

Abstract
Background & Aims:
Chromosomal instability (CIN) is a carcinogenesis event that promotes metastasis and resistance to therapy, by unclear mechanisms. Expression of the colon cancer associated transcript 2 gene (CCAT2), which encodes a long noncoding RNA (lncRNA), associates with CIN, but little is known about how CCAT2 lncRNA regulates this cancer enabling characteristic.
Methods:
We performed cytogenetic analysis of colorectal cancer (CRC) cell lines (HCT116, KM12C/SM, and HT29) overexpressing CCAT2 and colon organoids from C57BL/6N mice with the CCAT2 transgene and without (controls). CRC cells were also analyzed by immunofluorescence microscopy, γ-H2AX, and senescence assays. CCAT2 transgene and control mice were given azoxymethane and dextran sulphate sodium (DSS) to induce colon tumors. We performed gene expression array and mass spectrometry to detect downstream targets of CCAT2 lncRNA. We characterized interactions between CCAT2 with downstream proteins using MS2 pulldown, RNA immunoprecipitation, and SHAPE analyses. Downstream proteins were overexpressed in CRC cells and analyzed for CIN. Gene expression levels were measured in CRC and non-tumor tissues from 5 cohorts, comprising more than 900 patients.
Results:
High expression of CCAT2 induced CIN in CRC cell lines and increased resistance to 5-fluorouracil and oxaliplatin. Mice that expressed the CCAT2 transgene developed chromosome abnormalities, and colon organoids derived from crypt cells of these mice had a higher percentage of chromosome abnormalities compared to organoids from control mice. The transgenic mice given azoxymethane and DSS developed more and larger colon polyps than control mice given these agents. Microarray analysis and mass spectrometry indicated that expression of CCAT2 increased expression of genes involved in ribosome biogenesis and protein synthesis. CCAT2 lncRNA interacted directly with and stabilized BOP1 ribosomal biogenesis factor (BOP1). CCAT2 also increased expression of MYC, which activated expression of BOP1. Overexpression of BOP1 in CRC cell lines resulted in chromosomal missegregation errors, and increased colony formation, and invasiveness, whereas BOP1 knockdown reduced viability. BOP1 promoted CIN by increasing the active form of aurora kinase B (AURKB), which regulates chromosomal segregation. BOP1 was overexpressed in polyp tissues from CCAT2 transgenic mice, compared to healthy tissue. CCAT2 lncRNA and BOP1 mRNA or protein were all increased in microsatellite stable tumors (characterized by CIN), but not in tumors with microsatellite instability, compared with non-tumor tissues. Increased levels of CCAT2 lncRNA and BOP1 mRNA correlated with each other and with shorter survival times of patients.
Conclusions:
We found that overexpression of CCAT2 in colon cells promotes CIN and carcinogenesis, by stabilizing and inducing expression of BOP1 an activator of AURKB. Strategies to target this pathway might be developed for treatment of patients with microsatellite stable colorectal tumors.
Keywords: MSS, aneuploidy, tumorigenesis, non-coding RNA
Graphical Abstract
Introduction
Considered one of the initial molecular events and one of the most noticeable pathogenic feature of cancer, genomic instability 1, was predicted by Theodore Boveri, over 100 years ago 2. Chromosomal instability (CIN) is one of the most common forms of genomic instability, and is characterized by high rates of structural and numerical chromosomal aberrations 3. By constantly making new aneuploid genomes, CIN causes tumor cell heterogeneity, upon which clonal selection can act 4.
One of the main causes of CIN is the asymmetrical segregation of chromosomes during the metaphase as a consequence of abnormal spindle formation 5 that will also induce anaphase bridges during mitosis, which is a key feature of CIN 6. CIN is associated with poor survival, metastases, and therapy resistance in cancer 7. Yet, the complex molecular mechanism(s) underlying the process remains unclear. Tens of protein coding genes and only few non-coding RNAs (ncRNAs) have been associated with the occurrence of CIN 8. Nonetheless, all these genes are altered in a small fraction of patients and their involvement in CIN has been studied mainly in in vitro systems.
The long-non-coding RNA (lncRNA) colon cancer associated transcript 2 (CCAT2), is up-regulated in microsatellite stable (MSS) colorectal cancers (CRC), tumors characterized by CIN and in which CCAT2 promotes metastases 9 and influence glutamine metabolism 10. Additionally, CCAT2 lncRNA was also reported to be involved in the initiation of myelodysplastic syndrome (MDS) 11, a premalignant condition associated with abnormal chromosomes 12. These findings imply that CCAT2 plays a role in early stages of carcinogenesis. Despite its proven clinical value, the exact molecular mechanisms by which CCAT2 lncRNA induces CIN are unexplored. Here, we have analyzed, for the first time, the molecular pathways through which a lncRNA induces CIN both in vitro and in vivo, and we identify CCAT2 as a regulator of MYC-BOP1-AURKB pathway causing CIN.
Materials and Methods
Patient sample collection
For this study we used five different patient cohorts (Supplementary Tables 1–4): Cohort A (TCGA colorectal cancer cohort) was used for screening of the PES1, BOP1, and WDR12 (PeBoW) complex expression in MSS versus MSI subtypes of CRC; Cohort B for validating CCAT2 lncRNA and PeBoW complex mRNA overexpression in CRC; Cohort C for validating CCAT2 lncRNA and BOP1 mRNA role in MSS CRC subtype; Cohort D for further validation and prognosis analysis of CCAT2 lncRNA and PeBoW complex mRNA expression in CRC; and Cohort E (described previously in 10) for validation of the mechanism at the protein level. Written informed consent was obtained from each patient, and the study was approved by the institutional review boards of all the involved institutions.
Additional methods are available in Supplementary material and methods. Primers and antibody information are available in Supplementary Tables 5 and 6.
