigates TBI-induced disruption of AJC, mucosal barrier dysfunction, and endotoxemia.

Adult wild type mice were subjected to TBI (9.5 Gy). At 24 hours after irradiation, mice were injected with RP1 (0.5 mg/kg daily) or vehicle (Veh); the control group was sham-treated. At 24 and 48 hours after RP1 treatment, junctional integrity (A-F), gut permeability in vivo (G & H), and endotoxemia (I) were evaluated. Sections of the colon were co-stained for occludin, ZO-1, and nucleus (A), E-cadherin, β-catenin, and nucleus (B) or claudin-3, Claudin-2, and nucleus (C). Confocal fluorescence images were captured, and fluorescence densities at the junctions were measured. Fluorescence densities for ZO-1 (D) and β-catenin (E) and Claudin-3 (F) are presented. Mucosal permeability in vivo in the colon (G) and ileum (H) and plasma LPS levels (I) were measured. Values are Mean ± SEM (n = 4). The corresponding p-values (above bars) indicate significant differences between groups. NS indicates that the p-value is greater than 0.05. The experiment was repeated with similar results. Similar results were also produced in a similar experiment analyzed at seven days post-irradiation.
RP1 mitigates radiation-induced suppression of antioxidant gene expression
Our previous study demonstrated that radiation induces oxidative stress, and the antioxidant N-acetyl-L-cysteine protects and mitigates radiation-induced disruption of tight junctions and barrier dysfunction in mouse colons (11). In this study, we evaluated the effect of RP1 on radiation-induced effects on antioxidant gene expression to determine the reversibility of oxidative stress by RP1. RP1 (0.5 mg/kg daily; s.c.) administered at 24 hours post-irradiation reversed the radiation-induced reduction of mRNA for Gpx1 (Fig. 5A), SOD1 (Fig. 5B), and Prdx1 (Fig. 5C). However, RP1 failed to reverse radiation-induced reduction of CAT (catalase) mRNA (Fig. 5D). The mRNA levels for Nrf2, the transcription factor involved in antioxidant gene expression, were reduced by radiation; this effect of radiation was partially reversed by RP1 treatment (Fig. 5E).
Figure 5: RP1 alleviates TBI-induced downregulation of antioxidant gene expression.

Wild type (WT) mice were subjected to TBI (9.5 Gy). At 24 hours after irradiation, mice were injected with RP1 (0.5 mg/kg daily) or vehicle (Veh); the control group was sham-treated. At 24 hours after RP1 treatment, RNA extracted from colonic mucosa was analyzed for mRNA for Gpx1 (A), SOD1 (B), CAT (C), Prdx1 (D), and Nrf2 (E) by RT-qPCR. Values are Mean ± SEM (n = 4). The corresponding p-values (above bars) indicate significant differences between groups. NS indicates that the p-value is greater than 0.05.
Effects of RP1 on the tight junction and adherens junction integrity in the PBI-BM5 model
We evaluated the impact of PBI-BM5 on the intestinal epithelial tight junction integrity and its prevention by RP1 treatment. Unlike the rapid effect of TBI, PBI effects on tight junction disruption were slower. Therefore, the analyses were performed at 28–76 hours post-irradiation. Immunofluorescence confocal microscopy showed that PBI-BM5 induced a redistribution of occludin and ZO-1 from the colonic epithelial junctions when examined at 52 hours post-irradiation, and RP1 (3 mg/kg daily; s.c.) treatment alleviated this effect (Fig. 6A). Similarly, PBI induced redistribution of adherens junction proteins, E-cadherin, and β-catenin from epithelial junctions, and RP1 alleviated this effect (Fig. 6B). Densitometric analysis for ZO-1 fluorescence at the junctions indicated that PBI-BM5 did not affect tight junction integrity at 28 hours post-irradiation but did induce a severe disruption of tight junctions at 52 and 76 hours (Fig. 6C). RP1 treatment completely blocked PBI-induced ZO-1 redistribution at both 52 and 76 hours post-irradiation. RP1 also prevented PBI-induced reduction of junctional E-cadherin levels (Fig. 6D).
Figure 6: RP1 blocks PBI-induced disruption of AJC, barrier dysfunction, and endotoxemia.

