

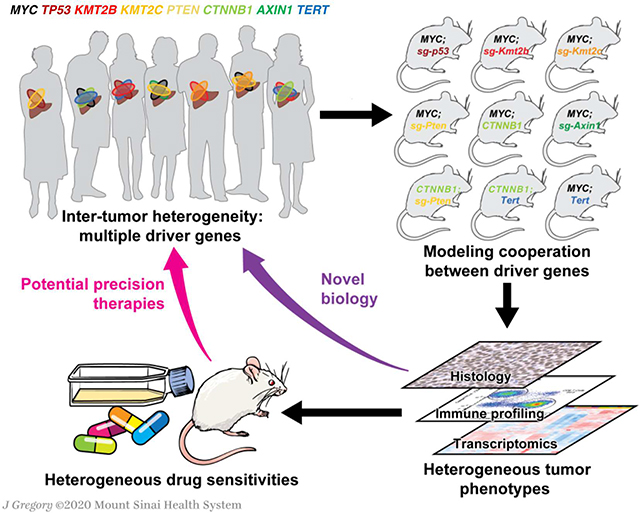
Abstract
BACKGROUND AND AIMS
The pattern of genetic alterations in cancer driver genes in patients with hepatocellular carcinoma (HCC) is highly diverse, which partially explains the low efficacy of available therapies. In spite of this, the existing mouse models only recapitulate a small portion of HCC inter-tumor heterogeneity, limiting the understanding of the disease and the nomination of personalized therapies. Here, we aimed at establishing a novel collection of HCC mouse models that captured human HCC diversity.
METHODS
By performing hydrodynamic tail-vein injections, we tested the impact of altering a well-established HCC oncogene (either MYC or β-catenin) in combination with an additional alteration in one of eleven other genes frequently mutated in HCC. Of the 23 unique pairs of genetic alterations that we interrogated, 9 were able to induce HCC. The established HCC mouse models were characterized at histopathological, immune, and transcriptomic level to identify the unique features of each model. Murine HCC cell lines were generated from each tumor model, characterized transcriptionally, and used to identify specific therapies that were validated in vivo.
RESULTS
Cooperation between pairs of driver genes produced HCCs with diverse histopathology, immune microenvironments, transcriptomes, and drug responses. Interestingly, MYC expression levels strongly influenced β-catenin activity, indicating that inter-tumor heterogeneity emerges not only from specific combinations of genetic alterations but also from the acquisition of expression-dependent phenotypes.
CONCLUSIONS
This novel collection of murine HCC models and corresponding cell lines establishes the role of driver genes in diverse contexts and enables mechanistic and translational studies.
Keywords: inter-tumor heterogeneity, mouse models, cooperation, cancer driver genes
Lay summary
This novel collection of murine hepatocellular carcinoma models and corresponding cell lines establishes the role of cancer driver genes in promoting inter-tumor heterogeneity and enables mechanistic and translational studies.
Graphical Abstract
Introduction
Liver cancer, whose most frequent type is hepatocellular carcinoma (HCC), is the fourth leading cause of cancer-related mortality worldwide, with more than 750,000 new cases annually1. Several therapies are now approved for the treatment of advanced HCC patients1; unfortunately, patient response rates fall below 20%, which can be partially explained by the high inter-tumor heterogeneity2. Several factors contribute to the unusually high diversity observed in this cancer. First, HCCs characteristically harbor genetic alterations in a variety of cancer drivers and their pattern of alteration is highly heterogeneous3–5. Moreover, 90% of HCCs arise in the context of a damaged liver, which can be caused by alcohol abuse, non-alcoholic fatty liver disease, viral hepatitis, or toxins that lead to varied tumor microenvironments6. Finally, other factors that are specific to each HCC patient, such as the patientś baseline genome or gut microbiota7, 8, are also likely to contribute to HCC heterogeneity.
To implement precision therapies for HCC and improve clinical outcomes it is crucial to accurately model and interrogate HCC heterogeneity using experimental systems. A recent study assessed how different etiologies affect the genetic and transcriptional profiles of HCC in mice by characterizing four mouse models of liver cancer and subsequently validating the data in patients9. Still, the mechanisms by which different combinations of alterations in driver genes affect tumorigenesis and inter-tumor heterogeneity have not been systematically investigated. Here, we established nine distinct mouse models of HCC, each presenting alterations in two driver genes, and demonstrated that cooperation between driver genes leads to unique histopathological features, immune landscapes, transcriptional profiles, and responses to therapies, recapitulating the inter-tumor complexity observed in HCC patients. Moreover, interrogations of gene function unveiled novel phenotypes that are context-dependent. We present a unique collection of mouse models and derived cell lines that enables in depth characterization of gene function, elucidation of molecular mechanisms, and preclinical testing for HCC.
Materials and Methods
Animal Studies
All the in vivo experiments, including tumor formation screen, screen validation, immune profiling, and drug treatment, were performed in 6–8 week old C57BL/6 mice purchased from Envigo. All mice were healthy and acclimated to the animal facility prior to experimental use and all procedures conducted on the mice were previously approved by the ISMMS Animal Care and Use Committee (IACUC-2014–0229). Animals were observed on a daily basis and sick mice were euthanized humanely in accordance with the Guidelines for Humane End Points for Animals used in biomedical research. HCC mouse models were generated performing hydrodynamic tail-vein injections (see Supplemental Methods).
Results
In vivo screen identifies cooperating driver genes in HCC
To address how cooperation between different driver genes affects liver tumorigenesis in mice, we tested the impact of altering a well-established HCC oncogene (either MYC or β-catenin, encoded by CTNNB1) in combination with an additional alteration in one of eleven other genes frequently mutated in HCC patients3, 4 (Figure 1A). We performed an in vivo screen based on the hydrodynamic tail-vein delivery of genetic elements engineered to mimic alterations in selected genes directly into hepatocytes10 (Figure 1B). We used CRISPR-based vectors with single-guide RNAs (sgRNAs) to delete genes affected by inactivating alterations, such as mutations or homozygous deletions, and transposon-based vectors to overexpress genes affected by activating alterations, such as mutations or amplifications (Figure 1A,B; Supplementary Figure 1A). In addition to combining the alteration of each selected gene with MYC overexpression or β-catenin activation, we also interrogated the cooperation between MYC and β-catenin (Figure 1B).
Figure 1. In vivo screen identifies cooperating driver genes in HCC.

(A) Frequency of alteration of selected genes in HCC patients from the TCGA cohort. (B) Schematic of experimental approach. The genes in the grey box were tested in combination with MYC overexpression or activation of β-catenin (encoded by CTNNB1). The combination of MYC overexpression and β-catenin activation was also tested. HDTVI, hydrodynamic tail-vein injection. (C) Number of C57BL/6 female and male mice that developed tumors within 6 months after HDTVI. (D) Survival graph of the corresponding conditions in C57BL/6 male mice. Median survival and number of mice per group are shown. D, days; undef, undefined. Single controls include MYC, CTNNB1, TERT, sg-p53, sg-Axin1, sg-Pten, sg-Kmt2c, and sg-Kmt2b alone models (5 male mice each). (E) Representative pictures of livers from mice from different conditions showing macroscopic tumors. Bar, 1 cm. (F) Number of tumors (left) and survival (right) in models with high (> 30%) or low (< 30%) penetrance. Median with interquartile range is shown. Mann-Whitney test. Mice from (C). (G) Survival graph of combined models with MYC overexpression (MYC;X), β-catenin activation (CTNNB1;X), and MYC;CTNNB1 model. Median survival and number of mice per group are shown. X denotes “other alteration”. D, days. Log-rank Mantel-Cox test. Mice from (C).