RESULTS
CCAT2 lncRNA induces CIN and activates pathways associated with ribosomal proteins
We decided to investigate the causal relationship between CCAT2 and CIN in cancer because of multiple lines of evidence. First, in multiple CRC and gastric cancer (GC) cell lines used, we observed that an aberrant chromosomal number is associated with high levels of CCAT2 lncRNA (Supplementary Figure 1A and Supplementary Table 7). Second, cytogenetic analysis suggested that HCT116 clones with exogenous CCAT2 over-expression (named here HCT116CCAT2) have a higher percentage of chromosome abnormalities (including breaks, fusion, and polyploidy) compared to HCT116 cells transduced with an empty vector (HCT116Empty) (31.0% versus 11.9%, P = .001). Similar results were found in the KM12SM cells (highly metastatic) compared to KM12C (poorly metastatic, from same patient) (44.5% versus 33.3%, P = .1099) (Figure 1A and 1B) KM12SM, with two times higher CCAT2 levels as KM12C, presents also higher degree of CIN (Supplementary Figure 1A and Supplementary Table 7). Third, by double strand breaks (DSB) analysis using γ-H2AX, the number of cells with DSBs was higher in HCT116CCAT2 (39/152, 25.7%) compared to HCT116Empty (2/162, 1.2%) (P < .0001) (Supplementary Figure 1B). Next, when we treated the cells with the DNA strand breaks inducer bleomycin, the number of DSBs was 42/126 in HCT116CCAT2 cells (33.3%) and 28/138 in HCT116Empty cells (20.3%) (P = .0165) (Supplementary Figure 1C). Additionally, we observed that HCT116CCAT2 showed significantly lower chemosensitivity to 5-Flurouracil (P = .003, P = .006 and P = .0001, respectively) (Supplementary Figure 1D) and to oxaliplatin (P < .0001 for all three concentrations) (Supplementary Figure 1E) compared to HCT116Empty. One of the possible mechanisms through which cells tolerate DNA damage and stop CIN, is senescence 13. We observed that HCT116CCAT2 clones had ten times lower number of senescent cells compared to HCT116Empty (P = .0001) (Supplementary Figure 1F). Fourth, HCT116CCAT2 clones had a significant increase in abnormal spindles, along with a higher percentage of anaphase bridges, indirect indicators of CIN 14, compared with HCT116Empty (mean 13.9% versus 6.2%, P = .009 and 51.8% versus 24.7%, P = .0052, respectively) (Figure 1C and 1D).
Figure 1. CCAT2 lncRNA induces CIN and activates pathways associated with ribosomal proteins.

(A) Cytogenetic analysis showing chromosomal aberrations in HCT116CCAT2 cells (left) and KM12SM cells (right). Red arrows indicate breaks, blue arrows indicate fusions and green arrows indicate fragments. (B) The frequency of aberrant metaphases in HCT116Empty versus HCT116CCAT2 and KM12C versus KM12SM. At least 35 metaphases were analyzed for each clone. (C) Immunofluorescence images and (D) frequency of abnormal spindle (upper lane) and anaphase bridge (lower lane) in HCT116CCAT2 cells. At least 200 interphase cells were analyzed for each clone. (E) Cytogenetic analysis showing chromosomal aberrations in organoids from WT (left) and CCAT2 mice (right). Blue arrow indicates fusions. (F) The frequency of aberrant metaphases in organoids from WT versus CCAT2 mice. At least 35 metaphases were analyzed for each organoid. (G) Schematic illustration of the AOM/DSS colon cancer model. (H) Images of colon mucosa from WT and CCAT2 mice after treatment with AOM/DSS. Red delineation indicates polyps’ area. (I) Total number of polyps, total surface area of colon polyps, and average polyp diameter size in WT and CCAT2 transgenic mice at the end of the AOM/DSS treatment (n = 7 per group). (J) H&E images of the colon from WT and CCAT2 transgenic mice after treatment with AOM/DSS. Black arrows indicate polyps. (K) Largest size polyp according to H&E analysis in WT and CCAT2 transgenic mice. (L) The percentage of WT and CCAT2 transgenic mice with grade 1–2 versus grade 3–4 hyperplasia, and (M) with normal glands versus dysplastic glands. (N) IPA analysis showing significantly enriched pathways in CCAT2 transgenic mouse model (left Y axis represents negative log P values; right Y axis represents the ratio of molecules in the dataset mapping to the number of molecules in the canonical pathways) (left panel). Venn diagram showing overlapping genes related to the ribosomal proteins from the canonical pathways (right panel). Mean ± SD. (**P < .01).
Fifth, our recently developed mouse model 11 indicates that CCAT2 lncRNA can trigger CIN in vivo (Supplementary Figure 1G). We found that the karyotypes from the bone marrow tissue of CCAT2 mice showed more chromosomal abnormalities compared to WT littermates (Supplementary Figure 1H). Finally, we developed normal colon organoid cultures from the crypt cells of CCAT2 and WT mice. The CCAT2 lncRNA expression remained significantly higher in CCAT2 organoids compared to WT organoids (Supplementary Figure 1I). No important morphology differences were observed between CCAT2 and WT mice derived organoids (Supplementary Figure 1J), but a more rapid growth rate was measured for the CCAT2 organoids (P = .001) (Supplementary Figure 1K). After 3 passages we performed cytogenetic analysis, which showed that normal colon organoids from CCAT2 mice had a significantly higher percentage of chromosome abnormalities compared to organoids from WT mice (68.6% versus 51.4%, P = .0094) (Figure 1E and 1F), denoting that CIN induced by CCAT2 preceded tumor development.
To find out if CCAT2 lncRNA plays a role in the acceleration and progression of colon tumors we used the azoxymethane/dextran sulphate sodium (AOM/DSS) model, which was previously reported to induce CIN 15. CCAT2 transgenic and WT mice were injected with AOM and then subjected to four rounds of DSS (Figure 1G). Macroscopic analysis revealed that CCAT2 mice had significantly higher number of polyps (P = .0143), the surface of colon mucosa covered by polyps was significantly larger (P = .0088), and the average polyp diameter was significantly longer (P = .0009) than WT mice (Figure 1H and 1I). Next, we performed Hematoxylin and Eosin (H&E) staining and histopathological evaluation of the colon of WT and CCAT2 mice (Figure 1J). The size of the largest polyp was significantly greater in CCAT2 mice compared to WT mice (P = .0093) (Figure 1K). Importantly in CCAT2 mice the grade of hyperplasia of the colon glands was higher in comparison with WT mice (Grade 3–4: CCAT2 – 71.4% versus WT - 50.0%, P = .0024) (Figure 1L) and the mice had a higher incidence of dysplastic colon glands in comparison with the control group (71.4% versus 50.0%, P = .0024) (Figure 1M).
We used microarray gene expression analysis (GEA) to identify downstream targets of CCAT2 on BM cells obtained from WT mice and CCAT2 transgenic mice (GSE106581). Ingenuity Pathway Analysis (IPA) revealed that EIF2, mTOR, and regulation of eIF4 and p70S6K pathways were all significantly up-regulated in the CCAT2 mouse model when compared to WT mice (Figure 1N). All these pathways are related to protein synthesis 16. Among the common genes involved in these pathways, those related to ribosomal proteins and ribosome biogenesis were enriched (Figure 1N). The deregulation of proteins involved in ribosome biogenesis or ribosomal proteins have been previously linked to CIN 17.
