

Abstract
Astrocytes are key homeostatic regulators in the central nervous system and play important roles in physiology. After brain damage caused by e.g., status epilepticus, traumatic brain injury, or stroke, astrocytes may adopt a reactive phenotype. This process of reactive astrogliosis is important to restore brain homeostasis. However, persistent reactive astrogliosis can be detrimental for the brain and contributes to the development of epilepsy. In this review, we will focus on physiological functions of astrocytes in the normal brain as well as pathophysiological functions in the epileptogenic brain, with a focus on acquired epilepsy. We will discuss the role of astrocyte-related processes in epileptogenesis, including reactive astrogliosis, disturbances in energy supply and metabolism, gliotransmission, and extracellular ion concentrations, as well as blood-brain barrier dysfunction and dysregulation of blood flow. Since dysfunction of astrocytes can contribute to epilepsy, we will also discuss their role as potential targets for new therapeutic strategies.
Keywords: glia, astrogliosis, seizures, epilepsy, treatment, gliotransmission, blood-brain barrier, neuroinflammation
Introduction
Epilepsy is a common neurological disease that is estimated to affect roughly 1–2% of the population (1). Despite the fact that quite some anti-epileptic drugs (AEDs) have been developed in the last decades, a large number of patients still fail to respond to these AEDs. This is associated with increased morbidity and mortality and since these patients need life-long care this is also an economic burden for society. Furthermore, patients feel stigmatized and report a reduced quality of life (2). Therefore, it is of crucial importance to find novel drug targets in order to develop novel therapeutic strategies. Moreover, disease-modifying therapies are currently not available and require a better understanding of the underlying disease processes. In the past two decades, astrocytes have been increasingly acknowledged as key players in the etiology and pathogenesis of epilepsy. Therefore, astrocytes should be considered as promising targets for new therapeutic strategies.
The human brain is comprised of ~100 billion cells, classically divided into neurons and glial cells, although new types of brain cells are still being discovered up to date (3, 4). Glia cells in the central nervous system are typically classified into four cell types: (1) astrocytes, (2) microglia, (3) oligodendrocytes, and (4) their progenitors, neuron-glial antigen 2(NG2)-glia (5). For almost a century it was believed that glial cells outnumbered neurons 10:1 (6). However, it has been shown that the actual ratio of glial cells compared to neurons is closer to 1:1 and may in fact be lower than 1 (6, 7). Nevertheless, the remarkably conserved numerical relationship between glia and neurons over 90 million years of evolution supports the notion that glial cells are crucial for normal brain functioning (8). These numbers suggest a far more prominent role for astrocytes in the brain than long considered. In addition to its vast number, it is estimated that a single astrocyte touches and interacts with up to 2 million synapses with its processes (9).
Although all four glial cell types play a pivotal role in normal brain function, in this review we will focus on astrocytes which are key homeostatic regulators in the central nervous system and play important roles in the pathophysiology of epilepsy (10).
For many years, astrocytes were regarded as “glue” that bound neuronal elements together, providing mere structural support for the brain. In fact, astrocytes are playing a pivotal role in brain homeostasis. From recent transcriptome studies it became clear that different subtypes of astrocytes exist that are not only anatomically and spatiotemporally restricted, but also show varying degrees of heterogeneity of morphology and physiology in distinct brain regions (11, 12). The relevance of astrocytic heterogeneity is also evident in its distinct subpopulations and cortical layer-specific gene signatures that underline the comprehensive involvement of astrocytes in physiology (13–15). More sophisticated research strategies paired with a systemic evaluation and comparison of different glial markers will lead to a better understanding of the role of astrocytes in the central nervous system under physiological and pathophysiological conditions.
Astrocytes have been shown to be involved in important processes such as brain inflammation (16, 17) and oxidative stress (18), energy supply and metabolism (19–21), support of synaptic function and plasticity (22, 23), extracellular balance of neurotransmitters (24, 25), extracellular water and ion homeostasis (26, 27), blood-brain barrier (BBB) maintenance (28, 29), and regulation of blood flow [(30, 31); Figure 1].
Figure 1.
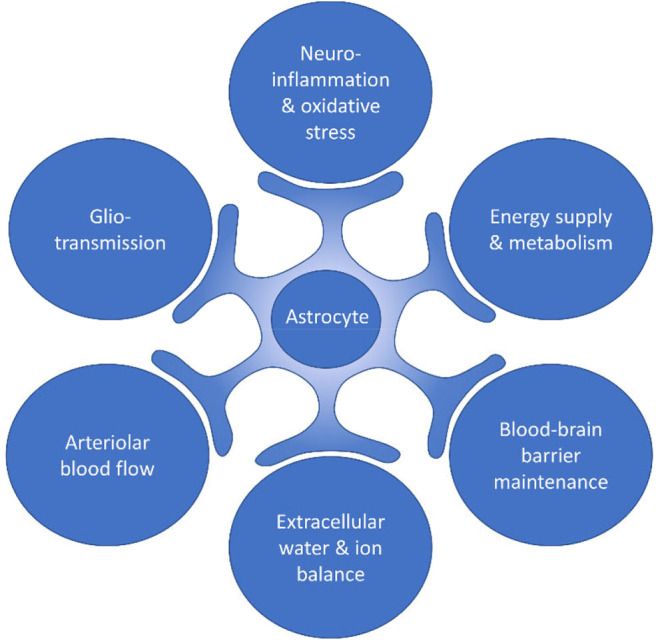
Processes within the brain in which astrocytes are involved. Astrocytes have been shown to be involved in important processes such as neuroinflammation and oxidative stress, energy supply and metabolism, blood-brain barrier maintenance, extracellular water and ion balance, arteriolar blood flow, and gliotransmission.
Although astrocytes employ many processes that protect the brain from hyperexcitability, dysregulation of glial functions may cause hyperexcitability or promote the development of epilepsy by a multitude of mechanisms. In the following paragraphs, we will focus on the underlying processes that can promote epileptogenesis, including astrogliosis, disturbed energy metabolism and gliotransmission, alterations in extracellular ion concentrations, as well as dysfunction of the BBB and dysregulation of blood flow (Figure 2).
Figure 2.

Astrocytic processes involved in epileptogenesis. Dysregulation of astrocyte functions can lead to epileptogenesis via disturbed energy metabolism and gliotransmission, alterations in extracellular ion concentrations, as well as dysfunction of the blood-brain barrier and dysregulation of blood flow. These mechanisms are discussed in detail in the main text.
Astrogliosis
Due to brain injury induced by status epilepticus, stroke or traumatic brain injury, astrocytes receive “instructions” from their environment (Figure 3A) and in response to these molecular signals, the number of astrocytes increases and the astrocyte expression profile as well as its morphology, biochemistry and functionality changes, a process called reactive astrogliosis (32, 33). In turn, reactive astrocytes can send “instructions” to their environment (Figure 3B). The term reactive astrogliosis has been introduced in the nineteenth century to characterize morphological and behavioral changes within astrocytes upon pathophysiological conditions caused by various central nervous system diseases. In the beginning, efforts were focused on the morphological changes astrocytes experience during reactive astrogliosis, but over the past three decades a body of evidence has been collected that support astrogliosis heterogeneity and acknowledges a spectrum of molecular, cellular and functional changes within astrocytes upon reactive astrogliosis (32, 34).
Figure 3.

Factors involved in astrogliosis. (A) After brain injury, astrocytes can receive “instructions” from their environment and respond to a plethora of signaling molecules. (B) In turn, astrocytes send “instructions” to their environment by releasing a variety of factors, including pro-inflammatory cytokines, growth factors, neurotransmitters, as well as vascular mediators. This vicious cycle may lead to persistent activation of astrocytes which can contribute to epileptogenesis. Adapted from Sofroniew (32).
The existence of spatiotemporal and anatomically localized subtypes of astrocytes needs to be taken into account when evaluating astrogliosis in the context of experimental epilepsy models, including the consequential effects on epileptogenesis and related neurobehavioral comorbidities, by employing genetic targeting studies and pharmacological therapies.
Cell-specific transcriptomics have revealed that astrocytes undergo massive changes in gene expression when they switch to a reactive phenotype (33). One of the most prominent changes during reactive astrogliosis is characterized by cell hypertrophy and upregulation of glial fibrillary acidic protein (GFAP), vimentin, nestin, and/or inducible nitric oxide synthase (iNOS) (35, 36). In addition, reactive astrocytes may produce and release a variety of factors, including pro-inflammatory cytokines (37), complement factors (38), gliotransmitters (39–41), reactive oxygen species (ROS) (42), trophic factors (43), and vascular endothelial growth factor (VEGF) [(44); Figure 3].
In particular, pro-inflammatory cytokines may affect astrocytes profoundly and cause changes that perpetuate astrogliosis and promote epileptogenesis (45, 46). Cytokines are widely studied in the context of reactive astrogliosis (47) and epilepsy (48). In this paragraph, we will focus on cytokines that exacerbate epilepsy progression and may therefore be interesting for therapeutic intervention. The most studied cytokines regarding astrogliosis and epilepsy are interleukin-1 beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α); pro-inflammatory cytokines that can be released by reactive astrocytes (49, 50) and activated microglia (51). In a complex pathology such as epilepsy, more cytokines are playing roles in the alleviation and exacerbation of the disease. Figure 3 shows a fraction of cytokines involved in astrocytosis. For further reading into cytokine involvement in epilepsy the reader is directed to the following reviews (45, 46, 52).
Numerous studies have shown upregulation of IL-1β, IL-6, and TNF-α in animals with (recurrent) seizures (53–59) and patients with epilepsy (60–63). IL-1β can affect neurotransmitter receptors (64, 65), induce calcium influx by N-methyl-D-aspartate (NMDA) and 3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-mediated mechanisms in neurons (66, 67), lead to alterations in expression of microRNAs in astrocytes (68–70), as well as potassium channels (71), metalloproteinases (72), altered astrocytic glutamate uptake (73, 74) and calcium uptake (75), and induces astrocytic release of other pro-inflammatory cytokines (50). IL-6 activates the Grp130/JAK/STAT pathway and thereby induces the release of additional pro-inflammatory cytokines, further endorsing inflammation (76). In addition, high levels of IL-6 lead to decreased astrocytic glutamate uptake via excitatory amino acid transporter 2 (EAAT2; formerly glutamate transporter 1) and even promote the release of glutamate by improving the activity of the astrocytic cystine/glutamate antiporter (Xc−) (76). Finally, IL-6 increases BBB permeability (77).
Similar to IL-6, TNF-α decreases astrocytic uptake of glutamate (78, 79) via purinergic signaling, thereby activating presynaptic NDMA receptors (80). Furthermore, TNF-α increases excitatory strength of AMPA receptors and at the same time decreases the amount of γ-aminobutyric acid (GABA)A receptors in neurons, impairing inhibitory signaling (81). Finally, release of pro-inflammatory cytokines often stimulate additional release of cytokines (50), and it is suggested that this perpetual exacerbation of inflammatory signaling contributes to epilepsy (16, 82, 83). VX-765, a small molecule inhibitor of interleukin-converting enzyme (ICE)/caspase-1, thereby inhibiting biosynthesis of IL-1β, has been shown to reduce the number and duration of seizures in rats (84) and mice (85) and has even been tested in Phase II clinical trials (ClinicalTrials.gov Identifier: NCT01501383). In a Phase IIa randomized double blind placebo-controlled study in drug-resistant focal onset epilepsy, VX-765 had delayed beneficial effects (subjects with ≥50% reduction in seizure frequency) that persisted after drug discontinuation (86). Furthermore, the IL-1 receptor antagonist Kineret (anakinra) showed a drastic improvement of seizure control in patients with super-refractory status epilepticus secondary to febrile infection-related epilepsy syndrome (FIRES) (87–90), as well as in patients with drug-resistant epilepsy (91, 92).
To our knowledge, there is no therapy that directly targets IL-6 or its receptor IL-6R, but it has been shown that the antiepileptic drug valproate affects IL-6 serum levels, hinting at a possible mechanistic involvement of IL-6 (93).
Another example is Adalimumab, a TNF-α monoclonal antibody that has been tested in Rasmussen's encephalitis, leading to seizure improvement in a small cohort of patients (94). Furthermore, n-3 docosapentaenoic acid-derived protectin D1 is a pro-resolving mediator that was administered to epileptic mice, showing subsequent downregulation of IL-1β and TNF-α mRNA and consequently a 50% decrease of seizure activity and a 40% decrease in seizure duration (95). Finally, 1400 W, an inhibitor of inducible nitric oxide synthase (iNOS/NOS-II) showed promising results in a rat model of kainic acid-induced epilepsy, since it suppressed astrogliosis, microgliosis, neurodegeneration, mossy fiber sprouting, and had disease modifying effects (96).
Reactive astrogliosis is implicated in acquired and genetic types of epilepsy, including neurodevelopmental diseases [i.e., tuberous sclerosis complex; (17, 97–99)]. Changes of activity and gene expression of key proteins that are involved in epilepsy pathology such as glutamine synthetase (GS) (100), adenosine kinase (ADK) (101, 102), Aquaporins (AQPs) including AQP4 (103, 104), inward rectifying potassium (Kir) channels including Kir4.1 (105, 106), and monocarboxylate transporters (MCTs) (107, 108) have been observed in resected brain tissue of patients with temporal lobe epilepsy (TLE). Initially, the astrocytic response can be beneficial for the brain, promoting restoration of brain homeostasis. However, a vicious cycle may lead to persistent astrogliosis which can affect metabolic activity (109–111), ion buffering (112), gap junction (GJ) connectivity (113, 114), neurotransmitter uptake (115, 116), and promotes neuronal death, BBB dysfunction (44), and onset of seizures (117, 118). In the following chapters we will elaborate how these changes can affect neuronal excitability and contribute to epileptogenesis.