Among the 23 pairs of genetic alterations that were independently tested in 6-week-old C57BL/6 mice, 9 yielded liver tumors in more than 20% of their respective cohort within ∼6 months: MYC;sg-p53, MYC;sg-Kmt2b, MYC;sg-Kmt2c, MYC;sg-Pten, MYC;CTNNB1, MYC;sg-Axin1, CTNNB1;sg-Pten, CTNNB1;Tert, and MYC;Tert (Figure 1C). The genetic alterations modeled in the nine models accounted for 30% of HCC patients (considering MYC amplification) or 43% (considering MYC overexpression) (Supplementary Table 1). In subsequent validation studies, each genetic alteration on its own was not associated with mortality within 8 months, demonstrating that cooperation between two cancer driver genes was required for efficient liver tumorigenesis in C57BL/6 mice (Figure 1D). We observed a near-complete penetrance in 6 of the models while penetrance was incomplete (< 75% of mice with tumors) in MYC;sg-Kmt2b and null in MYC;Tert, and CTNNB1;Tert in this second experiment (Figure 1D). Models with incomplete penetrance (Figure 1C) had significantly fewer tumors and longer survival (Figure 1E,F; Supplementary Figure 1B,C) compared to those with high penetrance. These results suggest that in those models with incomplete penetrance the cooperation between the two driver genes may not be sufficient to initiate tumorigenesis and the spontaneous acquisition of additional alterations may be required. Interestingly, the frequency of co-alteration of each pair of genetic alterations in HCC patients was not associated with tumor penetrance or tumor development in mice (Supplementary Figure 1D,E; Supplementary Table 2).
MYC was more effective at generating liver tumors (7/9 models) than β-catenin (3/9) (Figure 1C,D). Similarly, tumor latency was shorter in MYC-driven models than in β-catenin-driven models (Figure 1G) while the model with both alterations (MYC;CTNNB1) had the shortest median survival, confirming the dominant role of MYC in tumor cell proliferation (Figure 1G). As expected, those tumors expressing a given transposon-derived gene (MYC, CTNNB1, or Tert) showed significantly higher mRNA levels compared to the remaining tumors, and the sgRNAs employed successfully deleted the target genes (Supplementary Figure 1F,G). These results imply that MYC and β-catenin oncogenes differentially cooperate with specific genetic alterations to promote liver tumorigenesis.
Cooperation between distinct driver genes leads to HCC with unique histologies
To evaluate whether the tumors in our murine models indeed recapitulated human HCC, we performed a comprehensive histopathological analysis. We selected the largest tumor in each analyzed liver, representing the most advanced tumor of each mouse. The histology of murine tumors was consistent with that of human HCCs as tumor cells were arranged in solid, pseudoglandular, trabecular, or clear-cell pattern, and tumors frequently contained areas of necrosis and hemorrhage (Figure 2A,B; Supplementary Figure 2A–C). MYC;CTNNB1 tumors predominantly presented a solid/trabecular pattern while MYC;sg-Axin1, CTNNB1;sg-Pten and CTNNB1;Tert tumors mainly exhibited a trabecular pattern (Figure 2A,B). Interestingly, CTNNB1;Tert and CTNNB1;sg-Pten contained abundant clear cells (Figure 2A,B). As expected, most MYC-driven tumors harbored numerous mitotic figures and apoptotic bodies (Figure 2B; Supplementary Figure 2D).
Figure 2. Cooperation between distinct driver genes leads to HCC with unique histologies.

(A) Stainings for the indicated markers in tumors from representative models and mice. An illustrative normal liver is also included. HE, hematoxylin & eosin. The white bar represents 50 μm. (B) Heatmap summarizing different parameters and staining outcomes in tumor samples from different mice and models. Each column represents a mouse. Color code is on the right.
In MYC-driven tumors, MYC expression was nuclear and elevated compared to tumors without ectopic MYC expression or normal livers (Figure 2A,B). Similarly, the percentage of proliferating tumor cells (Ki67+) was significantly higher in MYC-driven tumors compared to non-MYC tumors or normal liver (Figure 2A,B; Supplementary Figure 2F). In β-catenin-driven tumors, β-catenin was localized in the nucleus at varying levels whereas in the remaining models, β-catenin was stained primarily in the membrane, where it is known to have a role in cell adhesion11 (Figure 2A,B). Glutamine synthetase (Glul), a β-catenin target that is expressed in normal hepatocytes surrounding the central vein (CV)12, was found in the cytoplasm of CTNNB1;sg-Pten and CTNNB1;Tert tumors but surprisingly much lower in MYC;CTNNB1 or MYC;sg-Axin1 tumors, suggesting that these two models may have distinct Wnt/β-catenin activation programs.
Only six tumors (all MYC-driven) showed positive staining (> 5% of cells) for cytokeratin 19 (CK19), a progenitor marker expressed in the cholangiocytes of the biliary tracts associated with the portal vein (PV), and which showed a patched pattern of staining (Figure 2B; Supplementary Figure 2E) suggesting that those tumors could be derived from progenitor cells instead of hepatocytes or could exemplify transdifferentiation associated with tumorigenesis. Staining for CD45 was variable in different models, with MYC;sg-Kmt2b, MYC;sg-Kmt2c, and CTNNB1;sg-Pten tumors showing the highest levels of staining (Figure 2B; Figure 3A). Finally, hepatocyte nuclear factor 4 alpha (Hnf4a), a marker of hepatocellular function, was found in the nucleus of hepatocytes in normal livers and in the nucleus of tumor cells from most models, except MYC;CTNNB1 tumors (Figure 2A,B). In general, MYC;CTNNB1 tumors were characterized as poorly and moderately differentiated while CTNNB1;Tert, CTNNB1;sg-Pten, and MYC;sg-Axin1 tumors were between well and moderately differentiated (Figure 2A,B). The rest of the tumors presented an intermediate phenotype between moderately and poorly differentiated suggesting that expression of MYC modifies the phenotype driven by β-catenin (Figure 2A,B; Figure 1). Taken together, the murine HCC tumors recapitulate histological features that are concordant with that of human HCC tumors13.
Figure 3. Cooperation between distinct driver genes leads to unique immune landscapes.

(A) Staining for immune cell marker CD45 in tumors from representative models and mice. The white bar represents 50 μm. (B-C), Frequency of different lymphoid (B) and myeloid (C) immune cell populations in tumors from different mouse models and normal liver. (D-E), Box and whisker plots representing different lymphoid (D) and myeloid (E) immune cell populations (% over CD45+ (lymphoid) or CD45+lin− (myeloid), respectively) in tumors and normal liver. Anova (Kruskal Wallis) test and multiple comparison to normal liver. (F) Heatmap summarizing the average of the % of immune cells (transformed to Z score) in the total CD45+ or CD45+lin− cells in different tumor models and normal liver. The black outline represents significant models when compared to normal liver (summary of D-E; Supplementary Figure 3A). Color code is shown under the heatmap and represents the Z score. (G) Correlation matrix between different models based on the immune profiles. Color code is shown under the heatmap. The correlation values that are higher than 0.6 and with a P-value lower than 0.05 are highlighted with the black outline. R, Pearson correlation coefficient. Mono, monocytes; neu, neutrophils; mac, macrophages; mDC2, myeloid dendritic cells type 2; mDC1, myeloid dendritic cells type 1; Treg, regulatory T cells; NK, natural killer cells.
Cooperation between distinct driver genes leads to unique immune landscapes
Histological analysis showed that MYC;CTNNB1 tumors presented a lower percentage of CD45+ cells (Figure 2B; Figure 3A), in agreement with the β-catenin-mediated immune exclusion previously shown in our laboratory14. To assess how the cooperation between distinct driver genes affects immune cell infiltration and composition we analyzed by flow cytometry the lymphoid and myeloid cell populations present in tumors from the seven most penetrant models (Figure 3B–F; Supplementary Figure 3A). Proportion of CD8+ T cells was significantly higher in MYC;sg-Kmt2b and CTNNB1;sg-Pten tumors compared to normal livers while the proportion of CD4+ T cells was decreased in all tumors except β-catenin-driven tumors (Figure 3D). B cells were significantly underrepresented in MYC;sg-Kmt2b, MYC;sg-Pten, and MYC;sg-Axin1 tumors (Figure 3D). Macrophages proportion was decreased in MYC;sg-Kmt2b and MYC;sg-Pten tumors (Figure 3E) while fraction of neutrophils, monocytes, and mDC2s (myeloid dendritic cells) was increased in most tumor types (Figure 3E; Supplementary Figure 3A). Finally, the proportions of NK cells, T regs, and γδT cells were predominantly unchanged (Supplementary Figure 3A). Regarding the different tumor models, MYC;sg-p53 tumors were characterized by a higher proportion of mDC2s (Figure 3F; Supplementary Figure 3A) while MYC;sg-Kmt2b tumors presented the highest proportion of CD8+ T cells and monocytes (Figure 3D,F). MYC;sg-Axin1 tumors were in general immunologically inert, with the exception of an enhanced proportion of neutrophils (Figure 3E,F; Supplementary Figure 3A). On the contrary, CTNNB1;sg-Pten tumors were immunologically similar to normal liver while the MYC-driven models (excluding MYC;CTNNB1) clustered together (Figure 3G). These results corroborate the influence of the genetic makeup of HCC tumors in the immune landscape and highlights that MYC;CTNNB1 is a singular model with exclusive histological and immune phenotypes.