CCAT2 lncRNA interacts with BOP1
Considering that the lncRNA CCAT2 is localized mostly in the nucleus (Supplementary Figure 2A), we performed Mass Spectrometry (MS) on HCT116 cells transiently transfected with CCAT2-MS2 vectors, and isolated CCAT2 lncRNA interacting nuclear proteins. Again, we found an enrichment in proteins involved in protein translation and ribosomal biogenesis (Supplementary Table 8). By screening the literature, we found that, one of these candidate proteins, BOP1 ribosomal biogenesis factor (BOP1), was previously reported to affect spindle formation and cause CIN in CRC 18, 19, therefore, we selected it for further analysis. BOP1 and CCAT2 genes are both located on chromosome 8q24, a region amplified in many cancers 20. BOP1 protein is one of the three components, which includes also PES1 and WDR12 proteins, of the PeBoW complex, a regulator of rRNA processing affecting ribosome biogenesis 21.
To explore the interaction between CCAT2 lncRNA and BOP1 protein, an in vivo MS2-pull down assay was conducted in COLO320 cells. BOP1 was retrieved through the CCAT2-MS2 construct and not by MS2-empty vector, while WDR12 and PES1 of the PeBoW complex, were only minimally retrieved (Figure 2A). To validate these results, RNA immunoprecipitation (RIP) using BOP1, WDR12, and PES1 antibodies was performed. CCAT2 was identified ~5 and ~11 times higher in the BOP1 immunoprecipitate than PES1 and WDR12 precipitates, respectively (Figure 2B and Supplementary Figure 2B). This suggests that BOP1 protein is a strong interactor of CCAT2 lncRNA, and the WDR12 and PES1 signals are probably identified due to indirect, low stability interactions with BOP1 protein. In vitro pull-down, was consistent with the previous results: the biotin-labeled CCAT2 lncRNA, but not biotin-labeled controls, pulled down the recombinant BOP1 (Figure 2C).
Figure 2. CCAT2 lncRNA interact with BOP1.

(A) Schematic illustration of MS2-pull down assay (left panel) and immuno-blotting results of BOP1, PES1, and WDR12 (right panel). (B) RIP assay was performed to check the enrichment of CCAT2 lncRNA in COLO320. (C) In vitro RNA pull-down using GST-tagged CCAT2 lncRNA and recombinant BOP1 protein. (D) Determination of the interaction between ΔBOP1 domains and CCAT2 lncRNA by in vitro RNA pull down. (E) In vitro RNA pull-down using multiple CCAT2 lncRNA segments (S1 to S10). (F) SHAPE assay showing the structure of the CCAT2 lncRNA region from nucleotide 207 to 492. Blue arrows indicate the start and the end of segment 3. Mean ± SD. (*P < .05), (**P < .01).
The BOP1 protein consists of seven WD40 repeats (from amino acids 411 to 746, Supplementary Figure 2C), which are discreet domains that interact with proteins and RNAs, providing platforms to assemble functional complexes 22. To characterize the region(s) of BOP1 that directly interacts with CCAT2 lncRNA, we generated a series of BOP1 truncations with deletion of each WD repeats (Figure 2D), and one with a deletion of the nuclear localization signal (NLS). Using RIP assays, we observed that the abundance of CCAT2 in WD 1, 2, 3, 5, 6, and 7 - truncated BOP1 groups was decreased compared to other groups. This suggested that deletion of any individual WD repeats, except WD 4, abolishes the BOP1-CCAT2 interaction (Figure 2D).
In order to map the RNA sequence of CCAT2 that interacts with BOP1 protein, RNA pull-down was performed using a set of 10 CCAT2 segments. Results indicated that segment 3 (nucleotides 333 to 435) interacted directly with BOP1 protein (Figure 2E). Although the conservation of CCAT2 gene is high in mammals (75.4% ± 19.9, n = 66 species) (Supplementary Figure 2D), the interacting segment 3 is mostly conserved in primates (more than 90% conservation), than in other mammals (30% - 70%) (Supplementary Figure 2E). The secondary structure of the CCAT2 lncRNA region that spans nucleotide 207 to 492, which includes segment 3, was determined experimentally by SHAPE. Consistent with the results from Figure 2E, some active regions were determined, suggesting that segment 3, provides a platform for protein interactions (Figure 2F and Supplementary Figure 2F and 2G).
CCAT2 lncRNA up-regulates BOP1 in vitro and in vivo
To investigate the effect of CCAT2 lncRNA on the PeBoW complex, we verified the expression of the PeBoW complex components in CCAT2 overexpression clones. In the cell lines with high levels of CCAT2, HCT116CCAT2, and KM12SM, BOP1 mRNA levels were four and two times higher while protein levels were eleven and three times higher compared to HCT116Empty and KM12C (Figure 3A). In both HCT116 and KM12SM, 50–60% knock-down of CCAT2 resulted in down regulation of BOP1 protein and mRNA levels (Figure 3B).
Figure 3. CCAT2 lncRNA up-regulates BOP1 in vitro and in vivo.

(A) Expression of PeBoW complex components in HCT116Empty and HCT116CCAT2 (left panel), and in KM12C and KM12SM (right panel). (B) Expression of PeBoW complex components in HCT116 WT (left panel) and KM12SM (right panel) after CCAT2 knock-down. (C) The half-life of BOP1 protein in KM12SM cells with transient CCAT2 overexpression or empty vector (left panel). Data from three experiments were quantified and are depicted as a graphic (right panel). (D) The nuclear and cytoplasmic localization of PeBoW complex in HCT116Empty and HCT116CCAT2. (E) Expression of CCAT2 lncRNA and PeBoW complex components in the colon of CCAT2 mice. (F) The expression of BOP1 in normal colon tissues and colon polyps of CCAT2 mice after AOM/DSS treatment (n = 7). Mean ± SD. (ns, not significant), (*P < .05), (**P < .01); (***P < .001), (****P < .0001).
These results indicated that CCAT2 lncRNA mainly regulates BOP1 by a post-transcriptional mechanism. To test if CCAT2 lncRNA affects the stability of BOP1 protein, cyclohexamide (CHX) chase assay was conducted on KM12SM cells with transient CCAT2 over-expression. Four hours after CHX addition, BOP1 protein expression started decreasing more rapidly in the Empty clone than in the CCAT2 overexpressing clone (Figure 3C). We then checked the intracellular localization of BOP1 protein: as expected, in HCT116CCAT2 cells, BOP1 was enriched in the nuclear fraction compared to the HCT116Empty clone, while there was no difference in the cytoplasm (Figure 3D). These data were confirmed in DLD-1Empty and DLD-1CCAT2 (Supplementary Figure 3A).