Besides affecting molecular pathways, astrocytes participate in bilateral signaling with microglia (119, 120). This interglial crosstalk has implications on both physiological and pathological processes (121, 122). Astrocytes influence microglial behavior by releasing molecules that regulate microglial functions. In turn, microglia are able to drive astrocytes from a neuroprotective to a neurotoxic phenotype (123), thereby potentially affecting the ability of astrocytes to protect against neuronal excitability. This bidirectional crosstalk may induce a persistent inflammatory environment under pathological conditions and may therefore exacerbate disease severity. Recent studies have shown that activated microglia induce neurotoxic phenotypes in astrocytes by secretion of pro-inflammatory mediators such as IL-1α, TNF-α, and complement component subunit 1q (C1q) (119, 124). Crosstalk between astrocytes and microglia may also be involved in epileptogenesis and should be taken into account when conducting studies into the mechanisms that drive epilepsy. Although attention has been primarily focused on astrocyte interactions with other central nervous system cell types, there is recent evidence that astrocyte functionality is influenced by the gut microbiome, and that this cross-talk between gut microbiota and brain, involving astrocytes, may have crucial implications in the development and progression of central nervous system disorders (125, 126). For instance, different types of gut bacteria may positively or negatively modulate the astrocytic inflammatory response (126–128).
Energy Supply and Metabolism
Under physiological conditions, glucose is the primary metabolic substrate of the brain and is required to maintain the transmembrane potential of neurons (21). Glucose is transferred from the blood into the brain by glucose-transporters (GLUTs). Then, glucose is metabolized into glucose-6-phosphate (G-6P) by hexokinase [(129); Figure 2]. Subsequently, it can undergo two types of metabolization: glycolysis or metabolization by the pentose phosphate pathway. During glycolysis, pyruvate is formed, producing ATP. Pyruvate can then be oxidized in mitochondria by the tricarboxylic acid (TCA) cycle or by oxidative phosphorylation, producing 30–34 molecules of ATP at the cost of oxygen (129). In addition to neuronal energy supply, astrocytes are also equipped with a glucose-metabolism to meet the local energy demand. In fact, in times of hypoglycaemia and during periods of high neuronal activation, astrocytes take over the energy supply completely (130, 131). Astrocytes are able to process glucose by a mechanism similar to that of neurons. Astrocytes express glucose transporter 1 (GLUT1) allowing for glucose uptake (Figure 2). Glucose is then metabolized into G-6P by hexokinase (HK) and further into lactate, via pyruvate by an isoenzyme of lactate dehydrogenase (LDH) 5 (20). Subsequently, lactate is exported from astrocytes into the extracellular space by monocarboxylate transporters (MCTs) 1 and 4 (132) and taken up into neurons, which convert lactate into pyruvate. This alternative pathway constitutes the astrocyte-neuron lactate shuttle (21). Alternatively, G-6P is converted into G-1P by phosphoglucomutase (PGM) and then into uridine triphosphate (UDP)-glucose by UDP-glucose pyrophosphate (UDPGPP) (130). Finally, UDP-Glucose is converted into glycogen by glycogen synthase (GYS). Glycogen can then be stored in glycogen granules, usually clustered in places of great synaptic density (133). When required, glycogen can be metabolized back to G-6P via the same route in reverse or mediated by glycogen phosphatase (GP) (134).
Glucose Sustains Synaptic Activity During Seizures
During seizures, excessive synaptic activity causes a rapid drop of glucose and a corresponding rise in lactate. Surrounding tissue responds to this by increasing glucose-administration to neurons by increasing blood perfusion and volume (135). At the same time, glucose can be distributed by astrocytes via by gap junctions (GJs) to reach distal neurons [(136); Figure 2]. During the excessive energy demands of seizures, astrocyte-derived lactate becomes an essential energy source for neurons (21). Furthermore, astrocytes can store glycogen which can supply energy to neurons via the lactate shuttle to sustain neuronal activity during seizures. Therefore, reducing brain glucose levels is considered anticonvulsive (137).
One way of achieving this is by the ketogenic diet, which is a low-carbohydrate and high-fat and adequate protein diet (138, 139). Thereby, the brain switches from a glucose-sustained metabolism to ketosis during which ketones, such as β-hydroxybutyrate, acetoacetate, and acetone are formed, which are thought to be important mediators for the suppression of seizures during the diet (140). In addition to the ketogenic diet, other antiepileptic diets have been proposed such as the modified Atkins diet, the medium-chain triglyceride ketogenic diet, and the low glycaemic index treatment (141, 142). Ketogenic diets are quite efficient in the alleviation of seizures in children, but also in adults with refractory epilepsy (138, 143, 144). However, it is difficult to adhere to the diet since it is not palatable. Furthermore, weight loss, constipation, high level of low-density lipoprotein, and elevated total cholesterol are most frequently reported as adverse effects (143, 145). Therefore, alternative approaches to inhibit glycolysis or interfere with lactate formation are studied. For instance, the use of the glycolysis inhibitor such as 2-deoxy-2-glucose has been proposed as a direct mechanism of lowering brain glucose, which has acute anticonvulsant and chronic antiepileptic actions in various epilepsy models (146–148). Furthermore, inhibition of LDH suppresses pilocarpine and kainic acid-induced seizures. Interestingly, LDH is also inhibited by the AED stiripentol (149). Another approach is to utilize GJ blockers that impair astrocytic intercellular glucose trafficking, thereby partially reducing the required energy for epileptiform activity (150). Taken together, these data imply that targeting specific brain glucose-pathways is an ambitious and challenging, but also a promising approach to interfere with epileptogenesis. Reducing glucose levels may be achieved by specific diets, local glycolysis-inhibition or by inhibition of GJs.
Gap Junctions
Astrocytic GJs are comprised of two “hemichannels” which are made up of 6 subunits or connexins (Cx) (151). Astrocytes predominantly express connexins Cx43, but also Cx30, Cx26, Cx40, Cx45, and Cx46 (152, 153). One of the functional properties of GJs is to facilitate inter-astrocyte transportation of glucose and glucose-metabolites (150). In addition, GJs are able to propagate intercellular Ca2+ signaling through release of ATP (153, 154). Furthermore, GJs permit potassium transport between astrocytes, allowing K+ influx to redistribute to sites of lower concentration, supporting spatial K+ buffering (discussed in detail in a following paragraph). GJs reduce the threshold for seizures by facilitating spatial K+ buffering and glutamate transport. The involvement of GJs in spatial K+ buffering is reflected in the AQP4−/− mouse model in which increased GJ coupling compensates for the loss of K+ uptake assisted by AQP4 (155). In line with this, mice with GJ-coupling deficiencies were shown to develop seizures and have problems with glutamate and K+ clearance (156).
Neuroprotective properties of GJs have been reported and therapeutically interfering with GJ functionality may introduce side effects (157). Moreover, uncoupling (loss of connectivity through loss of GJs) of astrocytic endfeet has been found to precede neurodegeneration and spontaneous seizure generation in a mouse model of TLE (158). Different expression patterns have been reported in studies on animal models and human tissue (159, 160). In astrocytes of sclerotic human hippocampal tissue, expression of connexins appears unchanged (161). It has been proposed that instead subcellular reorganization or post-translational modification of connexins accounts for the loss of GJ functionality in TLE. This could explain the variability in connexin expression in TLE found in earlier studies (159).
On the other hand, GJs may fuel synaptic and epileptiform activity by intracellular trafficking of metabolites to sustain neuronal activity at sites of high demand. GJs facilitate the spread of Ca2+ waves contributing to epileptogenesis by introducing a feedback loop from neurons to astrocytes (162). Furthermore, neuronal GJs are thought to be involved in the synchronous discharges during seizure activity (163, 164). These data suggest that inhibiting GJs has anticonvulsive effects.
Indeed, it has been shown that GJ blockers such as carbenoxolone (165–167), mefloquine (168), quinine (166, 167, 169), and quinidine (170) alleviate seizure severity in various animal models of epilepsy, although a general consensus on the exact mode of action and the efficacy of these compounds is still lacking. Anandamide and oleamide are fatty acids of the endocannabinoid family that have been demonstrated to inhibit intercellular GJs from glial cells (171, 172). Both anandamide (173, 174) and oleamide (175, 176) have been shown to have anticonvulsant effects in vivo, hinting at the involvement of glial networks in seizures.
Gliotransmission
The concept of “gliotransmission” remains one of the most controversial topics in astrocyte biology. The term gliotransmitter is loosely defined as chemically active transmitters that origin from glial cells which may participate in or affect the excitatory or inhibitory network of neurons. Numerous studies have been performed showing a plethora of astrocytic released gliotransmitters: (1) amino acids including glutamate (177–179), D-serine (180, 181), GABA (182–184) and glycine (185–187), (2) nucleotides, such as adenosine 5′-triphosphate (ATP) (188–190), (3) organic acids including lactate (191–193), taurine (194, 195), and homocysteic acid (196, 197), and (4) peptides such as atrial natriuretic peptide (ANP) (198) and brain-derived neurotrophic factor (BDNF) (199, 200). Some argue that cytokines are in a way also gliotransmitters as they are chemically active too and may affect neuronal excitability, albeit mainly via indirect mechanisms. However, in this review we will further focus on the most studied gliotransmitters: glutamate, D-serine, and ATP and give an insight on how these gliotransmitters affect neuronal excitability.
Ca2+-Dependent Gliotransmitter Release
In the early 1990s the first Ca2+ imaging studies were performed, showing increased astrocytic intracellular Ca2+ concentrations after local synaptic activity (201–203). A general consensus developed stating that astrocytes are in fact “excitable” cells and may respond to a wide range of neuronal factors and synaptically released spill-over neurotransmitters, and at the same time release so-called gliotransmitters that can communicate to neurons (24). In addition, newer imaging techniques showed that astrocytes appear to facilitate spontaneous focal Ca2+ oscillations or transients (204–206) and may even propagate Ca2+ signals to adjacent astrocytes (207–209). Unfortunately, due to limitations in experimental approaches required to understand the complexity of gliotransmission, it proved difficult to replicate findings in different models, or translate data from in vitro to in vivo. A heated debate followed in which contrasting evidence from various studies raised the question whether or not astrocytes contribute to information processing within the neural circuitry under physiological conditions (210). In addition, the dependence on Ca2+ signaling has been challenged time and again and is under critical review (211). To go further into this debate is beyond the scope of this review and the reader is directed to excellent literature on the topic (210, 212, 213).
Nowadays, a strong foundation of evidence that supports the bidirectional communication between neurons and astrocytes established the concept of a tripartite system that was originally proposed in the late 1990s (214). Progress on research neuron-glia crosstalk showed that the central role of astrocytes, besides regulation of brain homeostasis, is information processing. A body of evidence supports the existence of coordinated neuron-astrocyte network signaling, in which astrocytes are able to modulate neuronal excitability and synaptic transmission (206, 215–217). In turn, neuronal communication to astrocytes influences astrocytic signaling which may have implications in epilepsy (215, 218).
Two types of astrocyte “excitation” are well documented: neuron-dependent excitation and spontaneous excitation (24). There is evidence of Ca2+-dependent astrocytic release of different types of gliotransmitters including glutamate (204, 219–221), D-serine (222–224), and ATP (225, 226). To what extent these mechanisms are in fact dependent on Ca2+ or how they may or may not play a role in synaptic transmission under physiological conditions is discussed elsewhere (211, 227, 228).
Ca2+-Independent Mechanisms of Gliotransmitter Release
In addition to Ca2+-dependent mechanisms of gliotransmitters, several Ca2+-independent mechanisms have been identified for some, but not all gliotransmitters. Astrocytes facilitate glutamate release by targeting the two-pore domain K+ channel (TREK-1) (220), through the pannexin-1 (panx-1) (229), and Cx43 hemichannels (230, 231), by volume-regulated anion channels (VRACs) (194), reversible glutamate transporters (232–234), and in vitro via the (Xc−) (235, 236) and the ionotropic P2X purinoceptor 7 (P2X7R) [(237); Figure 2]. Astrocytic ATP is released through GJ channels such as panx-1 (229, 238) and Cx43 hemichannels (231, 239), and in culture via mechanically-induced release of ATP by P2X7R (240, 241).
The relevance of these mechanisms is demonstrated by the changes that occur under pathophysiological circumstances such as in the epileptogenic brain (242–244), during astrogliosis (245), or upon swelling of astrocytes (246). Reactive astrocytes display increased expression and activation of hemichannels such as Cx43 (247) and panx-1 (248, 249), which is generally believed to result in increased gliotransmitter release (234). Moreover, during epilepsy, the opening probability of both astrocytic and neuronal hemichannels is increased, augmenting local excitotoxicity (250).
Using transgenic mouse models, ATP release through panx-1 channels has been shown to enhance neuronal excitability (251, 252). Furthermore, panx-1 in conjunction with P2X7R potentiates seizure activity in an animal model of epilepsy as well as in brain slices of patients with TLE (252). Panx-1 channels are exciting new targets as global panx-1 inhibition has anticonvulsive effects in animal models of epilepsy (168, 251).
Similar to panx-1, global inhibition of P2X7R reduces epilepsy severity after kainic acid-induced epilepsy (253), although additional in vivo data indicates that this is mainly due to neuronal effects (244, 254). Inhibiting P2X7R presumably affects astrocytes and other glial cells indirectly as well, by blocking P2X7R-mediated excitotoxic IL-1β release (255). P2X7R inhibitors such as Brilliant Blue G, A438079, AFC-5128, and JNJ-47965567 could attenuate chemically-induced kindling but did not possess remarkable effects in acute screening tests when administrated alone (256, 257).
Taken together, these data indicate that modulating astrocytic gliotransmitter release pathways may affect neuronal excitability. Interestingly, in a recent review several experimental pharmacological agents were highlighted as tools to control astrocyte signaling (258). These agents were tested in preclinical models, but some antiepileptic drugs may exert similar effects. This needs to be studied in more detail, as well as the use of these agents as novel therapeutic approaches. In the following paragraphs we will further elaborate on ways that gliotransmitters influence the pathophysiology of epilepsy.
