

Summary
Differentiation therapy utilizes our understanding of the hierarchy of cellular systems to pharmacologically induce a shift towards terminal commitment. While this approach has been a paradigm in treating certain hematological malignancies, efforts to translate this success to solid tumors have met with limited success. Mammary-specific activation of PKA in mouse models leads to aberrant differentiation and diminished self-renewing potential of the basal compartment, which harbors mammary repopulating cells. PKA activation results in tumors that are more benign, exhibiting reduced metastatic propensity, loss of tumor-initiating potential and increased sensitivity to chemotherapy. Analysis of tumor histopathology revealed features of overt differentiation with papillary characteristics. Longitudinal single cell profiling at the hyperplasia and tumor stages uncovered an altered path of tumor evolution whereby PKA curtails the emergence of aggressive subpopulations. Acting through the repression of SOX4, PKA activation promotes tumor differentiation and represents a possible adjuvant to chemotherapy for certain breast cancers.
Keywords: Tumor differentiation, mammary development, epithelial-mesenchymal transition, tumor metastasis, tumor evolution
eTOC Blurb
The mammary basal compartment harbors the majority of repopulating cells in gland. Ognjenovic et al. demonstrate that activation of PKA signaling inhibits the self-renewing potential of these basal cells, leading to aberrant mammary development. Functioning through the inactivation of Sox4, PKA curtails the emergence of multiple aggressive tumor subpopulations.
Introduction
The process of differentiation involves a complex multi-step journey of cells from a state of multipotency towards one of commitment to a cell state that is, typically, post-mitotic. The process of tumorigenesis involves the acquisition of limitless proliferative capacity, the evasion of growth suppression, and the enabling of replicative immortality (Hanahan and Weinberg, 2011), all of which are characteristics of the circumvention of terminal differentiation. Differentiation therapy was first championed in hematological malignancies in which the use of all-trans-retinoic acid led to the terminal differentiation of acute promyelocytic leukemias (APL) (Huang et al., 1988; Warrell et al., 1993). Despite these advances, barring a few notable examples (Saha et al., 2014; Storm et al., 2016), differentiation therapy in solid tumors has largely been either unsuccessful or exploratory.
3',5'-cyclic adenosine monophosphate (cAMP) is a widely used second messenger that regulates multiple downstream signaling cascades upon G-protein coupled receptor activation and subsequent Gαs or Gαi-mediated regulation of adenylate cyclase (Tasken and Aandahl, 2004). One of the major routes of cAMP action is via activation of the ubiquitous enzyme Protein Kinase A (PKA). While in certain contexts, mutant Gαs acts in an oncogenic fashion (Coles et al., 2020; Patra et al., 2018; Wu et al., 2011), multiple lines of evidence suggest a role for Gαs-cAMP-PKA signaling in tumor-suppressive properties that could be mediated through the induction of differentiation (He et al., 2014; Iglesias-Bartolome et al., 2015; Pattabiraman et al., 2016).
The presence of heterogeneous tumor subpopulations has been a major hurdle in the clinical management of various cancer types. Indeed, recent studies have pointed to specific epigenetic mechanisms by which alterations in lineage restriction can contribute to tumor heterogeneity and progression of disease (Flavahan et al., 2017; LaFave et al., 2020; Pomerantz et al., 2020). Breast cancers are a heterogeneous group of diseases that have been classified based on their histopathological features, or molecularly, based on their gene expression profiles (Perou et al., 2000). These modes of classification have been useful in highlighting the cellular and molecular composition of tumors, while also providing prognostic information that can be leveraged for therapeutic decision-making. While these analyses have been instrumental in our ability to comprehend inter-tumoral heterogeneity by binning tumors into subtypes, they have not been able to shed light on the specific subpopulations within tumors that drive tumor growth, their differentiation state, and the relationship between the composition of cells in a tumor and the histopathological features that they manifest.
Here we show that activation of PKA impairs mammary differentiation by specifically reducing the self-renewal potential of the basal compartment. Mammary tumors with constitutively active PKA exhibit features of papillary differentiation with a reduced propensity for metastasis and increased susceptibility to chemotherapy. Longitudinal single cell profiling of mammary tumors at early and late stages of tumorigenesis revealed their inherent heterogeneity through the emergence of multiple, transcriptionally-distinct, subpopulations. Firstly, the emergence of a lumino-basal subpopulation that co-expresses both luminal and basal markers in MMTV-PyMT tumors, marking the relinquishing of their developmental lineage-of-origin. Additionally, the emergence of an EMT-like sub-population that signals loss of key epithelial markers.
Both these subpopulations are severely diminished in PKA-activated tumors, which exhibit reduced metastatic capacity and increased therapeutic sensitivity. These observations imply that the induction of a more differentiated cell state may favorably alter the representation of tumor cell subpopulations that are responsible for progression. Our single-cell analyses revealed Sox4 as a potential candidate in the regulation of the lumino-basal subpopulation, whereby its direct phosphorylation by PKA at S56 and S103 impair its ability to act as a transcription factor and promote tumor progression.
Results
Constitutively Active PKA Signaling Impairs Mammary Differentiation
Previous studies have shown the ability of PKA to induce MET in some breast cancer cell lines (Pattabiraman et al., 2016). These data do not, however, reveal the biological effects of PKA activation in normal mammary development and its impact on the subsequent in vivo development of tumors. To address this, we employed a mouse model harboring a floxed constitutively active mutant allele of the catalytic subunit of PKA (Niswender et al., 2005), and expressed this allele in the mouse mammary epithelium by crossing it to a mouse mammary tumor virus-driven Cre (MMTV-Cre; line D) model (Wagner et al., 2001)(Fig. 1A). The mammary glands of these PrkacaCαR/fl mice at eight weeks consistently contained fewer epithelial cells than littermate Prkacafl/fl controls, with a reduction of at least 50% in the EpCAMmedCD49fhi basal/myoepithelial cells (Fig. 1B,C), the compartment that harbors the highest proportion of mammary repopulating units. This reduction was not a result of differences in Cre recombination between the luminal and basal lineages as 70-85% of cells from both compartments underwent recombination when crossed with a R26-mTmG reporter (Fig S1A,B). Postnatal mammary gland development in the PrkacaCαR/fl mice was aberrant displaying a severe defect in ductal morphogenesis (Fig. 1D). Histological analyses of the mammary ducts revealed intact luminal and basal layers (Fig 1E), however, the overall number of terminal ductal lobular units (TDLUs) per gland was 3-fold lower in the PrkacaCαR/fl mice compared to Prkacafl/fl controls (Fig 1F,S1C), likely a result of impaired ductal outgrowth. In order to test whether the decrease in basal cells was merely quantitative, or whether activation of PKA led to a quantitative alteration of their self-renewal ability, we sorted for EpCAMmedCD49fhi basal cells from PrkacaCαR/fl mice and control Prkacafl/fl littermates, and carried out organoid-forming assays (Guo et al., 2012). EpCAMmedCD49fhi cells from PrkacaCαR/fl mice were impaired in their organoid-forming potential, indicating a loss of self-renewing potential (Fig. 1G,H). Additionally, when transplanted into the cleared fat pads of recipient mice, organoids from PrkacaCαR/fl mice were at least 100-fold less efficient at forming ductal trees (Fig. 1I), revealing a loss of stem-like repopulating ability. This lack of efficiency in repopulation was also observed upon transplantation of sorted EpCAMmedCD49fhi basal cells (Fig S1D). Notably, the basal cells from PrkacaCαR/fl mice were severely deficient in serial transplantation assays (Fig S1D), suggesting that the activation of PKA leads to the exhaustion of the repopulating ability of this compartment. This phenotype is reminiscent of previous observations whereby activation of Gαs-PKA signaling led to stem cell exhaustion and depletion (Iglesias-Bartolome et al., 2015).
Figure 1: Activation of PKA impairs mammary development and repopulating ability.

(A) PKA-CαR mice were crossed to MMTV-Cre mice to activate the CαR constitutively active allele. (B) FACS plots showing proportions of EpCAMhiCD49fmed luminal and EpCAMmedCD49fhi basal compartments upon constitutive activation of PKA and a summary of relative proportions of these subpopulations across multiple mice (C). Carmine alum stained whole mounts of mammary glands from and PrkacaCαR/fl control mice displaying ductal outgrowth (D). Bar, 3mm. (E) Fluorescence imaging of Krt8 (luminal) and Krt14 (basal) layers in mammary glands from PrkacaCαR/fl and Prkacafl/fl mice. Bar, 150μm. (F) Hematoxylin-eosin staining of FFPE sections of mammary glands depicting terminal ductal lobular units from PrkacaCαR/fl and Prkacafl/fl mice. Bar, 150μm. Organoid assays were carried out to assay for differences in self-renewal potential upon activation of PKA (G, H). Data are shown as mean ± SD (n =10 mice). p value was determined by Student’s two-tailed t test (unpaired). *p < 0.01. Limiting dilution transplantation into cleared fat pads estimated the frequency of mammary repopulating units in PrkacaCαR/fl and Prkacafl/fl controls (I). Schematic representation of breeding strategy to generate mammary-specific Gαs active mutants (J). FACS was carried out to capture differences in representation of luminal and basal subpopulations (K). Organoid assays carried out to test for differences in self-renewal potential (L) (n = 5 mice). Statistical significance was calculated by a Student t-test (two-tailed) to compare two groups (P < 0.05 was considered significant).
In order to ensure that the effects observed could be attributed specifically to activation of PKA signaling, we used a second model, GαsR201C/fl mice (Iglesias-Bartolome et al., 2015), that express constitutively-active Gαs under the control of a doxycycline-inducible promoter. Upon expressing this allele in the mammary gland (Fig. 1J), doxycycline treatment was carried out to activate the GαsR201C mutant from 3 weeks post-partum, at which point postnatal mammary development is initiated, to full maturity at 8 weeks. As observed in the PrkacaCαR/fl mice, the GαsR201C/fl mice also exhibited a reduction in the EpCAMmedCD49fhi basal cells, which were severely impaired in their organoid-forming potential (Fig. 1K,L) while also displaying severe defects in mammary ductal outgrowth (Fig. S1E). Importantly, organoid-forming potential of basal cells from control MMTV-Cre mice was unaffected by doxycycline treatment (Fig. S1F). Put together, these results indicate a role for activated PKA signaling in limiting the self-renewal and expansion of the mammary basal lineage.
Basal lineage-specific impairment upon activation of PKA
To further confirm the lineage-specific effects of activation of PKA, the CαR allele was expressed specifically in either luminal or basal lineages of the mammary gland. We crossed the Prkacafl/fl mice to a R26-mTmG reporter mouse (Muzumdar et al., 2007), and F1 mice were subsequently crossed to a Krt5-CreERT2 mouse to express the CαR allele in a basal-specific manner (Van Keymeulen et al., 2011)(Fig. 2A). Upon tamoxifen administration at 4 weeks of age and observation of mammary glands at 8 weeks, we noted an absence of GFP-positive basal cells in the PrkacaCαR/fl mice, present only in controls that lacked the CαR allele, or those that did not receive tamoxifen (Fig. 2B-D). Initial recombination following administration of tamoxifen led to 76% of basal cells undergoing Krt5-Cre recombination (Fig S2), whereas in 8-week old PrkacaCαR/fl mice, less than 1% of basal cells were GFP positive, compared to >30% in Prkacafl/fl controls (Fig 2B). These observations suggest that the expression of the PrkacaCαR allele inhibited the expansion of the basal compartment, leading to the observed defects in ductal outgrowth. We subsequently expressed the CαR allele specifically in luminal cells using the Krt8-CreERT2 mouse (Van Keymeulen et al., 2011)(Fig 2E). Upon tamoxifen administration at 5 weeks of age and observation of mammary glands at 8 weeks, the percentage of GFP-expressing luminal cells in PrkacaCαR/fl mice was comparable to those of Prkacafl/fl controls (Fig 2F), and indeed, the presence of GFP+ ducts was observed upon whole mount imaging and staining of sections (Fig 2G,H), indicating that activation of the PKA allele did not affect the luminal lineage. These results reiterate the ability of PKA signaling in inducing a qualitative and quantitative alteration of the mammary basal/myoepithelial compartment. We further explored the impact of these alterations on the development and progression of mammary tumors that arise from these aberrant glands.