Novel genetically-defined murine HCC models recapitulate human HCC transcriptional subclasses
To elucidate the cooperation between different driver genes and their combined impact on inter-tumor heterogeneity, we performed RNA-sequencing of 3–4 tumors per model as well as tissue from normal livers. Principal component analysis (PCA) of transcriptomic profiles of human HCCs, breast cancers, lung adenocarcinomas and squamous carcinomas, and colorectal adenocarcinomas showed that the murine HCCs (mHCCs) mainly overlapped with human HCCs, confirming them as bona-fide models of HCC (Figure 4A). PCA of transcriptomic profiles of murine HCCs showed a clear separation between MYC-driven and non-MYC-driven tumors which clustered closer to normal liver samples (Figure 4B). Interestingly, MYC;CTNNB1 tumors formed their own cluster, again supporting their unique biology (Figures 1–3).
Figure 4. Novel genetically-defined murine HCC models recapitulate human HCC transcriptional subclasses.

(A) PCA analysis of gene expression profiles of human HCCs (LIHC), breast cancers (BRCA), lung squamous carcinomas and lung adenocarcinomas (LUC), colorectal adenocarcinomas (COAD), normal human liver, and murine HCC (mHCCs). (B) PCA analysis of gene expression profiles of murine HCC (mHCCs) and normal murine livers. (C) Heatmap of the 2,500 most differentially expressed genes in murine HCCs (mHCCs). Z score is shown. Color code is shown under the heatmap and in (B) and (E). (D) Heatmap of 32 selected genes from the 2,500 most differentially expressed genes in murine HCCs (mHCCs) in (C). Z score is shown. Color code is shown under the heatmap and in (B) and (E). LS, liver-specific; EtOH, ethanol; compl, complement; ins, insulin; prol, proliferation; can, canonical; EMT, epithelial-to-mesenchymal transition. (E) Heatmap of ssGSEA values (shown as Z score) for significant pathways in the different murine HCCs. The P-value for each gene set is shown in the right (bar graph) (Anova). The dotted line indicates the threshold for significance after applying Benjamini-Hochberg multiple testing correction (p < 0.03). Color code is shown under the heatmap. For Hoshida and Boyault subclasses, the association Chi test value is shown.
K-means clustering algorithm also led to 3 subclasses ( mHCC1, mHCC2, and mHCC3) (Figure 4B,C; Supplementary Figure 4A; Supplementary Table 3). mHCC1 was composed of poorly-differentiated liver tumors (MYC;CTNNB1, one MYC;Tert, and one CTNNB1;Tert) and was enriched in MYC targets, genes related to cell proliferation and replication, canonical Wnt/β-catenin signaling pathway, epithelial to mesenchymal transition (EMT), and TGFβ and Hedgehog pathways (Figure 4C,D; Supplementary Table 3). mHCC2 was composed of moderately- to poorly-differentiated liver tumors (mainly MYC;sg-p53, MYC;sg-Pten, MYC;sg-Kmt2c, MYC;sg-Kmt2b, and MYC;Tert) and presented levels of expression of liver-specific genes that were intermediate between mHCC1 and mHCC3 tumors (Figure 4C,D; Supplementary Table 3). In addition, mHCC2 tumors were significantly enriched in MYC targets, genes related to cell proliferation and replication, and insulin-like growth factor 2 signaling. mHCC3 was composed of well-differentiated liver tumors (mainly CTNNB1;Tert, CTNNB1;sg-Pten, and MYC;sg-Axin1) and was significantly enriched in genes related to normal liver function (bile acid synthesis, ethanol and drug metabolism, complement and coagulation) and non-canonical liver-specific Wnt/β-catenin signaling pathway (Figure 4C,D; Supplementary Table 3).
Single-sample Gene Set Enrichment Analysis (ssGSEA) for “Hallmark” gene set collection15 proved that mHCC1 tumors were significantly enhanced in Wnt/β-catenin, TGFβ, Notch, and Hedgehog signaling, as well as EMT (Figure 4E; Supplementary Table 4). mHCC3 included tumors with intact liver function, presenting significant enrichment of cholesterol homeostasis, adipogenesis, and xenobiotic and bile acid metabolism (Figure 4E; Supplementary Table 4). The same gene sets were significantly augmented, although to a lower level, in mHCC2, which was significantly enriched in MTORC1 signaling and presented significantly lower levels of p53 pathway engagement (Figure 4E; Supplementary Table 4).
Finally, nearest template prediction (NTP) showed that murine HCC transcriptomic subclasses were significantly associated with the previously-established human HCC subclasses16, 17. mHCC1 tumors were significantly enriched in Hoshidás S1 subclass and Boyaultś G3, which in patients are associated with canonical Wnt/β-catenin signaling pathway activation, TGFβ signaling, poor differentiation, and tumor aggressiveness; mHCC2 tumors were distributed among the Hoshidás and Boyaultś subclasses; and mHCC3 tumors were significantly overrepresented in Hoshidás S3 subclass and Boyaultś G6, which in patients are characterized by CTNNB1 mutation, liver-specific Wnt/β-catenin signaling pathway, hepatocyte-like phenotype, well-differentiated tumors, and reduced aggressiveness (Figure 4E; Supplementary Table 4). Interestingly, the association between molecular subclasses (Hoshida17 or Boyault16) and tumor histology subtypes observed in HCC patients13, 18 was also found in murine HCC tumors (Supplementary Figure 4B,C). In addition, the “Immune class” of HCC19 was detected in around 40% of murine HCC tumors and was significantly enriched in the Hoshida S1 subclass17 and exclusion of Chiangś CTNNB1-mutant subclass20, similar to HCC patients19 (Supplementary Figure 4D; Supplementary Table 5). Collectively, the novel precision mouse models of HCC transcriptionally recapitulate the pathways and subclasses that are found in human HCCs.
Inter-tumor heterogeneity is shaped by oncogene expression levels and specific cooperating events
In the hydrodynamic model, gene expression could be influenced by the number of integrated copies of the transposon-based vectors. Therefore, we hypothesized that the expression levels of the driving oncogenes could influence the inter-tumor heterogeneity observed within each model. Indeed, for tumor PM148 (CTNNB1;Tert, mHCC1), CTNNB1 and Tert levels were substantially lower than for the other two CTNNB1;Tert tumors (mHCC3) (Supplementary Table 6). In the MYC;Tert model, each tumor belonged to a different subclass and presented variable levels of Tert: AL1679 (mHCC2) low, AL1678 (mHCC1) intermediate, and PM440 (mHCC3) higher levels (Supplementary Table 6). For AL608 (MYC;sg-Axin1, mHCC2), MYC levels were at least 50% higher than for the other two MYC;sg-Axin1 tumors (mHCC1) while Axin1 CRISPR deletion efficiency was very similar (Supplementary Table 6; Supplementary Figure 1F), suggesting that expression levels of the driver oncogenes in addition to the specific combinations of alterations in driver genes could shape the tumor phenotype and contribute to inter-tumor heterogeneity. Interestingly, AL608 (MYC;sg-Axin1) and PM148 (CTNNB1;Tert) presented a unique differentiation state when compared to the other tumors from the same model, suggesting an association between differentiation and transcriptional profiles (Supplementary Table 6). As anticipated, there was a significant positive correlation between MYC, CTNNB1, and Tert mRNA levels and the amount of transposon-based DNA from each corresponding vector (Supplementary Figure 4E), suggesting that the number of transposon-based vector integration influences expression levels of the corresponding cDNAs.