Glutamate
Astrocytes are able to influence extracellular concentrations of glutamate and an excess of extracellular glutamate is one of the mechanisms driving hyperexcitability (259, 260). Under physiological conditions, astrocytes restrict the diffusion of glutamate in the synaptic cleft and take up and recycle glutamate in a process called the glutamate/GABA-glutamine cycle [(25, 261); Figure 2]. In this cycle, glutamate is taken up by astrocytic glutamate-uptake channels such as the excitatory amino acid transporter 1 (EAAT1; formerly Na+-glutamate cotransporter) and EAAT2. Glutamate is then converted into glutamine by glutamine synthetase (GS) at the cost of ammonia and ATP. Interestingly, astrocyte subpopulations that express GS also co-express EAAT1 and EAAT2, emphasizing the link between the two mechanisms (262). After the conversion, glutamine is shuttled back to neurons through release by N system transporters (SN) 1 and 2 on the astrocytic membrane followed by neuronal uptake through system A transporters (SAT) 1 and 2 (263). It is then converted back into glutamate by neuronal glutaminase. In this cycle there are two steps by which astrocytes regulate glutamatergic excitability: (1) by removing excess glutamate from the extracellular space, and (2) by regulating the glutamine release from the astrocytic cytoplasm. In addition to glutamate uptake and conversion to glutamine, astrocytes are also able to synthesize glutamine de novo, by employing glycolytic enzymes and the TCA cycle, which produces glutamate from α-ketoglutarate and can then be converted to glutamine by GS (264).
Under pathophysiological circumstances, the regulation of the glutamate/GABA-glutamine cycle is perturbed, which can contribute to epileptogenesis. In vivo microdialysis experiments in the human brain showed that extracellular glutamate concentrations were chronically increased in the epileptogenic hippocampus compared to non-epileptic hippocampus (265, 266). This is likely the result of a failing glutamate uptake system from astrocytes in concert with a decreased ability to convert glutamate to glutamine.
Glutamate-Uptake Channels
Downregulation of glutamate-uptake channels such as the EAAT1 (267–269) and EAAT2 (267, 269, 270) has been frequently reported in animal models of epilepsy. Furthermore, EAAT1 deletion causes prolonged seizure activity (271) and EAAT2 knockout mice exhibit spontaneous and recurrent seizures (272). In patients with TLE, EAAT1, and EAAT2 are also downregulated and this is colocalized with GFAP and the proliferation marker Ki-67, suggesting that this is dependent on astrogliosis (273, 274). Transcriptional reactivation of EAAT2 by a small molecule reduced the frequency of spontaneous seizures by 50% in a mouse model of tuberous sclerosis complex, postulating that restoring glutamate-uptake channels is seizure ameliorating (275). The loss of EAAT2 is not only evident on mRNA expression level, but the protein itself is also internalized and subsequently degraded (276). Therefore, preventing the degradation of EAAT2 may pose as an effective treatment for epilepsy as was recently shown in a mice model of kainic acid-induced epilepsy (277).
Glutamine Synthetase
A growing body of evidence supports the notion that pathophysiological events such as epileptic seizures (278, 279) or astrogliosis (280, 281) result in a downregulation and corresponding decrease in immunoreactivity of GS (282). In accordance, chronic treatment with a GS-inhibitor caused spontaneous seizures in rats and increased local extracellular glutamate concentrations by 47%, showing that GS-deficiency alone is enough to evoke ictal events (260).
The exact mechanism of the lowered extracellular glutamate concentration due to GS-deficiency is still unknown, although several hypotheses have been proposed: (1) loss of GS leads to impaired clearance of glutamate because of a reduced conversion to glutamine, and (2) accumulating glutamate in astrocytes constitutes a concentration-dependent gradient that results in astrocytic glutamate release (282).
Indirectly, GS-deficiency may also contribute to hyperexcitability (282). Because glutamine is the precursor for the inhibitory neurotransmitter GABA, a reduction in astrocytic glutamine production evokes a local shortage of GABA. As the main inhibitory neurotransmitter of the brain, a local GABA shortage increases neuronal excitability and neuronal network synchronization (283). A second way that GS-deficiency affects local excitability is that a reduction in the glutamine metabolism consequently consumes less ammonia. Previously, it has been shown that high concentrations of local ammonia is neurotoxic and may even cause excitotoxicity by affecting chloride transport (284, 285). It is presently unclear to what extent these indirect mechanisms contribute to ictogenesis and research into this would certainly contribute to our understanding of how a GS-deficiency could cause epileptic seizures.
Evidence suggests that GS expression is dependent on neuronal survival as downregulation of GS in patients with TLE coincides with neuronal loss (100, 285). It has been proposed that neuronal loss precedes GS downregulation, and in turn, GS downregulation increases excitability (285). In addition, the resulting increase in extracellular glutamate may result in neuronal cell death (286), feeding a disease exacerbating cycle (285).
Other pathological changes within astrocytes such as swelling (194) and ischemia (233) may also affect glutamate release and extracellular concentrations. From all this data it becomes evident that the glutamatergic mechanisms that underlie excitability are exceedingly intricate and complex. Perturbations in any of the aforementioned glutamatergic mechanisms may lead to an increase of excitatory network activity, and eventually epilepsy (287).
D-Serine
Presently, all mechanisms regarding D-serine release from astrocytes appear to be either directly linked to intracellular Ca2+ concentration (i.e., vesicular release), or are receptor activation-dependent, which is indirectly linked to local Ca2+ concentration (288). Amongst these are the adenosine type 2A receptor (A2AR) (289), bradykinin-type2 (B2) receptor (290), ephrinB3 receptor (291), ionotropic (292), and metabotropic (223) glutamate receptors, transforming growth factor (TGF)-β receptor (293), as well as muscarinic (294) and nicotinic (295) acetylcholine receptors. Despite its extensive regulation, it was recently proposed that astrocytic D-serine is not available in sufficient amounts to modulate synaptic activity in vivo, under physiological conditions (296). Instead, astrocytic de novo synthesized L-serine that is required for the production of D-serine in neurons may affect synaptic activity after conversion to D-serine by neuronal serine racemase (SR) (297, 298). It is noteworthy however, that the profound effects of pathophysiological conditions such as epileptic seizures or astrogliosis dramatically change the behavior and expression profiles of astrocytes, which may in turn affect the dynamics of D-serine production. In culture (224) and in brain slices (222), astrocytes are able to express SR, and most notably, in an animal model of traumatic brain injury, it was shown that the switch from neuronal SR to astrocytic SR was in part responsible for traumatic brain injury-induced synaptic damage (299). Furthermore, increased release of D-serine may contribute directly to neuronal excitotoxicity by acting upon the NMDA receptor as a co-agonist together with glutamate (Figure 2). Indeed, it has been shown that lowering D-serine in epileptic rats by administering a competitive SR-inhibitor resulted in reduced seizure duration and severity, dependent on ERK signaling (300). These data suggest that modulating D-serine production could pose as a strategy for epilepsy treatment.
ATP
The actions of ATP and its metabolite adenosine arguably extend even further than that of glutamate or D-serine by acting upon purinergic receptors, influencing astrocytes, neurons, microglia, oligodendrocytes, and blood vessels (301). The complexity of ATP-mediated effects is demonstrated by studies that report both excitatory and inhibitory consequences from astrocytic ATP release. For instance, it has been shown that ATP activates the astrocytic ionotropic P2X and metabotropic P2Y receptors resulting in increased GABA release (302). Furthermore, ATP released by astrocytes may induce action potentials on inhibitory interneurons, thereby decreasing the excitatory network output (303). In contrast, astrocytic ATP negatively regulates GABAergic inhibitory transmission on post-synaptic neurons (226), suggesting that astrocytic ATP release may augment ictogenesis. Moreover, it has been shown that astrocytic ATP activates neuronal P2X receptors leading to pro-epileptic effects (304), including enhanced pre-synaptic release of glutamate (305). As argued earlier, purinergic signaling through P2X receptors is mediated by ATP release through panx-1 channels. However, it appears there is a clear distinction between astrocytic and neuronal panx-1, and surprisingly, astrocytic panx-1 may even be seizure alleviating (306) [for review see (234, 307)]. It is hypothesized that worsening of seizure activity in mice deficient of astrocytic panx-1 is likely connected to increased ADK levels in astrocytes.
Adenosine Kinase
ADK is a key metabolic enzyme of astrocytes that catalyses the conversion of adenosine into adenosine monophosphate. Therefore, modulation of ADK expression is of interest in the context of epilepsy. Adenosine is a potent anticonvulsant and is released during seizures (17). It is a substrate for the adenosine receptor family of which the A1AR and A2AR are the most studied. Anti-epileptic effects are mainly mediated by A1AR signaling which activates Kir channels and inhibit Ca2+ channels, but also exert astrocyte-function modulating effects by stimulatory coupling to K+ and Cl− ion channels (308–310). Since neuronal excitability is modulated by activation of A1A, A2A, A2B, and A3 receptors, the equilibrium of intra- and extracellular adenosine critically affects epilepsy severity (311).
Synaptic adenosine is mainly regulated by ADK, because uptake of adenosine into astrocytes is quickly equilibrated by nucleoside transporters (ENTs) (310). Upon brain injury, ADK is transiently downregulated for ca. 2 h, recovering to baseline levels over the course of 24 h (101). This acute response to stress results in increased adenosine levels, enhancing protective effects against brain injury, including status epilepticus and traumatic brain injury, through increased activation of A1AR (310). However, elevated synaptic adenosine levels also activate the A2AR, which signaling may in turn desensitize and downregulate the A1AR (312, 313). Indeed, it has been shown that in epileptogenic circuits, stimulation of A2AR downregulates A1AR (314, 315). Recently, it has been shown that a 3-fold induction of A2AR is present in astrocytes within the hippocampus of patients with TLE (316). Increased A2AR signaling promotes astrogliosis by various mechanisms including by increased stimulation of glutamate release, synaptic actions of BDNF in the hippocampus and through the Akt/NF-κB pathway (317–320). The shift in A1AR/A2AR signaling also causes a change from inhibition to promotion of cell proliferation and may contribute to the development of proliferative scar-forming astrocytes (310). Moreover, after the initial downregulation of ADK, its expression increased in reactive astrocytes (101). This is also confirmed in experimental animal models of epilepsy and human TLE brain slices (102). ADK inhibitors have since been developed (321–323) and tested in animal models of epilepsy (324, 325). Unfortunately, the first line of ADK inhibitors showed liver toxicity side effects, but recently efforts have been made to develop novel ADK inhibitors which may present a viable therapeutic strategy for epilepsy in the future (326).
Water and Ion Transport
One of the functions of astrocytes is to maintain homeostatic extracellular water and ion balance in the brain. Changes in ion or water balance affect local synaptic activity by modifying the concentration gradient upon which the electrochemical potential is based. Ultimately, this may result in hyperexcitability by mechanisms discussed below. To ensure homeostatic ion balance is preserved, astrocytes express a plethora of passive, ATP-, voltage-, and volume-gated ion channels (Figure 2 and Table 1).
Table 1.
Selection of ion and water transporters associated with homeostatic astrocyte function and epileptiform activity in disease.
| Ion | Transporter | Alteration | Expression in temporal lobe epilepsy |
|---|---|---|---|
| H2O | AQP1 AQP4 AQP9 EAAT1 |
Causing astrogliosis (327) Mislocalization (328) |
↑ (329) ↑ Overall (330, 331) ↓ Perivascular (331, 332) ↓ (329) No change (100) ↓ Hippocampus (274) |
| K+ | BK K2P Kir4.1 Kir5.1 Kir2.1 Kv Na+/K+-ATPase NKCC1 |
Transient upregulation (333, 334) |
↓ Mossy fibers (335) ↑ CA1 ↓ Dentate gyrus (336) ↓ Hippocampus, Perivascular (106) No change (337) ↑ CA1, CA3, Dentate gyrus (338) ↑ Hippocampus (339) ↓ (Suggested) (340) ↑ Subiculum, hippocampus (341) |
| Na+ | EAAT1 EAAT2 Na+/K+-ATPase NCX NKCC1 TRPA1 TRPCs TRPV1 |
Transient upregulation (333, 334) |
No change (100), ↓ Hippocampus (274) No change (100), ↓ Hippocampus (274) ↓ (Suggested) (340) ↓ Dentate gyrus (342) ↑ Subiculum, hippocampus (341) ↑ (343) ↑ (344–346) ↑ Cortex, Hippocampus (347) |
| Ca2+ | AMPA NMDA NCX P2X7 PMCA TRPA1 TRPCs TRPV1 |
Different splice variant (348) Subcellular relocation Transient upregulation |
↑ Hippocampus (349) ↑ Dentate gyrus (350) ↓ Dentate gyrus (342) No change (253) ↑ Dentate gyrus (342) ↑ (343) ↑ (344–346) ↑ Cortex, Hippocampus (347) |
| Cl− | NKCC1 | Transient upregulation (333, 334) | ↑ Subiculum, hippocampus (341) |
↑: upregulation, ↓: downregulation.
Intra- to extracellular water balance is of significance for epilepsy because it directly correlates to local osmolarity that plays a role in excitability. Astrocytes are particularly sensitive to changes in extracellular osmolarity (351). For instance, as a result of traumatic brain injury, up to ~30% of patients develop hyponatremia. This causes a decrease in osmolarity, after which astrocytes swell considerably (352, 353). As astrocytes swell up by water uptake, the extracellular space volume decreases. In turn, astrocytes respond by activating VRACs that work to restore the concentration equilibrium by expelling osmolytes and anions such as F− and Cl− [(351, 354, 355); Figure 2]. These mechanisms are of interest, because the volume of the extracellular space affects synaptic activity (26). In addition, opening of VRACs is accompanied with substantial amounts of glutamate (356, 357). These VRACs open primarily in astrocytes with high concentrations of K+ or during hypo-osmolar conditions that often occur during ictal events, although it has been proposed that Ca2+ signaling may induce swelling and thereby open VRACs as well (39, 354). Due to lack of selectivity and inability to differentiate between astrocytic and neuronal channels, modulation of VRACs has not been tested in animal models of epilepsy, but may pose an interesting avenue for seizure treatment by potentially lowering extracellular glutamate levels. Care should be taken when following this approach as a tight regulation of osmolarity and the volume of the extracellular space is required for homeostatic brain function.