Using the CCAT2 mouse model 11, we observed that the stable overexpression of CCAT2 lncRNA in mice leads to the up-regulation of BOP1 mRNA and protein in healthy colon tissue of CCAT2 mice compared to WT mice, hence this phenomenon is preceding tumor formation (Figure 3E and Supplementary Figure 3B). Moreover, we detected a higher protein level of BOP1 in the bone marrow of CCAT2 mice, who we previously showed to develop MDS and display chromosomal abnormalities 11, compared to WT mice (Supplementary Figure 3C). We also checked BOP1 mRNA level in macroscopically unaffected colon tissues and in polyps from AOM/DSS treated CCAT2 transgenic mice. We observed that the mRNA level of BOP1 was further increased in polyps compared to normal colon in CCAT2 transgenic mice (P = .0095) (Figure 3F).
In our previous study 9, we reported that CCAT2 up-regulates MYC protein through TCF7L2. We found that MYC is also a predicted transcription factor (TF) for BOP1 gene (Supplementary Figure 3D and 3E). CHIP-Seq data (UCSC Genome Browser Assembly) showed that MYC binds to its specific sequence (CACGTG) located in the 5’ region of BOP1 gene and acts as a TF for BOP1 23 (Supplementary Figure 3F). Next, we verified whether MYC could regulate the expression of BOP1 using a doxycycline inducible HCT116 MYC tet-on system. BOP1 mRNA levels were about 3 times higher 12h after induction of MYC and remained stable until 24h. Accordingly, the protein level of BOP1 increased after 12h, and reached an even higher level 24h after induction (Supplementary Figure 3G). In summary, CCAT2 lncRNA regulates BOP1 mainly at the protein level by direct binding, but also positively regulates its transcription through MYC.
Overexpression of BOP1 promotes CIN
To confirm the role of BOP1 in triggering CIN, we stably overexpressed BOP1 in HCT116, KM12SM, and HT29 cell lines (Supplementary Figure 4A and 4B) and performed cytogenetic analysis. After few passages (5–10), no difference on genomic instability was observed (Supplementary Figure 4C). However, after a longer propagation time (>15 passages), BOP1 clones started to present chromosomal aberrations. HCT116 cells with BOP1 overexpression (HCT116BOP1) showed a higher frequency of chromosomal abnormalities (fusions, breaks, and fragmentation) (Figure 4A panels (i), (ii), and (iii)) compared to HCT116Empty (31.4% versus 8.3%, P = .0001) (Figure 4B). The KM12SMBOP1 clones showed a higher percentage of aberrant metaphases compared to control cells (38.0% versus 22.0%, P = .0136). Specifically, a higher percentage of polyploidy or tetraploidy was observed in the KM12SMBOP1, indicative of a greater chromosome segregation failure (Figure 4A panel (iv) and Figure 4B). Moreover, a significantly higher genomic vulnerability was found in HT29BOP1 (28.6% versus 2.9%, P < .0001), including c-anaphases (Figure 4A panel (v) and Figure 4B).
Figure 4. Overexpression of BOP1 promotes CIN.

(A) Cytogenetic analysis showing chromosomal aberrations in cells with overexpression of BOP1. In panels (i), (ii) and (iii) are representative images of HCT116BOP1, with fusion (blue arrow), break (red arrow), and fragments (green arrows). In panel (iv) is an image of KM12SMBOP1 with polyploidy and acentric chromosomes (black arrows) and fusion (blue arrow), and in panel (v) is an image of HT29BOP1 with c-anaphase morphology. (B) The frequency of cells exhibiting chromosome abnormalities in HCT116, KM12SM, and HT29 Empty versus BOP1 overexpressed clones. At least 35 metaphases were analyzed for each clone. (C, D) Images and frequencies of abnormal spindles in HCT116 (C) and KM12SM (D) with BOP1 overexpression. At least 200 interphase nuclei were analyzed for each clone. (E, F) Images and frequency of anaphase bridges in HCT116 (E) and KM12SM (F) with BOP1 overexpression. At least 200 cells were analyzed for each clone. Mean ± SD. (*P < .05), (**P < .01), (****P < .0001).
BOP1 has previously been reported to alter the spindle apparatus and to cause aberrant lagging chromosomes 18. As shown in Figure 4C, in HCT116, the percentage of cells with abnormal spindles increased from 12.1% in HCT116Empty clones to 19.7% in HCT116BOP1 clones, P = .0236 (Figure 4C). Results from KM12SM cells were consistent, 21.1% of KM12SMEmpty clones showed aberrant spindles, as compared to 40.3% of KM12SMBOP1 cells P < .0001 (Figure 4D). A greater percentage of cells with anaphase bridges was found in the HCT116BOP1 clones when compared to controls, 49.0% vs 70.2%, P = .0036 (Figure 4E). KM12SMBOP1 clones displayed a higher frequency of anaphase bridges compared to KM12SMEmpty 37.6% vs 63.0%, P < .0001 (Figure 4E). Therefore, the CIN phenotype induced by BOP1 reproduced the one of its regulator, CCAT2.
BOP1 plays an oncogenic role in CRC
Functional assays were conducted to explore the role of BOP1 in CRC. Knocking-down BOP1 impaired the proliferation of HCT116 and KM12SM (Figure 5A). Consistently, the cell viability was higher in HCT116BOP1 (P = .0024), and in KM12SMBOP1 (P = .0032) versus controls (Figure 5B). Cell colony formation was inhibited after knocking-down BOP1 in HCT116 (P = .0156 for siRNA 1 and P = .0047 for siRNA 2) (Figure 5C), whereas up-regulation of BOP1 promoted colony formation in HCT116 (P = .0497) (Figure 5D) and KM12SM (P = .0028) (Supplementary Figure 5A). Fewer cells migrated and invaded into the lower chamber in a transwell assay after BOP1 siRNAs, both in HCT116 (P = .0175 for siRNA 1 and P < .0001 for siRNA 2) and KM12SM (P = .0015 for siRNA 1 and P = .0038 for siRNA 2) (Figure 5E). Stable over-expression of BOP1 enhanced cell migration and invasion in HCT116 (P = .013) and KM12SM (P = .0339) (Figure 5F). Scratch assays indicated that BOP1 positively regulates cell migration in HCT116: knocking-down BOP1 decreased migration (P = .0122 for siRNA 1 and P = .0031 for siRNA 2) (Supplementary Figure 5B). Collectively, BOP1 protein plays an oncogenic role in CRC, confirming recent findings 24 and the BOP1 overexpression phenotype mirrors the CCAT2 overexpression effects 9.
Figure 5. BOP1 plays an oncogenic role in CRC.