To further explore the effect of oncogene expression level on tumor phenotypes we calculated the correlation between transposon-driven MYC and CTNNB1 mRNA levels and MYC and β-catenin targets, respectively. As expected, there was a significant positive correlation between transposon-driven MYC mRNA levels and the expression of MYC targets and genes involved in cell replication (Figure 5A; Supplementary Figure 5A). However, the correlation between transposon-driven CTNNB1 mRNA levels and the expression of β-catenin targets was negligible (Figure 5A; Supplementary Figure 5A), suggesting that β-catenin activity rather than expression levels is critical for the expression of its targets. MYC has been shown to be a target of β-catenin; however, as shown in a recent study21, there was no correlation between transposon-driven CTNNB1 mRNA levels and levels of MYC or MYC targets (Figure 5A; Supplementary Figure 5A). Surprisingly, there was a significant positive correlation between transposon-driven MYC mRNA levels and β-catenin canonical targets whereas there was no correlation with β-catenin liver-specific targets (Figure 5A; Supplementary Figure 5A). Interestingly, β-catenin canonical targets were barely expressed in CTNNB1;Tert and CTNNB1;sg-Pten tumors (Figure 4D), which present low MYC levels, suggesting that β-catenin activity could be influenced by MYC expression levels.
Figure 5. Inter-tumor heterogeneity is shaped by oncogene expression levels and specific cooperating events.

(A) Heatmap showing the Pearson R values for the correlation between transposon-driven MYC or CTNNB1 mRNA levels and expression of the indicated genes. Color code is shown under the heatmap. The correlation values that are higher than 0.5 and with a P-value lower than 0.01 are highlighted with the black outline. R, Pearson correlation coefficient. LS, liver specific; Prol, proliferation; rep, replication. (B) Box and whisker plot representing ssGSEA values for MYC targets and Wnt/β-catenin signatures in HCC patients stratified depending on the MYC mRNA levels (high, 1st quartile; low, 2nd-4th quartiles) and CTNNB1 mutational status (mut, mutated; WT, wild-type). Mann-Whitney test. MYC high CTNNB1 mutant n=15; MYC low CTNNB1 mutant n=34; MYC high CTNNB1 wild-type n=30; MYC low CTNNB1 mutant n=104. (C) Heatmap of gene expression values (shown as Z score) in the different murine HCCs expressing β-catenin. The P-value for each gene is shown in the right (bar graph) (Anova). The dotted line indicates the threshold for significance after applying Benjamini-Hochberg multiple testing correction (p < 0.019). Color code is shown under the heatmap and on the right. (D) Heatmap of ssGSEA values (shown as Z score) for representative pathways in the different murine HCCs and normal livers. Color code is shown under the heatmap. Black outlines indicate gene sets that are significantly different in the corresponding model compared to normal liver (Anova test). (E) Bar graphs showing the ssGSEA score in human HCC samples from TCGA with genetic alterations in the indicated gene and compared to normal livers from GTEX. Mann-Whitney test or Anova test. MYC OE (overexpression) n=31, normal liver n=136; MYC OE CTNNB1 mut (mutant) n=16; PTEN mut n=12; TERT prom.mut (promoter mutation) n=80; TP53 mut n=58; KMT2C mut n=16; KMT2B mut n=12.
In HCC patient samples from the The Cancer Genome Atlas (TCGA)4, tumors with high MYC expression (first quartile) showed higher levels of “MYC targets” gene set compared to samples with low MYC expression (remainder of the quartiles) in both CTNNB1-mutant and -wild-type (WT) tumors (Figure 5B, upper panel). Similar to our murine results, the Wnt/β-catenin signaling pathway gene set was significantly higher in MYC-high HCC samples (Figure 5B, lower panel), reinforcing the idea that MYC may modulate β-catenin activity. To functionally test this hypothesis, we generated MYC-CTNNB1 tumors expressing lower MYC levels by injecting mice with a lower amount of MYC-expressing transposon vector (MYClow;CTNNB1). Transcriptomic analysis showed that compared to MYC;CTNNB1 tumors, MYClow;CTNNB1 tumors had significantly decreased expression of genes related to the canonical Wnt/β-catenin pathway, similar to CTNNB1;Tert and CTNNB1;sg-Pten tumors (Figure 5C). The lack of activation of the canonical pathway was not due to deficient expression of β-catenin, which was found in the nucleus of MYClow;CTNNB1 tumor cells, but due to lower MYC levels (Supplementary Figure 5B) indicating that MYC expression levels can profoundly influence the activity of β-catenin and dictate β-catenin-driven transcriptional output in HCC. Interestingly, in HCC patients from the TCGA, those tumors with CTNNB1 mutation presented significantly higher levels of CTNNB1 mRNA than CTNNB1 wild-type tumors (upper panel, Supplementary Figure 5C). In addition, there was a significant correlation between Wnt/β-catenin pathway gene signature and MYC and CTNNB1 mRNA levels (lower panel, Supplementary Figure 5C), suggesting that in addition to MYC levels and CTNNB1 mutation, CTNNB1 mRNA levels may be critical for the phenotypic output of β-catenin. The lack of correlation observed in our murine models was probably due to similar CTNNB1 expression levels across tumors, which led to transcriptional programs that were comparable to those driven by endogenous activation of β-catenin in murine Ctnnb1exon3/exon3 tumors22 (Supplementary Figure 5D). Most importantly, these results highlight that tumors with the same genetic alterations may exhibit different phenotypes depending on expression levels, further increasing the phenotypic diversity of HCC tumors.
To test how cooperation of MYC or β-catenin with distinct driver genes affects tumor phenotypes, we checked the activation or inhibition of “Hallmark” gene sets in each model in comparison to normal liver samples. Those models with the highest expression of MYC targets (MYC;sg-Kmt2b, MYC;sg-Kmt2c, MYC;CTNNB1) demonstrated significantly lower levels of gene sets related to normal liver function (Figure 5D; Supplementary Table 7), further supporting the notion that MYC overexpression is driving tumor cell dedifferentiation. Loss of Axin1, a negative regulator of the Wnt/β-catenin pathway, had little impact on this pathway when compared to normal livers (Figure 5D; Supplementary Table 7), in agreement with a previous study23. As expected, loss of p53 did not lead to an increase in p53 pathway activation, which was significantly enriched in MYC;sg-Pten, MYC;CTNNB1, and MYC;Tert models, but promoted a significant rise in glycolysis and MTORC1 signaling (Figure 5D; Supplementary Table 7). Loss of Kmt2c significantly induced the estrogen response gene set while loss of Kmt2b significantly suppressed Hedgehog signaling (Figure 5D; Supplementary Table 7). Loss of Pten in the context of MYC overexpression led to a significant increase in protein secretion, MTORC1 signaling and PI3K/MTOR/AKT signaling while in the context of β-catenin activation only shared with MYC;sg-Pten the significant augmentation in protein secretion (Figure 5D; Supplementary Table 7). This gene set was also enriched in CTNNB1;Tert and MYC;Tert models; however, CTNNB1;Tert significantly upregulated KRAS signaling while MYC;Tert showed significant enrichment of p53 pathway, glycolysis, and allograft rejection (Figure 5D; Supplementary Table 7). Most of these observations were validated in HCC patient samples from TCGA that were stratified according to the same genetic alterations (Figure 5E), supporting that cooperation between different driver genes leads to the activation of unique pathways that are conserved between humans and mice. Interestingly, 95% of human HCCs had at least one murine tumor with a correlation higher than 0.5, 30% of human HCCs had at least one murine tumor with a correlation higher than 0.75, and 87% of human HCCs had a correlation higher than 0.5 with at least one of the mHCC subclasses (Supplementary Figure 5E), further supporting the relevance of our models.
