

Abstract
Background & Aims:
Severe injury to the lining of the stomach leads to changes in the epithelium (reprogramming) that protect and promote repair of the tissue, including development of spasmolytic polypeptide expressing metaplasia (SPEM) and tuft and foveolar cell hyperplasia. Acute gastric damage elicits a type-2 inflammatory response that includes production of type-2 cytokines and infiltration by eosinophils and alternatively activated macrophages. Stomachs of mice that lack interleukin 33 (IL33) or interleukin 13 (IL13) did not undergo epithelial reprogramming after drug-induced injury. We investigated the role of group 2 innate lymphoid cells (ILC2s) in gastric epithelial repair.
Methods:
Acute gastric injury was induced in C57BL/6J mice (wild type and RAG1 knockout) by administration of L635. We isolated ILC2s by flow cytometry from stomachs of mice that were and were not given L635 and performed single-cell RNA sequencing. ILC2s were depleted from wild-type and RAG1-knockout mice by administration of anti-CD90.2. We assessed gastric cell lineages, markers of metaplasia, inflammation, and proliferation. Gastric tissue microarrays from patients with gastric adenocarcinoma were analyzed by immunostaining.
Results:
There was a significant increase in the number of GATA3-positive ILC2s in stomach tissues from wild-type mice after L635-induced damage, but not in stomach tissues from IL33-knockout mice. We characterized a marker signature of gastric mucosal ILC2s and identified a transcription profile of metaplasia-associated ILC2s, which included changes in expression of Il5, Il13, Csf2, Pd1, and Ramp3; these changes were validated by quantitative PCR and immunocytochemistry. Depletion of ILC2s from mice blocked development of metaplasia after L635-induced injury in wild-type and RAG1-knockout mice and prevented foveolar and tuft cell hyperplasia and infiltration or activation of macrophages after injury. Numbers of ILC2s were increased in stomach tissues from patients with SPEM compared to patients with normal corpus mucosa.
Conclusions:
In analyses of stomach tissues from mice with gastric tissue damage and patients with SPEM, we found evidence of type 2 inflammation and increased numbers of ILC2s. Our results suggest that ILC2s coordinate the metaplastic response to severe gastric injury.
Keywords: ILC2, SPEM, metaplasia, GATA3
Graphical Abstract
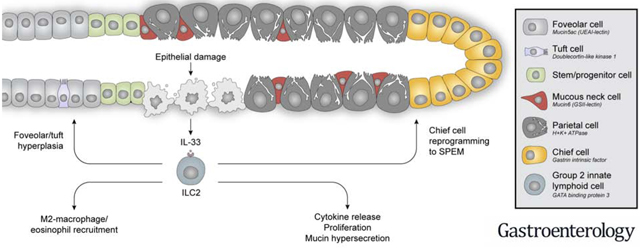
LAY SUMMARY
We have determined that intrinsic mucosal immune cells are integral for coordinated repair of the gastric lining following severe injury.
INTRODUCTION
The epithelial lining of the stomach is exposed to harsh conditions including ingested food and bacteria, as well as gastric acid and digestive enzymes. The stomach is protected from this extreme environment by the mucosal barrier which is lined with epithelial cells linked by tight junctions and a thick mucus layer to block stomach contents from penetrating to the underlying tissue layers. Severe damage to the lining of the stomach leads to reprogramming of the epithelium to recruit reparative cells to sites of mucosal injury. In the stomach, reprogramming is characterized by the transdifferentiation of digestive enzyme-secreting chief cells to mucin-secreting metaplasia (spasmolytic polypeptide-expressing metaplasia or SPEM), expansion of the foveolar and tuft cell lineages, and infiltration and activation of immune cells including M2 macrophages and eosinophils. Our previous investigations have suggested reprogramming is governed by a cytokine signaling cascade.1 The cascade begins with the alarmin interleukin-33 (IL-33) which is released from cells at sites of mucosal injury or infection.2 Mice lacking IL-33 or the IL-33 receptor (ST2) are blocked from epithelial reprogramming after drug-induced injury.3 Similarly, chronic treatment with recombinant IL-33 promotes oxyntic atrophy, metaplasia development, and inflammatory infiltration in the stomach.3 Downstream pathway analysis confirmed that IL-33 release in the stomach promotes the induction of type II cytokines, including IL-4, IL-5, IL-9, and IL-13. Mice lacking IL-13, but not other type II cytokines, are inhibited from the development of mucin-secreting metaplasia after gastric damage. Furthermore, recombinant IL-13 treatment to ST2 null mice restored metaplasia development following acute parietal cell loss.1
Recent advances in our understanding of immune responses to tissue damage or infection have indicated a crucial role for innate lymphoid cells (ILCs).4 Derived from lymphoid progenitors, ILCs generate potent levels of cytokines that were previously thought to be primarily produced by T helper cells.5 However, unlike T helper cells, ILCs have no antigen-specific receptors and cause antigen-independent immune responses.6 They are prevalent at mucosal surfaces where they respond to factors derived from the epithelium that indicate damage or infection.7 A subtype of the ILC family, known as group 2 innate lymphoid cells or ILC2s, are regulated by the transcription factors RORα and GATA3.8, 9 They are stimulated by epithelial cell-derived stress signals such as IL-33, IL-25, and TSLP to provide an innate source of type 2 cytokines.10–12 While ILC2s are commonly studied in tissues such as: lung, intestine, and skin, few investigations have addressed the role of ILC2s in the stomach.13, 14 ILC2s are involved in tissue remodeling, mucus metaplasia, eosinophilia, and alternative macrophage activation through the production of type 2 cytokines in other tissues.15 We hypothesized that ILC2s similarly regulate epithelial reprogramming after damage to the stomach.
Reprogramming to recruit reparative cells to sites of injury has been described in a number of mucosal contexts including esophagus, stomach, small bowel, colon, and pancreas.16 While such processes are programmed to recede following the resolution of injury, chronic damage and inflammation promote the evolution of these reparative metaplastic lineages into dysplastic or pre-neoplastic lineages that are a risk factor for developing cancer. Gastric cancer remains one of the leading causes of cancer-related death worldwide. Therefore a more comprehensive understanding of the development of metaplasia and progression to intestinal-type gastric cancer is needed The most common cause of chronic damage to the stomach occurs as a result of infection with the bacterium Helicobacter pylori, which leads to the loss of acid-secreting parietal cells.17 Gastric pathology can take months to develop in Helicobacter infected mice, and years in humans. To facilitate an accelerated model of gastric damage and epithelial reprogramming, our laboratory has developed an acute model of parietal cell loss through the administration of the parietal cell toxic drug L635.18 Epithelial reprogramming is visualized within hours after the administration of L635.
We have now sought to evaluate the role of ILC2s in the regulation of the gastric mucosal response to significant injury. The present study reveals that there is a sizable accumulation of ILC2s in the stomach after damage that is blocked in mice lacking IL-33. We have performed single cell RNA sequencing of ILC2s isolated from the normal “healthy” stomach as well as the L635-induced metaplastic stomach of mice and characterized a unique marker signature of gastric mucosal ILC2s, as well as identified a distinct transcriptional profile of metaplasia-associated ILC2s. Sequencing revealed several genes that may inform how ILC2s contribute to gastric pathology including Il5, Il13, Csf2, Pd1, and Ramp3. Depletion of ILC2s blocked the development of metaplasia after drug-induced injury in wild-type and Rag1 knockout mice. Similarly, ILC2 depletion prevented foveolar and tuft cell hyperplasia and the infiltration/activation of macrophages and eosinophils after injury. Understanding the role of type 2 inflammation and ILC2s in the induction of metaplasia may link the damage response in the stomach to other type 2-mediated diseases and could pave the way to better detection methods and therapies for precancerous metaplasia in the stomach.
MATERIALS AND METHODS
Mouse Models
Wild-type and Rag1 knockout C57BL/6J mice approximately eight weeks old were purchased from Jackson Labs (Bar Harbor, Maine, USA). Each experimental group consisted of at least three to four mice. L635 (synthesized by the Chemical Synthesis Core of the Vanderbilt Institute of Chemical Biology), dissolved in dH2O was administered by oral gavage (350 mg/kg) once a day for 3 consecutive days. Rat IgG2b isotype control antibody (BioXCell BP0090) or anti-CD90.2 antibody (BioXCell BP0066) was administered intraperitoneally (300 μg) to wild-type and Rag1 knockout C57BL/6J mice every fourth day for 12 days for a total of four injections. IL-13-tdTomato reporter mice were generated as previously described.19 These mice were the kind gift of Dr. Andrew NJ McKenzie. Archival sections of stomach from wild-type, IL-33KO, and IL-13KO mice (n=6 per group) were obtained from previous investigations.1 The care, maintenance, and treatment of animals in these studies adhere to the protocols approved by the Institutional Animal Care and Use Committee of Vanderbilt University.