(A) Proliferation rate of HCT116 (left) and KM12SM (right) after siRNA knock-down of BOP1. (B) Proliferation rate of HCT116Empty and HCT116BOP1 (left) and KM12SMEmpty and KM12SMBOP1 (right). (C, D) Representative images of colony formation assay in HCT116 with BOP1 knock-down (C) and HCT116 with stable overexpression of BOP1 (D). Quantitative analysis of colony numbers (right side of panels C and D). (E) Invasion potential of HCT116 and KM12SM cells after transfection with BOP1 siRNA. Representative images of invasion assay for HCT116 (upper panel) and KM12SM (lower panel). Quantitative analysis of invading cell (right panel). (F) Invasion potential in cells with stable overexpression of BOP1. Representative images of invasion assay for HCT116 (upper panel) and KM12SM (lower panel). Quantitative analysis of invading cell (right panel). Mean ± SD. (*P < .05), (**P < .01); (***P < .001), (****P < .0001).
BOP1 modulates the function of AURKB
We hypothesized that high levels of CCAT2 lncRNA would increase the level of ribosomal subunits and subsequently this could affect genomic integrity 25. By performing polysome profiling, we concluded that the function of CCAT2 lncRNA via BOP1 is ribosome independent (Supplementary Figure 6A–6C).
In order to find down-stream targets of the CCAT2-BOP1 pathway, we used a genome-wide screen, CINdex analysis 26, on the TCGA CRC cohort. We found that the aurora kinase family and PeBoW complex genes, associated with CIN and positively correlate with each other (Supplementary Figure 6D and Supplementary Table 9). This assay directed us to investigate aurora kinase family genes.
No difference in the levels of aurora kinase A (AURKA) protein, and of two other proteins reported to induce CIN, CDC20, and BUB1B, were detected between scramble siRNA and CCAT2 knockdown in KM12SM (Supplementary Figure 6E) and BOP1 knockdown in HCT116 (Supplementary Figure 6F). We further checked for differences in the levels of aurora kinase B (AURKB) and phosphorylated aurora B (pAURKB) in HCT116CCAT2 versus HCT116Empty clones. We observed that the over-expression of CCAT2 induces higher pAURKB at Thr 232 (active form of AURKB), but no changes in AURKB mRNA and protein (Figure 6A left panel). In a second model, both total levels of AURKB mRNA and protein and pAURKB were higher in KM12SM compared to KM12C (Figure 6A right panel). AURKB is predominantly activated by autophosphorylation, and does not require the involvement of other kinase, while interaction with other molecules augments the phosphorylation 27. Next, we observed that high BOP1 induces the phosphorylation of AURKB, both in HCT116 and KM12SM (Figure 6B) and the knock-down of BOP1 induced the downregulation of AURKB mRNA and protein and of pAURKB (Figure 6C). As MYC was reported to be a regulator of AURKB 28, we checked if MYC is a TF for AURKB. CHIP-Seq data (UCSC Genome Browser on Human Feb. 2009 (GRCh37/hg19) Assembly) showed that MYC binds to CCACGCC located in the 5’ region of AURKB and acts as a TF for AURKB (Supplementary Figure 6G). This was confirmed in HCT116 MYC tet-on system: after induction of MYC, the levels of AURKB mRNA and protein and of pAURKB protein increased (Figure 6D).
Figure 6. BOP1 modulates the function of AURKB.

(A) Expression of AURKB and pAURKB in HCT116Empty and HCT116CCAT2 (left panel) and KM12C and KM12SM (right panel). (B) Expression of BOP1, AURKB, and pAURKB analyzed in HCT116Empty and HCT116BOP1 (left panel) and KM12SMEmpty and KM12SMBOP1 (right panel). (C) Expression of BOP1, AURKB, and pAURKB analyzed in HCT116 (left panel) and KM12SM (right panel) after BOP1 knock-down with siRNA. (D) Expression of BOP1 and AURKB at 0, 6, 12, 18, and 24 hours in HCT116 cells with inducible c-MYC expression system. (E) MS2-pull down assay to identify if AURKB interacts with MS2-labeled CCAT2 in HCT116. Mean ± SD. (ns, not significant), (**P < .01); (***P < .001), (****P < .0001).
Previously, it was shown that AURKB protein can bind mRNA molecules that stimulate AURKB activity during mitosis 29. To identify whether CCAT2 RNA forms a complex with AURKB protein, we performed MS2-pull down assay: AURKB protein was retrieved in the CCAT2-MS2 but not in the MS2-empty vector transduced HCT116 cells (Figure 6E). By using RIP, we confirmed this complex: CCAT2 was detected 20 times higher in the AURKB precipitate compared to IgG control (P < .0001) (Supplementary Figure 6H). In order to determine which CCAT2 lncRNA segment associates with AURKB protein we did RNase I treatment of the lysate before RIP, so that unbound fragments were digested. Two of the segments were enriched: S8 and S6 (Supplementary Figure 6I).
To appreciate if the dysregulation of CCAT2-BOP1-AURKB pathway is widespread in cancers, we checked for this pathway in GC, which has high CIN rates (50%) 30. We performed a non-coding GEA comparing normal gastric samples with peritoneal carcinomatosis samples of GC patients (GSE133590). One of the top up-regulated lncRNA in peritoneal carcinomatosis was CCAT2 (P = .0115) (Supplementary Figure 7A). We analyzed the CCAT2 lncRNA expression in two pairs of GC patients derived xenografts (PDXs) and one GC patient derived organoid (PDO) and in each of them CCAT2 was up-regulated compared to parental cells (Supplementary Figure 7B), showing that successful engraftment, a marker of poor prognosis 31, is associated with high CCAT2 levels. Next, we did CINdex analysis using the GC TCGA cohort and identified that aurora family and PeBoW complex genes positively correlated with CIN at chromosome level (Supplementary Figure 7B and Supplementary Table 10). Additionally, we used the primary GC cells, AGS, with euploid chromosomal number and KATO III with a tetraploid chromosome number. The RNA expression levels of CCAT2 and BOP1 and the protein levels of BOP1, the other components of the PeBoW complex and pAURKB were higher in KATO III compared to AGS (Supplementary Figure 7C). These data imply that the identified mechanism relates to CIN more generally.