Novel murine HCC cell lines recapitulate the most aggressive HCC subclasses
In order to further understand the impact of the cooperation of driver genes in response to therapies, we established murine cell lines (mCL) from our tumor-bearing mouse models. Cell lines from MYC;Tert tumors were not generated since tumor development in this model was inconsistent (Figure 1). PCA and hierarchical clustering of RNAseq data from tumors and corresponding cell lines revealed a clear difference between cell lines and tumors in gene expression profiles (Figure 6A; Supplementary Figure 6A). Compared to mHCC tumors, the derived genetically-defined cell lines showed a significant enrichment in genes related to extracellular matrix, focal adhesion, and stroma, consistent with adaptation to ex vivo growth (Supplementary Figure 6B; Supplementary Table 8). In addition, murine HCC cell lines significantly downregulated genes related to immune function, denoting the growth in an in vitro setting, and liver function, indicating that murine cell lines undergo dedifferentiation (Supplementary Figure 6B; Supplementary Table 8). Similarly, ssGSEA analysis revealed significant upregulation of gene sets related to cell-cell contact, EMT, proliferation, or PI3K pathway in cell lines, and significant downregulation of gene sets related to liver differentiation (Supplementary Figure 6C; Supplementary Table 9). Correlation between expression profiles of each tumor and its derived cell line was, in general, significantly positive, with the exception of MYC;CTNNB1 tumors/cell lines, which showed an even higher positive correlation, and CTNNB1;sg-Pten and CTNNB1;Tert tumors/cell lines, which were not correlated (Supplementary Figure 6D), probably due to the selection of the fastest-proliferating cells in culture.
Figure 6. Novel murine HCC cell lines recapitulate the most aggressive HCC subclasses.

(A) PCA analysis of gene expression profiles of murine HCC tumors and cell lines. (B) PCA analysis of gene expression profiles of murine HCC cell lines (mCLs). (C) Heatmap of the 2,500 most differentially expressed genes in murine HCC cell lines (mCLs). Z score is shown. Color code is shown under the heatmap and in (B) and (E). (D) Heatmap of selected genes from the 2,500 most differentially expressed genes in murine HCC cell lines (mCLs) in (C). Z score is shown. Color code is shown under the heatmap and in (B) and (E). EMT, epithelial-to-mesenchymal transition; can, canonical. (E) Heatmap of ssGSEA values (shown as Z score) for significant pathways in the different murine HCC cell lines (mCLs). The P-value for each gene set is shown in the right (bar graph) (Anova). The dotted line indicates the threshold for significance after applying Benjamini-Hochberg multiple testing correction (p < 0.03). Color code is shown under the heatmap.
As in tumor samples, analysis of the murine HCC cell lines revealed 3 well-defined groups: mCL1, mCL2, and mCL3 (Figure 6B,C; Supplementary Figure 6E). mCL1 was composed of the most differentiated cell lines (CTNNB1;sg-Pten, MYC;sg-Pten, MYC;sg-Axin1, and 1 × MYC;sg-Kmt2c) with epithelial features and expression of hepatocyte and liver fetal/progenitor markers, mirroring human CL1 HCC cell line subclass described by Caruso et al. (Figure 6D; Supplementary Table 10). mCL2 (MYC;CTNNB1, MYC;sg-p53, 1 × MYC;sg-Kmt2c) displayed a mixed “epithelial-mesenchymal” pattern between mCL1 and mCL3 with an intermediate expression of hepato-specific genes and stem cell markers, similar to the human CL2 HCC cell line subclass24 (Figure 6D; Supplementary Table 10). The mCL2 subgroup was also enriched in genes from the canonical Wnt/β-catenin signaling and Hedgehog signaling pathways (Figure 6D; Supplementary Table 10). mCL3 included less differentiated murine cell lines (CTNNB1;Tert, MYC;sg-Kmt2b) with stromal, “mesenchymal-like”, and stem cell markers, and low levels of hepato-specific genes, mimicking human CL3 HCC cell line subclass24 (Figure 6D; Supplementary Table 10). Finally, NTP analysis showed that the new murine mHCC cell line transcriptomic subgroups were associated with the most aggressive HCC primary tumor subclasses (Figure 6E; Supplementary Table 9): mCL2 and mCL3 were enriched in Hoshidás S1 subclass and Boyaultś G3 while mCL1 consisted of a mixture of S1 and S2 Hoshida subclasses and of G1, G2, and G3 Boyault subclasses. The “non-proliferative,” most differentiated, and less aggressive HCC classes (G4-G6 and S3) were not represented in our panel of mCLs and indicated that Hoshida S3 mHCC tumors mainly gave rise to S1 cell lines, probably through the selection of highly-proliferative cells in culture. Similarly, mCL1 cell lines were significantly enriched in gene sets related to normal liver function, compared to the 2 other subgroups; mCL3 cell lines were enriched in gene sets related to the apical complex and inflammatory response; and mCL2 cell lines were enriched in MYC targets and Wnt/β-catenin signaling pathway (Figure 6E; Supplementary Table 10), showing again an association between MYC and canonical Wnt/β-catenin signaling pathway. Taken together, the novel murine HCC cell lines transcriptionally recapitulate the most aggressive human HCC subclasses.
Cooperation between distinct driver genes leads to unique drug responses
To assess whether cooperation between driver genes can also impact response to therapies, we screened 45 FDA-approved drugs, in representative cell lines from our novel panel (n = 12) and additional murine HCC cell lines generated in our laboratory harboring MYC overexpression, p53 deletion, and one additional genetic alteration (Tert, Ccne1, or Ccne2 overexpression; Axin1, Apob/Alb, Kmt2b, or Cdkn2a deletion; Molina-Sánchez et al, in preparation; n = 12). The most potent drugs, defined as having low AUC, were those that target general processes, such as proteasome (bortezomib) and DNA synthesis (methotrexate, topotecan); in contrast, the four HCC-approved compounds had little activity, with the exception of lenvatinib (Figure 7A,B; Supplementary Table 11). Of note, inhibitors targeting mTOR alone (n = 3) were among the eleven most effective drugs (Figure 7A,B; Supplementary Table 11). Unsupervised hierarchical clustering of drug responses on the 4 mCLs showed common sensitivity profiles for drugs with similar mechanism of action, such as mTOR inhibitors (everolimus, temsirolimus, and sirolimus) or EGFR inhibitors (gefinitib, erlotinib, and osimertinib) (Supplementary Figure 7A; Supplementary Table 11), thus validating the reliability of our screening platform.
Figure 7. Cooperation between distinct driver genes leads to unique drug responses.

(A) Schematic of the inhibitors used in the drug screen. The number indicates the number of inhibitors in each class, and the name, the target of the inhibitors. On the right, area under the curve (AUC) is used as a measure of drug activity (high AUC, less activity) in the panel of 24 murine HCC cell lines. Box and whisker plot of AUC per each drug in the panel of cell lines. (B) Heatmap representing the AUC of each drug in each cell line. Color code is shown in the left. The coefficient of variance (CV) is shown in the right. (C) Survival graphs of the corresponding mouse models treated with sorafenib, methotrexate, and everolimus. The number of mice per group and median survival is shown. Mantel-Cox test. **p < 0.01, ***p < 0.001. Ns, not significant. Below, gain in median survival (in days) between each treatment and the control group for each model. Green, more survival gain; yellow, less survival gain.
To identify model-specific therapies, we reasoned that those compounds with higher variation between models (measured by the coefficient of variation) would have differential specificities (Figure 7B; Supplementary Table 11). Methotrexate was particularly effective in the p53-deleted cell lines while mTOR inhibitors showed the strongest efficacy in the MYC;sg-Pten cell lines. Trametinib and cobimetinib, two selective MEK inhibitors, were also more efficient in the MYC;sg-Pten model. To investigate the activity of these compounds in vivo in models with a defined genetic makeup, we treated MYC;sg-p53, MYC;CTNNB1, and MYC;sg-Pten mice with methotrexate, everolimus, and the standard-of-care sorafenib (Figure 7C). While methotrexate significantly increased median survival in all three models, the effects were more pronounced in the MYC;sg-p53 and MYC;CTNNB1 models based on days gained in median survival, similar to the in vitro studies. Sorafenib and everolimus significantly improved survival in MYC;sg-p53 and MYC;sg-Pten models; however, the effects were more pronounced in the MYC;sg-Pten model, which was also sensitive in vitro to the MAPK pathway inhibitors cobimetinib and trametinib, suggesting that sorafenib may act through inhibition of this pathway. On the other hand, deletion of Pten conferred sensitivity to mTOR inhibitors (Supplementary Figure 7B,C), a result that has been validated in HCC patients25. Our work shows that cooperation between distinct driver genes leads to heterogeneous responses to therapy and demonstrates that our novel collection of HCC models could be effectively used to identify specific drug sensitivities in liver cancer.