Single Cell Isolation
Mouse stomachs were harvested, opened along the greater curvature and washed in ice cold 1X PBS without calcium and magnesium. The antrum was removed with a razor blade and the oxyntic mucosa was harvested with a cell scraper to separate the mucosa from the serosa. The corpus mucosa was then finely minced using a razor blade and collected in ice cold Advanced DMEM/F12 cell culture medium. Tissue was allowed to settle, and the top layer of media was removed leaving a few milliliters of media containing gastric mucosa. The solution with tissue was transferred to 50 mL of pre-warmed (37°C) digestion buffer (Advanced DMEM/F12 + 5% FBS + 1 mg/mL collagenase type 1a + 1/100 DNAse I) in a round bottom flask with stir bar. The flask containing digestion buffer and tissue was placed in a 37°C water bath with stir plate and stirred at low speed for 30 minutes. To stop the reaction, 50 mL of Advanced DMEM/F12 supplemented with Y-27632 (1:1000) and 1mM DTT was added. The mixture was centrifuged at 300 × g for 5 minutes to pellet glands/cells. The supernatant was aspirated and the pellet was resuspended in 1 mL of Advanced DMEM/F12 supplemented with Y-27632. The solution was drawn up into a 1 mL syringe with 25-gauge needle attached. The solution was pushed through needle and strained through 70 μm cell strainer to disrupt the glandular structures and achieve a single cell suspension. The cell strainer was washed with 9 mL of Advanced DMEM/F12 supplemented with Y-27632. Cells were centrifuged at 300 × g for 5 minutes. Supernatant was removed and cells were resuspended in 1 mL of Advanced DMEM/F12 supplemented with Y-27632 and stored on ice.
Flow Cytometry
Three million cells per sample were added to BD Falcon 5 mL polypropylene round-bottom tubes. Cells were washed with 2 mL of 1X PBS and centrifuged at 1200 rpm for 5 minutes at 4°C. Supernatant was removed and cells were washed in 2 mL of FACS buffer (1X PBS + 3% FBS). Supernatant was removed and 3 μL of Fc Receptor Block (BD) was diluted in 50 μL of FACS buffer per sample and incubated for 15 minutes at 4°C. Cells were washed with 2 mL of FACS buffer and centrifuged at 1200 rpm for 5 minutes at 4°C. Supernatant was removed and antibodies were diluted in 50 μL of FACS buffer and incubated for 30 minutes at 4°C. Cells were washed with 2 mL of FACS buffer and centrifuged at 1200 rpm for 5 minutes at 4°C. If biotinylated antibody was used, streptavidin was diluted in 100 μL FACS buffer per sample and incubated for 30 minutes at 4°C. Viability staining was performed by adding Tonbo Ghost Dye UV450 (1:500) or DAPI (1:1000). Cells were analyzed on the 5-Laser BD LSR II or sorted on the 5-Laser FACS Aria III in the VUMC Flow Cytometry Shared Resources.
Immunohistochemical Staining
Mouse stomachs were fixed in 4% PFA overnight at 4°C and were transferred into 70% ethanol for subsequent paraffin embedding. Five-micrometer sections were used for all immunohistochemistry studies. Deparaffinization, rehydration, and antigen retrieval were performed as previously described.20 The Zeiss Axio Imager M2 microscope with Axiovision digital imaging system or the Leica Aperio Versa 200 Fluorescent Slide Scanner in the Vanderbilt Digital Histology Shared Resource was used to image sections.
Human Tissue Studies
Tissue microarrays (TMA) containing human gastric tissue from two cohorts of patients were analyzed. The first cohort included samples of normal and metaplastic mucosa from 33 gastric adenocarcinoma resections performed at the University of Tokyo.21 The second cohort included samples from 18 patients (age ranging from 39 to 81 years) who underwent curative subtotal or total gastrectomy at Jeju National University Hospital, Jeju, Korea from 2015 to 2020. Formalin-fixed and paraffin-embedded blocks were obtained from each patient. Through histologic examination, one to seven corpus intestinal metaplasia (IM) and/or SPEM regions were identified in each case, and cores (4 mm in diameter) were obtained and arranged in a recipient paraffin block using a trephine apparatus (SuperBioChips Laboratories, Seoul, Korea) Additionally, cores of normal antrum and normal corpus were included. Altogether, multiple tissue microarrays were generated, containing 48 corpus metaplasia cores with IM (n=45) and/or SPEM (n=28), 9 normal corpus cores, and 9 normal antrum cores. TMA construction was approved by the Institutional Review Board (IRB) of JNUH (IRB No. 2016-10-001). TMAs were immunostained for CD3, ICOS, and CD44v9. Spatial quantification of ICOS-positive CD3-negative cells was performed as previously described on well-oriented cores containing CD44v9-negative normal corpus (n=12) and cores containing CD44v9-positive SPEM (n=12).22
Data Analysis
Experimental groups contained three to six mice. Images were analyzed using CellProfiler to quantify objects (nuclei, cells) and verified manually.23 At least five representative images (>150 glands) of proximal stomach corpus were taken from each mouse at 20X objective for quantification. Spatial quantification of GATA3-positive cells was performed as previously described.22 All graphs and statistics were completed in GraphPad Prism using unpaired Student’s t-test or one-way ANOVA with Bonferroni’s post-hoc multiple comparisons test to determine significance.
RESULTS
Gastric ILC2s express the transcription factor GATA3.
To evaluate the presence of ILC2s in the gastric mucosa, we performed flow cytometry on isolated single cells from the stomachs of wild-type C57BL6/J mice. Single cells were stained for cell-surface proteins and ILC2s were defined as CD45+Lin−CD127+CD90+ICOS+, where Lin included CD3, CD5, CD11b, CD45R, Anti-7–4, FcεRI, Ly6G/C, Anti-Ter-119.12, 24, 25 The cells were fixed/permeabilized and stained for GATA3, the transcription factor that is critical for the development and maintenance of ILC2s. Greater than 93-percent of ILC2s collected were positive for GATA3 when compared to an isotype control antibody (Figure 1A). Furthermore, we determined that GATA3-positive cells in the stomach were negative for other immune cell lineage markers by both flow cytometry and immunostaining (Supplemental Figure 1). These findings verified that GATA3 is an efficient marker for ILC2s in the gastric mucosa under homeostatic conditions and following L635-induced acute gastric damage.
Figure 1. GATA3-positive ILC2s accumulate in the stomach after injury and secrete IL-13 in an IL-33 dependent manner.
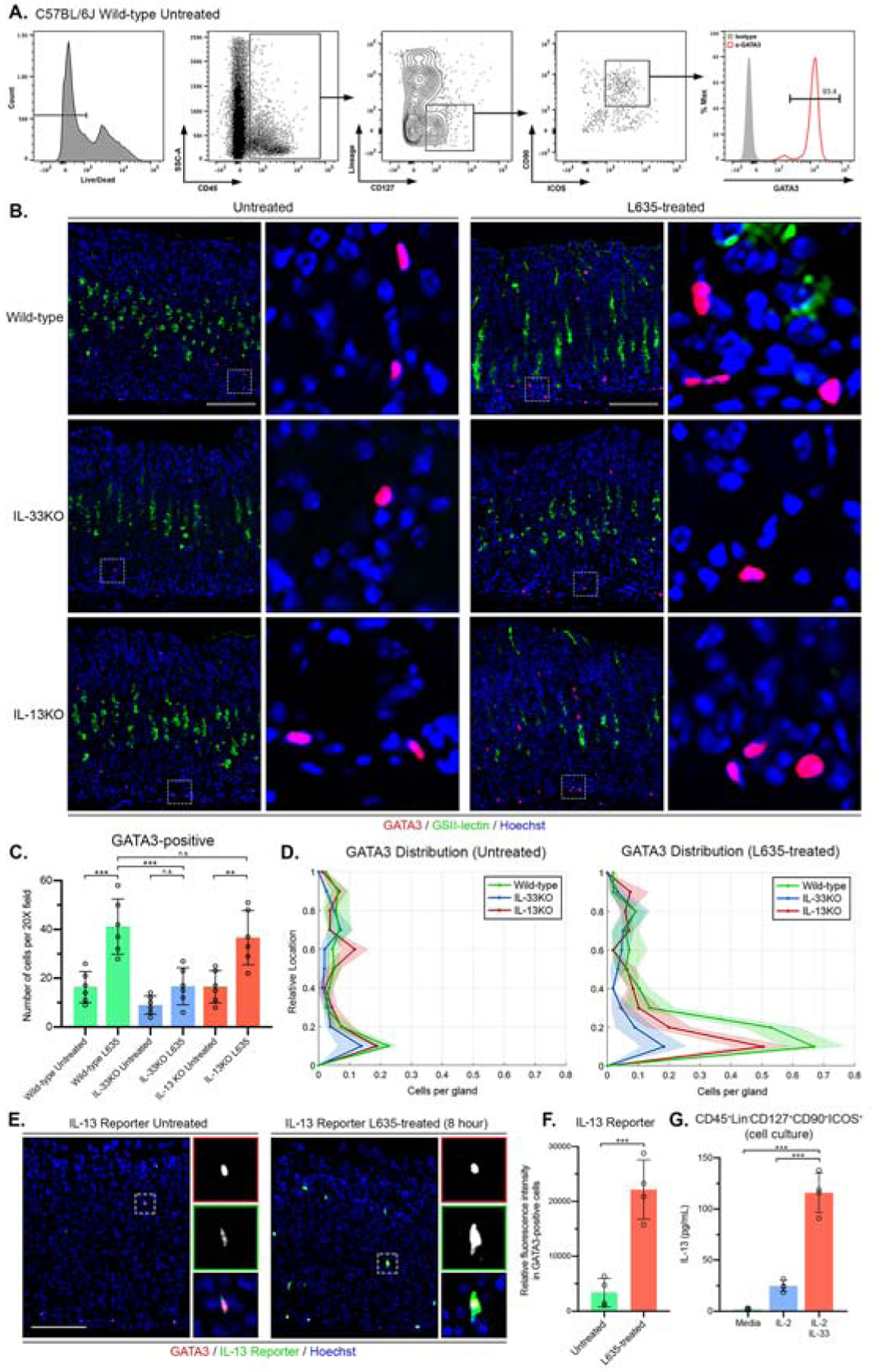
(A) Flow cytometric analysis of isolated fixed cells from wild-type C57BL/6J mice. Greater than 93-percent of CD45⁺Lin-CD127+CD90+ICOS+ ILC2s are positive for GATA3. Rat IgG2b kappa antibody utilized as isotype control. (B) Representative images of immunostaining for GATA3 (red), mucin-6 containing granule marker GSII-lectin (green), with nuclear counter stain Hoechst (blue) in both untreated and L635-treated wild-type, IL-33KO, and IL-13KO mice (scale bars = 100 μm). Magnified inset of GATA3-positive ILC2s (right). (C) Quantification of GATA3-positive ILC2s per 20X objective field. (D) Location of GATA3-positive ILC2s from panel B is shown in distribution histograms with the y-axis representing the relative location within the gastric gland divided into 10% increments (1 = lumen and 0 = base) and the x-axis depicting the number of cells per gland. (E) Immunofluorescence staining for GATA3 (red), IL13 reporter (tdTomato, green), with nuclear counterstain Hoechst (blue) (scale bars = 100 μm). Magnified inset of GATA3-positive ILC2 (right). (F) Relative fluorescence intensity of the IL-13 reporter in GATA3-positive ILC2s. (G) Effects of IL-2 or IL-2 + IL-33 on IL-13 secretion from sorted primary gastric ILC2s (CD45⁺Lin-CD127+CD90+ICOS+). IL-13 production assayed by ELISA in media after 24 hours of incubation. Statistical significance determined by unpaired Student t test or one-way ANOVA with Bonferroni’s post-hoc multiple comparisons test. N.S. for not significant p ≥ 0.05, ** for p < 0.01, and *** for p < 0.001. Error bars represent mean ± SD.