Adult wild type mice were injected with RP1 (3 mg/kg) or vehicle (Veh) daily starting one day after partial body irradiation (PBI; 15.6 Gy); the control group was sham-treated. At varying times after irradiation, colonic sections were co-stained for occludin, ZO-1, and nucleus (A) or E-cadherin, β-catenin, and nucleus (B). Confocal fluorescence images were captured, and the fluorescence densities of ZO-1 (C) and E-cadherin (D) at the junctions were measured. Values are Mean ± SEM (n = 4). The corresponding p-values (above bars) indicate significant differences between groups. NS indicates that the p-value is greater than 0.05. Similar results were produced in a similar experiment when examined at 72 and 96 hours post-irradiation.
RP1 attenuates PBI-BM5-induced oxidative stress in colonic mucosa
Protein thiol oxidation was examined by fluorescence staining of reduced-protein thiols and oxidized-protein thiols at 52 hours post-irradiation, the earliest time at which RP1 effects were recorded. Fluorescence images (Fig. 7A) and densitometric analyses (Fig. 7B) indicated that PBI-BM5 depleted the reduced-protein thiols with a corresponding increase in oxidized-protein thiols and that RP1 treatment (3 mg/kg daily; s.c.; beginning 24 hours after irradiation) reversed PBI-induced protein thiol oxidation. The expression of Nrf2 was examined by immunofluorescence staining (Fig. 7C), immunoblot analysis (Fig. 7D), and Nrf2-specific mRNA measurement (Fig. 7E). All of these analyses indicated that PBI-BM5 significantly reduced Nrf2 expression, while RP1 blocked this effect of radiation. PBI-BM5 significantly reduced SOD1 (Fig. 7F), Gpx1 (Fig. 7G), CAT (catalase) (Fig. 7H), Trx1 (Fig. 7I), and Prdx1 (Fig. 7J) mRNA levels, while RP1 attenuated these effects of PBI-BM5.
Figure 7: RP1 mitigates PBI-induced oxidative stress.

Adult mice were injected with RP1 (3 mg/kg) or vehicle (Veh) daily starting one day after partial body irradiation (PBI-BM5; 15.6 Gy); the control group was sham-treated. At 48 hours after irradiation (or 24 hours after RP1), colonic sections were stained for reduced-protein thiols, oxidized-protein thiols, and NRF2. Antioxidant gene expression was analyzed by RT-qPCR. A & B: Confocal images for protein thiols were captured (A), and the fluorescence densities were measured (B). C & D: Colonic sections were co-stained for F-actin (green), NRF2 (red) and nucleus (blue) (C). The protein extracts were immunoblotted for NRF2, and the band densities were measured. E-J: RNA isolated from colonic mucosa was analyzed for mRNA for Nrf2 (E), SOD1 (F), Gpx1 (G), CAT (H), Trx1 (I), and Prdx1 (J) by RT-qPCR. Values are Mean ± SEM (n = 4). The corresponding p-values (above bars) indicate significant differences between groups. NS indicates that the p-value is greater than 0.05. Similar results were produced in a similar experiment when examined at 72 and 96 hours post-irradiation.
RP1 attenuates PBI-BM5-induced F-actin remodeling and its association of apical junctional complexes
The association of tight junction and adherens junction proteins with the actin cytoskeleton was assessed by Western blot analysis of the actin-rich, detergent-insoluble fraction for tight junction and adherens junction proteins. The analyses were performed at 52 hours post-irradiation, the earliest time when the RP1 effect was recorded. Densitometric analysis of immunoblot bands (Fig. 8A) for E-cadherin (Fig. 8B), β-catenin (Fig. 8C), and Claudin-3 (Fig. 8D) indicated that PBI-BM5 significantly reduced the detergent-insoluble fraction of these proteins, which was blocked by RP-1 treatment. Although the molecular weight of occludin is around 60 kDa, it usually appears in multiple bands in Western blots due to phosphorylation at multiple sites (6). PBI-BM5 or RP1 did not significantly alter occludin (Fig. 8E) and β-actin (Fig. 8F) levels. Immunofluorescence analysis of F-actin showed that PBI-BM5 reduced F-actin levels, and RP1 treatment attenuated this effect of radiation (Fig. 8G). Immunofluorescence staining (Fig. 8H) and immunoblot analysis (Fig. 8I) indicated that PBI-BM5 reduced the levels of cofilinpS3 in the colonic mucosa, and RP1 blocked this effect of radiation.
Figure 8: RP1 mitigates PBI-induced F-actin remodeling and its association with apical junctional proteins.