Discussion
We have generated multiple mouse models of HCC, each one harboring genetic alterations in two driver genes, to recapitulate and investigate the diversity of HCC tumors and interrogate the contribution of different driver genes alone or in combination. Furthermore, we have generated murine cell lines derived from these genetically-defined mouse models of HCC and used them to seamlessly identify targeted therapies that can be validated in vivo.
In tumors with low levels of MYC, β-catenin promotes activation of the liver-specific Wnt/β-catenin signaling pathway while in those with high MYC levels, β-catenin stimulates the canonical pathway. It has been shown that MYC overexpression leads to LEF1 transcription, which in turn promotes β-catenin translocation to the nucleus, where it is active28. However, in our mouse models, β-catenin could be found in the nucleus, even in the context of low levels of MYC, which suggests a different mechanism, possibly involving suppression of β-catenin-mediated transcription. Expressed independently, MYC promotes proliferation and dedifferentiation, which is observed at the transcriptional and histological level while β-catenin is associated with differentiation. Expressed together and depending on relative MYC levels, the liver-specific or canonical Wnt/β-catenin pathway is activated. In a recent study, MYC and β-catenin were shown to cooperate in HCC through the Hippo pathway21. We have previously shown that cooperation between MYC and β-catenin promotes immune escape and resistance to anti-PD-1 immunotherapy in an immunogenic mouse model of HCC14, a result that has been validated in patients19, 25. In the current study, several of the models, including MYC;CTNNB1 with high levels of MYC, presented tumors that were enriched in the “Immune class” signature19, which is present in human HCC tumors that could potentially respond to immunotherapies. Therefore, we could possibly utilize the models that present the “Immune class” signature to test immunotherapies while the models that lack the signature could be used to study strategies that can turn “cold” tumors into “hot” ones.
Here, we have demonstrated that different combination of genetic alterations can contribute uniquely to HCC formation and progression. This contribution is not only attributed to the specific genes that are altered but also to their expression levels (e.g. MYC), which further increases HCC diversity, even in theoretically similar genetic contexts, a result validated in patient samples. Furthermore, we provide an innovative platform for the study of liver cancer and the understanding of its molecular mechanisms. By coupling the in vitro studies with in vivo validation, we have discovered unexpected mechanisms and identified vulnerabilities driven by specific genetic alterations that could guide precision therapies. Our collection of HCC mouse models recapitulates MYC or β-catenin-driven human HCCs, which account for around one third of all the cases when studied in combination with other genetic alterations. While this number may seem low, it also highlights the high inter-tumor heterogeneity present in HCC and reinforces the need of using multiple mouse models to study this diversity. While the murine models recapitulate quite closely histological, transcriptional, and immune features of human HCC, human tumors are more complex as they present additional mutations that can modify phenotypic outcomes, something to consider when using these models. In addition, the models presented in this study lack the liver damage that is characteristic of human HCC tumors, which could provide an additional layer of heterogeneity and could hamper drug activity. Nevertheless, our models could be used for immuno-oncology studies14 and made more complex by inducing liver damage due to environmental cues. Taken together, our models represent a unique resource that is available for the scientific community to generate and test hypotheses, to dissect the mechanisms underlying cooperation between different driver genes and inter-tumor heterogeneity, and to test personalized therapies.
Supplementary Material
Supplementary Fig. 1. In vivo screen identifies cooperating driver genes in HCC. (A) Schematic of vectors used in the hydrodynamic injections. LOF, loss of function; GOF, gain of function. Luc, luciferase. (B) Number of tumors per mouse in different models. Median with interquartile range is shown. (C) Survival graph of the corresponding conditions (high or low penetrance). Median survival and number of mice per group is shown. D, days. Log-rank Mantel-Cox test. **p < 0.01. Mice from Figure 1C. (D) Correlation between the penetrance in mice (% of mice that develop tumors in a given condition) and frequency of co-alteration in HCC patients1 of the pair of genes tested in mice. R, Pearson correlation coefficient. Mice from Figure 1C. Related to Supplementary Table 2. (E) Left, frequency of co-alteration of each pair of genetic alteration in HCC patients1 and the presence or not of tumors in mouse models. Median with interquartile range is shown. Mann-Whitney test. ns, not significant. Right, number of models showing positivity or for tumors depending on their association in HCC patients (co-occurrence or mutual exclusivity). Fisher test. ns, not significant. Related to Supplementary Table 2. (F) Relative levels of mRNA of the corresponding genes in tumor samples from mice from different conditions injected or not with the indicated oncogenes. Median with interquartile range is shown. Mann-Whitney test. ****p < 0.0001. (G) Percentage of deletion found in different models for each indicated single-guide RNA (sgRNA). Median with interquartile range is shown.
Supplementary Fig. 2. Cooperation between distinct driver genes leads to HCC with unique histologies. (A-E) Representative stainings depicting pseudoglandular (A) and trabecular (B) structures, necrosis (C), mitotic figures (yellow arrow) and apoptotic bodies (white arrow) (D), and cytokeratin 19 (E). HE, hematoxylin & eosin. The white bars represent 50 μm. (F) Average percentage of Ki67 positive cells in murine tumor samples from conditions with or without MYC overexpression. Median with interquartile range is shown. Mann-Whitney test. ****p < 0.0001.
Supplementary Fig. 3. Cooperation between distinct driver genes leads to unique immune landscapes. (A) Box and whisker plots representing different lymphoid (NK, Tregs, γδT) and myeloid (mDC1, mDC2) immune cell populations (% over CD45+ and CD45+lin− cells, respectively) in tumors and normal liver. Anova (Kruskal Wallis) test and multiple comparison to normal liver. *p < 0.05, **p < 0.01. (B) Gating strategy and illustrative plots of myeloid and lymphoid panels of a representative sample.
Supplementary Fig. 4. Novel genetically-defined murine HCC models recapitulate human HCC transcriptional subclasses. (A) Silhouette score depending on the number of groups of equal variance (k), calculated with K-means clustering algorithm, for murine HCC cell lines. (B) Table summarizing the histological features of each murine tumor as well as the Hoshida and Boyault subclasses. Average indicates the most frequent phenotype of each category. 50/50 indicates two phenotypes are equally represented in a given subclass. (C) Table summarizing the percentage of human tumors showing a given feature (YES) in different subclasses and studies. In green, the percentage of human tumors that are represented in mice belonging to the same subclass and showing the same feature. In bold, associations that are significant in HCC patients in the corresponding studies. (D) Heatmap of transcript levels and ssGSEA values for significant immune genes and gene sets in the different murine HCCs classified as “Immune class” (red) or rest (blue). The P-value for each gene and gene set shown is significant after applying Benjamini-Hochberg multiple testing correction (p < 0.0123). Red indicates high; blue, low. Z scores are shown. (E) Correlation between mRNA expression levels in tumors and transposon DNA integration levels. R, Pearson correlation coefficient. Color code is shown under the heatmap. The correlation values that are higher than 0.6 and with a P value lower than 0.05 are highlighted with the black outline.