Figure 2: Specific activation of PKA in basal, but not luminal cells, impairs ductal outgrowth.

Schematic of breeding strategy to generate Krt5-PrkacaCαR/fl mice that specifically express activated PKA in basal cells (A). FACS plots (B) and fluorescence imaging of ducts (C) show the extent of GFP-expressing cells within the basal compartment and ducts that have undergone Cre recombination in Krt5-PrkacaCαR/fl mice and Krt5-Prkacafl/fl controls at 8 weeks. Bar, 500μm (D) Fluorescence imaging of a cross-section of a mammary duct of Krt5-mTmG-PrkacaCαR/fl mice to assess histology and extent of tdTomato vs GFP recombination. Bar, 150μm. Schematic of breeding strategy to generate Krt8-PrkacaCαR/fl mice that specifically express activated PKA in luminal cells (E). FACS plots (F) and fluorescence imaging of ducts (G) show the extent of GFP-expressing that have undergone Cre recombination in Krt8-PrkacaCαR/fl mice and Krt8-Prkacafl/fl controls at 8 weeks. Bar, 500μm. (H) Fluorescence imaging of a cross-section of mammary ducts of Krt8-mTmG-PrkacaCαR/fl mice to assess histology and extent of tdTomato vs GFP recombination. Bar, 150μm.
Human Breast Cancers Harbor Recurrent Genomic Alterations in the PKA locus
Given the previously identified role for PKA in the induction of mesenchymal-epithelial transition (MET)(Pattabiraman et al., 2016), we interrogated the clinical significance of the PKA genes in human breast cancer. Analysis of the breast cancer genomic data from The Cancer Genome Atlas (TCGA)(Cancer Genome Atlas, 2012) revealed amplifications in the genomic loci encoding PKA subunits in 11.2% (121/1085) of breast cancers (Fig. 3A). PRKAR1A encodes one of four negative regulatory subunits that sequester the catalytic subunits, including that encoded by PRKACA, which carry out enzymatic functions downstream (Skalhegg and Tasken, 2000). The frequency of amplifications in the other regulatory subunits PRKAR1B, PRKAR2A and PRKAR2B were significantly lower than that of PRKAR1A at 1.1%, 0.1% and 0.6%, respectively (Fig 3B). We, thus, focused on the effects of PRKAR1A amplifications and resulting PRKACA catalytic subunit activity for further interrogation. Both of these subunits were also the highest to be expressed across breast tumors (Fig 3C). Mining the METABRIC datasets (Curtis et al., 2012; Pereira et al., 2016) also revealed PRKAR1A amplifications in 11.9% (82/684) of tumors, with amplifications being more frequent in the Luminal B subtype of tumors across both TCGA (13.03%) and METABRIC (14.32%) datasets (Fig. 3D). PRKAR1A amplification was correlated with higher expression levels of its mRNA (Fig. 3E), while also inversely correlating with expression levels of the catalytic subunit PRKACA (Fig. S3A,B) in a subtype-independent fashion. Higher PRKAR1A expression conferred better prognosis in tumors of the basal-like (Fig. 3F), but not other subtypes (Fig S3C). These observations were, however, only observed in TCGA, but not other, datasets (Saal et al., 2015)(Fig. S3D,E), or for other or for other PKA subunits (Fig 3G).
Figure 3: Recurrent genomic amplification of the PKA locus in human breast cancer.
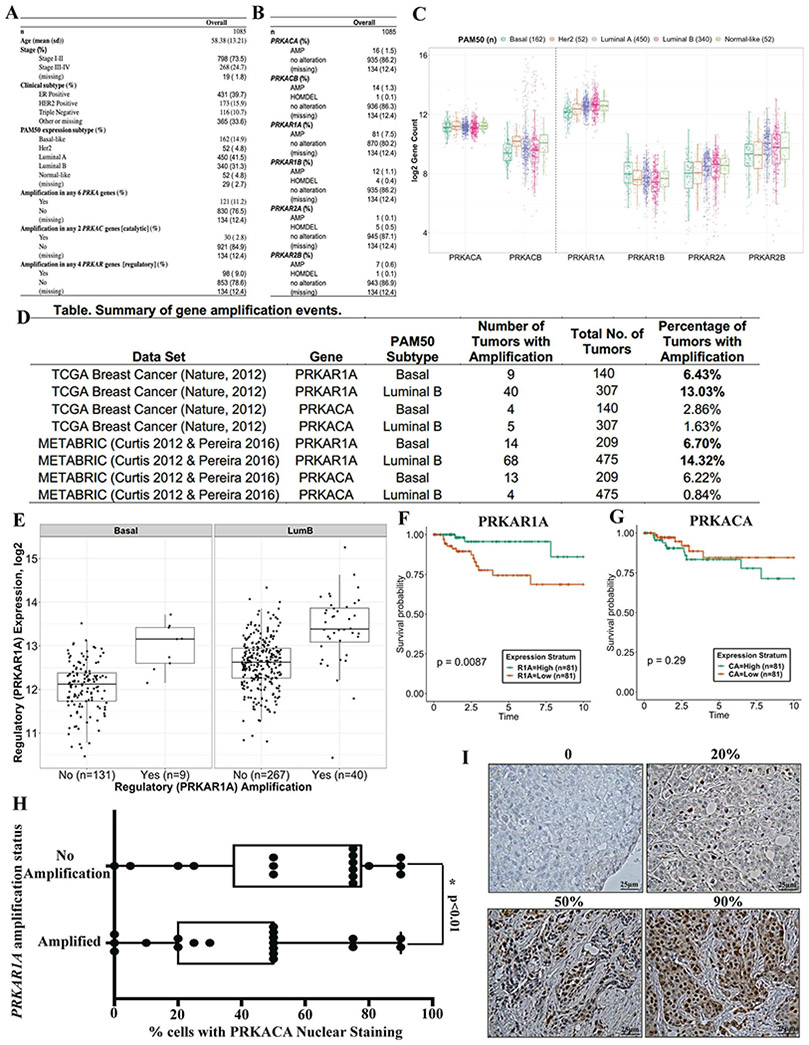
Analysis of 1085 breast tumors from the TCGA dataset revealing amplifications in the regulatory (PRKAR) and catalytic (PRKAC) genes that encode PKA subunits (A, B). (C) Box plots show the mRNA expression levels of genes encoding each PKA subunit and their relative expression levels across human breast cancer subtypes. Summary of PRKAR1A and PRKACA amplifications in the TCGA and METABRIC datasets in basal and luminal B subtypes (D). mRNA expression of PRKAR1A, the most amplified PRKA gene, across tumors with and without amplifications in the PRKAR1A locus (E). Kaplan-Meier curves outline the ability of high or low levels of PRKAR1A (F) or PRKACA (G) mRNA levels to stratify prognosis of patients over ten years. PRKACA activation was measured by immunohistochemistry (IHC) using a p-PKA substrate antibody and quantifying the percentage of cells with nuclear signal in primary breast cancer patient samples that harbor amplifications in PRKAR1A compared to controls (H). Statistical significance was calculated by a Student t-test (two-tailed) to compare two groups (P < 0.05 was considered significant) (n=35 patient tumors). IHC using p-PKA substrate antibody showing samples that express different levels of PRKACA activation (I). Bar, 25μm.
These recurrent genomic alterations in the PKA loci point to possible clinical implications for breast tumorigenesis, particularly of the luminal B subtype. We explored the direct connection between PRKAR1A amplification in human breast tumors and the corresponding activation of the PRKACA catalytic subunit signaling. In line with a prognostic role for the PKA locus, tumors that harbored amplifications in the PRKAR1A locus had lower percentages of cells that exhibited PKA activation compared to controls that harbored no amplifications (Fig 3H,I). This result indicates that genomic amplifications in the PRKAR1A locus play a direct role in regulating the activation status of PKA catalytic subunit signaling in human breast cancer. We, thus, proceeded to further study the role of PKA catalytic subunit activation and its downstream effectors in mammary tumorigenesis.
PKA-induced Differentiation leads to Attenuation of Metastatic Progression and Improved Response to Therapy
Given the high rate of recurrent PRKAR1A and PRKACA amplifications in the luminal B and basal subtypes of breast cancer (Fig. 3D), we chose to model PKA activation in the MMTV-PyMT mouse model of mammary tumorigenesis (Guy et al., 1992), which exhibit luminal B features in the early stages of tumor development, but acquire a more basal-like phenotype as tumors progress (Lin et al., 2003). Upon crossing the PrkacaCαR/fl mice to the MMTV-PyMT mice (Fig. 4A), we observed that tumors in the control Prkacafl/fl PyMT mice took, on average, 142 days to reach the tumor volume endpoint of 2cm2, whereas those in the PrkacaCαR/fl PyMT were slower to develop, taking 173 days (Fig. 4B), although overall tumor volume was comparable between the two genotypes (Fig. 4C). As expected, activation of the PKA signaling pathway was higher in the PrkacaCαR/fl PyMT tumors compared to the Prkacafl/fl PyMT controls (Fig. S4A,B).
Figure 4: Induction of tumor differentiation upon activation of PKA signaling.