Gastric injury leads to an IL-33-dependent increase in ILC2s that secrete IL-13.
To visualize ILC2s in situ we performed immunohistochemical analysis for GATA3 in tissue sections from wild-type, IL-33 knockout (IL-33KO), or IL-13 knockout (IL-13KO) mice that were untreated or treated for three days with L635 (Figure 1B). Analysis revealed an accumulation of GATA3-positive ILC2s after damage that is contingent on IL-33, but not IL-13 (Figure 1C). In the untreated mice, rare GATA3-positive ILC2s were observed scattered throughout the glands. After L635 treatment to induce parietal cell loss in wild-type mice, a significant increase in the number of ILC2s was detected specifically located near the base of the glands (Figure 1D). Basal accumulation of ILC2s coincides with the location of chief cell reprogramming to SPEM and macrophage/eosinophil recruitment to the gastric mucosa. The significant increase of GATA3-positive ILC2s after L635 treatment was blocked in IL-33KO mice. However, similar to wild-type mice, immunostaining revealed a significant increase in ILC2s in IL-13KO mice treated with L635 (Figure 1C and D). These results indicate that the activation and recruitment of ILC2s to the gastric mucosa after damage is contingent on IL-33, but not IL-13.
To evaluate the responsiveness of gastric ILC2s, IL-13-tdTomato reporter mice were treated with L635 for 8 hours. In the IL-13 reporter mice one of the copies of the IL-13 gene is replaced with tdTomato, a fluorescent reporter, and the amount of fluorescent reporter is proportional to the amount of IL-13 that is produced. To visualize IL-13 production in GATA3-positive ILC2s, tissue sections from untreated and L635-treated IL-13 reporter mice were immunostained with tdTomato and GATA3 antibodies (Figure 1E). A significant increase in the relative fluorescence of the IL-13 reporter was observed in GATA3-positive ILC2s after 8 hours of L635-induced injury (Figure 1F). These results indicate that L635-induced injury rapidly stimulates the production of IL-13 in gastric ILC2s. Additionally, flow sorted ILC2s from the stomachs of wild-type C57BL6/J mice were cultured with media supplemented with IL-2 alone or IL-2 and IL-33. IL-13 secretion by cultured ILC2s was detected using an enzyme-linked immunosorbent assay. The ILC2s treated with IL-2 and IL-33 secreted greater than four times the amount of IL-13 compared to ILC2s treated with IL-2 alone (Figure 1G). Thus, gastric ILC2s respond to IL-33 and are likely a source of type II cytokines including IL-13 after acute gastric damage.
Single-cell RNA sequencing of gastric ILC2s.
To characterize ILC2s from the normal and metaplastic gastric mucosa, we performed single cell RNA-sequencing on ILC2s from the stomachs of wild-type C57BL6/J mice that were untreated (ILC2 Control) or treated for one day with L635 (ILC2 L635). Individual ILC2s were sorted by fluorescence activated cell sorting (FACS) directly into a 96-well plate containing lysis buffer and bar-coded primers (Figure 2A). By flow sorting, ILC2s accounted for 0.47% of viable isolated cells from the untreated gastric mucosa. However, ILC2s increased to 0.88% of viable isolated cells after one day of L635 treatment. Single cell RNA sequencing was performed as previously described using Smart-seq2.26 Both principle component and t-distributed stochastic neighbor embedding (t-SNE) analysis revealed that the L635-treated ILC2s have a distinct phenotype from the control ILC2s (Figure 2B–C). We have identified a distinctive transcriptional profile of gastric mucosal ILC2s that includes the transcription factors Gata3 and Rorα, the immune checkpoint protein Icos, and the cell surface receptors Il1rl1, Il17rb, Il7r, Il2ra, and Il2rg. Not surprisingly, several secreted factors involved in ILC2 function are increased in ILC2 L635 cells compared to control ILC2s including Il4, Il5, Il9, Il13, and Areg. Unsupervised analysis identified a unique signature of the L635-treated, metaplasia-associated ILC2s that includes the secreted factor Csf2,27 lipid mediator Dgat2,28 adhesion molecule Icam1,29 cell surface receptors Il2rb30 and Il4ra,31 immune checkpoint protein Pd1,32 and receptor (calcitonin) activity modifying proteins Ramp333–35 (Figure 2D). These single-cell RNA sequencing results were validated by qPCR and by immunostaining when antibodies were available (Supplemental Figure 2–3). Interestingly, immunostaining for the proliferation marker PCNA and GATA3 revealed that more than thirty-percent of GATA3-positive ILC2s proliferate after L635-induced injury (Supplemental Figure 3B).
Figure 2. Single cell RNA-sequencing of gastric ILC2s.
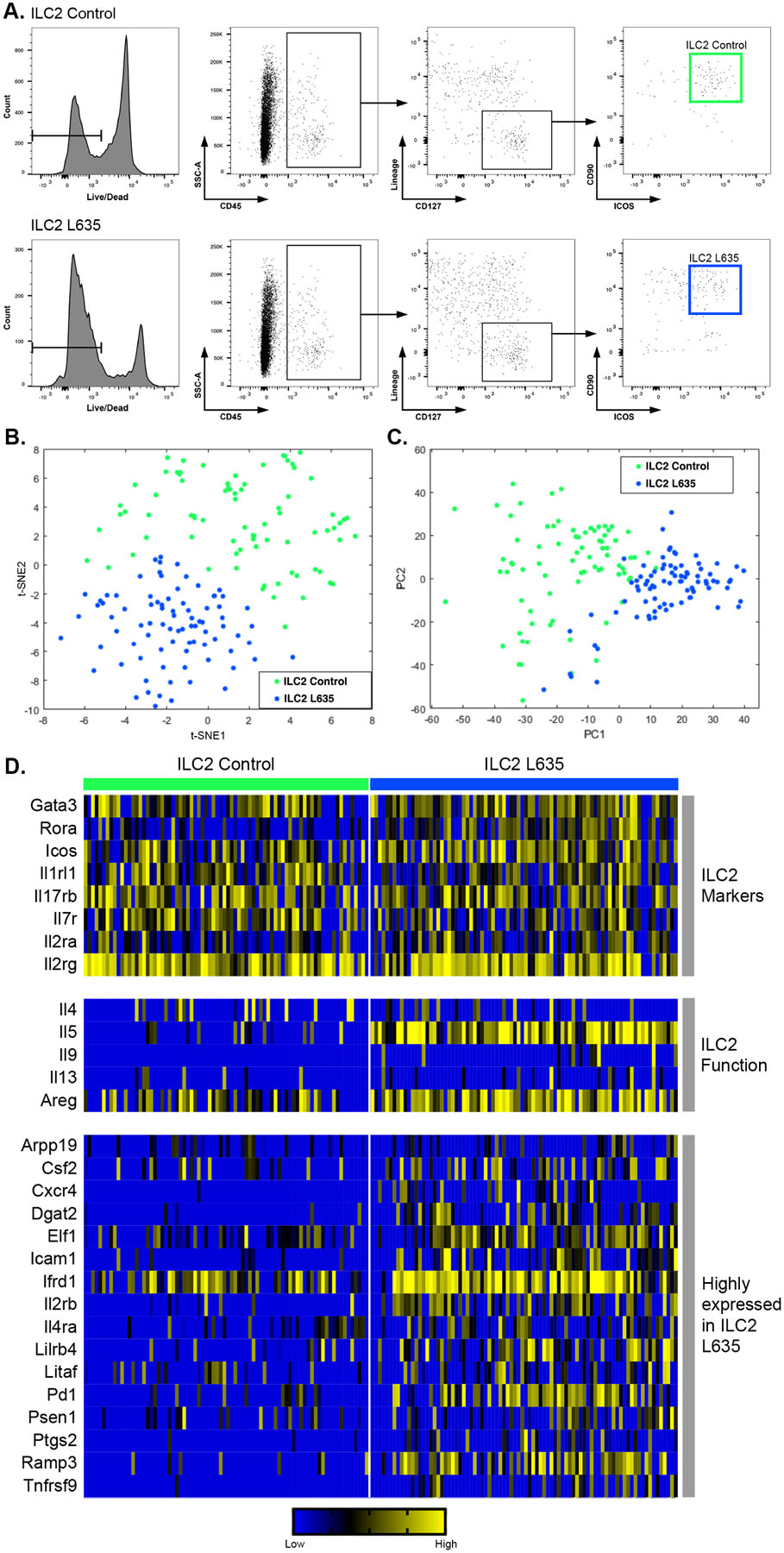
(A) Flow cytometric analysis of sorted single cells from wild-type untreated C57BL/6J mice (ILC2 Control) and wild-type C57BL/6J mice treated with one dose of L635 and sacrificed 24 hours after administration (ILC2 L635). (B) t-SNE plots and (C) principle component analysis of ILC2 Control (green) and ILC2 L635 (blue). (D) A heatmap of single cell RNA-seq data for eight ILC2 marker genes, five genes involved in ILC2 function, and sixteen genes that were identified as being highly expressed in ILC2 L635.