Adult mice were injected with RP1 (3 mg/kg) or vehicle (Veh) daily starting one day after partial body irradiation (PBI-BM5; 15.6 Gy); the control group was sham-treated. A-E: At 48 hours after irradiation (or 24 hours after RP1), detergent-insoluble fractions of colonic mucosa were immunoblotted for tight junction and adherens junction proteins as well as NRF2 and β-actin (A). Band densities for E-cadherin (B), β-catenin (C), CLDN-3 (D), ZO-1 (E), and β-actin (F) were measured. G: Cryosections of the colon were fixed and stained for F-actin (red) and nucleus (blue). H & I: Colonic sections were co-stained for cofilinpS3 (red), F-actin (green) and nucleus (blue) (H). Mucosal extracts were immunoblotted for cofilinpS3 and β-actin (I). Band densities were measured, and the cofilinpS3 band densities presented by values normalized to corresponding actin band densities (I). In all graphs, values are Mean ± SEM (n = 3). The corresponding p-values (above bars) indicate significant differences between groups. NS indicates that the p-value is greater than 0.05.
Discussion
A significant body of evidence indicates that LPA plays a crucial role in the growth and differentiation of intestine as well as the protection of intestinal mucosa from a variety of noxious conditions (36, 38, 39, 41, 42). The endogenous LPA and synthetic analogs such as RP-1 protect the gut from the genotoxic stress-triggered sequelae of pathophysiological events that underlie the acute gastrointestinal radiation syndrome (30). However, the role of LPA in the protection of the colonic mucosal barrier function is poorly understood. In this new study, we present the evidence that LPAR2 receptor activation is radioprotective in the colon by demonstrating that the LPAR2 receptor agonists, LPA and RP1) protect the intestinal epithelium against radiation-induced disruption of apical junctional complexes and prevents or alleviates barrier dysfunction.
In vitro studies in transformed Caco-2 and non-transformed m-ICC12 epithelial monolayers showed that radiation induces a rapid disruption of intestinal epithelial tight junctions in a dose-dependent manner. In a previous study, we have reported that radiation causes disruption of mouse colonic epithelial tight junctions in vivo as early as 2 hours post-irradiation (11). The present in vitro data demonstrate that radiation directly affects the intestinal epithelium without the systemic influences. The primary cause of radiation-induced cell injury is due to the generation of free radicals and oxidative stress (43). In the later stages, radiation-induced DNA damage may affect gene expression and contribute to long-term tissue damage (44). In the current study, tight junction disruption at two hours post-irradiation was likely caused by oxidative stress. Our previous study demonstrated that radiation-induced tight junction disruption in the colon could be blocked and restored by the antioxidant N-acetyl cysteine (11).
The protective effect of LPAR2 in irradiated Caco-2 and m-ICC12 cell monolayers indicates that LPA directly interacts with epithelial cells to attenuate radiation-induced tight junction disruption. Irradiation of Caco-2 cell monolayers was associated with a reduction in levels of the inactive form of cofilinpS3. This decrease in cofilinpS3 levels without a change in total cofilin indicates that radiation causes cofilin activation. Disruption of the actin cytoskeleton was previously shown to disrupt tight junctions, a common mechanism involved in tight junction disruption mediated by various injurious factors (45). Therefore, activation of cofilin, an actin severing protein, is likely involved in radiation-induced tight junction disruption and disruption of the actin cytoskeleton. Cofilin is known to be inactivated by LIM kinase-mediated phosphorylation on S3 residue (46), and LPA activates the Rho-ROCK-LIM kinase pathway (47). Abrogation of LPA-mediated protection of tight junctions by the Rho-GTPase inhibitor Toxin-B suggests that LPA likely promotes cofilin phosphorylation by the Rho-Rock-LIM kinase pathway.
Previous studies have indicated that the intestinal mucosal protective effects of LPA are mediated by activation of the LPAR2 receptor (12, 15, 16, 19, 28, 36, 39, 48–50). Data from the present study show that radiation-induced disruption of tight junction and adherens junction is more severe in LPAR2 deficient-mice compared to wildtype mice, suggesting that LPAR2 activity exerts a protective effect on intestinal epithelial tight junctions and adherens junctions in vivo. The LPAR2 receptor’s role in the protection of tight junction was further determined by evaluating the impact of an LPAR2-selective agonist, RP1 (13), on the radiation-induced disruption of intestinal epithelial junctions. The prevention of radiation-induced redistribution of tight junction and adherens junction proteins from the intercellular junctions by RP1-pretreatment indicates that RP1 and LPAR2 receptor activation blocks radiation-induced disruption of AJC in the mouse colon. Radiation-induced disruption of the tight junction was associated with an increase in mucosal permeability to inulin, indicating radiation-induced barrier disruption in the mouse colon in vivo.