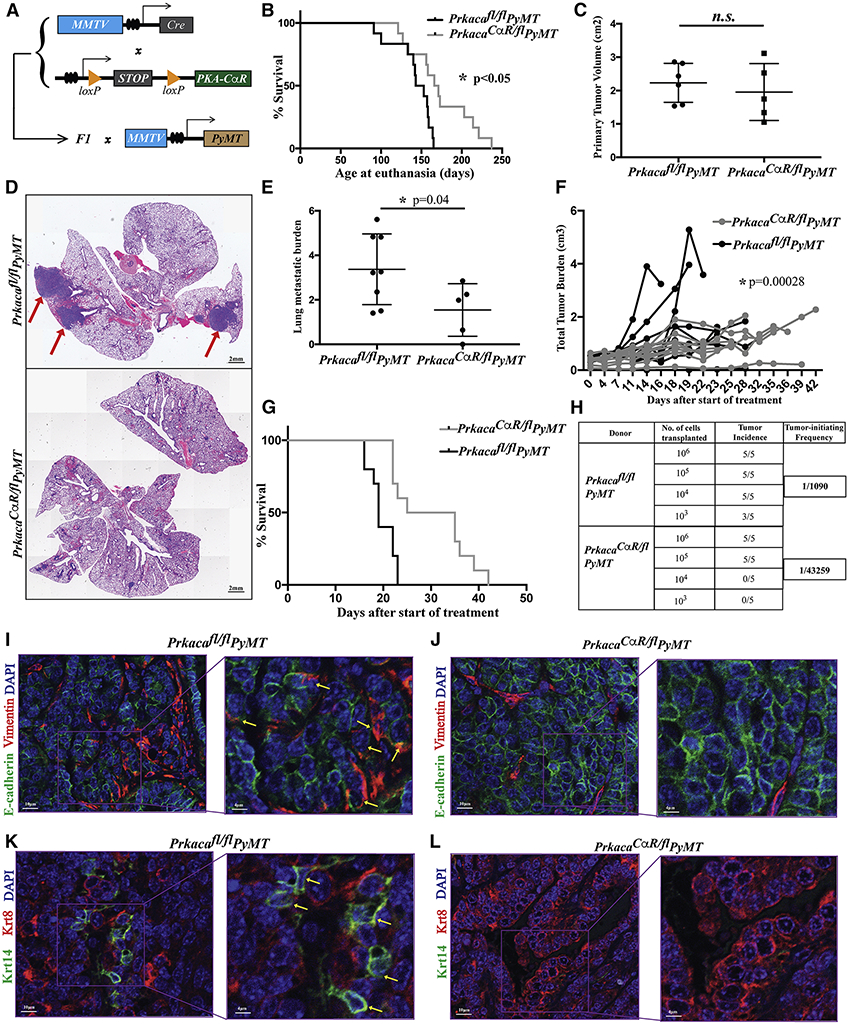
(A) Schematic of breeding strategy to generate MMTV-PyMT mice with active PKA. Differences in survival (B; n = 24 mice) and primary tumor volumes (C; n = 11 mice) of PrkacaCαR/fl PyMT mice compared to Prkacafl/fl PyMT controls. Differences in lung metastatic burden was captured from H&E stained FFPE lung sections (D, E), red arrows indicate macrometastases. Bar, 2mm (n = 13 mice). Treatment with Adriamycin showing differences in response to chemotherapy (F, G) between PrkacaCαR/fl PyMT mice and Prkacafl/fl PyMT controls (n = 20 tumors). Tumor initiating potential of PrkacaCαR/fl PyMT mice and Prkacafl/fl PyMT controls was assessed by limiting dilution transplantation analyses (H). Fluorescence imaging of tumor sections stained with antibodies against E-cadherin, Vimentin, Krt8 and Krt14 revealing epithelial-mesenchymal (I,J) and luminal-basal heterogeneity (K,L). Yellow arrows highlight Vimentin (I) - and Krt14 (K)-expressing tumor cells. Bar, 10μm and 4μm. Error bars represent ± standard deviations of the mean. Statistical significance was calculated by a Student t-test (two-tailed) to compare two groups (P < 0.05 was considered significant) except for survival analyses where significance was determined using a Log-rank (Mantel-Cox) test (P < 0.05 was considered significant) and chemotherapy treatment where a Wald Z-test was used to compute the p-value for the difference of slopes in two treatment groups.
In order to quantify the metastatic burden of the PrkacaCαR/fl PyMT, we enumerated the number of lung macrometastatic foci from H&E stained sections and normalized this value to the primary tumor volume at the time of sacrifice. Thus, despite harboring primary tumors of comparable volumes (Fig. 4C) and having spent longer periods developing in the mice, the PrkacaCαR/fl PyMT mice contained only half the metastatic burden of the Prkacafl/fl PyMT controls (Fig. 4D,E). Importantly, PrkacaCαR/fl PyMT tumors were also more sensitive to treatment with adriamycin (Fig. 4F), a commonly used chemotherapeutic agent in the neoadjuvant treatment of human triple negative breast cancer. Treatment with Adriamycin significantly improved the survival of mice bearing PrkacaCαR/fl PyMT when compared to controls (Fig. 4G).
Observation of hematoxylin-eosin (H&E) stained tumor sections revealed significant differences in their histopathology. The control Prkacafl/fl PyMT tumors resembled human invasive ductal carcinomas (IDC) of high grade comprised of solid sheets of pleomorphic cells with abundant mitotic activity (Fig. S4C). In contrast, tumors from PrkacaCαR/fl PyMT mice exhibited a more differentiated phenotype with gland formation and blander nuclear characteristics, akin to an intermediate grade invasive ductal carcinoma. Additionally, these tumors contained a rich vascularized stroma reminiscent of papillary differentiation (Fig. S4C). Activation of PKA also resulted in a large reduction in tumor cells co-expressing Vimentin and E-cadherin (Fig. 4I,J) indicating a partial loss of EMT traits. The wild-type tumors, also harbored a significant number of Krt14-positive cells basal cells as previously reported (Cheung et al., 2013), a feature that was less prominent in PrkacaCαR/fl PyMT tumors (Fig. 4K,L).
Upon transplantation in limiting dilution into secondary hosts, PrkacaCαR/fl tumors exhibited a 40-fold reduction in their tumor-initiating potential when compared to control tumors (Fig. 4H). To further understand the reduction in tumor-initiating potential we assessed the loss of stemness markers that have previously been uncovered in MMTV-PyMT tumors. We carried out immunofluorescence for Zeb1, which has been shown to enrich for tumor-initiating cells in this model (Ye et al., 2015) and observed that PrkacaCαR/fl PyMT tumors exhibit reduced expression of Zeb1 compared to Prkacafl/fl PyMT controls (Fig S5A,B). In another study that characterized the heterogeneity of EMT states in MMTV-PyMT tumors, the CD106neg CD51neg subpopulation was found to contain the most metastatic subpopulation (Pastushenko et al., 2018). We observe that PrkacaCαR/fl PyMT tumors have a 50% reduction in this compartment compared to Prkacafl/fl PyMT controls (Fig S5C). Together, these data point to a reduction in the EMT traits and decreased stemness potential in mammary tumors as a result of PKA activation.
Activation of PKA induces a more differentiated histology in multiple models
The histological differences in PrkacaCαR/fl PyMT tumors compared to Prkacafl/fl PyMT control tumors, specifically that of increased differentiation, could have arisen as a result of aberrant development of the mammary gland prior to oncogenic transformation. To test whether induction of PKA in already formed tumors would have a similar effect, we used the inducible GαsR201C/fl model in which activation of PKA signaling would occur following treatment with doxycycline (Fig. 5A). Upon doxycycline induction at 8-9 weeks of age until sacrifice, GαsR201C/fl tumors also exhibited characteristics of papillary differentiation (Fig. 5B) indicating that the altered histological features were a result of activation of Gαs-PKA signaling and not disproportionate cell-type representation in the pre-transformed mammary glands.
Figure 5: Differentiated histology induced by PKA across tumor models.

(A) Schematic of mouse crossing strategy to express the doxycycline-inducible GαsR201C/fl allele in the mammary glands of MMTV-PyMT mice following which tumors were harvested and FFPE sections were stained with hematoxylin and eosin (B) to study their histopathology and differentiation status. (C) Schematic of mouse crossing strategy to express the PKA-CαR allele in the mammary glands of C3(1)-Tag mice following which tumors were harvested and FFPE sections stained with hematoxylin and eosin to study their histopathology (D). Fluorescence imaging of PrkacaCαR/fl C3(1)-Tag and control Prkacafl/fl C3(1)-Tag tumors using Vimentin and E-cadherin to assess EMT status (E). Kaplan-Meier curve comparing the survival of PrkacaCαR/fl C3(1)-Tag and control Prkacafl/fl C3(1)-Tag mice (F). Bar values as shown. Statistical significance was determined using a Log-rank (Mantel-Cox) test (P < 0.05 was considered significant; n = 22 mice).
The induction of differentiation was also observed in a second model of mammary tumorigenesis, C3(1)-Tag (Maroulakou et al., 1994)(Fig. 5C), which develops basal-like tumors with features of metaplasia. PrkacaCαR/fl C3(1)-Tag tumors also show signs of differentiation with a reduction in metaplastic and sarcomatoid features, accompanied by a loss of Vimentin and gain in E-cadherin, when compared to control Prkacafl/fl C3(1)-Tag tumors (Fig. 5D,E). Importantly, PrkacaCαR/fl C3(1)-Tag tumor-bearing mice also exhibit extended survival compared to Prkacafl/fl C3(1)-Tag controls (Fig S5F). Thus, the ability to induce differentiation is observed across both Luminal B and basal-like models of breast cancer upon induction of PKA signaling. The resulting tumor pathology that manifests following PKA activation appears to depend on the nature of the tumor and its cellular origins.
Differentiation Results from Altered Evolution of Tumor Subpopulations
In order to understand the molecular and cellular basis for the histopathological properties of the Prkacafl/fl PyMT and PrkacaCαR/fl PyMT tumors, we longitudinally harvested cells at the early and late stages of tumorigenesis. At 9 weeks of age, the mammary glands of Prkacafl/fl PyMT mice are hyperplastic, while still largely maintaining their ductal architecture. We surgically resected one of two inguinal (no. 4) glands without euthanizing the animals, allowing them to develop tumors, which were later resected and sequenced. Using single-cell mRNA sequencing (scRNA-Seq) at both these time points, we were able to track the evolution of tumors at the hyperplasia and tumor stages (Fig. 6A). Based on expression of well-known marker genes, clusters were annotated into basal, mature luminal, luminal progenitor, lumino-basal and EMT-like (Fig. S6A-D).
Figure 6: Altered evolution of tumor cell subpopulations upon activation of PKA.
(A) Schematic of experimental set up to carry out sequential scRNA-Seq of hyperplastic and overt tumor samples from matched mice. tSNE plots show similar cellular constituents in hyperplastic glands of PrkacaCαR/fl PyMT mice and Prkacafl/fl PyMT controls (B, D) but divergent evolution of subpopulations during the transition from hyperplasia to tumor stages of development (C, E). (F) tSNE plots highlighting the expression levels of the Lalba (left) and Sox4 (right) genes in tumors from Prkacafl/fl PyMT (top) and PrkacaCαR/fl PyMT tumors (bottom) The lumino-basal subpopulation is highlighted by the red ellipse. Pseudotime analyses enabled the capture of the directionality of evolution from hyperplasia to tumor in the Prkacafl/fl PyMT tumors (G) that is altered upon PKA activation in PrkacaCαR/fl PyMT tumors (H).
Dimensionality reduction and clustering of cells from Prkacafl/fl PyMT and PrkacaCαR/fl PyMT mice yielded similar populations at the hyperplastic stage, with both exhibiting an expansion of the luminal progenitor compartment, while still retaining a portion of their normal basal and mature luminal subpopulations (Fig. 6B,D). There were, however, significant differences in the patterns of evolution from the hyperplasia to tumor stage. The Prkacafl/fl PyMT tumors harbored a luminal progenitor (LP) population that constituted the largest subpopulation within the tumor, but also contained two additional subpopulations. Firstly, lumino-basal (LB) subpopulation that expressed high levels of Sox4, which co-expressed LP and basal markers (Fig. 6C,E). Secondly, an EMT-like subpopulation, expressed relatively lower levels of a number of cytokeratins (Krt8, Krt14, Krt5, Krt79, Krt80), claudins (Cldn4, Cldn6, Cldn10), and markers of mammary differentiation (Prlr, Pgr, Lalba) (Fig. S6B,D). Notably, both these subpopulations were evident only at the tumor stage, indicating that they evolved between the hyperplasia and tumor stages (9-22 weeks of age). Importantly, the PrkacaCαR/fl PyMT tumors harbored significantly reduced populations of both the lumino-basal and EMT-like clusters (Fig. 6F).