Anti-CD90.2 treatment decreases GATA3-positive ILC2s and ILC2-related genes in the stomach.
To elucidate if ILC2s are essential for the injury response in the stomach, we depleted ILC2s using a previously described antibody depletion method.36–38 Treatment with anti-CD90.2 antibody effectively removes ILC2s from peripheral tissues. Mice were treated with Rat IgG2b antibody as an isotype control immunoglobulin. We treated wild-type C57Bl/6J mice with anti-CD90.2 or Rat IgG2b antibody every fourth day for 12 days. Following the final antibody administration, mice were treated with L635 for three days. Stomachs were harvested from four experimental groups: 1) Rat IgG2b only, 2) anti-CD90.2 only, 3) Rat IgG2B + L635-treated, and 4) anti-CD90.2 + L635-treated mice for histological analysis and flow cytometry (Figure 3A). Treatment with either Rat IgG2b only or anti-CD90.2 only did not alter the gastric mucosa. Flow cytometry and immunostaining for GATA3 revealed significant depletion of ILC2s with anti-CD90.2 treatment (Figure 3B–E). Similarly, we found that anti-CD90.2 depletion of ILC2s led to significant reductions in transcripts elevated in ILC2s following L635 treatment including IL4, IL5, IL9, IL13, Areg, Csf2, Pd1 and Ramp3 (Figure 3F). Furthermore, PD1 positive cells were identified in the gastric mucosa of anti-CD90.2 + L635-treated mice, however none of the cells were GATA3-positive ILC2s (Supplemental Figure 4).
Figure 3. Anti-CD90.2 treatment effectively depletes ILC2s and reduces expression of ILC2-related genes.
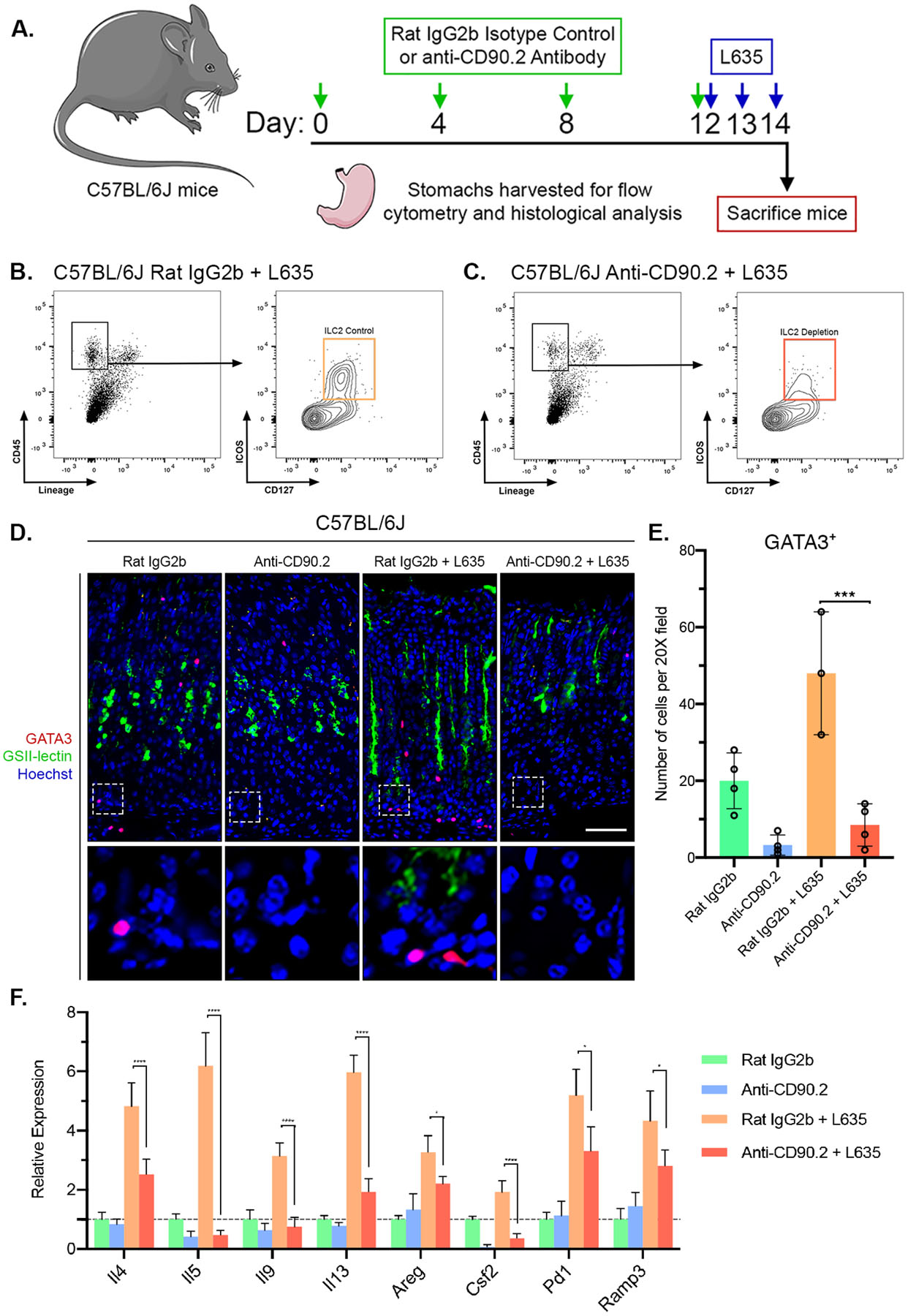
(A) Diagram of ILC2 depletion and drug treatments. Rat IgG2b isotype control antibody or Anti-CD90.2 antibody was administered intraperitoneally to wild-type C57BL/6J mice every fourth day for 12 days. Following the final antibody administration, mice were treated with the parietal cell toxic drug L635 by oral gavage daily for three days. Mice were sacrificed 2 hours after final dose of L635, and stomach tissue from Rat IgG2b only mice (n=4), Anti-CD90.2 only mice (n=4), Rat IgG2b + L635 mice (n=3), and Anti-CD90.2 + L635 mice (n=4) was harvested for flow cytometry or histological analysis. Flow cytometric analysis of isolated single cells from (B) Rat IgG2b + L635 and (C) Anti-CD90.2 + L635 stomach tissue. Depletion of CD45+Lin−CD127+ICOS+ ILC2 population with Anti-CD90.2 treatment. (D) Representative images of immunostaining for GATA3 (red), mucin-6 containing granule marker GSII-lectin (green), with nuclear counter stain Hoechst (blue) (scale bars = 100 μm). Magnified inset of gland base (bottom). (E) Quantification of GATA3-positive ILC2s per 20X objective field. (F) Relative mRNA expression of ILC2 related genes (Il4, Il5, Il9, Il13, Areg, Csf2, Pd1, and Ramp3) in each group. Expression values normalized to Rat IgG2b only group. Statistical significance determined by one-way ANOVA with Bonferroni’s post-hoc multiple comparisons test. * for p < 0.05, *** for p < 0.001, and **** for p < 0.0001. Error bars represent mean ± SD.
Depletion of ILC2s inhibits development of metaplasia following injury.
To visualize parietal cells and L635-induced parietal cell loss, we performed immunostaining for the proton pump H+/K+ ATPase, an integral membrane protein responsible for gastric acid secretion by parietal cells (Figure 4A). In the Rat IgG2b only and anti-CD90.2 only groups, a large number of H+/K+ ATPase-positive parietal cells were detected throughout the oxyntic glands. Both the Rat IgG2b and anti-CD90.2 groups had comparable levels of L635-induced parietal cell loss (Figure 4C). Parietal cell loss promotes the reprogramming of zymogenic chief cells to mucin-secreting metaplastic cells (SPEM) in order to protect and fuel repair of the stomach. During this process, SPEM cells contain both gastric intrinsic factor (GIF)-positive zymogenic granules and mucin granules that can be detected with Griffonia simplicifolia (GSII)-lectin which binds to sugar modifications on Mucin 6. To detect chief cell reprogramming in ILC2 depleted mice, we immunostained with GIF and GSII-lectin (Figure 4A). Anti-CD90.2 treatment decreased the number of GIF and GSII dual-positive SPEM cells by greater than sixty-percent after L635-induced parietal cell loss (Figure 4D). We also validated these experiments in Rag1 knockout C57BL/6J mice, which do not produce any mature B or T lymphocytes (Supplemental Figure 5), and we observed a similar pattern of decreased SPEM development with ILC2 depletion.