CCAT2 and BOP1 are overexpressed in MSS CRC
To assess the clinical relevance of our findings, we examined multiple patient cohorts. Firstly, we used the TCGA CRC cohort (Cohort A) as a screening cohort and identified significantly higher expression of BOP1, PES1, and WDR12 mRNAs in tumor versus normal tissue (P < .0001) (Figure 7A). It is known that MSS/MSI-L CRC cancers (analyzed together as MSS) are CIN positive and MSI-H (referred to as MSI) are CIN negative 32. Therefore, we compared the mRNA expression of PeBoW complex components in MSS versus MSI CRC. Only the mRNA level of BOP1 was significantly higher in MSS versus MSI (P < .0001) and PES1 and WDR12 showed no differences between the subgroups (P = .8115 and P = .2333) (Figure 7B). We were not able check for CCAT2 lncRNA in this cohort, as CCAT2 is not polyadenylated.
Figure 7. CCAT2 and BOP1 are overexpressed in MSS CRC.

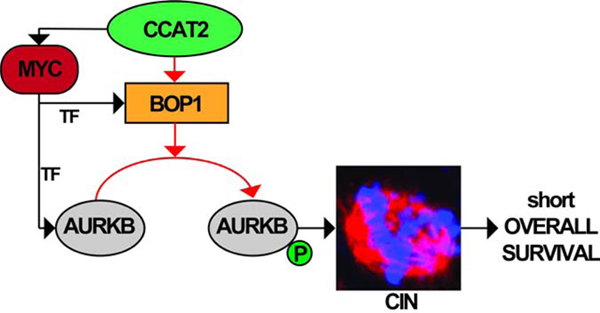
(A) The expression levels of PeBoW complex in Cohort A. (B) The expression levels of the PeBoW complex in MSI and MSS primary CRC in Cohort A. (C) The expression of CCAT2 lncRNA and BOP1 mRNA in tumor and adjacent normal tissues from Cohort C. (D) The expression of CCAT2 lncRNA and BOP1 mRNA in MSI and MSS CRC from Cohort C. (E) The expression of CCAT2 lncRNA and BOP1 mRNA in MSI and MSS CRC from Cohort D. (F) Correlation between the RNA expression of CCAT2 and BOP1 in patients from Cohort D. (G) Kaplan–Meier OS curves of CRC patients from Cohort D, CCAT2 lncRNA (left panel) and BOP1 mRNA (right panel). (H) Kaplan–Meier RFS curves of CRC patients from Cohort D, of CCAT2 lncRNA (left panel) and BOP1 mRNA (right panel). Time is expressed in days. (I) Western blot analysis of BOP1, AURKB, and pAURKB protein expression in paired CRC samples (Cohort E). N=normal tissue, T=tumor tissue, N/A=not available microsatellite status. The samples in which both BOP1 and pAURKB proteins are up-regulated in tumor versus normal tissues are marked with red stars. (J) A model of CCAT2 involvement in CIN (red arrows – new interactions; black arrows – available data). Data are represented as violin plots. (ns) not significant, (*P < .05), (**P < .01); (***P < .001), (****P < .0001).
In a second cohort of CRC tumors and paired adjacent normal tissues (Cohort B) we measured the RNA expression of CCAT2, BOP1, PES1, and WDR12: all were significantly overexpressed in tumor versus normal tissues (Supplementary Figure 8A). We also identified a significant positive correlation between CCAT2 lncRNA and BOP1 mRNA in tumor tissues (r = 0.6296) (Supplementary Figure 8B). Regarding MSS versus MSI comparison, there were insufficient patients in the MSI group for this analysis. We identified that CCAT2 and BOP1 expression levels remained unchanged in MSI tumors versus paired normal tissues (P = .7422 and P = .5649, respectively), but significantly increased in MSS versus paired normal tissues (P < .0001 for both genes) (Supplementary Figure 8C).
In a third group, Cohort C, we confirmed these data: CCAT2 lncRNA and the mRNA level of all three PeBoW components were highly expressed in tumor versus adjacent normal tissues (P < .0001) (Figure 7C and Supplementary Figure 8D). We also checked for the correlation between BOP1 mRNA and CCAT2 lncRNA expression in normal and tumor tissues; the correlation between the two transcripts increased from r = 0.3050 in normal tissues to an r = 0.5252 in cancer (Supplementary Figure 8E and 8F). Because of an ample MSI sub-group, we compared the expression of CCAT2 lncRNA and PeBoW mRNAs between MSI and MSS. Only CCAT2 and BOP1, but not PES1 or WDR12, were up-regulated in MSS (P = .001 and P = .03, respectively) (Figure 7D and Supplementary Figure 8G).
Additionally, we used Cohort D, with ample MSI subgroup, and we established that only CCAT2 and BOP1 were up-regulated in MSS versus MSI (P < .0001 and P = .03, respectively) (Figure 7E and Supplementary Figure 8H). The RNA expression levels of CCAT2 and BOP1 were positively correlated in tumor tissues (r = 0.6263), implying co-regulation (Figure 7F). High levels of CCAT2 or BOP1 were associated with worse overall survival (OS) (P < .0001 and P = .003, respectively) (Figure 7G). Additionally, high levels of PES1 and WDR12 mRNAs were prognostic for shorter OS (P = .0007 and P = .0004, respectively) (Supplementary Figure 8I). Similarly, increased transcription levels of CCAT2 and BOP1 predicted shorter recurrence free survival (RFS) (P < .0001 for both) (Figure 7H). PES1 and WDR12 proved to have a comparable prognostic value (P < .0001 and P = .0002, respectively) (Supplementary Figure 8J).
Cohort D had follow-up data for all patients. Univariate analysis revealed that high CCAT2 lncRNA and BOP1 mRNA levels were significantly associated with shorter OS and RFS (for CCAT2: HR: 6.1, 95%CI: 2.79–13.31, P < .0001 and HR: 4.78, 95%CI: 2.74–8.09, P < .0001, respectively; for BOP1: HR: 3.01, 95%CI: 1.38–6.55, P = .006 and HR: 2.77, 95%CI: 1.65–4.60, P = .0002, respectively) (Supplementary Table 11). Multivariate analysis revealed that high CCAT2 and BOP1 were independent factors for predicting poor OS and RFS (for CCAT2: HR: 5.51, 95%CI: 2.48–12.27, P < .0001 and HR: 4.85, 95% CI: 2.76–8.28, P < .0001, respectively; for BOP1: HR: 3.12, 95%CI: 1.42–6.84, P = .005 and HR: 2.67, 95%CI: 1.59–4.45, P = .0003, respectively) (Supplementary Table 12).
Finally, to assess if our findings are present at the protein level, we used Cohort E. We previously showed that the expression of CCAT2 is higher in tumor tissues of each of these patients compared to their normal tissue 10. We assessed the protein expression of BOP1, AURKB, and pAURKB and identified high protein levels of BOP1 and pAURKB in tumor tissue compared to normal mucosae for 60% (6/10) of the pairs, all six being MSS CRC (Figure 7I). We also identified that CCAT2 lncRNA and BOP1 protein are upregulated in MSS PDX compared to MSI PDX (Supplementary Figure 8K). Collectively, these results prove that CCAT2 and the PeBoW complex are oncogenes and only CCAT2 and BOP1 are specifically overexpressed in MSS CRC.