Pseudotime kinetics (Trapnell et al., 2014) of Prkacafl/fl PyMT tumors point to evolution from a hyperplastic signature to eventually acquiring a tumor signature (Fig. 6G). In contrast, the PrkacaCαR/fl PyMT tumors exhibited an altered evolution trajectory whereby gene kinetics for tumor cells preceded that of hyperplastic cells in PrkacaCaR/fl PyMT, with the tumor displaying a more differentiated profile than the hyperplasia (Fig. 6H). This provides a transcriptional basis for the PrkacaCαR/fl PyMT tumors adopting a more differentiated histopathology, which likely drives the observed phenotypic and functional differences. The scRNA-Seq profiles reveal that the major cellular differences between the PrkacaCαR/fl PyMT and controls lie in the evolution of the transcriptionally-distinct lumino-basal and EMT-like subpopulations. The presence of these cells, ostensibly, drives the metastatic and therapy-resistant traits of the wild-type tumors, which are diminished upon activation of PKA signaling. The emergence of these two subpopulations could mark the point at which wild-type tumors initiate metastatic spread.
Sox4 is a Key Downstream Target of PKA-induced Differentiation
To understand the mechanism by which PKA could induce tumor differentiation, we assessed the activation of the Hedgehog (Hh) pathway, which has previously been shown to act downstream of PKA (Iglesias-Bartolome et al., 2015). Upon treatment with forskolin for 30 minutes, nuclear translocation of the PKA catalytic subunit was observed, whereas no change in nuclear localization of Gli1 was observed (Fig S7B). Interestingly, a decrease in nuclear localization of β-catenin was observed after 6 hours of PKA activation, suggesting that repression of Wnt is likely an indirect downstream effect. We thus resorted to a more objective approach to uncovering possible mechanistic insights into pathways acting downstream of PKA.
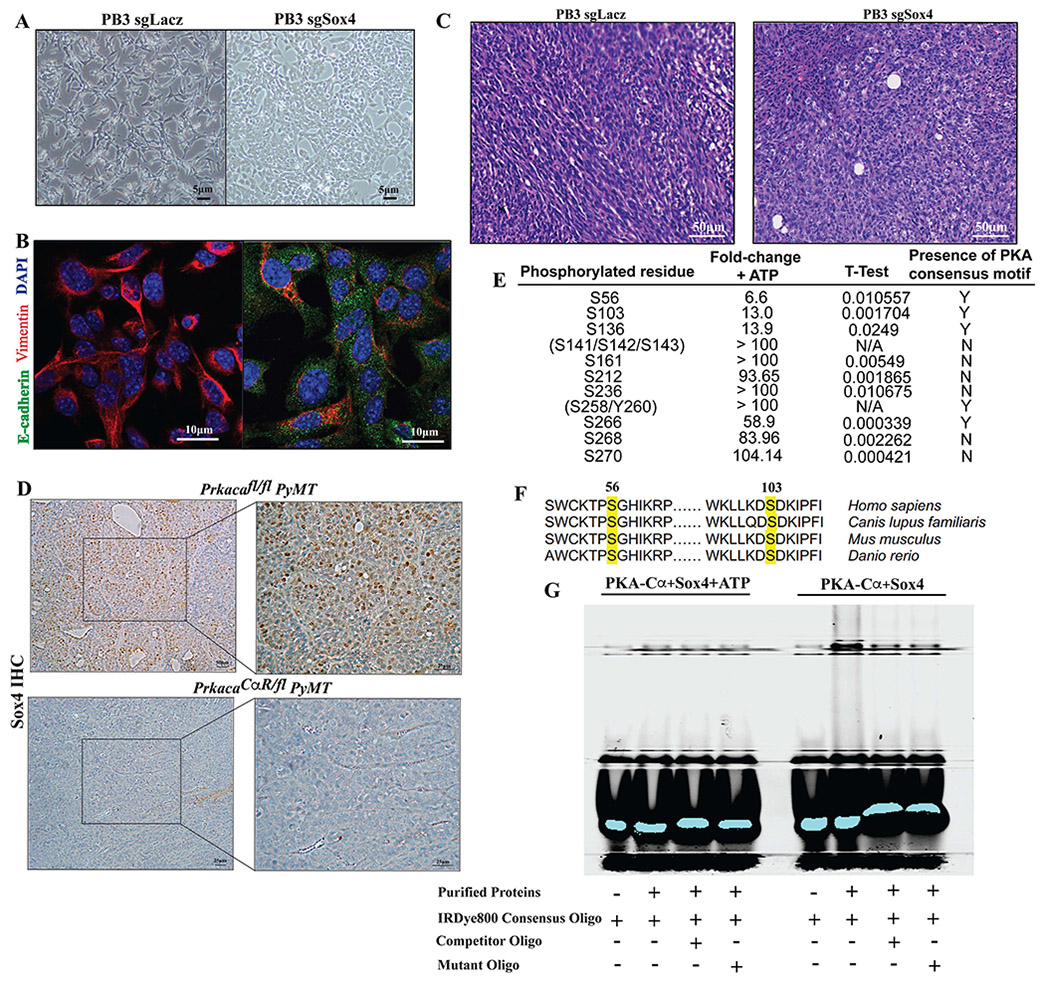
To further understand the emergence of the lumino-basal subpopulation, we focused on Sox4, one of the key transcription factors that is highly expressed in this cluster (Fig. 6F). Other Sox family members have been shown to have profound impacts on mammary development and tumorigenesis (Dravis et al., 2015; Guo et al., 2012), whereas Sox4 has previously been demonstrated to play a role in EMT (Tiwari et al., 2013). Based on these studies, we hypothesized that PKA could be repressing Sox4 activity and affecting its transcriptional output. We first tested whether Sox4 loss could recapitulate the effects of PKA activation by carrying out CRISPR-Cas9 mediated knockout in PB3, a cell line derived from an MMTV-PyMT tumor (Dongre et al., 2017) (Fig S7A). Loss of Sox4 induced a partial MET in PB3 cells, resulting in the acquisition of a more epithelial-like morphology (Fig 7A) as well as upregulation of the epithelial marker E-cadherin and loss of the mesenchymal marker Vimentin (Fig. 7B). Importantly, mammary fat pad transplantation of Sox4−/− PB3 cells resulted in the formation of tumors that exhibited characteristics of increased differentiation, with the presence of more epithelioid cells in comparison with controls that exhibit a more sarcomatoid profile (Fig. 7C). In line with these observations, PrkacaCαR/fl PyMT tumors exhibit reduced levels of Sox4 nuclear localization when compared to wild-type controls (Fig 7D).
Figure 7: Suppression of Sox4 activity by PKA leads to tumor differentiation.
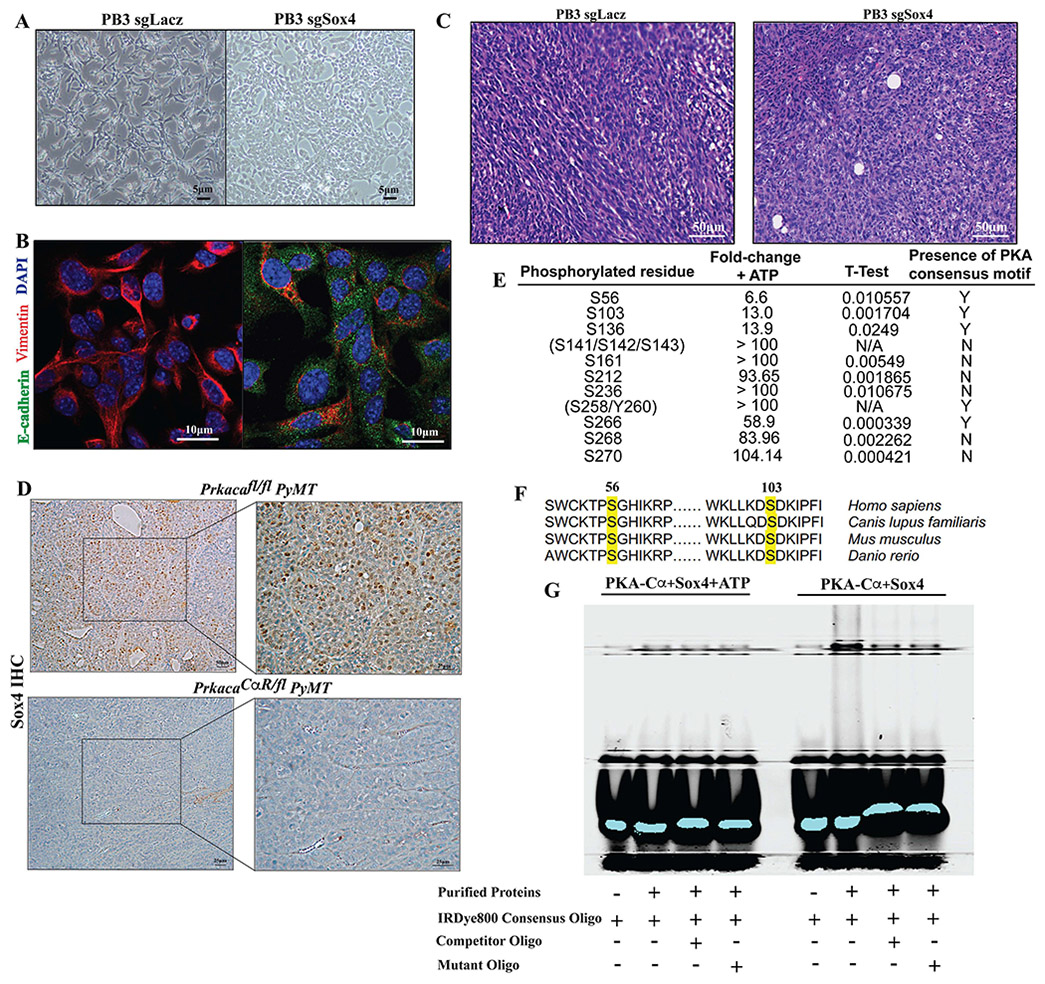
CRISPR-Cas9 mediated knockout of Sox4 in PB3 cells to assess differences in cell morphology as captured by bright-field images (A) and the expression of epithelial-mesenchymal markers by immunofluorescence (B). Bar, 5, 10μm. H&E stained FFPE sections of transplanted PB3 tumors were carried to estimate changes in histopathology which would reflect altered differentiation state (C). Bar, 50μm (D) Brightfield images of immunohistochemical staining of Sox4 in FFPE sections from Prkacafl/fl PyMT (top) and PrkacaCαR/fl PyMT (bottom) tumors. Bar, 25μm. PKA phosphorylation sites on Sox4 identified by in-vitro kinase assays (E) including sites in the HMG-box containing DNA binding domain that are conserved across evolution (F). Electrophoretic mobility shift assays carried out in the presence of purified PKA-Cα and Sox4 (right) or PKA-Cα, Sox4 and ATP (left) to test for retardation of mobility upon phosphorylation of Sox4 by PKA (G).
To test whether Sox4 is a direct substrate of PKA, we carried out an in vitro kinase assay with purified PKA catalytic subunit and Sox4. A number of residues were found to be directly phosphorylated upon addition of ATP to the reaction, including S56, S103, S136 and S266 (Fig 7E, Table S1). The first two phosphorylated residues reside in the highly conserved HMG box-containing DNA binding domain (van de Wetering et al., 1993)(Fig 7F), suggesting that their post-translational modifications might have an effect on the DNA-binding capability of the protein. Indeed, as observed in an electrophoretic mobility shift assay (EMSA), the presence of Sox4 and PKA leads to a retardation of mobility of a fluorescence-labeled consensus oligo sequence, ostensibly due to the formation of Sox4-DNA complexes (Fig 7G). Addition of ATP to the reaction relieved the retardation, suggesting that PKA phosphorylation could impair the DNA binding ability of Sox4. These results implicate Sox4 as being a direct downstream substrate of PKA, phosphorylation of which impairs its ability to act as a transcription factor. Given the role that Sox4 plays in EMT and promoting tumor metastasis, it is likely to be activated during the progression of tumorigenesis as a result of PRKAR1A locus amplifications that result in a reduction in PKA catalytic subunit activity.