Spatial Potassium Buffering
The most critical ion flux governed by astrocytes in relation to epilepsy is that of potassium. In a process called spatial potassium buffering, astrocytes clear the extracellular space of excess K+ during neuronal repolarization. To ensure rapid uptake of K+ ions, astrocytes express different types of K+-channels, including Kir channels, Ca2+-sensitive potassium (BK) channels, voltage-gated potassium (Kv) channels, two-pore domain (K2P) channels and several co-transporters [(358); Figure 2 and Table 1]. Upon entering astrocytes, K+ is dispersed to areas of lower potassium concentration and travels intercellularly to adjacent astrocytes by GJs. The spatial buffering model is based on the fact that the low resting potential of astrocytes provide a driving force for K+ uptake in regions of high neuronal activity.
Perturbations in the astrocytic K+ buffering is therefore directly responsible for increased neuronal activity and excitability. In addition, high extracellular K+ concentrations may affect the activity of ion and water transporters such as AQP4, EAAT2, Na+/Ca2+-exchanger (NCX), sodium-potassium pump Na+/K+-ATPase, and Na+/K+/Cl−-cotransporter (NKCC) (355). Furthermore, increases in the extracellular K+ concentration induce opening of panx-1 channels, may cause seizure activity by release of ATP and glutamate (252, 307, 355).
Aquaporins
The integral membrane protein AQP4 is responsible for most of the water uptake by astrocytes (Figure 2), but a total of 13 human AQPs (0–12) have been characterized (26, 359). Of those, expression and protein levels of AQP1, 3, 4, 5, 8, 9, and 11 have been shown in rodent brains (329). Aside from aquaporins, there are additional mechanisms to transport water, for instance via co-transporters such as EAAT1 (360).
It is hypothesized that concomitant water uptake by AQP4 during spatial K+ buffering decreases the volume of the extracellular space, thereby inducing an increase of [K+]0, which in turn stimulates astrocytic K+ uptake (358, 361). In epileptic foci, elevated extracellular potassium concentrations due to impaired K+ uptake by astrocytes may cause hyperexcitability (following paragraphs) (362). Surprisingly, AQP4 expression is increased in the hippocampus of patients with TLE (329, 330). However, local expression of the protein at perivascular astrocytic endfeet is lost. This is due to downregulation of the dystrophin gene that encodes for the protein responsible for anchoring AQP4, which causes the AQP4 channel to be mislocalized (363, 364). Decrease of perivascular AQP4 channels has been shown to perturbate spatial potassium buffering (103, 365). In vivo models of acute epilepsy with AQP4−/− mice showed elevated seizure thresholds, which can be explained by the increase in extracellular space volume from impaired water uptake (366). In addition, prolonged seizure activity was measured, likely due to impaired K+ uptake (159, 331). Recently, it was shown that loss of perivascular AQP4 precedes seizure onset after kainic acid-induced epilepsy in rats, suggesting an involvement in epilepsy etiology (367). Taken together, these data suggest a that dysregulation of AQP4 plays an important role in epilepsy pathology.
Expression of other members of the AQP family such as AQP1 and AQP9 is also frequently reported to be altered in animal models of seizures (368), epilepsy (369), and traumatic brain injury (370, 371). In addition, expression changes in resected brain tissue of patients with TLE have been reported. Transcriptome and ELISA analysis showed that AQP1 expression is increased and AQP9 is decreased in human hippocampal sclerotic tissue compared to adjacent neocortex tissue (329). Moreover, AQP1 and AQP4 have been shown to play a role in cell growth and migration, and may be involved in glial scar formation (327, 372). Overexpression of AQP1 may therefore exacerbate disease progression by worsening astrogliosis. AQP9 is an aquaglyceroporin, meaning it is also able to shuttle glycerol, urea, and monocarboxylates such as lactate, suggesting that loss of AQP9 may disrupt local lactate levels which could affect neuronal excitability (327). Evidently, the functionality of aquaporins extends far beyond their primary function of channeling water molecules and it is important that the mechanisms behind these proteins are elucidated, to help understand their impact on epilepsy pathophysiology.
Inward Rectifying Potassium Channels
Under physiological conditions, the main inward rectifying potassium channel Kir4.1 is abundantly expressed in cortical astrocytes, as well as in the hippocampus and thalamus (373, 374). In addition, heteromeric channels of Kir4.1/5.1 are expressed in astrocytes of the forebrain. Furthermore, expression of several members of the Kir2 and Kir6 families have been reported in astrocytes (375, 376).
Kir4.1 has been shown to colocalize with AQP4, suggesting a functional role for water transport in relation to K+ buffering (104). In vitro experiments have shown that Kir4.1 is able to directly bind to α-syntrophin, a member of the dystrophin-complex that has been shown to assist in AQP4 localization (377, 378). However, expression and immunoreactivity of Kir4.1 is not altered in AQP4−/− mice, nor is AQP4 immunoreactivity altered in Kir4.1−/− mice, suggesting that functionality of neither transporter is fully dependent on the other (332, 379). Nevertheless, clearance of extracellular K+ by Kir4.1 is partially dependent on simultaneous water flux by AQP4, to enable proper osmolarity for K+ distribution and uptake. Recently, the synergy between AQP4 and Kir4.1 channel mediated K+ uptake has been validated by a mathematical model of neuroexcitation (380). Furthermore, in an experiment where heterologous AQP4 and Kir4.1/5.1 were co-expressed in Xenopus oocytes, cell shrinkage produced K+ currents, indicating another, more direct functional coupling between AQP4 and Kir channels (381).
During astrogliosis, proliferative astrocytes are shifted toward an immature phenotype in which they lose Kir4.1 and EAAT1 functionality, reducing spatial K+ buffering and impairing glutamate uptake (382, 383). Accordingly, Kir is often reported to be downregulated in animal models of epilepsy (71, 384). More specifically, loss of Kir immunoreactivity is located on astrocytic processes within epileptic foci, but not on astrocytes of the surrounding tissue (385). Furthermore, a significant loss of Kir4.1 immunoreactivity has been reported in resected hippocampal tissue of TLE patients (106). Interestingly, the loss of Kir4.1 was associated with loss of AQP4-associated proteins α-syntrophin and dystrophin, further emphasizing the link between Kir4.1, dystrophin-complex, and AQP4 localization. Decrease or loss of Kir4.1 or Kir4.1/5.1 channels undoubtedly cause perturbations in spatial K+ buffering, but functional mechanisms modulating epileptogenesis remain unidentified. Recently, it was shown that antagonism of Kir4.1 or suppression of Kir4.1 expression by siRNAs induces synthesis of BDNF (386). Expression of BDNF is upregulated in several animal models of epilepsy and in human epileptic disorders (387, 388). One way BDNF is proposed to promote seizures is by reduction of inhibitory synaptic transmission of GABAA receptor signaling (389). In addition, release of BDNF has been shown to downregulate expression of K+/Cl−-cotransporter (KCC2) (390, 391). Knock-out of KCC2 has been shown to induce hyperexcitability in mice (392). This shows that impaired Kir4.1 signaling may result in hyperexcitability by a multitude of mechanisms.
Other Potassium Channels
BK channels are expressed in astrocytic endfeet and they regulate vasodilation and vasoconstriction (Figure 2). BK channels are sensitive to calcium levels, membrane potential, and certain types of arachidonic acid (AA) metabolites which can lead to vasodilation or vasoconstriction (discussed in one of the following paragraphs) (358, 393). This is an important astrocytic property that supports the dynamic neuronal energy demand. In addition, calcium-dependent (BKCa) channels regulate K+ export from astrocytes, directly affecting local excitability (394).
Under physiological conditions, BK channels participate in the spatial K+ buffering that is required for normal brain function. A specific subunit of the protein (β4) prevents the channel to contribute to neuronal membrane repolarization, which allows the channel to protect the brain from hyperexcitability (395). A gain-of-function mutation or a loss of β4 subunit activity removes the protective function and is associated with epileptic seizures (396, 397). In animal models of epilepsy, the β4 subunit is downregulated and a switch is made in subtypes of the BK channels, resulting in faster gating (335, 398). BK blockers may reverse the adverse effects of β4 subunit loss and have been able to reduce action potential firing in brain slices of epileptic rats (399) and reduce seizure activity in vivo in mice (400). Recently, efforts were made to develop BK blockers, that selectively target BK channels with a gain-of-function mutation in the β4 subunit, posing as a new strategy for therapy aimed at patients with retractable epilepsy (397).
Specific roles for other K+ channels such as Kv or K2P channels (Figure 2) remain elusive. Downregulation of Kv channels in astrocytes upon seizure activity has been reported and agonists of Kv have been suggested as anticonvulsant therapies, but additional research is required to understand how Kv channels are involved in excitability (339, 401). K2P channels are open at rest and thereby aid Kir channels in driving the membrane potential of astrocytes to the K+ equilibrium, a feature thought to promote glutamate uptake (358, 402).
Sodium Channels
Although astrocytes are considered non-excitable cells (in the classical sense that they are unable to produce action potentials), they dynamically express all 9 isoforms of Nav sodium channels, with Nav1.5 as the main voltage-gated sodium channel (VGSC) (403). Expression of VGSCs is increased upon brain insult and during astrogliosis and appear to assist via a mechanism involving NCX (403–405). Little is known about the exact function of sodium channels in astrocytes, but it is believed that continuous Na+ influx is required to maintain [Na+]i for activity of Na+/K+-ATPase [(406); Figure 2]. Na+/K+-ATPase assists in extracellular K+ buffering by uptake of K+ while simultaneously releasing Na+ at the cost of ATP. In fact, Na+/K+-ATPase, rather than Kir4.1, is responsible for most of the net uptake of K+. Changes in the activity of Na+/K+-ATPase have been proposed as an underlying mechanism for epilepsy (340, 407). Mutations in the gene encoding Na+/K+-ATPase were shown to cause seizure activity in animals (408) and patients with epilepsy (409).
Another mechanism supporting spatial K+ buffering is through the Na+/K+/Cl− co-transporter (NKCC1; Figure 2), which has been shown to play a role in astrocytic swelling under conditions of high extracellular K+ (410–412). This is another example stressing the importance of the volume regulation of the extracellular space. Na+-transport can also be facilitated by members of the transient receptor potential (TRP) family, including “ankyrin” TRPA1, “canonical” TRPC1, TRPC4, TRPC5, and “vanilloid” TRPV4 receptors (413, 414). In addition to VGSCs and ion cotransporters, Na+ is transported over the membrane in conjunction with various other mechanisms such as glutamate uptake by EAAT1 (415), glutamine export by Na+/H+-coupled neutral amino acid transporters (SN1) and SN2 (416), and lactate shuttling by Na+/K+-ATPase (417).
Calcium Transporters
Many astrocytes functions occur in response to focal or global Ca2+ transients. Therefore, a tight regulation of intra- and extracellular levels of Ca2+ is vital for homeostatic astrocytic functionality. Ca2+ can permeate the membrane through a variety of channels, including plasmalemmal Ca2+-ATPase (PMCA), TRPA1, TRPC1, TRPC4, TRPC5, TRPV1, ionotropic glutamate receptors AMPA and NMDA, purinergic receptors (i.e., P2X7) and by several ion exchangers of which the NCX is the most relevant [(153); Table 1]. Of note, astrocytes express all 3 isoforms of NCX and it has been shown that NCX colocalizes with Na+/K+-ATPase and glutamate receptors (418).
One mechanism in which focal Ca2+ transients in astrocytes regulate brain homeostasis is mediated by TRPA1. Influx of Ca2+ by TRPA1 regulates GABAergic transmission via the astrocytic GABA3 transporter (419) and D-serine release (420).
TRPCs are involved in store-operated Ca2+ entry and have been shown to contribute to Ca2+-mediated glutamate release in astrocytes (413). On the other hand, glutamate can activate astrocytic NMDA receptors and thereby induce Ca2+ influx, although they are ~2 times less permeable than their neuronal counterparts (421, 422).
Chloride Transporters
Anions are also transported across astrocytic membranes. Astrocytes express different isoforms of potassium-chloride and cation-chloride cotransporters of the solute carrier 12 (SLC12) gene family, which include NKCC1, Na+/Cl−-cotransporter (NCC) and KCC1, KCC3, KCC4 (423–425). Mounting evidence suggests KCC2 is neuron-specific, but some experimental data shows that KCC2 may be present in astrocytes (424, 425). The main role of KCCs in astrocytes is volume regulation, whereas in neurons they regulate membrane potential by keeping intracellular Cl− levels low, to enable GABAergic transmission (425, 426).
Astrogliosis causes a downregulation of KCC2 and NKCC1 in cortical pyramidal neurons, thereby preventing the Cl− gradient required for GABAergic transmission (287). In contrast, increased expression of NKCC1 has been found in hippocampal sclerotic tissue of patients with TLE (427). During the development of neurons, the ratio between KCC2 and NKCC1 changes, as KCC2 is upregulated and NKCC1 is downregulated in mature neurons (428). Considering that astrocytes may express both KCC2 and NKCC1, and at the same time appear to differentiate to an immature state during astrogliosis, it is plausible that this change in expression is also reversed in astrocytes in the sclerotic hippocampus. The shift in expression of KCC2 and NKCC1 has been shown in the subiculum of TLE patients, but is yet to be confirmed in astrocytes specifically (341).
Antagonism of NKCC1 reduces seizure frequency in patients with TLE (429). Interestingly, inhibition of NKCC1 with the diuretic bumetanide does not influence K+ buffering post-stimulation (430). In this study it was found that neither Kir4.1 nor NKCC1 inhibition changed K+ buffering after neuronal activation, but that Na+/K+-ATPase was mostly responsible for the post-stimulation K+ uptake. Nevertheless, the development of selective NKCC1 inhibitors may prove rewarding in the clinic (431).
Blood-Brain Barrier Dysfunction
The BBB functions as a physical barrier to protect the brain from toxins, undesirable metabolites and ions that could permeate the brain from the blood stream. The BBB is comprised of endothelial cells that are connected via tight junctions [(28); Figure 4]. This physical barrier is considered the “first line of defence” for the brain. Astrocytes ensheath with their endfeet the endothelial cells (Figure 2) and serve as a “second line of defence.” Together with neurons, other glia cells and mural cells, they form the neurovascular unit. The main function of astrocytes at the BBB is the control of nutrient exchange with the bloodstream and maintaining BBB integrity (432).
Figure 4.