Supplementary Fig. 5. Inter-tumor heterogeneity is shaped by oncogene expression levels and specific cooperating events. (A) Correlation plots. R, Pearson correlation coefficient. Ns, not significant. *** p < 0.0001, **** p < 0.0001. (B) Staining for the indicated antibody (β-catenin) in a representative MYClow;CTNNB1 tumor. The white bar represents 50 μm. Dashed line indicates border between tumor tissue and adjacent tissue. (C) Upper panel, CTNNB1 mRNA levels in CTNNB1-mutant (n = 49) and wild-type (WT, n = 134) human HCCs. Student t test. * p < 0.05. Lower panel, correlation between ssGSEA score for Hallmarks_Wnt_beta-catenin_signaling and MYC and CTNNB1 mRNA levels in human HCC (n = 183). R, Pearson correlation coefficient. **** p < 0.0001. (D) Correlation between expression profiles of each murine tumor and murine Ctnnb1exon3/exon3 tumors. Color code is shown on the right. R, Pearson correlation coefficient. The average correlation per model is also shown. (E) Correlation between expression profiles of each murine tumor and human HCC tumors. Color code is shown on the right. R, Pearson correlation coefficient. Purple, 95% of human HCCs have at least one murine tumor with a correlation higher than 0.5; grey, 87% of human HCCs have at least one of the mHCC subclasses with a correlation higher than 0.5 with; pink, 30% of human HCCs have at least one murine tumor with a correlation higher than 0.75.
Supplementary Fig. 6. Novel murine HCC cell lines recapitulate the most aggressive HCC subclasses. (A) Silhouette score depending on the number of groups of equal variance (k), calculated with K-means clustering algorithm, for murine HCC cell lines and tumors. (B) Volcano plot representing the log2 of the fold change (FC) between the expression in cell lines versus tumors and the −log10 of the p value. Selected genes are highlighted. Related to Supplementary Table 8. (C) Bar graph showing the p values for student t test in murine HCC cell lines. The dotted line indicates the threshold for significance after applying Benjamini-Hochberg multiple testing correction (p < 0.042). (D) Correlation between expression profiles of each tumor and its corresponding derived cell line. Color code is shown under the heatmap. The correlation values that are higher than 0.5 and with a P value lower than 0.05 are highlighted with the black outline. R, Pearson correlation coefficient. (E) Silhouette score depending on the number of groups of equal variance (k), calculated with K-means clustering algorithm, for murine HCC cell lines.
Supplementary Fig. 7. Cooperation between distinct driver genes leads to unique drug responses. (A) Heatmap showing the Pearson R values for the correlation between different compounds. The compounds are organized by proximity. Color code is shown under the heatmap. The correlation values of some compounds with high correlation are highlighted with the black outline. R, Pearson correlation coefficient. (B) Volcano plot showing linear regression between Pten deletion status and cell viability under various drug treatments in murine HCC cell lines. Effective drugs showing significant association are labeled (log-fold-change [logFC] < 0; p.value < 0.05). (C) Bar plots comparing the AUC of mTOR inhibitors in Pten knock-out (Pten-KO) and Pten wild-type (WT) murine cell lines. Mean and standard deviation are shown. * p < 0.05.
What You Need to Know.
BACKGROUND AND CONTEXT
Hepatocellular carcinoma patients present high inter-tumor heterogeneity, yet existing HCC mouse models fail to capture this high diversity, limiting mechanistic and translational studies.
NEW FINDINGS
Cooperation between distinct cancer driver genes fuels inter-tumor heterogeneity, which is affected not only by specific combinations of genetic alterations but also by driver gene expression levels.
LIMITATIONS
Our novel collection of murine HCC models does not capture the whole diversity of HCC tumors in patients; therefore, complementary models recapitulating additional combinations of genetic alterations will be needed.
IMPACT
This novel collection of murine HCC models constitutes a unique and valuable resource for the liver cancer community as it will enable both mechanistic and translational studies.
Acknowledgements
We thank the Center for Comparative Medicine and Surgery, the Tisch Cancer Institute Flow Cytometry Shared Resource Facility, the Icahn School of Medicine at Mount Sinai (ISMMS) Oncological Sciences Histology Share Resource Facility, ISMMS Genomics Core Facility, the Translational and Molecular Imaging Institute Imaging Core, and the ISMMS Biorepository and Pathology Core. We also thank Magali Guffroy for support with histological characterization. We thank Jill Gregory for the artistic work.
Funding/Grant support
This work was supported by the following grants: Fundación Alfonso Martín Escudero Fellowship (M.R.G.), Philippe Foundation Inc (T.C.M.), Damon Runyon-Rachleff Innovation Award (DR52-18; A.L.), Pfizer Emerging Science Fund (P.M-S, A.L.), NIH/NCI R37 Merit Award (R37CA230636; A.L.), Asociación Española para el Estudio del Hígado (M.B-V.), Department of Defense (DoD) Career Development Award (CA150178; A.L.), DoD Translational Team Science Award (CA150272P2; A.L.) and ISMMS. The Tisch Cancer Institute and related research facilities are supported by P30 CA196521.
Conflicts of interest
A. Lujambio has received grant support from Genentech for unrelated projects. D.J.S., R.A.R., Z.K., S.C., J.G., and Y.D. are/were employees of Pfizer Inc. and hold shares in the company. No potential conflicts of interest were disclosed by the rest of the other authors.
Abbreviations
Abbreviations used in this paper
- CK19
cytokeratin 19
- CV
central vein
- EMT
epithelial to mesenchymal transition
- Glul
Glutamine synthetase
- HCC
hepatocellular carcinoma
- Hnf4a
hepatocyte nuclear factor 4 alpha
- mCL
murine cell lines
- mHCCs
murine HCCs
- NTP
nearest template prediction
- PV
portal vein
- SD
standard deviation
- sgRNAs
single-guide RNAs
- ssGSEA
single-sample Gene Set Enrichment Analysis
- PCA
principal component analysis
- TCGA
the Cancer Genome Atlas
Footnotes
Transcript profiling
The RNA-seq files are available at GEO (GSE148379).
To review GEO accession GSE148379: Go to https://urldefense.proofpoint.com/v2/url?u=https-3A__www.ncbi.nlm.nih.gov_geo_query_acc.cgi-3Facc-3DGSE148379&d=DwIBAg&c=shNJtf5dKgNcPZ6Yh64b-A&r=BH3UNPUNhOc-niJT9d00EAx3ah5AHfQlhQQ_SHtjigA&m=sTfdL6rTRofGOUNdqePF08V4vPMWKhaYdstZUJY4_−0&s=kEDQNb16oXxpaoS4ZZHauEf8msNDiS1v_D2LbyWoIGQ&e=Enter token gboramegnboltsb into the box.