Figure 4. ILC2 depletion blocks chief cell reprogramming to mucin-secreting SPEM after L635-induced injury.
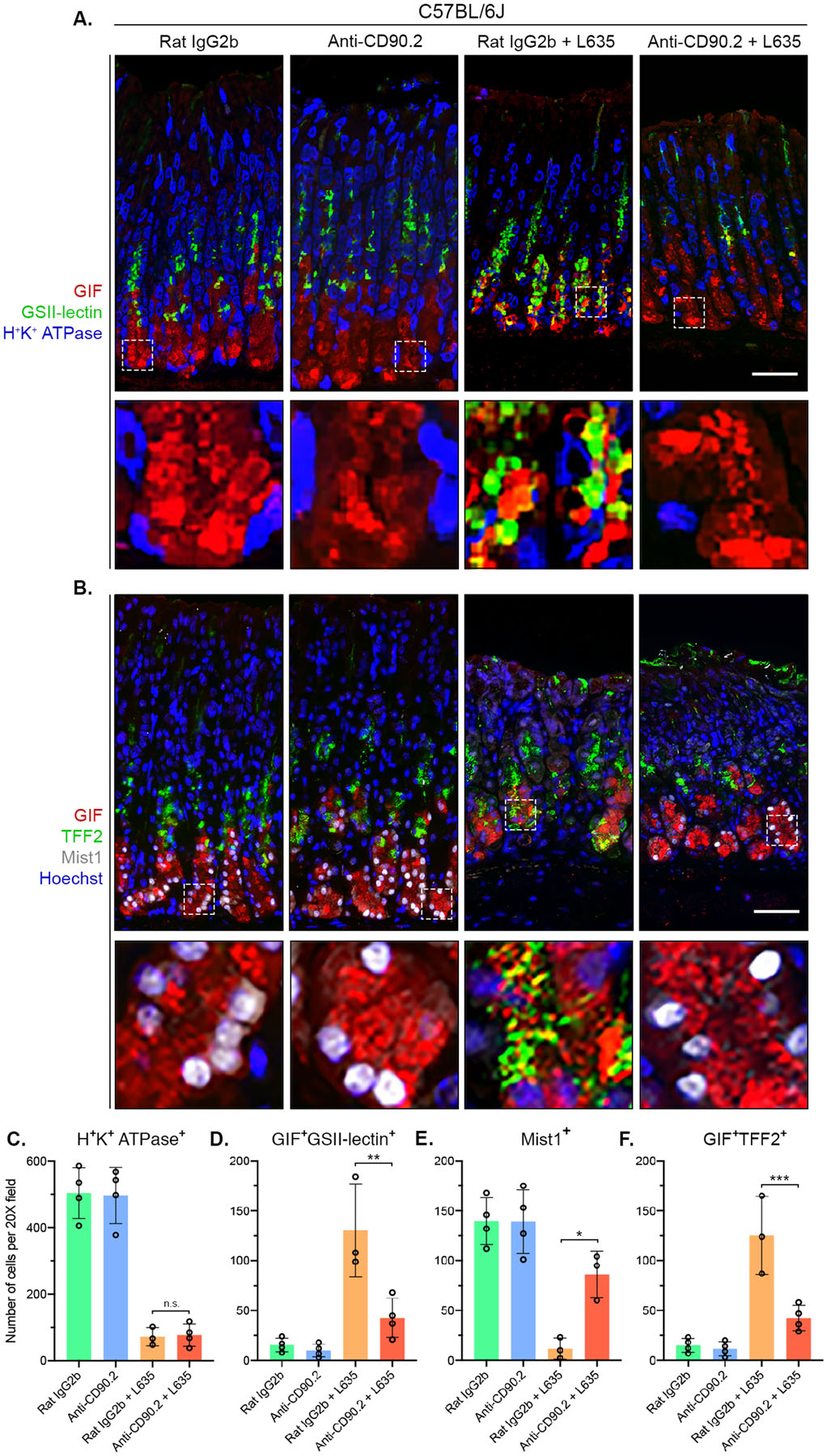
Immunostained sections from Rat IgG2b (n=4), Anti-CD90.2 (n=4), Rat IgG2b + L635 (n=3), and Anti-CD90.2 + L635 (n=4) treated C57BL/6J mice. (A) Representative images of zymogenic granule marker GIF (red), mucin-6 containing granule marker GSII-lectin (green), and parietal cell marker H+/K+ ATPase (blue) (scale bars = 100 μm). Magnified inset of chief cell region (bottom). (B) Representative images of zymogenic granule marker GIF (red), mucin granule marker TFF2 (green), chief cell transcription factor Mist1 (white) with nuclear counter stain Hoechst (blue) (scale bars = 100 μm). Magnified inset of chief cell region (bottom). Quantification of (C) H+K+ ATPase-positive parietal cells (D) GIF and GSII-lectin dual-positive (SPEM) cells (E) Mist1-positive cells and (F) GIF and TFF2 dual-positive (SPEM) cells per 20X objective field. Statistical significance determined by one-way ANOVA with Bonferroni’s post-hoc multiple comparisons test. N.S. for not significant, * for p < 0.05, ** for p < 0.01, and *** for p < 0.001. Error bars represent mean ± SD.
The basic helix-loop-helix transcription factor Mist1 (Bhlha15) controls the secretory architecture of chief cells including cellular organization and production of large protein-containing zymogenic granules.39 Loss of Mist1 is a distinct feature of chief cell reprogramming into SPEM. Dual immunofluorescence staining for GIF and Mist1 allows for monitoring of Mist1 loss in zymogenic granule-containing chief cells (Figure 4B). Immunostaining revealed that chief cells from anti-CD90.2 + L635-treated mice retained expression of Mist1 (Figure 4E). In the normal oxyntic mucosa, the expression of TFF2 is restricted to mucous neck cells found in the neck region of oxyntic glands. In L635-treated mice, TFF2 is up-regulated in chief cells at the base of glands as they reprogram into SPEM. In anti-CD90.2 + L635-treated mice, significantly fewer TFF2 and GIF dual-positive cells at the base of glands were observed compared to Rat IgG2b + L635-treated mice (Figure 4F). Collectively, these results suggest that ILC2 depletion prevents the initiation of chief cell reprogramming to mucin-secreting SPEM after acute parietal cell loss.
ILC2s are responsible for alteration in tuft cell abundance after oxyntic atrophy.
Tuft cells represent an unusual type of chemosensory epithelial cell present in multiple organs of the digestive system, including the stomach and the intestine.40, 41 Loss of parietal cells leads to the reversible expansion of Dclk1-expressing tuft cells in the mouse gastric mucosa.42 Interestingly, ILC2-derived IL-13 promotes tuft cell hyperplasia following helminth infection in the small intestine.43, 44 To determine if ILC2s are required for tuft cell hyperplasia following L635-induced parietal cell loss, we performed immunostaining for Dclk1 (Figure 5A). Anti-CD90.2 treatment blocked expansion of the tuft cell lineage following L635-induced parietal cell loss (Figure 5C). Thus, ILC2s coordinate tuft cell hyperplasia after gastric damage.
Figure 5. ILC2 depletion inhibits L635-induced foveolar/tuft cell hyperplasia and proliferation.