Furthermore, barrier dysfunction was associated with an increase in plasma LPS in irradiated mice, which demonstrated radiation-induced endotoxemia. Prophylactic administration of RP1 blocked radiation-induced colonic mucosal permeability and endotoxemia. Therefore, data from this part of the study indicates that LPAR2 activation via prophylactic RP1-treatment prevents radiation-induced disruption of intestinal epithelial AJC, mucosal barrier dysfunction, and endotoxemia. Our previous study indicated that TBI disrupts colonic epithelial tight junction and causes barrier dysfunction as early as 2 hours post-irradiation and that this damage is sustained for at least 24 hours (11). Radiation-induced apoptosis in the intestine takes 6–8 hours (24, 25), and therefore the barrier dysfunction recorded in the first part of the current study was exclusively caused by tight junction disruption.
To determine whether RP1 can mitigate radiation-induced intestinal barrier dysfunction, we evaluated the effect of RP1 when administered at 24 hours post-irradiation in mice in vivo. The results of this study showed that RP1 completely reverses radiation-induced redistribution of occludin, ZO-1, claudin-3, E-cadherin and β-catenin, indicating the reversibility of radiation-induced disruption of tight junctions and adherens junctions, and the presence of functioning LPAR2 in the irradiated enterocytes. RP1 also reversed the radiation-induced increase in ileal and colonic mucosal permeability to inulin and endotoxemia. The permeability changes in these tissues are likely due to tight junction disruption. However, we cannot rule out the contribution by apoptosis of epithelial cells, as the permeability analyses were performed at 48 hours post-irradiation. Additionally, a previous study indicated that pathologic removal of epithelial cells by apoptosis does not result in loss of intestinal barrier function (51). This result demonstrates that the LPAR2 is a useful target for the treatment of radiation injury to the gut and that RP1 is effective in alleviating radiation-induced intestinal damage.
Our previous study indicated that radiation caused oxidative stress in the colonic mucosa and that N-acetylcysteine, the antioxidant, effectively blocked radiation-induced disruption of colonic epithelial tight junctions and barrier dysfunction (11). In the present study, we examined the effect of RP1 on radiation-induced oxidative stress by measuring levels of mRNA for antioxidant genes in the colonic mucosa. A significant reduction in mRNA for Gpx1, SOD1, Prdx1, CAT (catalase), and Nrf2 indicated that radiation suppressed antioxidant gene expression in colonic mucosa. Data also showed that RP1 completely reversed radiation-induced suppression of Gpx1, SOD1, and Prdx1 expression, and partially reversed the effect of radiation on Nrf2 expression; RP1 showed no significant impact on catalase expression. Therefore, RP1-mediated activation of the LPAR2 receptor provided substantial protection against oxidative damage by irradiation.
All of the studies described above applied the TBI model to evaluate the effects of RP1. To mimic the GI-ARS conditions, we assessed the impact of RP1 in the PBI-BM5 model in which the shielded bone marrow expands and allows for the survival of the animal proviso protection of the gut mucosa. At 28 hours post-irradiation, the junctional distribution of tight junction and adherens junction proteins were unaffected, indicating that PBI-BM5 did not affect intestinal tight junctions and adherens junctions until 28 hours post-irradiation. This PBI effect is in contrast to TBI effects, which disrupted tight junctions within 2 hours post-irradiation. However, by 52 hours post-irradiation, PBI-BM5 caused a severe loss in the junctional distribution of occludin, ZO-1, E-cadherin, and β-catenin that was sustained for at least 76 hours. RP1 (3 mg/kg daily, s.c.) treatment beginning 24 hours post-irradiation completely alleviated PBI-induced disruption of tight junctions and adherens junctions at 52 and 76 hours post-irradiation. These data demonstrate that LPAR2 receptor activation via RP1 administered 24 hours post-irradiation can alleviate PBI-induced damage to the intestinal epithelial junctions. Conventionally, PBI-BM5 is characterized by higher dose (>5 Gy) radiation exposure and sparing of 2.5–5.0% of bone marrow; this is considered the ideal model for GI-ARS. TBI, characterized by the ablation of 100% of bone marrow, is regarded as hematopoietic acute radiation syndrome (H-ARS). Interestingly, in the present study, we showed that PBI-BM5 does not affect colonic epithelial tight junction for at least 28 hours post-irradiation; tight junction disruption was observed at 52 hours and sustained for at least 100 hours post-irradiation. This is distinctly different from the effect of TBI. TBI caused tight junction disruption in less than 2 hours post-irradiation. This observation suggests that 5% of bone marrow and bone marrow-derived factors may have delayed the damage to colonic tight junction disruption and mucosal barrier dysfunction. A new avenue of studies is necessary to understand the protective role of bone marrow in the intestinal epithelium.