Discussion
This study reveals a role for PKA as a differentiation-promoting pathway in some breast tumors. The ability of PKA to promote stem or progenitor cell differentiation has previously been shown in epidermal and hair follicle stem cells (Iglesias-Bartolome et al., 2015), in the adrenal cortex (Drelon et al., 2016), and granule neural progenitors (He et al., 2014). Wnt and Hh pathways have been previously implicated in stem cell self-renewal and maintenance in different tissue types (Briscoe and Therond, 2013; Clevers et al., 2014). Although PKA has previously been shown to inhibit β-catenin (Drelon et al., 2016), our results point to a reduction in nuclear translocation only after 4 hours of PKA activation (Fig S7B), suggesting a secondary mechanism of repression of the Wnt pathway. In line with this assumption, Sox4 has previously been shown to play a role in stabilizing β-catenin (Lee et al., 2011; Sinner et al., 2007), pointing to an alternate mechanism by which the latter could be de-stabilized and inactivated upon PKA activation. The reduction in nuclear localization of β-catenin also correlates with increased cytoplasmic accumulation of Sox4 (Fig S7B).
Previous studies identified a subpopulation in MMTV-PyMT tumors that expresses basal markers and is responsible for collective invasion (Cheung et al., 2013). Our scRNA-Seq results also point to the emergence of a subpopulation of cells, the lumino-basal (LB) fraction, that is correlated with an increase in metastatic burden. We observe that these cells still retain the expression of luminal markers, including Krt8 and CD14, while also expressing basal markers. Our scRNA-Seq analyses reveal that activation of PKA suppresses the acquisition of basal traits that appear to be necessary to generate LB cells, mirroring phenotypes observed in the normal mammary gland. In drawing parallels between normal development and tumorigenesis, Sox4 expression has also been correlated with the process of mammary gland involution (Shibayama et al., 2019). Assessing the activation state of PKA during involution could provide further clues on how this kinase-substrate duo regulate the balance between self-renewal and differentiation. We observed features of papillary differentiation in tumors that arise in two separate models of PKA activation, one from glands that expressed constitutive activation of PKA (Fig. S4C), as well as upon activation of PKA following initiation of tumorigenesis (Fig. 5B). Interestingly, the lack of an intact myoepithelial layer appears to be a feature of malignant papillary carcinomas (Pal et al., 2010), indicating that the resulting papillary features could be a direct effect of a decrease in basal/myoepithelial cells. Moreover, the activation of Gαs-PKA signaling appears to confer papillary features in other tumor-types (Wu et al., 2011), although the overall tumorigenic or tumor suppressive effects appear to be tissue-type dependent.
The presence of recurrent genomic amplifications in PRKAR1A suggests a role for suppression of this pathway in tumorigenesis. Our analyses looked at genomic alterations in each regulatory and catalytic subunit and we decided to pursue PRKAR1A amplifications owing to their presence in greater proportions of Luminal B and basal-like breast cancers (Fig 3B,D). Indeed, the major downstream effectors of the PKA complex are the catalytic subunits, whose enzymatic activity is known to regulate numerous cellular processes (Skalhegg and Tasken, 2000; Tasken and Aandahl, 2004). Our efforts were, thus, focused on PRKACA, for which have uncovered a critical role in the induction of differentiation. This suggests that higher levels of PRKACA could confer better prognosis in breast cancer patients, whereas the negative regulators of the catalytic subunit, encoded by PRKAR1A and other genes, could confer poorer prognosis.
It must also be noted that, while the effects of PKA reported here, and by others (Drelon et al., 2016; He et al., 2014; Iglesias-Bartolome et al., 2015; Klutzny et al., 2018; Nadella et al., 2008; Pattabiraman et al., 2016), may be more akin to those of a tumor suppressor, there are contexts in which PKA signaling acts in a growth-promoting fashion. It is known to play oncogenic roles in pancreatic and small cell lung cancers (Coles et al., 2020; Patra et al., 2018) induces EMT in certain contexts (MacPherson et al., 2010; Shaikh et al., 2012), and promotes cell proliferation through downstream activation of Src and MAPK (Beristain et al., 2015; Stork and Schmitt, 2002). These seemingly inconsistent observations are reflective of highly context-specific roles for PKA in different tissue types. Moreover, shifts in balance of proteins that maintain the tightly regulated network of Gαs-PKA signaling including adenylyl cyclases, phosphodiesterases, A-kinase anchoring proteins (AKAPs) and PRKAR proteins all play a role in dysregulation of this pathway. In the subset of breast tumors that we focus on here, the amplifications and increased expression of PRKAR proteins are likely what triggered the suppression of the catalytic subunit and its downstream effects.
Activation of PKA results in increased chemosensitivity of PyMT tumors to adriamycin, likely a result of a more differentiated histopathology. We have previously observed increased sensitivity to both adriamycin and paclitaxel in-vitro upon PKA-induced mesenchymal-epithelial transition (Pattabiraman et al., 2016). Here we extend these observations to show that in-vitro effects of MET induction manifest in vivo in the form of enhanced differentiation. Given that the effects are observed with multiple chemotherapeutic drugs acting via different mechanisms, it is likely that the increased sensitivity is a result of differentiation rather than any specific interactions between adriamycin and PKA. Indeed, along these lines, others have previously shown that inducing a more differentiated cell state renders tumor cells more sensitive to a range of chemotherapeutic drugs in breast cancers (Bhola et al., 2013; Gupta et al., 2009; Qu et al., 2014).
Basal cells in mammary development, while contributing to the regenerative capacity of the gland during pre-pubertal development, also have other heterotypic signaling functions. Specifically, basal-to-luminal cell signaling has been shown to be critical for luminal cell maturation (Forster et al., 2014). Upon activation of PKA, which impairs basal cell self-renewal, we observe no overt effects on the proportions (Fig 1B), the histological phenotype (Fig 2D), or maturation (Fig 6E) of luminal cells, suggesting that basal-to-luminal cell heterotypic signaling are still active. This is conceivable since the basal cells that persist following PKA activation, while defective in self-renewal, may still carry out other normal functions. Alternatively, it is plausible that PKA signaling may also diminish the ability of basal cells to signal to luminal cells, ultimately preventing their maturation. Currently available Cre models are, however, insufficient to distinguish these two scenarios as the percentage of recombination invariably leaves a portion of cells unlabeled or in the wild-type state. Along these lines, previous studies have highlighted the polyclonal nature of metastasis in the PyMT model (Cheung et al., 2016), suggesting a role for basal-luminal heterotypic interactions in the establishment of metastatic colonies. We observe significantly reduced metastases in PrkacaCaR/fl PyMT tumors (Fig 4D), which we interpret as a result of reduction in the lumino-basal subpopulation of the tumor. The reduction in metastasis could, however, also occur as a consequence of impaired basal-luminal cell interactions.
This study provides rationale for the targeting of PKA signaling in at least certain breast tumor types, although there are a number of challenges associated with the translation of such a strategy. Molecular therapeutics have traditionally relied on inhibition of enzymatic activity e.g., the use of kinase inhibitors, whereas our results point to a requirement for activation, rather than inhibition of PKA signaling, which would present a unique set of challenges. Additionally, since PKA is a ubiquitous kinase that carries out numerous context-specific roles in different tissue types, it is likely that a strategy to target it will be met with issues of toxicity and untoward side-effects. On the flip side, identifying key downstream molecules could help alleviate some of these concerns i.e., targeting Sox4 could be a more attractive avenue, especially since its expression pattern appears to be confined only to certain tissue types and lacking in others such as the liver, pancreas and small intestine (Dy et al., 2008). Our studies show that PKA directly phosphorylates Sox4 at S56 and S103, both residues falling within its HMG box that is critical for its DNA binding activity (Dy et al., 2008). Strategies that interfere with Sox4 DNA-binding or transactivation might have clinical utility in the progression of Luminal B and basal-like breast cancers that harbor PRKAR amplifications.
Our study provides insights into how the induction of differentiation could help derive therapy responsiveness in solid tumors, while curtailing their aggressive traits and metastatic spread. The ability of constitutively active PKA signaling to deplete self-renewal of basal cells in multiple tissue types suggests that careful calibration of its activity is essential for the maintenance of the stem cell state and the regulation of cellular differentiation. While targeted and immunotherapies have shown immense success in a number of cancer types, the inherent cellular heterogeneity of some breast cancers are a major barrier to the identification of targetable recurrent genomic alterations. For such cancer types, a more feasible approach could be the reversal of a trait that is common to all cancers: the evasion of terminal differentiation.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Diwakar Pattabiraman (raman@dartmouth.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data Availability
scRNA-Seq data generated in this study have been deposited in GEO and are accessible under the accession number GSE158257.
Experimental Model and Subject Details
Plasmids
pLentiCRISPRv2 was a gift from the Feng Zhang via Addgene (#52961, http://n2t.net/addgene:52961; RRID:Addgene_52961). sgRNAs for Sox4 were cloned into the construct using protocols from the Zhang lab. The Sox4 sgRNA sequences that were used were:
sgSox4_2: CAACAACGCGGAGAACACTG
sgSox4_4: CGACAAGATTCCGTTCATCC
sgLacZ: TGCGAATACGCCCACGCGAT
Cells
PyMT tumor-derived PB3 cells were obtained from the Robert A. Weinberg lab (Whitehead Institute for Biomedical Research) and were cultured in DMEM/F12 medium containing 5% adult bovine serum with 1× penicillin–streptomycin and 1× nonessential amino acids. For the Sox4 knockout studies, cells were cultured for 14 days in the presence of puromycin for complete CRISPR-Cas9 mediated loss of Sox4, following which they were used for subsequent studies including immunoblotting, immunofluorescence and transplantation.
Animal studies
Research involving animals complied with protocols approved by the Dartmouth College Committee on Animal Care. All animals were housed in specific pathogen-free (SPF) barrier facility in ventilated microisolator cages containing 5 mice each. The PKA-CαR (Niswender et al., 2005) mice were a gift from Stanley McKnight (UWashington). Tg(MMTV-cre)4Mam/J line D (Wagner et al., 2001) was obtained from JAX (Stock No. 003553). The GαsR201C mice (Iglesias-Bartolome et al., 2015) were a gift from J. Silvio Gutkind. The B6.Cg-Gt(ROSA)26Sorrtm1(rtTA,EGFP)Nagy/J mice (Belteki et al., 2005) (Stock No. 005670), mTmG reporter mice - B6.129(Cg)-Gt(ROSA)26Sorrtm4(ACTB-tdTomato,-EGFP)Luo/J mice (Muzumdar et al., 2007) (Stock No. 007676), Tg(KRT5-cre/ERT)Blh/J mice (Stock No. 029155)(Van Keymeulen et al., 2011), Tg(KRT8-cre/ERT)Blpn/J mice (Stock No. 017947)(Van Keymeulen et al., 2011), Tg(MMTV-PyVT)634Mul/LellJ mice (Guy et al., 1992) on a C57Bl/6J background (Stock No. 022974) and Tg(C3-1-TAg)cJeg/JegJ mice (Maroulakou et al., 1994) (Stock No. 013591) were obtained from The Jackson Laboratory. For transplantation studies, cells suspended in DMEM containing 30% Matrigel (GFR)/PBS mix (BD Biosciences; 356230) were injected into the inguinal mammary gland fat pads of age-matched female NOD/SCID or C57Bl6/J mice (Jackson Laboratory). Mice were euthanized after 10 weeks or when tumors reached a diameter of 2 cm. For chemotherapy studies, mice were administered adriamycin intraperitoneally at 2mg/kg twice a week for up to 40 days or until tumors reach the endpoint of 2cm in diameter. For studies of mammary development, female mice at 3 weeks, 6 weeks and 8 weeks were used to study the initiation of postnatal development, puberty and adulthood, respectively. For tumor studies, 9 week-old female mice were used for the hyperplasia stage and 18-23 week-old mice were used for the tumor stage. For the study of metastasis and therapy resistance, female tumor-bearing mice that were 23-40 weeks-old were used.