Writing assistance
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Villanueva A Hepatocellular Carcinoma. N Engl J Med 2019;380:1450–1462. [DOI] [PubMed] [Google Scholar]
- 2.Zhu S, Hoshida Y. Molecular heterogeneity in hepatocellular carcinoma. Hepat Oncol 2018;5:HEP10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Network. CGAR. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Totoki Y, Tatsuno K, Yamamoto S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet 2011;43:464–9. [DOI] [PubMed] [Google Scholar]
- 6.Nault JC, Villanueva A. Intratumor molecular and phenotypic diversity in hepatocellular carcinoma. Clin Cancer Res 2015;21:1786–8. [DOI] [PubMed] [Google Scholar]
- 7.Sawai H, Nishida N, Khor SS, et al. Genome-wide association study identified new susceptible genetic variants in HLA class I region for hepatitis B virus-related hepatocellular carcinoma. Sci Rep 2018;8:7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwabe RF, Greten TF. Gut microbiome in HCC - Mechanisms, diagnosis and therapy. J Hepatol 2020;72:230–238. [DOI] [PubMed] [Google Scholar]
- 9.Dow M, Pyke RM, Tsui BY, et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc Natl Acad Sci U S A 2018;115:E9879–E9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nusse R, Clevers H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017;169:985–999. [DOI] [PubMed] [Google Scholar]
- 12.Cadoret A, Ovejero C, Terris B, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002;21:8293–301. [DOI] [PubMed] [Google Scholar]
- 13.Calderaro J, Couchy G, Imbeaud S, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol 2017;67:727–738. [DOI] [PubMed] [Google Scholar]
- 14.Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, et al. beta-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liberzon A, Birger C, Thorvaldsdottir H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyault S, Rickman DS, de Reynies A, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007;45:42–52. [DOI] [PubMed] [Google Scholar]
- 17.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer research 2009;69:7385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan PS, Nakagawa S, Goossens N, et al. Clinicopathological indices to predict hepatocellular carcinoma molecular classification. Liver Int 2016;36:108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sia D, Jiao Y, Martinez-Quetglas I, et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017. [DOI] [PubMed] [Google Scholar]
- 20.Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer research 2008;68:6779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bisso A, Filipuzzi M, Gamarra Figueroa GP, et al. Cooperation between MYC and beta-catenin in liver tumorigenesis requires Yap/Taz. Hepatology 2020. [DOI] [PubMed] [Google Scholar]
- 22.Dong B, Lee JS, Park YY, et al. Activating CAR and beta-catenin induces uncontrolled liver growth and tumorigenesis. Nat Commun 2015;6:5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abitbol S, Dahmani R, Coulouarn C, et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of beta-catenin activation. J Hepatol 2018;68:1203–1213. [DOI] [PubMed] [Google Scholar]
- 24.Caruso S, Calatayud AL, Pilet J, et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019;157:760–776. [DOI] [PubMed] [Google Scholar]
- 25.Harding JJ, Nandakumar S, Armenia J, et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin Cancer Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nature genetics 2010;42:715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu Z, Li H, Zhang Z, et al. A Pharmacogenomic Landscape in Human Liver Cancers. Cancer Cell 2019;36:179–193 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao YH, Lafita-Navarro MC, Zacharias L, et al. Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun Signal 2019;17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. In vivo screen identifies cooperating driver genes in HCC. (A) Schematic of vectors used in the hydrodynamic injections. LOF, loss of function; GOF, gain of function. Luc, luciferase. (B) Number of tumors per mouse in different models. Median with interquartile range is shown. (C) Survival graph of the corresponding conditions (high or low penetrance). Median survival and number of mice per group is shown. D, days. Log-rank Mantel-Cox test. **p < 0.01. Mice from Figure 1C. (D) Correlation between the penetrance in mice (% of mice that develop tumors in a given condition) and frequency of co-alteration in HCC patients1 of the pair of genes tested in mice. R, Pearson correlation coefficient. Mice from Figure 1C. Related to Supplementary Table 2. (E) Left, frequency of co-alteration of each pair of genetic alteration in HCC patients1 and the presence or not of tumors in mouse models. Median with interquartile range is shown. Mann-Whitney test. ns, not significant. Right, number of models showing positivity or for tumors depending on their association in HCC patients (co-occurrence or mutual exclusivity). Fisher test. ns, not significant. Related to Supplementary Table 2. (F) Relative levels of mRNA of the corresponding genes in tumor samples from mice from different conditions injected or not with the indicated oncogenes. Median with interquartile range is shown. Mann-Whitney test. ****p < 0.0001. (G) Percentage of deletion found in different models for each indicated single-guide RNA (sgRNA). Median with interquartile range is shown.
Supplementary Fig. 2. Cooperation between distinct driver genes leads to HCC with unique histologies. (A-E) Representative stainings depicting pseudoglandular (A) and trabecular (B) structures, necrosis (C), mitotic figures (yellow arrow) and apoptotic bodies (white arrow) (D), and cytokeratin 19 (E). HE, hematoxylin & eosin. The white bars represent 50 μm. (F) Average percentage of Ki67 positive cells in murine tumor samples from conditions with or without MYC overexpression. Median with interquartile range is shown. Mann-Whitney test. ****p < 0.0001.
Supplementary Fig. 3. Cooperation between distinct driver genes leads to unique immune landscapes. (A) Box and whisker plots representing different lymphoid (NK, Tregs, γδT) and myeloid (mDC1, mDC2) immune cell populations (% over CD45+ and CD45+lin− cells, respectively) in tumors and normal liver. Anova (Kruskal Wallis) test and multiple comparison to normal liver. *p < 0.05, **p < 0.01. (B) Gating strategy and illustrative plots of myeloid and lymphoid panels of a representative sample.
Supplementary Fig. 4. Novel genetically-defined murine HCC models recapitulate human HCC transcriptional subclasses. (A) Silhouette score depending on the number of groups of equal variance (k), calculated with K-means clustering algorithm, for murine HCC cell lines. (B) Table summarizing the histological features of each murine tumor as well as the Hoshida and Boyault subclasses. Average indicates the most frequent phenotype of each category. 50/50 indicates two phenotypes are equally represented in a given subclass. (C) Table summarizing the percentage of human tumors showing a given feature (YES) in different subclasses and studies. In green, the percentage of human tumors that are represented in mice belonging to the same subclass and showing the same feature. In bold, associations that are significant in HCC patients in the corresponding studies. (D) Heatmap of transcript levels and ssGSEA values for significant immune genes and gene sets in the different murine HCCs classified as “Immune class” (red) or rest (blue). The P-value for each gene and gene set shown is significant after applying Benjamini-Hochberg multiple testing correction (p < 0.0123). Red indicates high; blue, low. Z scores are shown. (E) Correlation between mRNA expression levels in tumors and transposon DNA integration levels. R, Pearson correlation coefficient. Color code is shown under the heatmap. The correlation values that are higher than 0.6 and with a P value lower than 0.05 are highlighted with the black outline.
Supplementary Fig. 5. Inter-tumor heterogeneity is shaped by oncogene expression levels and specific cooperating events. (A) Correlation plots. R, Pearson correlation coefficient. Ns, not significant. *** p < 0.0001, **** p < 0.0001. (B) Staining for the indicated antibody (β-catenin) in a representative MYClow;CTNNB1 tumor. The white bar represents 50 μm. Dashed line indicates border between tumor tissue and adjacent tissue. (C) Upper panel, CTNNB1 mRNA levels in CTNNB1-mutant (n = 49) and wild-type (WT, n = 134) human HCCs. Student t test. * p < 0.05. Lower panel, correlation between ssGSEA score for Hallmarks_Wnt_beta-catenin_signaling and MYC and CTNNB1 mRNA levels in human HCC (n = 183). R, Pearson correlation coefficient. **** p < 0.0001. (D) Correlation between expression profiles of each murine tumor and murine Ctnnb1exon3/exon3 tumors. Color code is shown on the right. R, Pearson correlation coefficient. The average correlation per model is also shown. (E) Correlation between expression profiles of each murine tumor and human HCC tumors. Color code is shown on the right. R, Pearson correlation coefficient. Purple, 95% of human HCCs have at least one murine tumor with a correlation higher than 0.5; grey, 87% of human HCCs have at least one of the mHCC subclasses with a correlation higher than 0.5 with; pink, 30% of human HCCs have at least one murine tumor with a correlation higher than 0.75.
Supplementary Fig. 6. Novel murine HCC cell lines recapitulate the most aggressive HCC subclasses. (A) Silhouette score depending on the number of groups of equal variance (k), calculated with K-means clustering algorithm, for murine HCC cell lines and tumors. (B) Volcano plot representing the log2 of the fold change (FC) between the expression in cell lines versus tumors and the −log10 of the p value. Selected genes are highlighted. Related to Supplementary Table 8. (C) Bar graph showing the p values for student t test in murine HCC cell lines. The dotted line indicates the threshold for significance after applying Benjamini-Hochberg multiple testing correction (p < 0.042). (D) Correlation between expression profiles of each tumor and its corresponding derived cell line. Color code is shown under the heatmap. The correlation values that are higher than 0.5 and with a P value lower than 0.05 are highlighted with the black outline. R, Pearson correlation coefficient. (E) Silhouette score depending on the number of groups of equal variance (k), calculated with K-means clustering algorithm, for murine HCC cell lines.
Supplementary Fig. 7. Cooperation between distinct driver genes leads to unique drug responses. (A) Heatmap showing the Pearson R values for the correlation between different compounds. The compounds are organized by proximity. Color code is shown under the heatmap. The correlation values of some compounds with high correlation are highlighted with the black outline. R, Pearson correlation coefficient. (B) Volcano plot showing linear regression between Pten deletion status and cell viability under various drug treatments in murine HCC cell lines. Effective drugs showing significant association are labeled (log-fold-change [logFC] < 0; p.value < 0.05). (C) Bar plots comparing the AUC of mTOR inhibitors in Pten knock-out (Pten-KO) and Pten wild-type (WT) murine cell lines. Mean and standard deviation are shown. * p < 0.05.