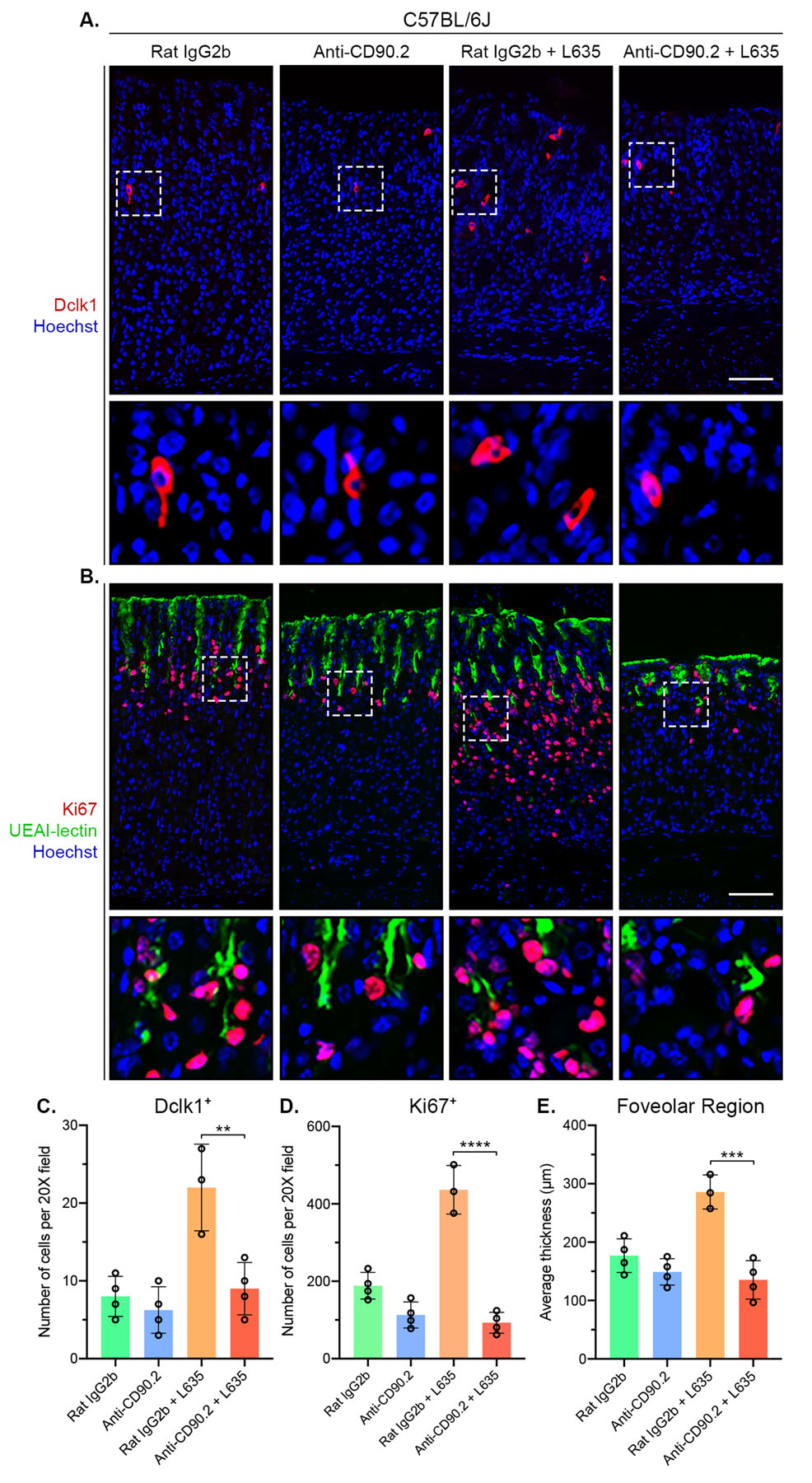
Immunostained sections from Rat IgG2b (n=4), Anti-CD90.2 (n=4), Rat IgG2b + L635 (n=3), and Anti-CD90.2 + L635 (n=4) treated C57BL/6J mice. (A) Representative images of tuft cell marker Dclk1 (red) with nuclear counter stain Hoechst (blue) (scale bars = 100 μm). Magnified inset of Dclk1-positive tuft cells (bottom). (B) Representative images of proliferation marker Ki67 (red), foveolar cell marker UEAI-lectin (green), with nuclear counter stain Hoechst (blue) (scale bars = 100 μm). Magnified inset of gland isthmus (bottom). Quantification of (C) Dclk1-positive tuft cells and (D) Ki67-positive proliferating cells per 20X objective field. (E) Average thickness (μm) of UEAI-lectin-positive foveolar region. Statistical significance determined by one-way ANOVA with Bonferroni’s post-hoc multiple comparisons test. ** for p < 0.01, *** for p < 0.001, and **** for p < 0.0001. Error bars represent mean ± SD.
ILC2s are required for proliferation and foveolar hyperplasia after injury.
ILC2s have been implicated in maintaining the gastric stem cell niche, so we sought to determine the effect of ILC2 depletion on proliferation in the gastric mucosa.36 To do this, we immunostained for the proliferation marker Ki67 (Figure 5B). In the normal oxyntic mucosa, Ki67 labelled stem/progenitor cells are located in the gland isthmus about a third of the way down the gland. Upon gastric injury, chief cells at the base of the glands reprogram and are capable of re-entering into the cell cycle and proliferating.18, 45, 46 Additionally, mucin-producing foveolar cells located near the lumen of gastric glands also expand in response to injury, a gastric lesion referred to as foveolar hyperplasia.47 Foveolar cells produce Muc5ac, a mucin recognized by Ulex Europaeus Agglutinin I (UEAI)-lectin in the gastric mucosa. Anti-CD90.2 + L635-treated mice had significantly less proliferation than Rat IgG2b + L635-treated mice (Figure 5D). To evaluate foveolar hyperplasia after L635 treatment, we measured the average thickness of the UEAI-positive foveolar region. The average thickness of the foveolar region of anti-CD90.2 + L635-treated mice was significantly shorter than Rat IgG2b + L635-treated mice (Figure 5E).
ILC2s promote infiltration of macrophages and eosinophils into the gastric mucosa.
Our previous investigations determined that L635-induced parietal cell loss results in F4/80-positive macrophage infiltration into the gastric mucosa. RNA sequencing and immunohistochemical data revealed macrophages associated with advanced mucin-secreting metaplasia in L635-treated mice have an M2 polarized phenotype and are considered alternatively activated.20 Alternatively-activated M2 macrophages are typically associated with wound healing and tissue repair. To visualize macrophage infiltration and polarization in ILC2 depleted mice we performed co-immunostaining for the macrophage marker F4/80 with the M2 polarization marker CD163 (Figure 6A). Anti-CD90.2 + L635-treated mice showed significantly decreased macrophage infiltration into the gastric mucosa and very few M2 polarized macrophages were detected (Figure 6B). These data suggest a role for ILC2s in macrophage recruitment and activation after acute gastric damage.
Figure 6. ILC2 depletion blocks M2-macrophage and eosinophil infiltration after gastric damage.
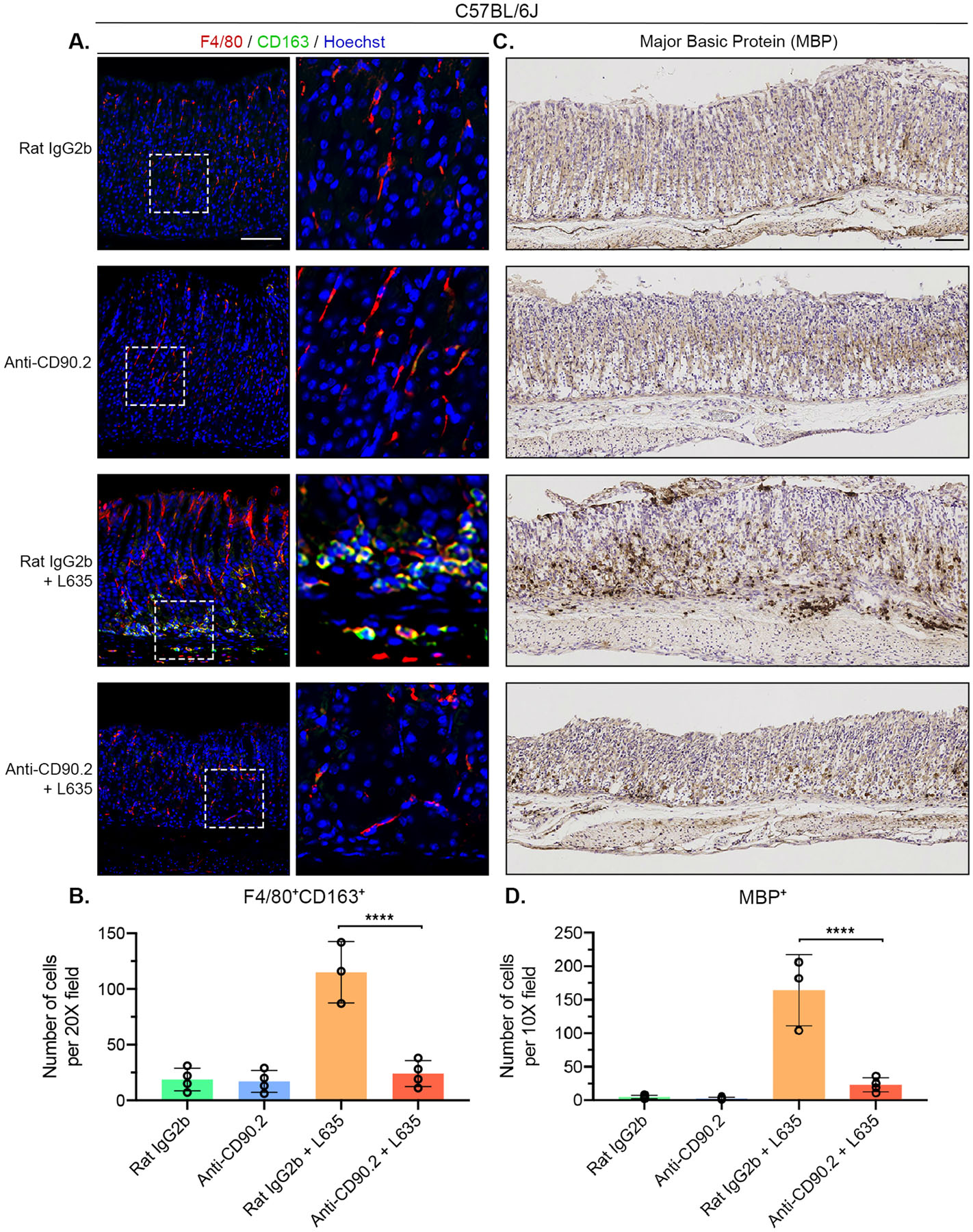
Immunostained sections from Rat IgG2b (n=4), Anti-CD90.2 (n=4), Rat IgG2b + L635 (n=3), and Anti-CD90.2 + L635 (n=4) treated C57BL/6J mice. (A) Representative images of macrophage/ dendritic cell marker F4/80 (red), alternatively activated macrophage marker CD163 (green), with nuclear counter stain Hoechst (blue) (scale bars = 100 μm).