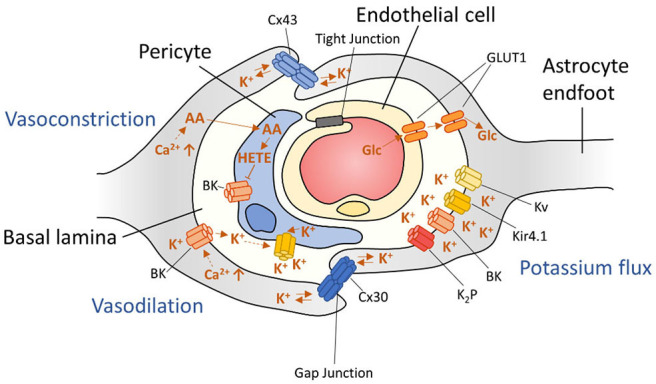
Schematic representation of arteriolar regulation at the neurovascular unit. Astrocyte signaling is able to influence neuronal vascularity by inducing both vasoconstriction and vasodilation through mechanisms that involve BK channels.
Endothelial cells at the BBB express several transporter proteins and channels such as GLUT1, several amino acid carriers including EAAT1, EAAT2, EAAT3, and L-system for large neutral amino acids (LAT1), specific transporters (i.e., for nucleosides, nucleobases), non-specific transporters, such as multidrug transporters (MDTs), and organic ion transporters (28, 433). Importantly, astrocytes are able to alter the expression or activity of endothelial transporters including GLUT1 (434, 435) and MDTs such as P-glycoprotein (P-gp) (436, 437). Moreover, astrocytes may affect BBB permeability directly by changing the density of tight junctions (438, 439), for instance through release of angiopoietin 1 and 2, ATP, endothelin-1, fibroblast growth factor, glial cell line-derived neurotrophic factor (GDNF), glutamate, retinoic acid, nitric oxide or VEGF (440, 441).
Astrogliosis and neuroinflammation can lead to BBB dysfunction. Under inflammatory conditions, bradykinin is released in the blood, increasing BBB permeability by acting on endothelial (B2) receptors (442). In addition, bradykinin induces IL-6 release from astrocytes through activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (443) resulting in modulation of endothelial tight junctions (77). Moreover, following brain injury astrocyte-mediated inflammation causes transient opening of the BBB (444). BBB dysfunction is common in epilepsy and can contribute to the development and progression of epilepsy (365, 445–448). In the following paragraphs we will discuss several mechanisms by which BBB dysfunction contributes to epilepsy pathophysiology.
VEGF Signaling
Downregulation or loss of the proteins that make up tight junctions, such as zonula occludens (ZO-1), occludin, and claudins results in opening of the BBB. Loss of tight junctions is shown to be caused by increased expression of the VEGF receptor 1 and 2 in a rat model of pilocarpine-induced epilepsy (449). In addition, in an animal model of kainic acid-induced epilepsy it was shown that upregulation of VEGF-R1 and VEGF-R2 caused downregulation of ZO-1 (450). Furthermore, astrocyte-released VEGF has been shown to downregulate tight junction proteins claudin-5 and occludin (44). Several studies report increased VEGF release and receptor expression in patients suffering from refractory TLE (449, 451). The primary role of VEGF is to induce angiogenesis, which is correlated with seizure frequency (449). Angiogenesis and down-regulation of ZO-1 could be reversed by neutralization of VEGF, suggesting that VEGF signaling is involved in BBB dysfunction. This was demonstrated by oral administration of the VEGF pathway inhibitor sunitinib, which prevented seizures and epilepsy development in pilocarpine-induced seizures in rats, showing the potential of anti-angiogenesis therapies.
Albumin Leakage
In epilepsy, angiogenesis is spatially correlated to leakage of serum proteins into the brain parenchyma (452). Under pathophysiological conditions, BBB leakage exposes the brain to plasma proteins such as IgG and albumin (445, 449, 453). Subsequently, astrocytes are able to internalize serum albumin by binding to the TGF-β receptors, inducing epileptogenesis via a mechanism similar to TGF-β1 induced TGF-β signaling (453). Importantly, TGF-β1 was upregulated during gliosis in periods after SE (454). Furthermore, TGF-β1 has been shown to downregulate Kir2.3 in reactive astrocytes (455). In turn, albumin-induced TGF-β signaling causes impaired GJ coupling and down-regulates Kir4.1 (453). This shows that TGF-β signaling interferes with potassium buffering in at least two distinct mechanisms involving inward rectifying channels. In addition, albumin extravasation into the brain has been shown to (1) transiently affect GJ coupling (456), (2) induce GFAP expression (457), (3) upregulate pro-inflammatory cytokine IL-6 (458), (4) reduce astrocyte potassium and glutamate clearance (459), and (5) induce excitatory, but not inhibitory synaptogenesis, contributing to potential hyperexcitability (460). Together, these data show that BBB dysfunction can promote epileptogenesis.
Multidrug Transporters
BBB opening is associated with increased expression of MDTs (448, 461). Several studies have reported upregulation of genes encoding for MDTs, including P-gp (MDR1 gene), multidrug resistance protein (MRP) 1, MRP2, MRP5, and breast cancer resistance protein (BRCP) in the epileptogenic brain (448, 462–465). Overexpression of MDTs is shown in endothelial cells, but also in astrocytic endfeet and neurons. These transporters have been shown to affect drug transport in the brain and it has been proposed that drug resistance in patients with refractory epilepsy may be due to changes in MDTs (466–468). For instance, increased expression of P-gp causes enhanced efflux of AEDs, impairing AED entry into the brain. Indeed, P-gp blockers can increase AED levels in the brain and overcome pharmacoresistance in animal models of epilepsy (469–471), suggesting co-administration of antiepileptic drugs and a P-gp blocker may prove useful in patients as well. In contrast, recent experiments based on measurements of extracellular fluid unbound drug concentrations and mathematical models predicting drug target site concentrations, suggest that P-gp expression does not translate to BBB permeability for all AEDs, as other factors may affect target-site concentration more profoundly, such as brain tissue binding (472). Moreover, it is unclear whether changes in MDT expression are different between various animal models or correlate to different types of disease progression. Patient data confirms these speculations as it appears that therapeutic success appears to be influenced by the heterogeneity of the etiology of the seizures (473, 474). On the other hand, mounting evidence from patients that were co-administered with AEDs and P-gp blockers (i.e., Verapamil) show improved clinical outcome compared to AED only treatment (475–478).
Arteriolar Blood Flow Regulation
Astrocytes also aid in the local regulation of vasoconstriction and vasodilation. Changes in intracellular Ca2+ at astrocytic endfeet can induce two major arteriolar regulating pathways: (1) The cytoplasmic phospholipase A2 (PLA2) pathway and (2) BK channel mediated vascular control [(393, 479); Figure 4]. Increased PLA2 activity results in production of AA, which is metabolized into various vasoactive compounds and is also able to pass the cell membrane to pericytes. Inside pericytes, AA is then metabolized into the vasoconstrictive 20-hydroxyeicosa-tetraenoic acid (20-HETE).
Different concentrations of Ca2+ in astrocytic endfeet are also able to regulate arteriolar dilation or vasoconstriction by induction of BK-channels that release K+ in the perivascular space—a space formed by the envelopment of astrocytic processes around arterioles (31, 393). In addition, BK channels respond to components of the PLA2 pathway, such as 20-hydroxyeicosatetraenoic acid (20-HETE), epoxy-eicosatetraenoic acids (EET)s, and prostaglandin E2 (PGE2). Importantly, these mechanisms are not mutually exclusive, and even overlap. Efforts of blocking either pathway individually did not result in total impairment of vascular control, emphasizing the extent of vascular control for homeostatic brain function (393). Together, these mechanisms can regulate cerebral blood-flow in the brain.
During epileptic seizures cerebral blood-flow and also cerebral blood volume are transiently increased as a response to the high energy demand of neurons (135). However, these increases are not sufficient to meet metabolic demands of synchronously activated neurons during ictal events. Several studies have found impaired neurovascular coupling in epilepsy (135, 480, 481). In some studies, this is correlated to hypoxia-induced tissue damage. Others suggest the possibility to predict ictal events based on increased cerebral perfusion preceding seizure onset in the clinic (482). Vast Ca2+ waves at astrocytic endfeet recorded during ictal events have been shown to regulate local arteriole responses, and these effects could be blocked by pharmacological inhibition of the Ca2+ signals in astrocytic processes (483). Although the underlying mechanisms behind the regulation of cerebral microcirculation in epilepsy are poorly defined, these data emphasize how astrocytes may control the neuronal micro environment during seizures.
Concluding Remarks
Under physiological conditions, astrocytes protect neurons from becoming hyperexcitable. However, under pathophysiological conditions found before and during epilepsy, the evident and complex involvement of astrocytes in the neuronal network is perturbed. In this review we showed how aberrant astrocytic signaling and changes in astrocyte function contribute to the development and aggravation of epilepsy.
Despite an abundance of clues in the vast literature on the mechanistic involvement of astrocytes in epilepsy, there are presently no drugs in the clinic that target these mechanisms. In the near future it is imperative that we continue the development of drugs that specifically target mechanisms that are underlying the etiology of epilepsy and also focus on astrocytes as novel therapeutic targets. So far, pioneering preclinical studies have shown promising results.
Interestingly, recently it was suggested that astrocytes may also be used as biomarkers for epileptogenesis (484, 485). In this review the recently published evidence was reported, supporting the utility of measuring astrocyte activation, the soluble molecules they release, and the associated cognitive deficits during epileptogenesis for early stratification of animals developing epilepsy. Whether this may also be of clinical use needs to be investigated.
Author Contributions
QV and LH developed the concept and prepared the first draft. EV and EA provided feedback and contributed to the writing of the manuscript together with QV and LH. All authors read, revised, and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie Grant Agreement No. 722053 (EU-GliaPhD; EA and EV).
References
- 1.Hesdorffer DC, Rochester A, Katri N, Cascino G, Hauser W. Estimating risk for developing epilepsy. Neurology. (2010) 76:23–7. 10.1212/WNL.0b013e318204a36a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baulac M, De Boer H, Elger C, Glynn M, Kälviäinen R, Little A, et al. Epilepsy priorities in Europe: a report of the ILAE-IBE epilepsy advocacy Europe task force. Epilepsia. (2015) 56:1687–95. 10.1111/epi.13201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Am Soc Exp Neurother. (2010) 7:338–53. 10.1016/j.nurt.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boldog E, Bakken TE, Hodge RD, Novotny M, Aevermann BD, Baka J, et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat Neurosci. (2018) 21:1185–95. 10.1038/s41593-018-0205-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jäkel S, Dimou L. Glial cells and their function in the adult brain : a journey through the history of their ablation. Front Cell Neurosci. (2017) 11:24. 10.3389/fncel.2017.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bahney J, Herculano-Houzel S, Biology C. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. (2017) 524:3865–95. 10.1002/cne.24040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herculano-Houzel S, Miller DJ, Kaas J, Biology C. How to count cells: the advantages and disadvantages of the isotropic fractionator compared with stereology. Cell Tissue Res. (2016) 360:29–42. 10.1007/s00441-015-2127-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herculano-Houzel S. The glia/neuron ratio : how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia. (2014) 62:1377–91. 10.1002/glia.22683 [DOI] [PubMed] [Google Scholar]
- 9.Oberheim NA, Takano T, Han X, He W, Lin JHC, Wang F, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. (2009) 29:3276–87. 10.1523/JNEUROSCI.4707-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robel S, Sontheimer H. Glia as drivers of abnormal neuronal activity. Nat Neurosci. (2016) 19:28–33. 10.1038/nn.4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, et al. Molecular architecture of the mouse nervous system. Cell. (2018) 174:999–1014. 10.1016/j.cell.2018.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batiuk MY, Martirosyan A, Wahis J, de Vin F, Marneffe C, Kusserow C, et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun. (2020) 11:1220. 10.1038/s41467-019-14198-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.John Lin CC, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci. (2017) 20:396–405. 10.1038/nn.4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanjakornsiripan D, Pior BJ, Kawaguchi D, Furutachi S, Tahara T, Katsuyama Y, et al. Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat Commun. (2018) 9:1623. 10.1038/s41467-018-03940-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duran RCD, Wang CY, Zheng H, Deneen B, Wu JQ. Brain region-specific gene signatures revealed by distinct astrocyte subpopulations unveil links to glioma and neurodegenerative diseases. eNeuro. (2019) 6:1–19. 10.1523/ENEURO.0288-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vezzani A, Masa L, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. (2011) 7:31–40. 10.1038/nrneurol.2010.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. (2013) 36:174–84. 10.1016/j.tins.2012.11.008 [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Qin C, Huang J, Tang X, Liu C, Huang K, et al. The role of astrocytes in oxidative stress of central nervous system: a mixed blessing. Cell Prolif. (2020) 53:1–13. 10.1111/cpr.12781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellerin L, Bouzier-Sore A, Aubert A, Serres S, Merle M, Costalat R, et al. Activity-dependent regulation of energy metabolism by astrocytes an update. Glia. (2007) 55:1251–62. 10.1002/glia.20528 [DOI] [PubMed] [Google Scholar]