Discussion
CIN correlates with patient survival in CRC 33 and multiple other cancers 34. Therefore, understanding the mechanisms underpinning CIN is essential to discover new therapies. Dozens of proteins are involved in chromosome segregation 35. Many of them have recently been evaluated as therapeutic targets, but none of the proposed treatments have reached approval. We propose that targeting ncRNAs involved in chromosomal segregation errors might provide an alternative approach to inhibiting CIN. We present compelling new data supporting the involvement of the lncRNA CCAT2 in the development of CIN.
First, we showed that high CCAT2 lncRNA is sufficient to induce early premalignant modifications. CCAT2 mice, after AOM/DSS treatment, have significantly more polyps and the degree of colon glands’ hyperplasia and dysplasia is higher compared to WT mice. Additionally, a high CCAT2 background induces CIN in organoids established from healthy mouse colon, before tumor formation. We demonstrated that CCAT2 lncRNA induces CIN via BOP1 in two ways: through an “indirect transcriptional mechanism”, probably via c-MYC, a TF promoting BOP1 gene expression, and by a “direct post-transcriptional mechanism”, by binding the mature BOP1 protein, prolonging its half-life. The genes within this pathway (CCAT2, MYC, and BOP1) are located on the well-studied, oncogenic chromosomal amplicon 8q24. In support of our experimental data, we showed that BOP1 mRNA and CCAT2 lncRNA expression correlate positively, in CRC patient cohorts. These results led us to the conclusion that CCAT2 lncRNA is an important factor that induces the up-regulation of BOP1 in cancer and promotes CIN.
Second, we discovered that CCAT2 lncRNA and BOP1 are abnormal in three cancer models, CRC, GC, and in the in vivo CCAT2 transgenic mice treated with AOM/DSS. These results suggest that the new mechanism identified could represents a general model of CIN initiation.
Third, by using one independent genome-wide screening method – CINdex in TCGA CRC cohort, we hypothesized that CCAT2 lncRNA via BOP1 induces CIN through aurora kinase family proteins. Indeed, the expression of CCAT2 lncRNA or BOP1 correlated with the phosphorylation of AURKB suggesting that CCAT2 lncRNA acts as an adaptor, promoting interaction of BOP1 and AURKB, bringing them in physical proximity. Together, our findings concerning the CCAT2 – BOP1 – AURKB pathway expression and function describe a new mechanism of CIN (Figure 7J).
NcRNAs and ribosomal biogenesis proteins have rarely been studied in the context of CIN. Several studies have emerged reporting BOP1 as an oncogene that can induce CIN. Killian et al. showed that the depletion of BOP1 in CRC cells increased the number of aberrant mitotic cells 19. The same group analyzed the expression of BOP1 in CRC patients and observed that high BOP1 was tumor specific. Other, studies revealed that BOP1 overexpression increased the number of multipolar spindles 18. In rectal cancer patients, BOP1 was shown to be overexpressed in samples with gain of the 8q chromosome arm and steadily increased from adenoma to carcinoma, implying a tumorigenic role 36. None of the studies explored the upstream pathways that regulate BOP1, or the precise mechanism by which BOP1 induces CIN. On the other hand, AURKB, is one of the key regulators of mitosis 37, increased activation of AURKB is present in multiple cancer types and correlates with CIN 38. The variety of interactions that facilitate the post-transcriptional modifications that activate AURKB are not fully deciphered. Hence, it is crucial to understand new mechanisms that regulate AURKB function and CCAT2 via BOP1 appears to be a novel pathway activating AURKB.
In conclusion, this study presents a new mechanism in which the lncRNA, CCAT2, induces CIN, an early tumorigenic event. This pathway reveals new potential therapeutic targets for CIN.
Supplementary Material
What you need to know:
Background and Context:
High expression of the colon cancer associated transcript 2 gene (CCAT2), which encodes a long noncoding RNA (lncRNA), associates with chromosome instability, but little is known about how CCAT2 lncRNA regulates this cancer enabling characteristic.
New Findings:
Colorectal cancer cells overexpress CCAT2 lncRNA, which promotes chromosome instability by stabilizing and inducing expression of BOP1 and activation of aurora kinase B.
Limitations:
In this study pharmacological targeting of CCAT2 lncRNA was not assessed. Further studies are needed to analyze the targetability of CCAT2 lncRNA and associated toxicity.
Impact:
Strategies to target this pathway might be developed for treatment of patients with microsatellite stable colorectal tumors.
Lay Summary:
The authors identified a gene product that causes chromosomes to become unstable in colon cells, promoting development of colorectal cancer.
Acknowledgements:
We thank the MDACC Cytogenetics and Cell Authentication Core for technical assistance in the cytogenetic analysis.
Grant Support: Dr. Calin is the Felix L. Haas Endowed Professor in Basic Science. Work in Dr. Calin’s laboratory is supported by National Institutes of Health (NIH/NCATS) grant UH3TR00943–01 through the NIH Common Fund, Office of Strategic Coordination (OSC), the NCI grants 1R01 CA182905–01 and 1R01CA222007–01A1, an NIGMS 1R01GM122775–01 grant, a U54 grant #CA096297/CA096300 – UPR/MDACC Partnership for Excellence in Cancer Research 2016 Pilot Project, a Team DOD (CA160445P1) grant, a Chronic Lymphocytic Leukemia Moonshot Flagship project, the UT MD Anderson Cancer Center Duncan Family Institute for Cancer Prevention and Risk Assessment, a Sister Institution Network Fund (SINF) 2017 grant, and the Estate of C. G. Johnson, Jr. Dr. Pardini is recipient of a Fulbright Research Scholarships (year 2018). Work at the University of Washington was supported by NIGMS R35 GM121487. The work of Dr. Baoqing Chen is supported by National Natural Science Foundation of China (No. 81902462). The work in Dr. Goel’s lab is supported by the grants CA72851, CA181572 and CA202797 from the National Cancer Institute, National Institute of Health. The work of Dr. Parker-Thornburg was supported by the grants 5R50CA211121–03 and CCSG for the GEMF core.