Method Details
Mammary Gland Preparation
Mouse mammary fat pads (no. 3 and 4) were harvested and dissociated as per previously published protocols (Prater et al., 2013) with minor modifications. Briefly, glands were digested with Collagenase A and Hyaluronidase for 2-3 hours at 37°C in a rotator followed by mild trituration with a 10ml pipette. Following red cell lysis, cells were treated with trypsin, and subsequently with dispase to aid dissociation. Cells were then stained with antibodies (see table below) to analyze and sort luminal and basal populations
Carmine Alum Staining
Harvested mammary glands are spread out on a slide and allowed to dry for 1 hour at RT. Slides are fixed overnight in Carnoy’s fixative (6 parts 100% EtOH, 3 parts CHCL3, 1 part glacial acetic acid) at RT. On day 2 slides were rehydrated by incubating in 70% ethanol twice for 10 min each, twice in 50% ethanol for 10 min each, twice in 30% ethanol for 10 minutes each, twice in 10% ethanol for 10 minutes each and in distilled water for 5 min. Slides are then stained for 1-2 days in Carmine Alum (Stem Cell Technologies; Catalog No. 07070) at RT. Slides are dehydrated by incubating in 70% ethanol twice for 10 min each, twice in 95% ethanol for 10 min each and twice in 100% ethanol for 10 minutes each. Slides were then cleared in Xylene for 3-4 days, with the solution being changed every day after which they are rehydrated and dehydrated again as above before mounting using Permount (Fisher Chemical; SP15). Slides were scanned on a Perkin Elmer© Vectra 3 slide scanner and analyzed on Phenochart©.
Organoid Assays & Transplantation
Dissociated mammary epithelial cells were dissociated into single cells and cultured with advanced DMEM (Gibco; 12491015) containing 5% Matrigel, 5% heat-inactivated FBS, 10 ng/ml EGF, 20 ng/ml bFGF, 4 mg/ml heparin, and 5 mM Y-27632. Cells were seeded at 1,000 per well in 96-well ultralow attachment plates (Corning; 29443-034). The number of organoids was counted 7–14 days after seeding.
1000 or 500 cells from dissociated organoids were resuspended in 10 μl 75% DMEM/ 25% matrigel were injected into the number 4 glands of 4w old NSG female mice that had been cleared of endogenous epithelium. Recipient glands were harvested at 12wk, dissected, fixed and stained with carmine alum. An outgrowth was defined as an epithelial structure comprising ducts and branching and the percentage of outgrowth was quantified by proportion of ducts and branches in relation to the fat pad.
Tumor Dissociation
Tumors from MMTV-PyMT mice and C3(1)-Tag mice were resected and minced using a razor blade in DMEM containing 2 mg/mL collagenase and 100 U/mL hyaluronidase (Fischer Sci) in a rotator at 37°C for 30 minutes. Following incubation, tumors were minced further and re-incubated for another 30 minutes. Dissociated cells were washed twice in PBS and filtered through a 70- and 40-μm cell strainer to obtain single-cell suspensions.
Immunofluorescence (cultured cells)
Cells were cultured on dishes containing coverslips for 2 to 3 days, after which coverslips were washed in cold PBS, fixed in 5% Neutral Buffered Formalin for 10 min at 4°C and permeabilized in 0.2% TritonX in PBS for 2 min. Cells were then washed in PBS, blocked for 1 hour at room temperature in PBS containing 3% normal horse serum (Vector Labs, USA; S-2000). Fixed cells were then incubated with the primary antibody in PBS containing 1% bovine serum albumen (BSA) solution overnight at 4°C. Cells were washed in PBS three times, and secondary antibody was added in PBS containing 1%BSA solution for 1 to 2 hours at room temperature in the dark. Cells were washed three times in PBS and were incubated for 2min in DAPI solution, after which they were washed in PBS and mounted with a drop of Prolong Diamond antifade reagent (Thermo; P36962) and placed on coverslips. Slides were viewed on a Zeiss© LSM800 microscope and analyzed using the Zen© Digital Imaging software.
Immunofluorescence (FFPE tissue sections)
Slides were rehydrated by incubating in Histoclear solution twice for 5 min each, followed by incubation in 100% ethanol twice for 5 min each, in 95% ethanol twice for 5min each, 70% ethanol twice for 5 min each, once in 35% ethanol for 5 min, and in water for 5 min. Pressure cooker– mediated heat-induced epitope retrieval was carried out in 250 ml of unmasking buffer containing sodium citrate at pH 6. After retrieval, slides were blocked for 30 min in PBS containing 3% normal horse serum after which they were incubated with primary antibody in blocking solution overnight at 4°C. Slides were washed twice with PBS and incubated with secondary antibody at room temperature for 1 hour in the dark. After two PBS washes, 20 ml of mounting medium was added, then slide contents were topped with coverslips, and stored in the dark for 24 hours before imaging on a Zeiss LSM800 microscope and analyzed using the Zen Digital Imaging software.
| Antibody | Vendor | Catalog No. | Application | Dilution |
|---|---|---|---|---|
| CD45 PECy7 | BioLegend | 103114 | FACS | 1:100 |
| CD31 PECy7 | BioLegend | 102418 | FACS | 1:100 |
| EpCAM APC | BioLegend | 118214 | FACS | 1:100 |
| CD49f PE | BioLegend | 313612 | FACS | 1:100 |
| Vimentin | Cell Signaling | 5741 | IF/IHC | 1:100 |
| E-cadherin | BD | 610182 | IF/IHC, WB | 1:200 |
| Keratin 8 | Thermo Fisher | PA5-29607 | IF/IHC | 1:400 |
| Keratin 14 | Thermo Fisher | MA5-11599 | IF/IHC | 1:1000 |
| Sox4 | Thermo Fisher | MA5-31424 | WB | 1:1000 |
| p-PKA substrate | Cell Signaling | 9624 | IHC | 1:200 |
Analysis of publicly available datasets
The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas, 2012) and METABRIC (Curtis et al., 2012; Pereira et al., 2016) breast cancer gene expression, copy number, clinical and survival data, were accessed from the cBioPortal database (www.cbioportal.org). The Swedish Cancerome gene expression and clinical data were publicly available in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/) under accession GSE96058 (Saal et al., 2015). All statistical analyses were performed in the R statistical computing environment (v3.5.1). Specifically, the correlation between mRNA expression of two genes were assessed by ordinary least-square regression. The univariate Kaplan-Meier analyses and related data visualizations were implemented in R packages survival (v.2.43.3) and survminer (v.0.4.3)(Therneau and Grambsch, 2000).
Inferring copy number alterations
Copy number alteration profiles were inferred relative to normal breast samples (n=5, Oltra et al., 2018). MethylationEPIC array intensities were used to estimate segmented copy number alterations, defined as the base 2 logarithmic ratio of tumor-to-control copy number (log2R) (Feber et al., 2014). The frequency of copy number gains or losses was determined using a log2R threshold of +0.1 and −0.1, respectively.
Single-cell RNA-seq library construction and sequencing
Single Cell Capture and Library Preparation: Immediately following dissociation, single cell suspensions were placed on ice and counted on a Luna automated cell counter. Cell concentrations from each sample were be normalized to 1000 cells/ul and loaded onto a Chromium Single Cell A Chip (10x Genomics Inc.) targeting a capture rate of 5,000 cells per sample. Single cell RNA-seq libraries were be prepared using the Chromium Single Cell 3’ v2 kit (10x Genomics) following the manufacturer’s protocol. Libraries were quantified by qubit and peak size determined on a fragment analyzer instrument. All libraries were pooled and sequenced on an Illumina NextSeq500 High Output 26bp x 98bp run to generate an average of 50,000 reads/cell. Data Analysis: Raw sequencing were processed using the 10x Genomics Cell Ranger to generate quality metrics and primary data visualizations as well as gene expression matrices for downstream analysis in R using Seurat and other open-source packages.
Single-cell RNA-seq data processing
To investigate the transcriptional evolution of tumors at single cell level, we performed of matched hyperplasia and tumor samples was performed on the 10X genomics platform, and generated data at an average of ~78M reads per sample with cell numbers ranging from ~2600 to 5000. Single-cell transcriptome sequencing raw reads were quality filtered using Fqtrim (v0.9.7) tool [10.5281/zenodo.1185412]. Reads were trimmed and filtered for low quality bases, poly-A/T tails and N bases while retaining the paired-end integrity of the reads. Read1 of the pair containing barcodes was not considered for trimming but allowed to be filtered to maintain paired-end integrity. Sequencing and PCR errors in cell barcodes can convolute the process of differentiating reads per cell barcode; hence we used UMItools (v0.5.4) to distinguish the reads per cell (barcode) accommodating for technical errors(Smith et al., 2017). A knee density method-based approach is used in UMItools to estimate the number of acceptable cell barcodes and then reads were assigned for each cell.
Clean and barcode classified reads were aligned against Mouse genome reference (GRCm38) using STAR aligner (v2.5.3a)(Dobin et al., 2013) and output was restricted to uniquely aligned reads. De-duplication of transcripts with the same UMIs arising from PCR amplification were removed. Reads were assigned with position based annotation of genomic features using featureCounts module(Liao et al., 2014) in the subread package (v1.6.0). Taking advantage of the UMI information, read counts were extrapolated to quantify molecular level count for each transcript using directional-adjacency method-based count module in UMItools. Reads were grouped per cell (based on barcode) and then a gene expression matrix was generated with RNA molecule count of genes in rows for each cell in columns represented in GxC matrix format (where G is gene in rows and C is cells in columns).
Cell-type classification and Pseudotime analysis
Gene expression matrix was filtered for cells with <500 genes expressed and <500 total UMI counts genes and >0.25 percentage of reads aligned to Mitochondrial genome using Monocle2 (v2.6.4) R package(Qiu et al., 2017a; Qiu et al., 2017b; Trapnell et al., 2014). Further, outliers of total mRNAs count for each cell was removed from the downstream analysis. Expression counts were normalized using negative binomial distribution of library sizes. Genes with mean expression value of 0.1 across cells were used for PCA based dimensionality reduction and then clustered using an unsupervised densityPeaks algorithm in Monocle2 and projected using t-SNE method.
To classify the population into various cell-types, we investigated the expression of curated and well-established cell-type specific marker genes’ expression in our data and selected a specific list of markers that are expressed and/or not expressed for each cell-type. Following are the cell-type specific markers used for the classification in this study,
| Cell types | Higher expression |
|---|---|
| Basal cells | Krt14, Krt5, Snai2 |
| Luminal Mature cells | Pgr, Prlr, Foxa1, Gpx3, Esr1 |
| Luminal Progenitor cells | CD14, Kit, Lalba |
| Lumino-basal | Krt14, Krt8, CD14 |
| EMT-like | Trim29, Vim, Cldn6, Sox4 |
We identified and classified cell-types using Monocle2’s classifyCells function. First, each cell was annotated into a cell-type based on the presence and absence of positive and negative cell-type marker genes respectively. Then cells were re-clustered using added cell-type information to bring cells with similar gene-expression pattern and cell-type annotation into closer clusters, and to segregate un-annotated cells. Ambiguous and Un-annotated clusters were investigated manually for other known marker genes using find_cluster_markers function in Monocle3 alpha (v2.99.3) and re-annotated manually into different sub-types based on GO enrichment analysis of selected marker genes and expression of other known cell-type specific marker genes.