- 20.Falkowska A, Gutowska I, Goschorska M, Nowacki P. Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int J Mol Sci. (2015) 16:25959–81. 10.3390/ijms161125939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boison D, Steinhäuser C. Epilepsy and astrocyte energy metabolism. Glia. (2018) 66:1235–43. 10.1016/j.physbeh.2017.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunel N, Volterra A, Pitta MDE. Astrocytes: orchestrating synaptic plasticity? Neuroscience. (2016) 323:43–61. 10.1016/j.neuroscience.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 23.Hussaini SMQ, Jang MH. New roles for old glue : astrocyte function in synaptic plasticity and neurological disorders. Int Neurourol J. (2018) 22:106–14. 10.5213/inj.1836214.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. (2005) 6:626–40. 10.1038/nrn1722 [DOI] [PubMed] [Google Scholar]
- 25.Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. (2006) 98:641–53. 10.1111/j.1471-4159.2006.03913.x [DOI] [PubMed] [Google Scholar]
- 26.Haj-Yasein NN, Jensen V, Ostby I, Omholt SW, Voipio J, Kaila K, et al. Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation A gene deletion study in mouse hippocampus. Glia. (2012) 60:867–74. 10.1002/glia.22319 [DOI] [PubMed] [Google Scholar]
- 27.Vasile F, Dossi E, Rouach N. Human astrocytes : structure and functions in the healthy brain. Brain Struct Funct. (2017) 222:2017–29. 10.1007/s00429-017-1383-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. (2006) 7:41–53. 10.1038/nrn1824 [DOI] [PubMed] [Google Scholar]
- 29.Serlin Y, Shelef I, Knyazer B, Friedman A, Biology C, Sciences B. Anatomy and physiology of the blood-brain barrier yonatan. Semin Cell Dev Biol. (2015) 38:2–6. 10.1016/j.semcdb.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon GRJ, Mulligan SJ, Vicar BAMAC. Astrocyte control of the cerebrovasculature. Glia. (2007) 1221:1214–21. 10.1002/glia.20543 [DOI] [PubMed] [Google Scholar]
- 31.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. (2007) 10:1369–76. 10.1038/nn2003 [DOI] [PubMed] [Google Scholar]
- 32.Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. (2015) 7:1–16. 10.1101/cshperspect.a020420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Escartin C, Guillemaud O, Carrillo-de Sauvage MA. Questions and (some) answers on reactive astrocytes. Glia. (2019) 67:2221–47. 10.1002/glia.23687 [DOI] [PubMed] [Google Scholar]
- 34.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kettenmann H, Ransom BR. Neuroglia. 3rd ed New York, NY: Oxford University Press; (2013). [Google Scholar]
- 36.Robel S. Astroglial scarring and seizures : a cell biological perspective on epilepsy. Neurosci. (2017) 23:152–68. 10.1177/1073858416645498 [DOI] [PubMed] [Google Scholar]
- 37.Brambilla R, Bracchi-Ricard V, Hu W, Frydel B, Bramwell A, Karmally S, et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. (2005) 202:145–56. 10.1084/jem.20041918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with alzheimer's disease. Neuron. (2015) 85:101–15. 10.1016/j.neuron.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, et al. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci USA. (2005) 102:16466–71. 10.1073/pnas.0506382102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med. (2014) 20:886–96. 10.1038/nm.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu Z, Guo Z, Gearing M, Chen G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer's disease model. Nat Commun. (2014) 5:4159. 10.1038/ncomms5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamby ME, Hewett JA, Hewett SJ. TGF-β1 potentiates astrocytic nitric oxide production by expanding the population of astrocytes that express NOS-2. Glia. (2006) 54:566–77. 10.1002/glia.20411 [DOI] [PubMed] [Google Scholar]
- 43.Chou SY, Weng JY, Lai HL, Liao F, Sun SH, Tu PH, et al. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J Neurosci. (2008) 28:3277–90. 10.1523/JNEUROSCI.0116-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tadesse A, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. (2009) 106:1977–82. 10.1073/pnas.0808698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocr. (2012) 33:116–25. 10.1016/j.yfrne.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. (2015) 96:70–82. 10.1016/j.neuropharm.2014.10.027 [DOI] [PubMed] [Google Scholar]
- 47.Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. (2014) 20:160–72. 10.1177/1073858413504466 [DOI] [PubMed] [Google Scholar]
- 48.Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. (2019) 15:459–72. 10.1038/s41582-019-0217-x [DOI] [PubMed] [Google Scholar]
- 49.Lau LOKT, Yu AC. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. (2001) 18:351–9. [DOI] [PubMed] [Google Scholar]
- 50.Hyvärinen T, Hagman S, Ristola M, Sukki L, Vijula K, Kreutzer J, et al. Co-stimulation with IL-1β and TNF-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci Rep. (2019) 9:16944 10.1038/s41598-019-53414-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bianco F, Pravettoni E, Colombo A, Möller T, Matteoli M, Bianco F, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol. (2005) 174:7268–77. 10.4049/jimmunol.174.11.7268 [DOI] [PubMed] [Google Scholar]
- 52.Terrone G, Balosso S, Pauletti A, Ravizza T, Vezzani A. Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology. (2019) 167:107742. 10.1016/j.neuropharm.2019.107742 [DOI] [PubMed] [Google Scholar]
- 53.Minami M, Kuraishi Y, Satoh M. Effects of kainic acid on messenger RNA levels of IL-1b, IL-6, TNFa and LIFE in the rat brain. Biochem Biophys Res Commun. (1991) 176:593–8. 10.1016/s0006-291x(05)80225-6 [DOI] [PubMed] [Google Scholar]
- 54.Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, et al. Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci. (1999) 19:5054–65. 10.1523/jneurosci.19-12-05054.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simoni MG, De Perego C, Ravizza T, Moneta D, Conti M, Marchesi F, et al. Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci. (2000) 12:2623–33. [DOI] [PubMed] [Google Scholar]
- 56.Merbl Y, Sommer A, Chai O, Aroch I, Zimmerman G, Friedman A, et al. Tumor necrosis factor- a and interleukin-6 concentrations in cerebrospinal fluid of dogs after seizures. J Vet Intern Med. (2014) 28:1775–81. 10.1111/jvim.12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patel DC, Wallis G, Dahle EJ, McElroy PB, Thomson KE, Tesi RJ, et al. Hippocampal TNFα signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. Disord Nerv Syst Hippocampal. (2017) 4:1–20. 10.1523/ENEURO.0105-17.20171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Semple BD, Brien TJO, Gimlin K, Wright DK, Kim SE, Casillas-Espinosa PM, et al. Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J Neurosci. (2017) 37:7864–77. 10.1523/JNEUROSCI.0982-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kostic D, Carlson R, Henke D, Rohn K, Tipold A. Evaluation of IL-1β levels in epilepsy and traumatic brain injury in dogs. BMC Neurosci. (2019) 20:29. 10.1186/s12868-019-0509-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ichiyama T, Nishikawa M, Yoshitomi T, Furukawa S. Tumor necrosis factor-alfa, interleukin-1beta, and interleukin-6 in cerebrospinal fluid from children with prolonged febrile seizures comparison with acute encephalitis/encephalopathy. Neurology. (1998) 50:407–12. [DOI] [PubMed] [Google Scholar]
- 61.Peltola J, Palmio J, Korhonen L, Suhonen J, Miettinen A. Interleukin-6 and interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. (2000) 41:205–11. [DOI] [PubMed] [Google Scholar]
- 62.Uludag I, Duksal T, Tiftikcioglu B, Zorlu Y, Ozkaya F, Guldal K. IL-1β, IL-6 and IL1Ra levels in temporal lobe epilepsy. Seizure Eur J Epilepsy. (2015) 26:22–5. 10.1016/j.seizure.2015.01.009 [DOI] [PubMed] [Google Scholar]
- 63.Alapirtti T, Lehtimäki K, Nieminen R, Mäkinen R, Raitanen J, Moilanen E, et al. The production of IL-6 in acute epileptic seizure: a video-EEG study. J Neuroimmunol. (2017) 316:50–55. 10.1016/j.jneuroim.2017.12.008 [DOI] [PubMed] [Google Scholar]
- 64.Wang SAM, Cheng Q, Malik S, Yang JAY. Interleukin-1beta Inhibits gamma-aminobutyric acid type A (GABA A) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther. (2000) 292:497–504. [PubMed] [Google Scholar]
- 65.Yang S, Liu Z, Wen L, Qiao H, Zhou W, Zhang Y. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. (2005) 1034:172–9. 10.1016/j.brainres.2004.11.018 [DOI] [PubMed] [Google Scholar]
- 66.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. (2003) 23:8692–700. 10.1523/jneurosci.23-25-08692.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1β. Brain. (2008) 131:3256–65. 10.1093/brain/awn271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scheppingen J, Van Iyer AM, Prabowo AS, Muhlebner A, Giordano F, Genitori L, et al. Expression of MicroRNAs miR21, miR146a, and miR155 in tuberous sclerosis complex cortical tubers and their regulation in human astrocytes and SEGA-derived cell cultures. Glia. (2016) 64:1066–82. 10.1002/glia.22983 [DOI] [PubMed] [Google Scholar]
- 69.Scheppingen J, Van Mills JD, Zimmer TS, Broekaart DWM, Iori V, Bongaarts A, et al. miR147b: a novel key regulator of interleukin 1 beta-mediated inflammation in human astrocytes. Glia. (2018) 66:1082–97. 10.1002/glia.23302 [DOI] [PubMed] [Google Scholar]
- 70.Korotkov A, Baayen JC, Aronica E, Banchaewa L, Anink JJ, Vliet EA, et al. microRNA-132 is overexpressed in glia in temporal lobe epilepsy and reduces the expression of pro-epileptogenic factors in human cultured astrocytes. Glia. (2020) 68:60–75. 10.1002/glia.23700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zurolo E, de Groot M, Iyer A, Anink J, van Vliet EA, Heimans JJ, et al. Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: a role for interleukin-1 β. J Neuroinflamm. (2012) 9:1–17. 10.1186/1742-2094-9-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korotkov A, Broekaart DWM, Scheppingen J, Van Anink JJ, Baayen JC, Idema S, et al. Increased expression of matrix metalloproteinase 3 can be attenuated by inhibition of microRNA-155 in cultured human astrocytes. J Neuroinflam. (2018) 15:1–14. 10.1186/s12974-018-1245-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ye ZC, Sontheimer H. Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport. (1996) 7:2181–5. 10.1097/00001756-199609020-00025 [DOI] [PubMed] [Google Scholar]
- 74.Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. (2000) 7:153–9. 10.1159/000026433 [DOI] [PubMed] [Google Scholar]
- 75.Pita I, Jelaso AM, Ide CF. IL-1β increases intracellular calcium through an IL-1 type 1 receptor mediated mechanism in C6 astrocytic cells. Int J Devl Neurosci. (1999) 17:813–20. 10.1016/S0736-5748(99)00063-5 [DOI] [PubMed] [Google Scholar]
- 76.Sanz P, Garcia-Gimeno MA. Reactive glia inflammatory signaling pathways and epilepsy. Int J Mol Sci. (2020) 21:1–17. 10.3390/ijms21114096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J, Sadowska GB, Chen X, Park SY, Kim JE, Bodge CA, et al. Anti-IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus. FASEB J. (2015) 29:1739–53. 10.1096/fj.14-258822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fine SM, Angel RA, Seth W, Epstein LG, Rothstein JD, Dewhurst S, et al. Tumor necrosis factor α inhibits glutamate uptake by primary human astrocytes. J Biol Chem. (1996) 271:15303–6. 10.1074/jbc.271.26.15303 [DOI] [PubMed] [Google Scholar]
- 79.Clark IA, Vissel B. Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflamm. (2016) 13:1–16. 10.1186/s12974-016-0708-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bedner P, Steinhäuser C. TNFα-driven astrocyte purinergic signaling during epileptogenesis. Trends Mol Med. (2019) 25:70–2. 10.1016/j.molmed.2018.12.001 [DOI] [PubMed] [Google Scholar]
- 81.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. (2005) 25:3219–28. 10.1523/JNEUROSCI.4486-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aronica E, Crino PB. Inflammation in epilepsy: clinical observations. Epilepsia. (2011) 52:26–32. 10.1111/j.1528-1167.2011.03033.x [DOI] [PubMed] [Google Scholar]
- 83.Das A, Iv GCW, Holmes C, Mcdowell ML, Smith JA, Marshall JD, et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience. (2012) 220:237–46. 10.1016/j.neuroscience.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ravizza T, Lucas S, Balosso S, Bernardino L, Ku G, No F, et al. Inactivation of Caspase-1 in rodent brain : a novel anticonvulsive strategy. Epilepsia. (2006) 47:1160–8. 10.1111/j.1528-1167.2006.00590.x [DOI] [PubMed] [Google Scholar]
- 85.Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, et al. Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. J Am Soc Exp Neurother. (2011) 8:304–15. 10.1007/s13311-011-0039-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bialer M, Johannessen SI, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the eleventh eilat conference (EILAT XI). Epilepsy Res. (2013) 103:2–30. 10.1016/j.eplepsyres.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 87.Lafrance-Corey RG, Ho M, Muskardin TW. Super-refractory status epilepticus and febrile infection-related epilepsy syndrome treated with anakinra. Ann Neurol. (2016) 80:939–45. 10.1002/ana.24806.Super-refractory [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kenney-Jung DL, Vezzani A, Kahoud RJ, Lafrance-Corey RG, Ho M, Muskardin TW, et al. FIRES induced status epilepticus treated with anakinra. Ann Neurol. (2016) 80:939–45. 10.1002/ana.24806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dilena R, Mauri E, Aronica E, Bernasconi P, Bana C, Cappelletti C, et al. Therapeutic effect of Anakinra in the relapsing chronic phase of febrile infection – related epilepsy syndrome. Epilepsia Open. (2019) 4:344–50. 10.1002/epi4.12317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Westbrook C, Subramaniam T, Seagren RM, Tarula E, Co D, Furstenberg-Knauff M, et al. Febrile infection-related epilepsy syndrome (FIRES) treated successfully with anakinra in A 21-year-old woman. WMJ. (2019) 118:135–9. 10.1111/j.1528-1167.2011.03250.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jyonouchi H, Geng L. Intractable epilepsy (IE) and responses to anakinra, a human recombinant IL-1 receptor agonist (IL-1ra): case reports. J Clin Cell Immunol. (2016) 7:1–5. 10.4172/2155-9899.1000456 [DOI] [Google Scholar]