Dramatic reduction in levels of reduced-protein thiols accompanied by elevation of oxidized-protein thiols in the colon indicates that PBI-BM5 induces oxidative stress in colonic mucosa. Here we demonstrated for the first time that RP1-treatment significantly attenuated PBI-BM5-induced oxidative stress. PBI-BM5-induced oxidative stress was associated with modulation of Nrf2 and antioxidant gene expression. NRF2 is a transcription factor critical for the expression of antioxidant genes (52, 53). Immunofluorescence imaging, immunoblot analysis, and Nrf2 mRNA measurement by RT-qPCR indicated that PBI-BM5 caused a significant reduction of Nrf2 expression, which was prevented by RP1 at 24 hours post-irradiation. PBI-BM5-mediated decreases in the levels of Gpx1, SOD1, Prdx1, Trx1, and CAT mRNA and its prevention by RP1 indicate that PBI suppresses antioxidant gene expression and that RP1 alleviates this effect. Therefore, induction of oxidative stress is likely one of the mechanisms involved in PBI-BM5-induced intestinal barrier dysfunction. This effect underlines the antioxidant action of RP1-mediated activation of LPAR2 receptors in the mechanism of its protective effects.
To determine whether disruption of the actin cytoskeleton and loss of its interaction with tight junction and adherens junction are involved in the mechanism of PBI-BM5-induced disruption of the apical junctional complex, we measured levels of detergent-insoluble fractions of tight junction and adherens junction. The detergent-insoluble fraction predominantly consists of the actin cytoskeleton. In the intact epithelium and non-disrupted tight junctions, the junctional proteins are bound to the actin cytoskeleton; therefore, they are recovered in the actin-rich detergent-insoluble fractions (7). Disruption of the actin cytoskeleton leads to disruption of tight junctions and loss of detergent-insoluble fractions of tight junction proteins (45). The present study showed that PBI-BM5 significantly reduced the detergent-insoluble fractions of claudin-3, E-cadherin, and β-catenin, which was blocked by RP1-treatment. Data indicated that γ-irradiation disrupts the association between the actin cytoskeleton and the junctional proteins. Furthermore, it suggests that preservation of the actin cytoskeletal integrity may be involved in the mechanism of RP1-mediated protection of AJC in irradiated mice. The results of this study also show that γ-irradiation decreases F-actin levels, likely through activating cofilin in the colonic mucosa, and that RP1 treatment blocks this effect via LPAR2 receptor-mediated activation of the Rho-Rock-LIM kinase pathway. A working model for the potential mechanisms involved in radiation-induced tight junction disruption in colonic epithelium and its prevention by LPA agonists is presented in Figure 9.
Figure 9: Schematic outlining working model of the potential mechanisms associated with radiation-induced gut barrier dysfunction and its prevention by LPAR2 agonists.

Radiation induces oxidative stress by the production of reactive oxygen species (ROS) and down-regulation of antioxidant gene expression. Oxidative stress disrupts tight junctions by either signaling a cascade that targets tight junction directly or induces remodeling of the actin cytoskeleton via cofilin activation that leads to tight junction disruption. LPA and analogs that activate the LPAR2 receptor prevent and mitigate radiation-induced tight junction disruption, barrier dysfunction, and endotoxemia by blocking radiation-induced oxidative stress.
In summary, this study demonstrated that γ-irradiation disrupts AJC in the colonic epithelium, induces mucosal barrier dysfunction, and causes endotoxemia, likely by inducing oxidative stress and disrupting the actin cytoskeleton. Furthermore, our data demonstrate that activation of the LPAR2 receptor prevents and mitigates radiation-induced intestinal barrier dysfunction and endotoxemia. Therefore, by protecting the AJCin the colonic mucosa, LPAR2 agonists like RP1 could have therapeutic benefits in treating diseases associated with disruption of the intestinal barrier.