Magnified inset of F4/80-positive macrophages (right). (B) Quantification of F4/80 and CD163 dual-positive alternatively activated macrophages per 20X objective field. (A) Representative images of immunohistochemical staining for eosinophil specific marker Major Basic Protein (MBP). (B) Quantification of MBP-positive eosinophils per 10X objective field. Statistical significance determined by one-way ANOVA with Bonferroni’s post-hoc multiple comparisons test. **** for p < 0.0001. Error bars represent mean ± SD.
Our previous studies also revealed L635-treated mice have robust eosinophil infiltration.1 Eosinophil-derived mediators have wide-ranging effects on normal tissue homeostasis and tissue remodeling/repair.48 Eosinophil recruitment to the gastric mucosa is dependent on IL-5. Therefore, we sought to determine if ILC2s play a role in eosinophil infiltration by performing immunohistochemical staining for the eosinophil marker Major Basic Protein (MBP) (Figure 6C). Anti-CD90.2 treatment blocked eosinophil infiltration following L635-induced parietal cell loss (Figure 6D). These results provide evidence that ILC2s play a role in eosinophil recruitment to the gastric mucosa following acute injury, likely through the production of IL-5.
Metaplasia is associated with an increase of ILC2s in the human stomach.
Given the significant response of ILC2s in acute models of parietal cell loss in mice, we sought to analyze ILC2s in samples of SPEM from human patients by staining for ICOS and CD3 in sections of normal corpus and SPEM from patients who underwent gastrectomy for gastric cancer (Figure 7A). ICOS-positive, CD3-negative ILC2s were detected in each sample. Interestingly, sections containing CD44v9-positive SPEM lesions showed increased numbers of ILC2s. Distribution analysis revealed ICOS-positive CD3-negative ILC2s shift toward the base of the gland in metaplastic samples compared to normal corpus, coincident with CD44v9-positive SPEM cells (Figure 7B). These studies confirm that, similar to the response in mice, accumulation of ILC2s is associated with SPEM in the human stomach.
Figure 7. Accumulation of ICOS-positive ILC2s is associated with human SPEM.
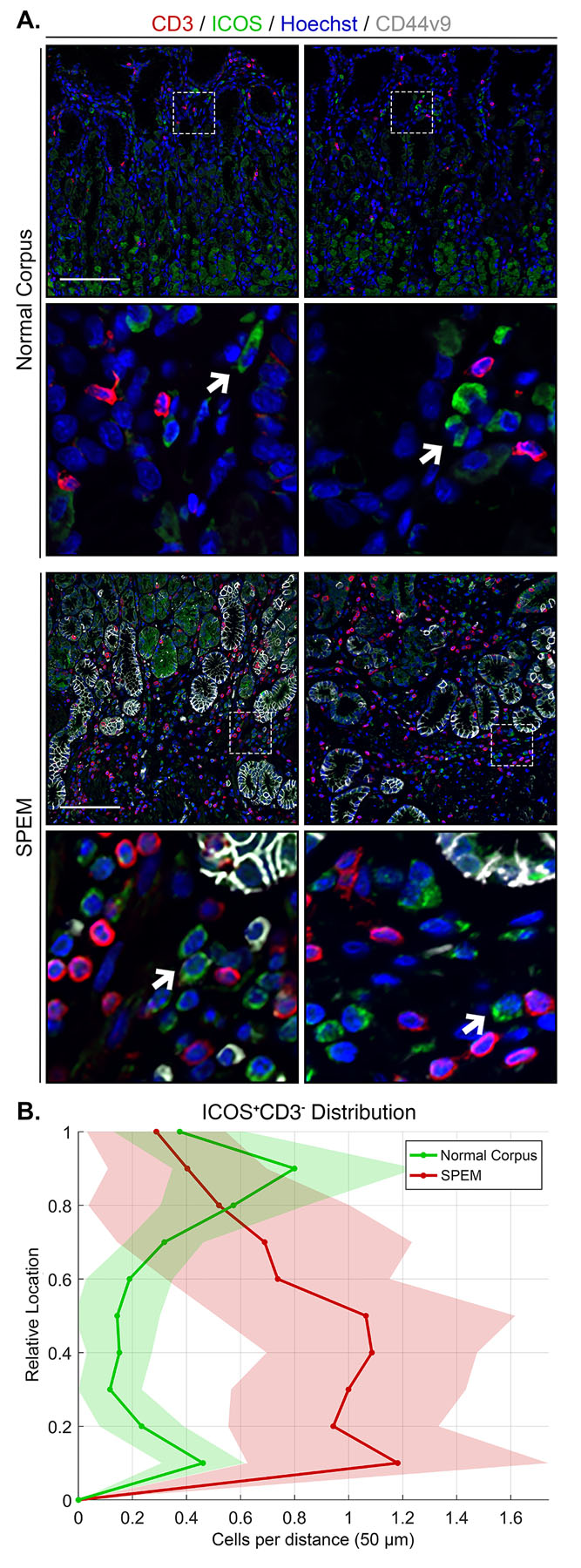
Immunostained sections of normal corpus (n=12) and SPEM (n=12) from human patients. (A) Representative images of T cell marker CD3 (red), ICOS (green), SPEM cell marker CD44v9 (white), with nuclear counter stain Hoechst (blue) (scale bars = 100 μm). Magnified inset of ICOS-positive CD3-negative ILC2s (bottom). Arrows indicate ICOS-positive CD3-negative ILC2s. Artifact from autofluorescent parietal cells is visible (green). (B) Location of ICOS-positive CD3-negative ILC2s from panel A is shown in distribution histograms with the y-axis representing the relative location within the gastric gland divided into 10% increments (1 = lumen and 0 = base) and the x-axis depicting the number of cells per distance (50 μm).
DISCUSSION
Following significant gastric injury, the epithelium recruits reparative lineages to sites of damage through both the reprogramming and expansion of mucin-secreting lineages. Chief cell reprogramming to SPEM is a necessary process for gastric epithelial repair. This process of SPEM development is triggered by damage to gastric epithelium including acute arietal cell loss,18 ulceration,49 or chronic infection with Helicobacter species.18 Previous investigations in the stomach have attempted to identify the mechanism of gastric repair following injury or chronic Helicobacter infection. We recently identified that an IL-33/IL-13 cytokine-signaling network is necessary and sufficient for the induction of epithelial reprograming following chemically-induced acute parietal cell loss.1 IL-33 and IL-13 knockout mice showed a blockade in SPEM development and have reduced M2-macrophage infiltration and polarization. Additionally, macrophage depletion attenuated advancement of metaplastic lesions.20 The present study identified that ILC2-derived factors are required for the reprogramming of the gastric mucosa after injury. Using in vivo mouse models, we have determined that, in the absence of ILC2s, the development of mucin-secreting metaplasia, expansion of foveolar and tuft cell lineages, and the infiltration and activation of macrophages and eosinophils are all attenuated. These results indicate that ILC2s perform a central role in the coordination of gastric epithelial repair after severe damage.
Tissue resident ILC2s are an intrinsic mucosal source of cytokines including IL-4, IL-5, IL-9, and IL-13.4, 5 Cytokine production and release are activated by epithelial stress signals including IL-33. We have previously noted that IL-13 deletion, but not loss of IL-4, IL-5 or IL-9 markedly inhibits epithelial reprogramming following acute parietal cell loss.1 The present studies indicate that gastric damage triggers a significant accumulation of ILC2s in the mucosa and induces their redistribution to the bases of glands co-incident with the reprogramming of chief cells into SPEM. This increase in ILC2s was not observed in IL-33KO mice but was found in IL-13KO mice following acute gastric injury. These results indicate that IL-33 release is required for activation of ILC2s. Gastric ILC2s produced IL-13 after L635-induced injury and showed strong secretion of IL-13 in response to IL-33 in vitro. ILC2 depletion in either wild-type or Rag1 knockout mice prominently attenuated chief cell reprogramming to SPEM following acute gastric damage. In fact, both foveolar hyperplasia and SPEM require ILC2 activation to develop. Additionally, we found that ablation of ILC2 cells blocked the expansion of tuft cells and the activation of infiltrating macrophages and eosinophils following injury. Collectively, these findings suggest that ILC2 cells are the major source of IL-13 and are required for the coordinated response to severe injury in the gastric epithelium.
Many of the putative roles for ILC2s in gastric mucosal healing demonstrated here are similar to those identified in asthma or allergic airway responses as well as infection-related intestinal repair.13, 50 In all cases, activation of ILC2s leads to mucin hypersecretion that is integral to mucosal repair and protection. In addition, a close relationship between ILC2s and populations of sensory tuft cells appear critical to the coordination of the response. In the intestinal epithelium, release of IL-25 from tuft cells is a critical activator of ILC2s.43, 44 While ILC2s can respond to tuft cell derived signals, in the present investigation the expansion of tuft cell numbers following oxyntic atrophy was dependent on ILC2s. Thus, there appears to be a reciprocal relationship for signaling between tuft cells and ILC2s in the gastric mucosa. Furthermore, ILC2 functional responses appear to have tissue specific characteristics.51 While many of the responses observed in activated ILC2 cells overlap with ILC2 responses noted in other tissues, including upregulation of IL-13, IL-4, IL-5 and Areg, other highly expressed transcripts appear to be more specific to the gastric ILC2s. In addition to the expansion of ILC2 numbers, single cell sequencing demonstrated that ILC2s markedly upregulate the expression of cytokines as well as a number of key regulators including Pd1, Dgat2 and Ramp3. How these specific regulators are related to the function of ILC2s in the coordinated response to gastric injury remains to be determined.