- 92.Desena AD, Do T, Schulert GS. Systemic autoinflammation with intractable epilepsy managed with interleukin-1 blockade. J Neuroinflamm. (2018) 15:1–6. 10.1186/s12974-018-1063-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Steinborn B, Zarowksi M, Winczewska-Wiktor A, Wójcicka M, Mlodzikowska-Albrecht J, Losy J. Concentration of Il-1β, Il-2, Il-6, TNFα in the blood serum in children with generalized epilepsy treated by valproate. Pharmacol Rep. (2014) 66:972–5. 10.1016/j.pharep.2014.06.005 [DOI] [PubMed] [Google Scholar]
- 94.Lagarde S, Villeneuve N, Lepine A, Mcgonigal A, Roubertie A, Barthez MJ, et al. Anti – tumor necrosis factor alpha therapy (adalimumab) in rasmussen's encephalitis: an open pilot study. Epilepsia. (2016) 57:956–66. 10.1111/epi.13387 [DOI] [PubMed] [Google Scholar]
- 95.Frigerio F, Pasqualini G, Craparotta I, Marchini S, Vliet EA, Van Foerch P, et al. n-3 Docosapentaenoic acid-derived protectin D1 promotes resolution of neuroinflammation and arrests epileptogenesis. Brain. (2018) 141:3130–43. 10.1093/brain/awy247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Puttachary S, Sharma S, Verma S, Yang Y, Putra M, Thippeswamy A, et al. 1400W, a highly selective inducible nitric oxide synthase inhibitor is a potential disease modifier in the rat kainate model of temporal lobe epilepsy. Neurobiol Dis. (2016) 93:184–200. 10.1016/j.nbd.2016.05.013 [DOI] [PubMed] [Google Scholar]
- 97.Boison D, Aronica E. Comorbidities in neurology: is adenosine the common link? Neuropharmacology. (2015) 97:18–34. 10.1016/j.gde.2016.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pekny M, Pekna M, Messing A, Steinhäuser C, Lee JM, Parpura V, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. (2016) 131:323–45. 10.1007/s00401-015-1513-1 [DOI] [PubMed] [Google Scholar]
- 99.Binder DK. Astrocytes: stars of the sacred disease. Epilepsy Curr. (2018) 18:172–9. 10.5698/1535-7597.18.3.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eid T, Thomas MJ, Spencer DD, Lai JCK, Malthankar GV, Kim JH, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus : possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. (2004) 363:28–37. 10.1016/s0140-6736(03)15166-5 [DOI] [PubMed] [Google Scholar]
- 101.Gouder N, Scheurer L, Fritschy J, Boison D. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci. (2004) 24:692–701. 10.1523/JNEUROSCI.4781-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Aronica E, Zurolo E, Iyer A, Groot M, De Anink J. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia. (2012) 52:1645–55. 10.1111/j.1528-1167.2011.03115.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, Lanerolle NC, De Nagelhus EA, et al. Delayed K+ clearance associated with aquaporin-4 mislocalization : phenotypic defects in brains of alfa-syntrophin-null mice. Proc Natl Acad Sci USA. (2003) 100:13615–20. 10.1073/pnas.2336064100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Binder DK, Nagelhus EA, Ottersen OLEP. Aquaporin-4 and epilepsy. Glia. (2012) 1214:1203–14. 10.1002/glia.22317 [DOI] [PubMed] [Google Scholar]
- 105.Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. (1998) 32:286–303. [DOI] [PubMed] [Google Scholar]
- 106.Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Tauboll E, et al. Loss of perivascular kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol Exp Neurol. (2012) 71:814–25. 10.1097/NEN.0b013e318267b5af [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lauritzen F, Heuser K, Lanerolle NCDE, Lee TW, Spencer DD, Kim JH, et al. Redistribution of monocarboxylate transporter 2 on the surface of astrocytes in the human epileptogenic hippocampus. Gila. (2012) 1181:1172–81. 10.1002/glia.22344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lauritzen F, Perez EL, Melillo ER, Roh J, Zaveri HP, Lee TW, et al. Neurobiology of disease altered expression of brain monocarboxylate transporter 1 in models of temporal lobe epilepsy. Neurobiol Dis. (2012) 45:165–76. 10.1016/j.nbd.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Escartin C, Pierre K, Colin A, Brouillet E, Delzescaux T, Guillermier M, et al. Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci. (2007) 27:7094–104. 10.1523/JNEUROSCI.0174-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gavillet M, Allaman I, Magistretti PJ. Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia. (2008) 56:975–89. 10.1002/glia.20671 [DOI] [PubMed] [Google Scholar]
- 111.Valenza M, Leoni V, Karasinska JM, Petricca L, Fan J, Carroll J, et al. Cholesterol defect is marked across multiple rodent models of huntington's disease and is manifest in astrocytes. J Neurosci. (2010) 30:10844–50. 10.1523/JNEUROSCI.0917-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rossi D, Volterra A. Astrocytic dysfunction: Insights on the role in neurodegeneration. Brain Res Bull. (2009) 80:224–32. 10.1016/j.brainresbull.2009.07.012 [DOI] [PubMed] [Google Scholar]
- 113.Escartin C, Rouach N. Astroglial networking contributes to neurometabolic coupling. Front Neuroenergetics. (2013) 5:4. 10.3389/fnene.2013.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pannasch U, Rouach N. Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci. (2013) 36:405–17. 10.1016/j.tins.2013.04.004 [DOI] [PubMed] [Google Scholar]
- 115.Escartin C, Brouillet E, Gubellini P, Trioulier Y, Jacquard C, Smadja C, et al. Ciliary neurotrophic factor activates astrocytes, redistributes their glutamate transporters GLAST and GLT-1 to raft microdomains, and improves glutamate handling in vivo. J Neurosci. (2006) 26:5978–89. 10.1523/JNEUROSCI.0302-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. (2007) 51:333–55. 10.1016/j.neuint.2007.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tian G, Azmi H, Takano T, Xu Q, Peng W, Lin J, et al. An astrocytic basis of epilepsy. Nat Med. (2005) 11:973–81. 10.1038/nm1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. (2010) 32:638–47. 10.1016/j.tins.2009.08.002.Molecular [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. (2017) 46:957–67. 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- 120.Jha MK, Jo M, Kim JH, Suk K. Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist. (2019) 25:227–40. 10.1177/1073858418783959 [DOI] [PubMed] [Google Scholar]
- 121.Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. (2016) 4:71 10.3389/fcell.2016.00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vainchtein ID, Molofsky AV. Astrocytes and microglia: in sickness and in health. Trends Neurosci. (2020) 43:144–54. 10.1016/j.tins.2020.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Burda JE, Sofroniew MV. Seducing astrocytes to the dark side. Cell Res. (2017) 27:726–7. 10.1038/cr.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heiss CN, Olofsson LE. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. J Neuroendocrinol. (2019) 31:1–11. 10.1111/jne.12684 [DOI] [PubMed] [Google Scholar]
- 126.Ma Q, Xing C, Long W, Wang HY, Liu Q, Wang RF. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J Neuroinflamm. (2019) 16:1–14. 10.1186/s12974-019-1434-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA. (2009) 106:3698–703. 10.1073/pnas.0812874106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflamm. (2018) 15:1–14. 10.1186/s12974-017-1052-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism : focus on astrocyte-neuron metabolic cooperation. Cell Metab. (2011) 14:724–38. 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 130.Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. (2007) 1271:1263–71. 10.1002/glia.20557 [DOI] [PubMed] [Google Scholar]
- 131.Suh SW, Bergher JP, Anderson CM, Treadway JL, Fosgerau K, Swanson RA. Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-Chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J Pharmacol Exp Ther. (2007) 321:45–50. 10.1124/jpet.106.115550 [DOI] [PubMed] [Google Scholar]
- 132.Pellerin L, Halestrap AP, Pierre K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res. (2005) 64:55–64. 10.1002/jnr.20307 [DOI] [PubMed] [Google Scholar]
- 133.Phelps CREIGHTONH. Barbiturate-induced glycogen accumulation in brain. an electron microscopic study. Brain Res. (1972) 39:225–34. 10.1016/0006-8993(72)90797-4 [DOI] [PubMed] [Google Scholar]
- 134.Pfeiffer-Guglielmi B, Fleckenstein B, Hamprecht B. Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J Neurochem. (2003) 85:73–81. 10.1046/j.1471-4159.2003.01644.x [DOI] [PubMed] [Google Scholar]
- 135.Schwartz TH. Neurovascular coupling and epilepsy: hemodynamic markers for localizing and predicting seizure onset. Epilepsy Curr. (2007) 7:91–4. 10.1111/j.1535-7511.2007.00183.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wong M. Astrocyte networks and epilepsy: when stars collide. Curr Lit Basic Sci. (2009) 9:113–5. 10.1111/j.1535-7511.2009.01310.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Greene AE, Todorova MT, Seyfried TN. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J Neurochem. (2003) 86:529–37. 10.1046/j.1471-4159.2003.01862.x [DOI] [PubMed] [Google Scholar]
- 138.Henderson CB, Filloux FM, Alder SC, Lyon JL, Caplin DA. Efficacy of the ketogenic diet as a treatment option for epilepsy : meta-analysis. J Child Neurol. (2006) 21:193–8. 10.2310/7010.2006.00044 [DOI] [PubMed] [Google Scholar]
- 139.Kossoff EH, Rho JM. Ketogenic diets : evidence for short- and long-term efficacy. Am Soc Exp Neurother. (2009) 6:406–14. 10.1016/j.nurt.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rho JM. How does the ketogenic diet induce anti-seizure effects? Neurosci Lett. (2017) 637:4–10. 10.1016/j.neulet.2015.07.034 [DOI] [PubMed] [Google Scholar]
- 141.Kossoff EH, Dorward JL. The modified atkins diet. Epilepsia. (2008) 49:37–41. 10.1111/j.1528-1167.2008.01831.x [DOI] [PubMed] [Google Scholar]
- 142.Pfeifer HH, Lyczkowski DA, Thiele EA. Low glycemic index treatment : implementation and new insights into efficacy. Epilepsia. (2008) 49:42–5. 10.1111/j.1528-1167.2008.01832.x [DOI] [PubMed] [Google Scholar]
- 143.Guzel O, Uysal U, Arslan N. Efficacy and tolerability of olive oil-based ketogenic diet in children with drug-resistant epilepsy: a single center experience from Turkey. Eur J Paediatr Neurol. (2019) 23:143–51. 10.1016/j.ejpn.2018.11.007 [DOI] [PubMed] [Google Scholar]
- 144.Masino SA, Rho JM. Metabolism and epilepsy: ketogenic diets as a homeostatic link. Brain Res. (2019) 1703:26–30. 10.1016/j.brainres.2018.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu H, Yang Y, Wang Y, Tang H, Zhang F, Zhang Y, et al. Ketogenic diet for treatment of intractable epilepsy in adults: a meta-analysis of observational studies. Epilepsia Open. (2018) 3:9–17. 10.1002/epi4.12098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Garriga-Canut M, Schoenike B, Qazi R, Bergendahl K, Daley TJ, Pfender RM, et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP–dependent metabolic regulation of chromatin structure. Nat Neurosci. (2006) 9:1382–7. 10.1038/nn1791 [DOI] [PubMed] [Google Scholar]
- 147.Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann Neurol. (2009) 65:435–47. 10.1002/ana.21603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ockuly JC, Gielissen JM, Levenick CV, Zeal C, Groble K, Munsey K, et al. Behavioral, cognitive, and safety profile of 2-deoxy-2-glucose (2DG) in adult rats. Epilepsy Res. (2012) 101:246–52. 10.1016/j.eplepsyres.2012.04.012 [DOI] [PubMed] [Google Scholar]
- 149.Sada N, Lee S, Katsu T, Otsuki T, Inoue T. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science. (2015) 347:1362–7. 10.1126/science.aaa1299 [DOI] [PubMed] [Google Scholar]
- 150.Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. (2008) 322:1551–6. 10.1126/science.1164022 [DOI] [PubMed] [Google Scholar]
- 151.Dermietzel R, Spray DC. Gap jundions in the brain: where, what type, how many and why? TINS. (1993) 16:186–92. [DOI] [PubMed] [Google Scholar]
- 152.Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, et al. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Rev. (2000) 32:45–56. 10.1016/s0165-0173(99)00067-3 [DOI] [PubMed] [Google Scholar]
- 153.Parpura V, Verkhratsky A. Homeostatic function of astrocytes: Ca2+ and Na+ signalling. Transl Neurosci. (2012) 3:334–44. 10.2478/s13380-012-0040-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stout CE, Costantin JL, Naus CCG, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. (2002) 277:10482–8. 10.1074/jbc.M109902200 [DOI] [PubMed] [Google Scholar]
- 155.Strohschein S, Uttmann KH, Gabriel S, Binder DK. Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia. (2011) 980:973–80. 10.1002/glia.21169 [DOI] [PubMed] [Google Scholar]
- 156.Wallraff A, Heinemann U, Theis M, Willecke K, Steinha C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. (2006) 26:5438–47. 10.1523/JNEUROSCI.0037-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huguet G, Joglekar A, Messi LM, Buckalew R, Wong S, Terman D. Neuroprotective role of gap junctions in a neuron astrocyte network model. Biophys J. (2016) 111:452–62. 10.1016/j.bpj.2016.05.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mu J, Herde MK, Bedner P, Dupper A, Hu K, Haas CA, et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. (2015) 138:1208–22. 10.1093/brain/awv067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Crunelli V, Carmignoto G, Steinhäuser C. Novel astrocyte targets : new avenues for the therapeutic treatment of epilepsy. Neurosci. (2015) 21:62–83. 10.1177/1073858414523320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li Q, Li Q-Q, Jia J-N, Liu Z-Q, Zhou H-H, Mao X-Y. Targeting gap junction in epilepsy: perspectives and challenges. Biomed Pharmacother. (2019) 109:57–65. 10.1016/j.biopha.2018.10.068 [DOI] [PubMed] [Google Scholar]
- 161.Deshpande T, Li T, Herde MK, Becker A, Vatter H, Schwarz MK, et al. Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia. (2017) 65:1809–20. 10.1002/glia.23196 [DOI] [PubMed] [Google Scholar]
- 162.Losi G, Chiavegato A, Zonta M, Brondi M, Vetri F, Uva L, et al. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. (2010) 8:e1000352. 10.1371/journal.pbio.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jacobson GM, Voss LJ, Melin SM, Mason JP, Cursons RT, Steyn-Ross DA, et al. Connexin36 knockout mice display increased sensitivity to pentylenetetrazol-induced seizure-like behaviors. Brain Res. (2010) 1360:198–204. 10.1016/j.brainres.2010.09.006 [DOI] [PubMed] [Google Scholar]