Pseudotime analysis attempts to reconstruct the transcriptional transitions of cells during the process of tumorigenesis. By predicting the transcriptional state of each cell, cells are ordered into an estimated pseudotime indicating their cell-state and transition across a time-series. Transition of gene expression kinetics was performed by integrative Pseudotime analysis using Monocle2. Given that the hyperplasia and tumor stage cells were extracted from the same mice, the hyperplasia and tumor stage samples of same mice were merged using Seurat R package (v2.3.0)(Butler et al., 2018) while retaining the cell-type and sample annotation. Merged data was processed again for dimensionality reduction using DDRTree algorithm and then pseudotime trajectories were calculated. Plots were generated using different color codes based on cell-type, sample and pseudotime to illustrate the gene expression kinetics transition across stages and samples. Data plots were generated using Monocle2 and Monocle3 alpha packages.
In-vitro kinase assay
Replicate samples from in vitro kinase assays were prepared for LC-MS/MS analysis by SP3 isolation and on-bead digestion exactly as described (Hughes et al., 2014; Hughes et al., 2019) and trypsin digestion overnight in 50mM ammonium bicarbonate buffer. In brief, kinase assay samples were diluted to 100ul final volume with 1% SDS/50mM Tris pH 8.1, followed by addition of DTT to 2mM and reduction at 45C for 15 minutes. Samples were allowed to cool to room temperature and alkylated with 7mM iodoacetamide (final concentration) for 1h at room temperature in the dark, followed by quenching with 2mM DTT (final concentration). SP3 was performed by addition of 1ul SP3 magnetic beads and vortexing thoroughly to mix, precipitation with 100ul 200 proof ethanol, shaking in a thermomixer at 24C for 5 minutes at 1000 RPM, and isolation of the beads in a magnetic tube rack. The supernatant was removed to waste, and the beads were washed 3 times with 80% ethanol/water solution with vortexing in between. After the final wash, the supernatant was removed, and the beads were additioned with 50ul 50mM ammonium bicarbonate solution containing 250ng sequencing grade (Promega) trypsin and digestion overnight at 28C in a thermomixer with shaking at 2000 RPM. Finally, the resulting peptides were separated from beads by magnetic tube rack, desalted over homemade STAGE-TIPs and loaded on to the LC-MS/MS system. Digested peptides were separated from paramagnetic beads by magnetic isolation and desalted on an OASIS uHLB (2mg) desalting plate (Waters-Millipore). The resulting desalted peptides were dried by vacuum centrifugation, resuspended in 1% formic acid/5% methanol/94% ultrapure water, transferred to limited volume inserts and injected on to a Proxeon EasyLC-1200 UPLC system couple to an Orbitrap Fusion Lumos mass spectrometer. Samples were analyzed by data-dependent analysis using a linear gradient from 0 - 39% B (Buffer A: 0.125% formic acid, 3% acetonitrile; Buffer B: 0.125% formic acid, 80% acetonitrile) over 45 minutes on an in-house manufactured microcapillary reverse-phase column (100um ID, 34cm long). Parent ion scans (R = 120,000, AGC target = 200,000 ions, charge states 2 - 4, dynamic exclusion +/− 10ppm for 20s) were collected for precursors from 350 to 1500 m/z, followed by child ion scans (R = 15,000, AGC target = 25,000 ions, quadrupole isolation = 1.0 m/z, HCD collision = 28%, maximum injection time = 50ms), all as centroided Orbitrap scans, for a 1.5s duty cycle (top speed mode). The resulting data were then data-searched using the COMET algorithm (Eng et al., 2013) against the human proteome (UniProt) using dynamic modifications for oxidation on methionine and phosphorylation on serine, threonine and tyrosine, and filtered to a 1% FDR using the target-decoy strategy (Elias and Gygi, 2010). These peptide identifications were then quantified in a "label-free" mode using in-house software across all six replicates. Quantified phosphopeptides were then collapsed into single identifiers as a function of modified residue ("ModSites") as done previously (Kettenbach et al., 2011). Quantification values were then averaged for each ModSite across the three replicates for control and + ATP samples; these averaged quantification values were then compared for significance using Student's T-test (Excel). ModSites that were identified in at least two of three replicates and exhibited a p-value < 0.05 were included in Table S1.
Preparation of oligonucleotides duplexes
Oligonucleotides end-labeled with or without IRDye 800 (IDT) infrared dye were annealed by combining the oligonucleotides at a 1:1 molar ratio with complementary oligonucleotides for probes or competitors, respectively in 25 mM HEPES, 1 mM MgCl2 and 50 mM NaCl. The duplex mixtures were heated at 95°C for 5 minutes and then at 70°C for 10 minutes, and samples ramped down to 25°C for 45 minutes to dissociate any intra-strand duplexes.
Electrophoresis Mobility Shift Assay
In vitro-kinase assays were performed with 500ng recombinant human cAMP Protein Kinase Catalytic subunit (Abcam, ab56268) and 1000ng recombinant Human SOX4 (Novus Biologicals, H00006659-Q01) proteins with and without ATP in kinase assay buffer (20mM HEPES pH 7.7, 5mM MgCl2, 0.1mM EGTA, 0.2mM DTT, 0.5mM ATP) for 6 hours. Labeled probes (50 fmol) were incubated with the in vitro kinase assay reaction in 20 mL of binding buffer (10 mM Tris-HCl, pH 7.5, 50 mM KCl, 3.5 mM DTT, 0.25% Tween 20, 5% glycerol, 5 mM MgCl2, and 50 mM EGTA) and 1 mg poly (dI-dC), in the presence or absence of non-labeled competitor (50 pmol) for 30 min at room temperature and then run on to 6% polyacrylamide gels (Thermo Fisher Scientific, EC6265BOX) in 0.5X TBE buffer at 4°C. The infrared dye signal was detected and digitized using an Odyssey CLx imaging system (LI-COR Biosciences, Lincoln, NE, USA).
Elf5_F_sense_1
CAAGCTCCTCATCTATCAGGGAACAAAGGCTAAACATGGCTGTCCACCAT
Elf5_R_antisense_1
GTTCGAGGAGTAGATAGTCCCTTGTTTCCGATTTGTACCGACAGGTGGTA
Elf5_F_sense_Mutant
CAAGCTCCTCATCTATCAGGGATCAGAGGCTAAACATGGCTGTCCACCAT
Elf5_R_sense_Mutant
GTTCGAGGAGTAGATAGTCCCTAGTCTCCGATTTGTACCGACAGGTGGTA
Quantification and Statistical Analysis
Data are presented as means ± SD. A Student’s t test (two-tailed) was used to compare two groups (P < 0.05 was considered significant) unless otherwise indicated. Numbers of animals (biological replicates) are indicated in figure legends.
Supplementary Material
Table S1: Identification of PKA phosphorylation sites on Sox4 by Mass Spectrometry. Related to Figure 7. LC-MS analysis of PKA-Sox4 in-vitro kinase assay. Table outlines raw data obtained from LC-MS showing peptides on Sox4 that are modified upon addition of ATP. Fold change is shown under the “Ratio” tab and significance was calculated by Student T-test.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| CD45 PECy7 | BioLegend | Cat# 103114 | ||
| CD31 PECy7 | BioLegend | Cat# 102418 | ||
| EpCAM APC | BioLegend | Cat# 118214 | ||
| CD49f PE | BioLegend | Cat# 313612 | ||
| Vimentin | Cell Signaling Technology | Cat# 5741 | ||
| E-cadherin | BD Biosciences | Cat# 610182 | ||
| Keratin 8 | Thermo Fisher | Cat# PA5-29607 | ||
| Keratin 14 | Thermo Fisher | Cat# MA5-11599 | ||
| Sox4 | Thermo Fisher | Cat# MA5-31424 | ||
| p-PKA Substrate | Cell Signaling Technology | Cat# 9624 | ||
| Chemicals, Peptides, and Recombinant Proteins | ||||
| Prolong Diamond antifade reagent | Thermofisher Scientific | Cat# P36962 | ||
| Normal Horse Serum | Vector Labs | Cat# S-2000 | ||
| Collagenase A | Sigma | Cat# 10103586001 | ||
| Hyaluronidase | Fisher Scientific | Cat# ICN10074091 | ||
| Carmine Alum | Stem Cell Technologies | Cat# 07070 | ||
| Permount | Fisher Chemicals | Cat# SP15 | ||
| Recombinant PRKACA | Abcam | Cat# ab56268 | ||
| Recombinant SOX4 | Novus Biologicals | Cat# H00006659 | ||
| Deposited Data | ||||
| scRNA-Seq data of cells from PrkacaCαR/fl PyMT and Prkacafl/fl PyMT mice at the hyperplasia and tumor stages. | GEO | GSE158257 | ||
| Experimental Models: Cell Lines | ||||
| PB3 cell line | Weinberg lab (WIBR) | Dongre et al. 2017 | ||
| Experimental Models: Organisms/Strains | ||||
| PKA-CαR mice | Stanley McKnight (UW) | Niswender et al. 2005 | ||
| GαsR201C mice | J. Silvio Gutkind (UCSD) | Iglesias-Bartolome et al. 2015 | ||
| Tg(MMTV-cre)4Mam/J line D | The Jackson Laboratory | Stock No. 003553 | ||
| B6.Cg-Gt(ROSA)26Sortm1(rtTA,EGFP)Nagy/J | The Jackson Laboratory | Stock No. 005670 | ||
| B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | Stock No. 007676 | ||
| Tg(KRT5-cre/ERT)Blh/J | The Jackson Laboratory | Stock No. 029155 | ||
| Tg(KRT8-cre/ERT)Blpn/J | The Jackson Laboratory | Stock No. 017947 | ||
| Tg(MMTV-PyVT)634Mul/LellJ | The Jackson Laboratory | Stock No. 022974 | ||
| Tg(C3-1-TAg)cJeg/JegJ | The Jackson Laboratory | Stock No. 013591 | ||
| Oligonucleotides | ||||
| CAACAACGCGGAGAACACTG | This paper | sgSox4_2 | ||
| CGACAAGATTCCGTTCATCC | This paper | sgSox4_4 | ||
| TGCGAATACGCCCACGCGAT | This paper | sgLacZ | ||
| CAAGCTCCTCATCTATCAGGGAACAAAGGCTAAACATGGCTGTCCACCAT | This paper | Elf5_F_sense_1 | ||
| GTTCGAGGAGTAGATAGTCCCTTGTTTCCGATTTGTACCGACAGGTGGTA | This paper | Elf5_R_antisense_1 | ||
| CAAGCTCCTCATCTATCAGGGATCAGAGGCTAAACATGGCTGTCCACCAT | This paper | Elf5_F_sense_Mutant | ||
| GTTCGAGGAGTAGATAGTCCCTAGTCTCCGATTTGTACCGACAGGTGGTA | This paper | Elf5_R_sense_Mutant | ||
| Software and Algorithms | ||||
| Fqtrim (v0.9.7) | https://ccb.jhu.edu/software/fqtrim/ | 10.5281/zenodo.1185412 | ||
| UMItools (v0.5.4) | https://github.com/CGATOxford/UMI-tools | Smith et al., 2017 | ||
| STAR aligner (v2.5.3a) | https://github.com/alexdobin/STAR | Dobin et al., 2013 | ||
| subread package (v1.6.0), featureCounts module | http://subread.sourceforge.net | Liao et al., 2014 | ||
| Monocle2 (v2.6.4) R package | http://cole-trapnell-lab.github.io/monocle-release/ | Qiu et al., 2017a | ||
Highlights.