In summary, our investigations have demonstrated the importance of ILC2 in the coordinated gastric epithelial response following severe injury. ILC2s elicit a cascade of events including the initiation of chief cell reprogramming, expansion of sensory tuft cells and mucin-secreting foveolar cells, and recruitment of immune cells to the gastric mucosa. All of these mechanisms serve as critical responses to coordinate the repair of the gastric epithelium after severe mucosal injury.
Supplementary Material
WHAT YOU NEED TO KNOW:
BACKGROUND AND CONTEXT
Severe injury to the lining of the stomach leads to changes in the epithelium (reprogramming) that protect and promote repair of the tissue. Foveolar hyperplasia and spasmolytic polypeptide-expressing metaplasia (SPEM) are integral to the restoration of the stomach after damage.
NEW FINDINGS
The studies detailed here suggest that ILC2s coordinate gastric repair. ILC2s recruit reparative cell lineages to protect the gastric epithelium after injury. ILC2s accumulate in regions of SPEM development in both mice and humans and show a unique metaplasia-associated transcriptome.
LIMITATIONS
These studies were performed in acute models of parietal cell loss, future studies are required to define how ILC2s contribute to gastric repair in the setting of chronic injury.
IMPACT
The present investigations focus on the fundamental mechanisms that drive metaplasia development and inflammation in response to gastric injury, and could provide important preclinical data that may lead to therapeutic strategies for reversal of metaplasia in the stomach.
Acknowledgements
These studies were supported by grants from Department of Veterans Affairs Merit Review Awards IBX000930 and a Cancer UK Grand Challenge Grant (JRG) and 2I01BX000624 (RSP), and NIH RO1 DK071590 RO1 DK101332 (JRG). ARM was supported by NIH T32 GM008554 and F31 DK117592. ACE was supported by NIH F32 DK111101 and KO1 DK121869. EB was supported by T35 DK 007383. JAW was supported by NIH National Cancer Institute K25 CA204599. This work was supported by core resources of the Vanderbilt Digestive Disease Center (NIH P30 DK058404), Translational Pathology Shared Resource (NCI/NIH Cancer Center Support Grant 2P30 CA068485-14) and imaging supported by the Vanderbilt Digital Histology Shared Resource supported by a VA Shared Instrumentation grant (1IS1BX003097).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: WJM is an employee and shareholder of 10x Genomics, Inc.
REFERENCES
- 1.Petersen CP, Meyer AR, De Salvo C, et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut 2017;67:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz C, O’Grady K, Lavelle EC, et al. Interleukin 33: an innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur J Immunol 2016;46:1091–100. [DOI] [PubMed] [Google Scholar]
- 3.Buzzelli JN, Chalinor HV, Pavlic DI, et al. IL33 Is a Stomach Alarmin That Initiates a Skewed Th2 Response to Injury and Infection. Cell Mol Gastroenterol Hepatol 2015;1:203–221 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010;464:1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo L, Junttila IS, Paul WE. Cytokine-induced cytokine production by conventional and innate lymphoid cells. Trends Immunol 2012;33:598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberl G, Colonna M, Di Santo JP, et al. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 2015;348:aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 2011;12:21–7. [DOI] [PubMed] [Google Scholar]
- 8.Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol 2015;15:415–28. [DOI] [PubMed] [Google Scholar]
- 9.Klein Wolterink RG, Serafini N, van Nimwegen M, et al. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc Natl Acad Sci U S A 2013;110:10240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li D, Guabiraba R, Besnard AG, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 2014;134:1422–1432 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salimi M, Barlow JL, Saunders SP, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 2013;210:2939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stier MT, Bloodworth MH, Toki S, et al. Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol 2016;138:814–824 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs A, Colonna M. Innate lymphoid cells in homeostasis, infection, chronic inflammation and tumors of the gastrointestinal tract. Curr Opin Gastroenterol 2013;29:581–7. [DOI] [PubMed] [Google Scholar]
- 14.Satoh-Takayama N, Kato T, Motomura Y, et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity 2020;52:635–49. [DOI] [PubMed] [Google Scholar]
- 15.Bernink JH, Germar K, Spits H. The role of ILC2 in pathology of type 2 inflammatory diseases. Curr Opin Immunol 2014;31:115–20. [DOI] [PubMed] [Google Scholar]
- 16.Goldenring JR. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J Pathol 2018;245:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burkitt MD, Duckworth CA, Williams JM, et al. Helicobacter pylori-induced gastric pathology: insights from in vivo and ex vivo models. Dis Model Mech 2017;10:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010;139:2028–2037 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barlow JL, Bellosi A, Hardman CS, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol 2012;129:191-8 e1-4. [DOI] [PubMed] [Google Scholar]
- 20.Petersen CP, Weis VG, Nam KT, et al. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology 2014;146:1727–38 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leys CM, Nomura S, Rudzinski E, et al. Expression of Pdx-1 in human gastric metaplasia and gastric adenocarcinoma. Hum Pathol 2006;37:1162–8. [DOI] [PubMed] [Google Scholar]
- 22.Weis VG, Petersen CP, Weis JA, et al. Maturity and age influence chief cell ability to transdifferentiate into metaplasia. Am J Physiol Gastrointest Liver Physiol 2017;312:G67–G76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones TR, Kang IH, Wheeler DB, et al. CellProfiler Analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics 2008;9:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tait Wojno ED, Beamer CA. Isolation and Identification of Innate Lymphoid Cells (ILCs) for Immunotoxicity Testing. Methods Mol Biol 2018;1803:353–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoyler T, Klose CS, Souabni A, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012;37:634–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picelli S, Faridani OR, Bjorklund AK, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014;9:171–81. [DOI] [PubMed] [Google Scholar]
- 27.Gour N, Smole U, Yong HM, et al. C3a is required for ILC2 function in allergic airway inflammation. Mucosal Immunol 2018;11:1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinette ML, Fuchs A, Cortez VS, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 2015;16:306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei AH, Xiao Q, Liu GY, et al. ICAM-1 controls development and function of ILC2. J Exp Med 2018;215:2157–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moro K, Yamada T, Tanabe M, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010;463:540–4. [DOI] [PubMed] [Google Scholar]
- 31.Kim BS, Wang K, Siracusa MC, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol 2014;193:3717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor S, Huang Y, Mallett G, et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J Exp Med 2017;214:1663–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sui P, Wiesner DL, Xu J, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 2018;360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagashima H, Mahlakoiv T, Shih HY, et al. Neuropeptide CGRP Limits Group 2 Innate Lymphoid Cell Responses and Constrains Type 2 Inflammation. Immunity 2019;51:682–695 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallrapp A, Burkett PR, Riesenfeld SJ, et al. Calcitonin Gene-Related Peptide Negatively Regulates Alarmin-Driven Type 2 Innate Lymphoid Cell Responses. Immunity 2019;51:709–723 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayakawa Y, Ariyama H, Stancikova J, et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015;28:800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelbertsen D, Foks AC, Alberts-Grill N, et al. Expansion of CD25+ Innate Lymphoid Cells Reduces Atherosclerosis. Arterioscler Thromb Vasc Biol 2015;35:2526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 2011;12:1045–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramsey VG, Doherty JM, Chen CC, et al. The maturation of mucus-secreting gastric epithelial progenitors into digestive-enzyme secreting zymogenic cells requires Mist1. Development 2007;134:211–22. [DOI] [PubMed] [Google Scholar]
- 40.Saqui-Salces M, Keeley TM, Grosse AS, et al. Gastric tuft cells express DCLK1 and are expanded in hyperplasia. Histochem Cell Biol 2011;136:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerbe F, Legraverend C, Jay P. The intestinal epithelium tuft cells: specification and function. Cell Mol Life Sci 2012;69:2907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi E, Petersen CP, Lapierre LA, et al. Dynamic expansion of gastric mucosal doublecortin-like kinase 1-expressing cells in response to parietal cell loss is regulated by gastrin. Am J Pathol 2015;185:2219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howitt MR, Lavoie S, Michaud M, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016;351:1329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Moltke J, Ji M, Liang HE, et al. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016;529:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leushacke M, Tan SH, Wong A, et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol 2017;19:774–86. [DOI] [PubMed] [Google Scholar]
- 46.Radyk MD, Burclaff J, Willet SG, et al. Metaplastic Cells in the Stomach Arise, Independently of Stem Cells, via Dedifferentiation or Transdifferentiation of Chief Cells. Gastroenterology 2018;154:839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nomura S, Yamaguchi H, Ogawa M, et al. Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild-type and gastrin-deficient mice. Am J Physiol Gastrointest Liver Physiol 2005;288:G362–75. [DOI] [PubMed] [Google Scholar]
- 48.Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol 2017;17:746–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engevik AC, Feng R, Choi E, et al. The Development of Spasmolytic Polypeptide/TFF2-Expressing Metaplasia (SPEM) During Gastric Repair Is Absent in the Aged Stomach. Cell Mol Gastroenterol Hepatol 2016;2:605–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scanlon ST, McKenzie AN. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol 2012;24:707–12. [DOI] [PubMed] [Google Scholar]
- 51.Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol 2018;19:1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.