Supplementary Material
Acknowledgements
This study was supported by the National Institute of Health grants DK55532, AA12307, CA013621, and 1UO-1AI107331. The authors thank Dr. Nathan G. Tipton (University of Tennessee Health Science Center) for his expert editing of the manuscript for language and scientific writing.
Abbreviations
- AJC
Apical junctional complex
- AJ
Adherens junction
- ARS
Acute radiation syndrome
- Cldn
Claudin
- FITC
Fluorescein isothiocyanate
- GI
Gastrointestinal
- GI-ARS
Gastrointestinal-Acute Radiation Syndrome
- HRP
Horseradish peroxidase
- LPA
Lysophosphatidic acid
- LPS
Lipopolysaccharide
- Lpar2/LPAR2
Lysophosphatidic acid receptor 2
- PBI-BM5
Partial body irradiation with 5% bone marrow shielded
- RP-1
Radioprotectin-1
- TBI
Total body irradiation
- TJ
Tight junction
- ZO-1
Zona occludens-1
Footnotes
Conflict of interest
GT is a founder and stockholder in RxBio Inc. that has licensed the intellectual property for RP-1.
Availability of data
Data supporting the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
References
- 1.Anderson JM, and Van Itallie CM (2009) Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 1, a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao R (2008) Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci 13, 7210–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Itallie CM, and Anderson JM (2006) Claudins and epithelial paracellular transport. Annu Rev Physiol 68, 403–429 [DOI] [PubMed] [Google Scholar]
- 4.Basuroy S, Seth A, Elias B, Naren AP, and Rao R (2006) MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J 393, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basuroy S, Sheth P, Kuppuswamy D, Balasubramanian S, Ray RM, and Rao RK (2003) Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and accelerates calcium-mediated reassembly of tight junctions in the Caco-2 cell monolayer. J Biol Chem 278, 11916–11924 [DOI] [PubMed] [Google Scholar]
- 6.Rao RK, Basuroy S, Rao VU, Karnaky KJ Jr, and Gupta A (2002) Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J 368, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seth A, Sheth P, Elias BC, and Rao R (2007) Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J Biol Chem 282, 11487–11498 [DOI] [PubMed] [Google Scholar]
- 8.Sheth P, Samak G, Shull JA, Seth A, and Rao R (2009) Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem J 421, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheth P, Seth A, Atkinson KJ, Gheyi T, Kale G, Giorgianni F, Desiderio DM, Li C, Naren A, and Rao R (2007) Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem J 402, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao RK, Seth A, and Sheth P (2004) Recent Advances in Alcoholic Liver Disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 286, G881–884 [DOI] [PubMed] [Google Scholar]
- 11.Shukla PK, Gangwar R, Manda B, Meena AS, Yadav N, Szabo E, Balogh A, Lee SC, Tigyi G, and Rao R (2016) Rapid disruption of intestinal epithelial tight junction and barrier dysfunction by ionizing radiation in mouse colon in vivo: protection by N-acetyl-l-cysteine. Am J Physiol Gastrointest Liver Physiol 310, G705–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, and Johnson LR (2002) Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology 123, 206–216 [DOI] [PubMed] [Google Scholar]
- 13.Patil R, Fells JI, Szabo E, Lim KG, Norman DD, Balogh A, Patil S, Strobos J, Miller DD, and Tigyi GJ (2014) Design and synthesis of sulfamoyl benzoic acid analogues with subnanomolar agonist activity specific to the LPA2 receptor. J Med Chem 57, 7136–7140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patil R, Szabo E, Fells JI, Balogh A, Lim KG, Fujiwara Y, Norman DD, Lee SC, Balazs L, Thomas F, Patil S, Emmons-Thompson K, Boler A, Strobos J, McCool SW, Yates CR, Stabenow J, Byrne GI, Miller DD, and Tigyi GJ (2015) Combined mitigation of the gastrointestinal and hematopoietic acute radiation syndromes by an LPA2 receptor-specific nonlipid agonist. Chem Biol 22, 206–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng W, Shuyu E, Tsukahara R, Valentine WJ, Durgam G, Gududuru V, Balazs L, Manickam V, Arsura M, VanMiddlesworth L, Johnson LR, Parrill AL, Miller DD, and Tigyi G (2007) The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology 132, 1834–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng W, Kimura Y, Gududuru V, Wu W, Balogh A, Szabo E, Emmons-Thompson K, Yates