Abbreviations:
- CIN
chromosomal instability
- ncRNA
non-coding RNA
- lncRNA
long non-coding RNA
- CCAT2
colon cancer associated transcript 2 gene
- CRC
colorectal cancer
- BOP1
BOP1 ribosomal biogenesis factor
- AURKB
aurora kinase B
- MSS
microsatellite stable
- MSI
microsatellite instable
- MDS
myelodysplastic syndrome
- GC
gastric cancer
- DSB
double strand breaks
- AOM
azoxymethane
- DSS
dextran sulphate sodium
- IPA
Ingenuity Pathway Analysis
- GEA
gene expression analysis
- MS
mass spectrometry
- RIP
RNA immunoprecipitation
- NLS
nuclear localization signal
- CHX
cycloheximide
- TF
transcription factor
- AURKA
aurora kinase A
- pAURKB
phosphorylated aurora B
- PDX
patient derived xenograft
- PDO
patient derived organoid
- OS
overall survival
- RFS
recurrence free survival
Footnotes
Disclosure: The authors declare no competing interests.
Transcript profiling: Gene expression Omnibus accession number from data used in this study (GSE106581, GSE133590).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012;13:189–203. [DOI] [PubMed] [Google Scholar]
- 2.Boveri M. Über Mitosen bei einseitiger Chromosomenbindung. Jenaische Zeitschrift für Naturwissenschaft 1903;37:401–443. [Google Scholar]
- 3.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010;11:220–8. [DOI] [PubMed] [Google Scholar]
- 4.Gronroos E, Lopez-Garcia C. Tolerance of Chromosomal Instability in Cancer: Mechanisms and Therapeutic Opportunities. Cancer Res 2018;78:6529–6535. [DOI] [PubMed] [Google Scholar]
- 5.Galimberti F, Thompson SL, Ravi S, et al. Anaphase catastrophe is a target for cancer therapy. Clin Cancer Res 2011;17:1218–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffelder DR, Luo L, Burke NA, et al. Resolution of anaphase bridges in cancer cells. Chromosoma 2004;112:389–97. [DOI] [PubMed] [Google Scholar]
- 7.Bakhoum SF, Cantley LC. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018;174:1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dragomir MP, Kopetz S, Ajani JA, et al. Non-coding RNAs in GI cancers: from cancer hallmarks to clinical utility. Gut 2020;69:748–763. [DOI] [PubMed] [Google Scholar]
- 9.Ling H, Spizzo R, Atlasi Y, et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res 2013;23:1446–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redis RS, Vela LE, Lu W, et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol Cell 2016;61:520–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah MY, Ferracin M, Pileczki V, et al. Cancer-associated rs6983267 SNP and its accompanying long noncoding RNA CCAT2 induce myeloid malignancies via unique SNP-specific RNA mutations. Genome Res 2018;28:432–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellagatti A, Boultwood J. The molecular pathogenesis of the myelodysplastic syndromes. Eur J Haematol 2015;95:3–15. [DOI] [PubMed] [Google Scholar]
- 13.Maslov AY, Vijg J. Genome instability, cancer and aging. Biochim Biophys Acta 2009;1790:963–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayani J, Selvarajah S, Maire G, et al. Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Seminars in Cancer Biology 2007;17:5–18. [DOI] [PubMed] [Google Scholar]
- 15.Gerling M, Glauben R, Habermann JK, et al. Characterization of chromosomal instability in murine colitis-associated colorectal cancer. PLoS One 2011;6:e22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol 2012;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim TH, Leslie P, Zhang Y. Ribosomal proteins as unrevealed caretakers for cellular stress and genomic instability. Oncotarget 2014;5:860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Killian A, Sarafan-Vasseur N, Sesboue R, et al. Contribution of the BOP1 gene, located on 8q24, to colorectal tumorigenesis. Genes Chromosomes Cancer 2006;45:874–81. [DOI] [PubMed] [Google Scholar]
- 19.Killian A, Le Meur N, Sesboue R, et al. Inactivation of the RRB1-Pescadillo pathway involved in ribosome biogenesis induces chromosomal instability. Oncogene 2004;23:8597–602. [DOI] [PubMed] [Google Scholar]
- 20.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohrmoser M, Holzel M, Grimm T, et al. Interdependence of Pes1, Bop1, and WDR12 controls nucleolar localization and assembly of the PeBoW complex required for maturation of the 60S ribosomal subunit. Mol Cell Biol 2007;27:3682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neer EJ, Schmidt CJ, Nambudripad R, et al. The ancient regulatory-protein family of WD-repeat proteins. Nature 1994;371:297–300. [DOI] [PubMed] [Google Scholar]
- 23.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi J, Yu Y, Akilli Ozturk O, et al. New Wnt/beta-catenin target genes promote experimental metastasis and migration of colorectal cancer cells through different signals. Gut 2016;65:1690–701. [DOI] [PubMed] [Google Scholar]
- 25.Grummt I. The nucleolus-guardian of cellular homeostasis and genome integrity. Chromosoma 2013;122:487–97. [DOI] [PubMed] [Google Scholar]
- 26.Song L, Bhuvaneshwar K, Wang Y, et al. CINdex: A Bioconductor Package for Analysis of Chromosome Instability in DNA Copy Number Data. Cancer Inform 2017;16:1176935117746637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yasui Y, Urano T, Kawajiri A, et al. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem 2004;279:12997–3003. [DOI] [PubMed] [Google Scholar]
- 28.den Hollander J, Rimpi S, Doherty JR, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010;116:1498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jambhekar A, Emerman AB, Schweidenback CT, et al. RNA stimulates Aurora B kinase activity during mitosis. PLoS One 2014;9:e100748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeRose YS, Wang G, Lin YC, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 2011;17:1514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simons CC, Hughes LA, Smits KM, et al. A novel classification of colorectal tumors based on microsatellite instability, the CpG island methylator phenotype and chromosomal instability: implications for prognosis. Ann Oncol 2013;24:2048–56. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe T, Kobunai T, Yamamoto Y, et al. Chromosomal instability (CIN) phenotype, CIN high or CIN low, predicts survival for colorectal cancer. J Clin Oncol 2012;30:2256–64. [DOI] [PubMed] [Google Scholar]
- 34.Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 2017;376:2109–2121. [DOI] [PubMed] [Google Scholar]
- 35.Carter SL, Eklund AC, Kohane IS, et al. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nature Genetics 2006;38:1043–1048. [DOI] [PubMed] [Google Scholar]
- 36.Lips EH, van Eijk R, de Graaf EJ, et al. Integrating chromosomal aberrations and gene expression profiles to dissect rectal tumorigenesis. BMC Cancer 2008;8:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmena M, Wheelock M, Funabiki H, et al. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 2012;13:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz-Barrera M, Monje-Casas F. Increased Aurora B activity causes continuous disruption of kinetochore-microtubule attachments and spindle instability. Proc Natl Acad Sci U S A 2014;111:E3996–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.