Activation of PKA attenuates self-renewal and expansion of the mammary basal compartment
This activation induces tumor differentiation, curtailing metastasis and chemoresistance
PKA activation attenuates the evolution of multiple aggressive tumor subpopulations
PKA directly phosphorylates and inactivates Sox4, contributing to tumor differentiation
Acknowledgements
We thank Dr. Stanley McKnight for the PKA-CαR mice and Dr. Silvio Gutkind for the GαsR201C mice; the genome technology core at the Whitehead Institute and the Genomics and Molecular Biology Shared Resource at the Norris Cotton Center for optimization of protocols for single cell RNA sequencing; Dartlab (Flow cytometry), Microscopy, Pathology and Mouse Modeling Shared Resources at the Norris Cotton Cancer Center. We thank Ferenc Reinhardt, Joana Liu Donaher, Jennifer Fields and Rebecca O’Meara for technical assistance and Drs. Yashi Ahmed and Alan Eastman for critical reading of the manuscript. Funding and resources for the shared resources were supported in part by a core grant (5P30CA023108-40; Norris Cotton Cancer Center). This work was supported by funding from the NIH R01GM122846 (to S.A. Gerber) R01CA216265 (to B.C. Christensen) and 5R00CA201574-05 (to D.R. Pattabiraman).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest
The lead author has a patent titled “Methods and Compositions for Targeting Cancer Stem Cells.” U.S. Patent No. 10,398,672 (issued September 3, 2019) for the activation of PKA as a means of targeting breast cancer stem cells.
References
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, and Nagy A (2005). Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res 33, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beristain AG, Molyneux SD, Joshi PA, Pomroy NC, Di Grappa MA, Chang MC, Kirschner LS, Prive GG, Pujana MA, and Khokha R (2015). PKA signaling drives mammary tumorigenesis through Src. Oncogene 34, 1160–1173. [DOI] [PubMed] [Google Scholar]
- Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, and Arteaga CL (2013). TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest 123, 1348–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, and Therond PP (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14, 416–429. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Gabrielson E, Werb Z, and Ewald AJ (2013). Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155, 1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS, et al. (2016). Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A 113, E854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Loh KM, and Nusse R (2014). Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346, 1248012. [DOI] [PubMed] [Google Scholar]
- Coles GL, Cristea S, Webber JT, Levin RS, Moss SM, He A, Sangodkar J, Hwang YC, Arand J, Drainas AP, et al. (2020). Unbiased Proteomic Profiling Uncovers a Targetable GNAS/PKA/PP2A Axis in Small Cell Lung Cancer Stem Cells. Cancer Cell 38, 129–143 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, and Weinberg RA (2017). Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 77, 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravis C, Spike BT, Harrell JC, Johns C, Trejo CL, Southard-Smith EM, Perou CM, and Wahl GM (2015). Sox10 Regulates Stem/Progenitor and Mesenchymal Cell States in Mammary Epithelial Cells. Cell Rep 12, 2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drelon C, Berthon A, Sahut-Barnola I, Mathieu M, Dumontet T, Rodriguez S, Batisse-Lignier M, Tabbal H, Tauveron I, Lefrancois-Martinez AM, et al. (2016). PKA inhibits WNT signalling in adrenal cortex zonation and prevents malignant tumour development. Nat Commun 7, 12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy P, Penzo-Mendez A, Wang H, Pedraza CE, Macklin WB, and Lefebvre V (2008). The three SoxC proteins--Sox4, Sox11 and Sox12--exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res 36, 3101–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol 604, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, and Hoopmann MR (2013). Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24. [DOI] [PubMed] [Google Scholar]
- Feber A, Guilhamon P, Lechner M, Fenton T, Wilson GA, Thirlwell C, Morris TJ, Flanagan AM, Teschendorff AE, Kelly JD, et al. (2014). Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol 15, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster N, Saladi SV, van Bragt M, Sfondouris ME, Jones FE, Li Z, and Ellisen LW (2014). Basal cell signaling by p63 controls luminal progenitor function and lactation via NRG1. Dev Cell 28, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, and Lander ES (2009). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, and Muller WJ (1992). Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- He X, Zhang L, Chen Y, Remke M, Shih D, Lu F, Wang H, Deng Y, Yu Y, Xia Y, et al. (2014). The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med 20, 1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, and Wang ZY (1988). Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 72, 567–572. [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, and Krijgsveld J (2014). Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CS, Moggridge S, Muller T, Sorensen PH, Morin GB, and Krijgsveld J (2019). Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc 14, 68–85. [DOI] [PubMed] [Google Scholar]
- Iglesias-Bartolome R, Torres D, Marone R, Feng X, Martin D, Simaan M, Chen M, Weinstein LS, Taylor SS, Molinolo AA, et al. (2015). Inactivation of a Galpha(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat Cell Biol 17, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, and Gerber SA (2011). Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 4, rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klutzny S, Anurin A, Nicke B, Regan JL, Lange M, Schulze L, Parczyk K, and Steigemann P (2018). PDE5 inhibition eliminates cancer stem cells via induction of PKA signaling. Cell Death Dis 9, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, Naranjo S, Westcott PMK, Duarte FM, Sankar V, et al. (2020). Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Ahn SG, Yoon JH, and Kim SA (2011). Sox4 stimulates ss-catenin activity through induction of CK2. Oncol Rep 25, 559–565. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, and Pollard JW (2003). Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol 163, 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson MR, Molina P, Souchelnytskyi S, Wernstedt C, Martin-Perez J, Portillo F, and Cano A (2010). Phosphorylation of serine 11 and serine 92 as new positive regulators of human Snail1 function: potential involvement of casein kinase-2 and the cAMP-activated kinase protein kinase A. Mol Biol Cell 21, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Anver M, Garrett L, and Green JE (1994). Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci U S A 91, 11236–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nadella KS, Jones GN, Trimboli A, Stratakis CA, Leone G, and Kirschner LS (2008). Targeted deletion of Prkar1a reveals a role for protein kinase A in mesenchymal-to-epithelial transition. Cancer Res 68, 2671–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Willis BS, Wallen A, Sweet IR, Jetton TL, Thompson BR, Wu C, Lange AJ, and McKnight GS (2005). Cre recombinase-dependent expression of a constitutively active mutant allele of the catalytic subunit of protein kinase A. Genesis 43, 109–119. [DOI] [PubMed] [Google Scholar]
- Oltra SS, Pena-Chilet M, Vidal-Tomas V, Flower K, Martinez MT, Alonso E, Burgues O, Lluch A, Flanagan JM, and Ribas G (2018). Methylation deregulation of miRNA promoters identifies miR124-2 as a survival biomarker in Breast Cancer in very young women. Sci Rep 8, 14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal SK, Lau SK, Kruper L, Nwoye U, Garberoglio C, Gupta RK, Paz B, Vora L, Guzman E, Artinyan A, et al. (2010). Papillary carcinoma of the breast: an overview. Breast Cancer Res Treat 122, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556, 463–468. [DOI] [PubMed] [Google Scholar]
- Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, Grossman EA, Ji F, Sadreyev RI, Liss AS, et al. (2018). Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol 20, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C, Reinhardt F, Tam WL, and Weinberg RA (2016). Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351, aad3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al. (2016). The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7, 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, Gusev A, Korthauer KD, Severson TM, Ha G, et al. (2020). Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prater M, Shehata M, Watson CJ, and Stingl J (2013). Enzymatic dissociation, flow cytometric analysis, and culture of normal mouse mammary tissue. Methods Mol Biol 946, 395–409. [DOI] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, and Trapnell C (2017a). Single-cell mRNA quantification and differential analysis with Census. Nat Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner H, and Trapnell C (2017b). Reversed graph embedding resolves complex single-cell developmental trajectories. bioRxiv, 110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C, Zhang W, Zheng G, Zhang Z, Yin J, and He Z (2014). Metformin reverses multidrug resistance and epithelial-mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Mol Cell Biochem 386, 63–71. [DOI] [PubMed] [Google Scholar]
- Saal LH, Vallon-Christersson J, Hakkinen J, Hegardt C, Grabau D, Winter C, Brueffer C, Tang MH, Reutersward C, Schulz R, et al. (2015). The Sweden Cancerome Analysis Network - Breast (SCAN-B) Initiative: a large-scale multicenter infrastructure towards implementation of breast cancer genomic analyses in the clinical routine. Genome Med 7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, et al. (2014). Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh D, Zhou Q, Chen T, Ibe JC, Raj JU, and Zhou G (2012). cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial-mesenchymal transition, migration, and invasion in lung cancer cells. Cell Signal 24, 2396–2406. [DOI] [PubMed] [Google Scholar]
- Shibayama H, Yamamoto T, Oshima K, Matsuda T, and Nadano D (2019). Transcription Factor Sox4 as a Potential Player in Mammary Gland Involution. DNA Cell Biol 38, 1125–1133. [DOI] [PubMed] [Google Scholar]
- Sinner D, Kordich JJ, Spence JR, Opoka R, Rankin S, Lin SC, Jonatan D, Zorn AM, and Wells JM (2007). Sox17 and Sox4 differentially regulate beta-catenin/T-cell factor activity and proliferation of colon carcinoma cells. Mol Cell Biol 27, 7802–7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalhegg BS, and Tasken K (2000). Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front Biosci 5, D678–693. [DOI] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork PJ, and Schmitt JM (2002). Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12, 258–266. [DOI] [PubMed] [Google Scholar]
- Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, Ye X, Chiu C, Pham T, Hongo JA, et al. (2016). Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 529, 97–100. [DOI] [PubMed] [Google Scholar]
- Tasken K, and Aandahl EM (2004). Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev 84, 137–167. [DOI] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling survival data : extending the Cox model (New York: Springer; ). [Google Scholar]
- Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schubeler D, van Nimwegen E, and Christofori G (2013). Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 23, 768–783. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Oosterwegel M, van Norren K, and Clevers H (1993). Sox-4, an Sry-like HMG box protein, is a transcriptional activator in lymphocytes. EMBO J 12, 3847–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, Sharma N, Dekoninck S, and Blanpain C (2011). Distinct stem cells contribute to mammary gland development and maintenance. Nature 479, 189–193. [DOI] [PubMed] [Google Scholar]
- Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, and Hennighausen L (2001). Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res 10, 545–553. [DOI] [PubMed] [Google Scholar]
- Warrell RP Jr., de The H, Wang ZY, and Degos L (1993). Acute promyelocytic leukemia. N Engl J Med 329, 177–189. [DOI] [PubMed] [Google Scholar]
- Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, et al. (2011). Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 3, 92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, and Weinberg RA (2015). Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525, 256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Identification of PKA phosphorylation sites on Sox4 by Mass Spectrometry. Related to Figure 7. LC-MS analysis of PKA-Sox4 in-vitro kinase assay. Table outlines raw data obtained from LC-MS showing peptides on Sox4 that are modified upon addition of ATP. Fold change is shown under the “Ratio” tab and significance was calculated by Student T-test.
Data Availability Statement
scRNA-Seq data generated in this study have been deposited in GEO and are accessible under the accession number GSE158257.