CR, Balazs L, Johnson LR, Miller DD, Strobos J, W.S. M, and Tigyi GJ (2015) Mitigation of the Hematopoietic and Gastrointestinal Acute Radiation Syndrome by Octadecylthiophosphate a Small Molecule Mimic of Lysophosphatidic Acid. Radiation Res. 183, 465–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiss GN, Lee SC, Fells JI, Liu J, Valentine WJ, Fujiwara Y, Thompson KE, Yates CR, Sumegi B, and Tigyi G (2013) Mitigation of radiation injury by selective stimulation of the LPA(2) receptor. Biochim Biophys Acta 1831, 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng W, Kimura Y, Gududuru V, Wu W, Balogh A, Szabo E, Thompson KE, Yates CR, Balazs L, Johnson LR, Miller DD, Strobos J, McCool WS, and Tigyi GJ (2015) Mitigation of the hematopoietic and gastrointestinal acute radiation syndrome by octadecenyl thiophosphate, a small molecule mimic of lysophosphatidic acid. Radiat Res 183, 465–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin FT, Lai YJ, Makarova N, Tigyi G, and Lin WC (2007) The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J Biol Chem 282, 37759–37769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bens M, Bogdanova A, Cluzeaud F, Miquerol L, Kerneis S, Kraehenbuhl JP, Kahn A, Pringault E, and Vandewalle A (1996) Trans-immortalized intestinal cells (m-ICcl2) that maintain a crypt phenotype. Am. J. Physiol. Cell Physiol 270, C1666–C1674 [DOI] [PubMed] [Google Scholar]
- 21.Sheth P, Samak G, Shull JA, Seth A, and Rao R (2009) Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem J 421, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendry JH, Potten CS, Chadwick C, and Bianchi M (1982) Cell death (apoptosis) in the mouse small intestine after low doses: effects of dose-rate, 14.7 MeV neutrons, and 600 MeV (maximum energy) neutrons. Int J Radiat Biol Relat Stud Phys Chem Med 42, 611–620 [DOI] [PubMed] [Google Scholar]
- 23.Potten CS (1977) Extreme sensitivity of some intestinal crypt cells to X and gamma irradiation. Nature 269, 518–521 [DOI] [PubMed] [Google Scholar]
- 24.Potten CS (1990) A comprehensive study of the radiobiological response of the murine (BDF1) small intestine. Int J Radiat Biol 58, 925–973 [DOI] [PubMed] [Google Scholar]
- 25.Potten CS, Merritt A, Hickman J, Hall P, and Faranda A (1994) Characterization of radiation-induced apoptosis in the small intestine and its biological implications. Int J Radiat Biol 65, 71–78 [DOI] [PubMed] [Google Scholar]
- 26.Booth C, Tudor G, Tonge N, Shea-Donohue T, and MacVittie TJ (2012) Evidence of delayed gastrointestinal syndrome in high-dose irradiated mice. Health Phys 103, 400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Booth C, Tudor G, Tudor J, Katz BP, and MacVittie TJ (2012) Acute gastrointestinal syndrome in high-dose irradiated mice. Health Phys 103, 383–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuo B, Szabo E, Lee SC, Balogh A, Norman D, Inoue A, Ono Y, Aoki J, and Tigyi G (2018) The LPA2 receptor agonist Radioprotectin-1 spares Lgr5-positive intestinal stem cells from radiation injury in murine enteroids. Cell Signal 51, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng W, Wang DA, Gosmanova E, Johnson LR, and Tigyi G (2003) LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am J Physiol Gastrointest Liver Physiol 284, G821–829 [DOI] [PubMed] [Google Scholar]
- 30.Tigyi GJ, Johnson LR, Lee SC, Norman DD, Szabo E, Balogh A, Thompson K, Boler A, and McCool WS (2019) Lysophosphatidic acid type 2 receptor agonists in targeted drug development offer broad therapeutic potential. J Lipid Res 60, 464–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shukla PK, Meena AS, Manda B, Gomes-Solecki M, Dietrich P, Dragatsis I, and Rao R (2018) Lactobacillus plantarum prevents and mitigates alcohol-induced disruption of colonic epithelial tight junctions, endotoxemia, and liver damage by an EGF receptor-dependent mechanism. Faseb J, fj201800351R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao RK, Basuroy S, Rao VU, Karnaky KJ Jr., and Gupta A (2002) Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J 368, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madara JL, Stafford J, Barenberg D, and Carlson S (1988) Functional coupling of tight junctions and microfilaments in T84 monolayers. Am J Physiol 254, G416–423 [DOI] [PubMed] [Google Scholar]
- 34.Vardouli L, Moustakas A, and Stournaras C (2005) LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. The Journal of biological chemistry 280, 11448–11457 [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Halasz E, and Townes-Anderson E (2019) Actin Dynamics, Regulated by RhoA-LIMK-Cofilin Signaling, Mediates Rod Photoreceptor Axonal Retraction After Retinal Injury. Invest Ophthalmol Vis Sci 60, 2274–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, Nelson DJ, Tigyi GJ, and Naren AP (2005) Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med 202, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singla A, Dwivedi A, Saksena S, Gill RK, Alrefai WA, Ramaswamy K, and Dudeja PK (2010) Mechanisms of lysophosphatidic acid (LPA) mediated stimulation of intestinal apical Cl-/OH- exchange. Am J Physiol Gastrointest Liver Physiol 298, G182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin S, Yeruva S, He P, Singh AK, Zhang H, Chen M, Lamprecht G, de Jonge HR, Tse M, Donowitz M, Hogema BM, Chun J, Seidler U, and Yun CC (2010) Lysophosphatidic acid stimulates the intestinal brush border Na(+)/H(+) exchanger 3 and fluid absorption via LPA(5) and NHERF2. Gastroenterology 138, 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson KE, Ray RM, Alli S, Ge W, Boler A, Shannon McCool W, Meena AS, Shukla PK, Rao R, Johnson LR, Miller MA, and Tigyi GJ (2018) Prevention and treatment of secretory diarrhea by the lysophosphatidic acid analog Rx100. Exp Biol Med (Maywood) 243, 1056–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moding EJ, Kastan MB, and Kirsch DG (2013) Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov 12, 526–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yun CC, and Kumar A (2015) Diverse roles of LPA signaling in the intestinal epithelium. Exp Cell Res 333, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konno T, Kotani T, Setiawan J, Nishigaito Y, Sawada N, Imada S, Saito Y, Murata Y, and Matozaki T (2019) Role of lysophosphatidic acid in proliferation and differentiation of intestinal epithelial cells. PLoS One 14, e0215255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Datta K, Suman S, Kallakury BV, and Fornace AJ Jr. (2012) Exposure to heavy ion radiation induces persistent oxidative stress in mouse intestine. PLoS One 7, e42224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerda H, Johanson KJ, and Rosander K (1979) Radiation-induced DNA strand breaks and their repair in the developing rat brain. Int J Radiat Biol Relat Stud Phys Chem Med 36, 65–73 [DOI] [PubMed] [Google Scholar]
- 45.Madara JL, Moore R, and Carlson S (1987) Alteration of intestinal tight junction structure and permeability by cytoskeletal contraction. Am J Physiol 253, C854–861 [DOI] [PubMed] [Google Scholar]
- 46.Chen X, and Macara IG (2006) Par-3 mediates the inhibition of LIM kinase 2 to regulate cofilin phosphorylation and tight junction assembly. J Cell Biol 172, 671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandey D, Goyal P, and Siess W (2007) Lysophosphatidic acid stimulation of platelets rapidly induces Ca2+-dependent dephosphorylation of cofilin that is independent of dense granule secretion and aggregation. Blood Cells Mol Dis 38, 269–279 [DOI] [PubMed] [Google Scholar]
- 48.Khurana S, Tomar A, George SP, Wang Y, Siddiqui MR, Guo H, Tigyi G, and Mathew S (2008) Autotaxin and lysophosphatidic acid stimulate intestinal cell motility by redistribution of the actin modifying protein villin to the developing lamellipodia. Exp Cell Res 314, 530–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida M, He P, and Yun CC (2016) Transgenic Expression of Human Lysophosphatidic Acid Receptor LPA2 in Mouse Intestinal Epithelial Cells Induces Intestinal Dysplasia. PLoS One 11, e0154527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elias BC, Suzuki T, Seth A, Giorgianni F, Kale G, Shen L, Turner JR, Naren A, Desiderio DM, and Rao R (2009) Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J Biol Chem 284, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, Montrose MH, Turner JR, and Watson AJ (2011) The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 140, 1208–1218 e1201–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cameron BD, Sekhar KR, Ofori M, and Freeman ML (2018) The Role of Nrf2 in the Response to Normal Tissue Radiation Injury. Radiat Res 190, 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jaiswal AK (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36, 1199–1207 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.