- 164.Curti S, Hoge G, Nagy JI, Pereda AE. Synergy between electrical coupling and membrane properties promotes strong synchronization of neurons of the mesencephalic trigeminal nucleus. J Neurosci. (2012) 32:4341–59. 10.1523/JNEUROSCI.6216-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hosseinzadeh H, Nassiri Asl M. Anticonvulsant, sedative and muscle relaxant effects of carbenoxolone in mice. BMC Pharmacol. (2003) 3:3. 10.1186/1471-2210-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ventura-Mejía C, Medina-Ceja L. Decreased fast ripples in the hippocampus of rats with spontaneous recurrent seizures treated with carbenoxolone and quinine. Biomed Res Int. (2014) 2014:282490. 10.1155/2014/282490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ran X, Xiang J, Song P, Jiang L, Liu B. Effects of gap junctions blockers on fast ripples and connexin in rat hippocampi after status epilepticus. Epilepsy Res. (2018) 146:28–35. 10.1016/j.eplepsyres.2018.07.010 [DOI] [PubMed] [Google Scholar]
- 168.Franco-Pérez J, Ballesteros-Zebadúa P, Manjarrez-Marmolejo J. Anticonvulsant effects of mefloquine on generalized tonic-clonic seizures induced by two acute models in rats. BMC Neurosci. (2015) 16:7. 10.1186/s12868-015-0145-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nassiri-Asl M, Zamansoltani F, Torabinejad B. Antiepileptic effects of quinine in the pentylenetetrazole model of seizure. Seizure. (2009) 18:129–32. 10.1016/j.seizure.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 170.Manjarrez-Marmolejo J, Franco-Pérez J. Gap Junction blockers : an overview of their effects on induced seizures in animal models. Curr Neuropharmacol. (2016) 14:759–71. 10.2174/1570159X14666160603115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Venance L, Plomellit D, Glowinski J, Giaume C. Inhibition by anandamide of gap junctions and intercellular calcium signalling in striatal astrocytes. Nature. (1995) 376:590–4. 10.1038/376590a0 [DOI] [PubMed] [Google Scholar]
- 172.Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, et al. The sleep-inducing lipid oleamide deconvolutes gap junction communication and calcium wave transmission in glial cells. J Cell Biol. (1997) 139:1785–92. 10.1083/jcb.139.7.1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lambert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR. Anticonvulsant activity of N-palmitoylethanolamide, a putative endocannabinoid, in mice. Epilepsia. (2001) 42:321–7. 10.1046/j.1528-1157.2001.41499.x [DOI] [PubMed] [Google Scholar]
- 174.Wallace MJ, Martin BR, DeLorenzo RJ. Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur J Pharmacol. (2002) 452:295–301. 10.1016/S0014-2999(02)02331-2 [DOI] [PubMed] [Google Scholar]
- 175.Wu C-F, Li C-L, Song H-R, Zhang H-F, Yang J-Y, Wang Y-L. Selective effect of oleamide, an endogenous sleepinducing lipid amide, on pentylenetetrazole-induced seizures in mice. J Pharm Pharmacol. (2003) 55:1159–62. 10.1211/0022357021431 [DOI] [PubMed] [Google Scholar]
- 176.Solomonia R, Nozadze M, Mikautadze E, Kuchiashvili N, Kiguradze T, Abkhazava D, et al. Effect of oleamide on pentylenetetrazole-induced seizures in rats. Bull Exp Biol Med. (2008) 145:225–7. 10.1007/s10517-008-0056-z [DOI] [PubMed] [Google Scholar]
- 177.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. (1994) 369:744–7. 10.1038/369744a0 [DOI] [PubMed] [Google Scholar]
- 178.Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. (2004) 24:6920–7. 10.1523/JNEUROSCI.0473-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fellin T, Carmignoto G. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J Physiol. (2004) 559:3–15. 10.1113/jphysiol.2004.063214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schell MJ, Brady RO, Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci. (1997) 17:1604–15. 10.1523/jneurosci.17-05-01604.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Beltrán-Castillo S, Olivares MJ, Contreras RA, Zúñiga G, Llona I, Von Bernhardi R, et al. D-serine released by astrocytes in brainstem regulates breathing response to CO2 levels. Nat Commun. (2017) 8:838. 10.1038/s41467-017-00960-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Barakat L, Bordey A. GAT-1 and reversible GABA transport in bergmann glia in slices. J Neurophysiol. (2002) 88:1407–19. 10.1152/jn.2002.88.3.1407 [DOI] [PubMed] [Google Scholar]
- 183.Jiménez-González C, Pirttimaki T, Cope DW, Parri HR. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets δ-subunit-containing GABAA receptors. Eur J Neurosci. (2011) 33:1471–82. 10.1111/j.1460-9568.2011.07645.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yoon BE, Lee CJ. GABA as a rising gliotransmitter. Front Neural Circuits. (2014) 8:141. 10.3389/fncir.2014.00141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Eulenburg V, Gomeza J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res Rev. (2010) 63:103–12. 10.1016/j.brainresrev.2010.01.003 [DOI] [PubMed] [Google Scholar]
- 186.Bardóczi Z, Pál B, Koszeghy Á, Wilheim T, Watanabe M, Záborszky L, et al. Glycinergic input to the mouse basal forebrain cholinergic neurons. J Neurosci. (2017) 37:9534–49. 10.1523/JNEUROSCI.3348-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Shibasaki K, Hosoi N, Kaneko R, Tominaga M, Yamada K. Glycine release from astrocytes via functional reversal of GlyT1. J Neurochem. (2017) 140:395–403. 10.1111/jnc.13741 [DOI] [PubMed] [Google Scholar]
- 188.Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. (2003) 23:1659–66. 10.1523/jneurosci.23-05-01659.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, et al. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. (2003) 40:971–82. 10.1016/S0896-6273(03)00717-7 [DOI] [PubMed] [Google Scholar]
- 190.Gordon GRJ, Baimoukhametova DV, Hewitt SA, Rajapaksha WRAKJS, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. (2005) 8:1078–86. 10.1038/nn1498 [DOI] [PubMed] [Google Scholar]
- 191.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. (1994) 91:10625–9. 10.1073/pnas.91.22.10625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tang F, Lane S, Korsak A, Paton JFR, Gourine AV, Kasparov S, et al. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat Commun. (2014) 5:3284. 10.1038/ncomms4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.DiNuzzo M. Astrocyte-neuron interactions during learning may occur by lactate signaling rather than metabolism. Front Integr Neurosci. (2016) 10:2. 10.3389/fnint.2016.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. (1990) 10:1583–91. 10.1523/jneurosci.10-05-01583.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Choe KY, Olson JE, Bourque CW. Taurine release by astrocytes modulates osmosensitive glycine receptor tone and excitability in the adult supraoptic nucleus. J Neurosci. (2012) 32:12518–27. 10.1523/JNEUROSCI.1380-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Do KQ, Benz B, Sorg O, Pellerin L, Magistretti PJ. β-Adrenergic stimulation promotes homocysteic acid release from astrocyte cultures: evidence for a role of astrocytes in the modulation of synaptic transmission. J Neurochem. (1997) 68:2386–94. 10.1046/j.1471-4159.1997.68062386.x [DOI] [PubMed] [Google Scholar]
- 197.Benz B, Grima G, Do KQ. Glutamate-induced homocysteic acid release from astrocytes: Possible implication in glia-neuron signaling. Neuroscience. (2004) 124:377–86. 10.1016/j.neuroscience.2003.08.067 [DOI] [PubMed] [Google Scholar]
- 198.Krzan M, Stenovec M, Kreft M, Pangršič T, Grilc S, Haydon PG, et al. Calcium-dependent exocytosis of atrial natriuretic peptide from astrocytes. J Neurosci. (2003) 23:1580–3. 10.1523/jneurosci.23-05-01580.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. (1999) 6:284–91. 10.1101/lm.6.3.284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jean YY, Lercher LD, Dreyfus CF. Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol. (2008) 4:35–42. 10.1017/S1740925X09000052 [DOI] [PubMed] [Google Scholar]
- 201.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. (1992) 8:429–40. 10.1016/0896-6273(92)90271-e [DOI] [PubMed] [Google Scholar]
- 202.Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. (1996) 16:5073–81. 10.1523/JNEUROSCI.16-16-05073.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: A highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. (1997) 17:7817–30. 10.1523/jneurosci.17-20-07817.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. (2006) 9:816–23. 10.1038/nn1703 [DOI] [PubMed] [Google Scholar]
- 205.Takata N, Hirase H. Cortical layer 1 and layer 2/3 astrocytes exhibit distinct calcium dynamics in vivo. PLoS ONE. (2008) 3:e2525. 10.1371/journal.pone.0002525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. (2009) 32:421–31. 10.1016/j.tins.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 207.Nimmerjahn A, Kirchhoff F, Kerr JND, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. (2004) 1:31–7. 10.1038/nmeth706 [DOI] [PubMed] [Google Scholar]
- 208.Hoogland TM, Kuhn B, Gobel W, Huang W, Nakai J, Helmchen F, et al. Radially expanding transglial calcium waves in the intact cerebellum. Proc Natl Acad Sci USA. (2009) 106:3496–501. 10.1073/pnas.0809269106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kuga N, Sasaki T, Takahara Y, Matsuki N, Ikegaya Y. Large-scale calcium waves traveling through astrocytic networks in vivo. J Neurosci. (2011) 31:2607–14. 10.1523/JNEUROSCI.5319-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Savtchouk I, Volterra A. Gliotransmission: beyond black and white. J Neurosci. (2018) 38:14–25. 10.1523/JNEUROSCI.0017-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Fiacco TA, McCarthy KD. Multiple lines of evidence indicate that gliotransmission does not occur under physiological conditions. J Neurosci. (2018) 38:3–13. 10.1523/JNEUROSCI.0016-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hubbard JA, Binder DK. (2010). Astrocytes and epilepsy. Neurotherapeutics. (2010) 7:424–38. 10.1016/j.nurt.2010.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Volterra A, Liaudet N, Savtchouk I. Astrocyte Ca2+ signalling: an unexpected complexity. Nat Neurosci. (2014) 15:327–35. 10.1038/nrn3725 [DOI] [PubMed] [Google Scholar]
- 214.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. (1999) 22:208–15. 10.1016/S0166-2236(98)01349-6 [DOI] [PubMed] [Google Scholar]
- 215.Santello M, Calì C, Bezzi P. Synaptic plasticity: dynamics, development and disease. In: Kreutz MR, Sala C. editors. Advances in Experimental Medicine and Biology. Vienna: Springer; (2012). p. 945–61. 10.1007/978-3-7091-0932-8_14 [DOI] [Google Scholar]
- 216.Arizono M, Inavalli VVGK, Panatier A, Pfeiffer T, Angibaud J, Levet F, et al. Structural basis of astrocytic Ca2+ signals at tripartite synapses. Nat Commun. (2020) 11:1906 10.1038/s41467-020-15648-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lorenzo J, Vuillaume R, Binczak S, Jacquir S. Spatiotemporal model of tripartite synapse with perinodal astrocytic process. J Comput Neurosci. (2020) 48:1–20. 10.1007/s10827-019-00734-4 [DOI] [PubMed] [Google Scholar]
- 218.Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. (2007) 13:54–63. 10.1016/j.molmed.2006.12.005 [DOI] [PubMed] [Google Scholar]
- 219.Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, et al. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. (2007) 10:331–9. 10.1038/nn1849 [DOI] [PubMed] [Google Scholar]
- 220.Woo DH, Han K, Shim JW, Yoon B, Kim E, Bae JY, et al. TREK-1 and best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell. (2012) 151:25–40. 10.1016/j.cell.2012.09.005 [DOI] [PubMed] [Google Scholar]
- 221.Park H, Han KS, Oh SJ, Jo S, Woo J, Yoon BE, et al. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol Brain. (2013) 6:1–9. 10.1186/1756-6606-6-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, et al. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci USA. (2003) 100:15194–9. 10.1073/pnas.2431073100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci USA. (2005) 102:5606–11. 10.1073/pnas.0408483102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Martineau M, Shi T, Puyal J, Knolhoff AM, Dulong J, Gasnier B, et al. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J Neurosci. (2013) 33:3413–23. 10.1523/JNEUROSCI.3497-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Pascual O, Casper KB, Kubera C, Zhang J, Revilla R, Sul J, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science. (2005) 310:113–6. 10.1126/science.1116916 [DOI] [PubMed] [Google Scholar]
- 226.Lalo U, Palygin O, Rasooli-Nejad S, Andrew J, Haydon PG, Pankratov Y. Exocytosis of ATP from astrocytes modulates phasic and tonic inhibition in the neocortex. PLoS Biol. (2014) 12:e1001747. 10.1371/journal.pbio.1001747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bazargani N, Attwell D. Astrocyte calcium signaling : the third wave. Nat Neurosci. (2016) 19:182–9. 10.1038/nn.4201 [DOI] [PubMed] [Google Scholar]
- 228.Xiong Y, Sun S, Teng S, Jin M, Zhou Z. Ca2+-Dependent and Ca2+-independent ATP release in astrocytes. Front Mol Neurosci. (2018) 11:224. 10.3389/fnmol.2018.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Heinrich A, Andõ RD, Túri G, Rõzsa B, Sperlágh B. K+ depolarization evokes ATP, adenosine and glutamate release from glia in rat hippocampus: a microelectrode biosensor study. Br J Pharmacol. (2012) 167:1003–20. 10.1111/j.1476-5381.2012.01932.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Orellana JA, Froger N, Ezan P, Jiang JX, Bennet MVL, Naus CC, et al. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem. (2011) 118:826–40. 10.1111/j.1471-4159.2011.07210.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Stehberg J, Moraga-Amaro R, Salazar C, Becerra A, Echeverría C, Orellana JA, et al. Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. (2012) 26:3649–57. 10.1096/fj.11-198416 [DOI] [PubMed] [Google Scholar]
- 232.Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. (1990) 348:443–6. 10.1038/348443a0 [DOI] [PubMed] [Google Scholar]
- 233.Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. (2000) 403:316–21. 10.1038/35002090 [DOI] [PubMed] [Google Scholar]
- 234.Abudara V, Retamal MA, Del Rio R, Orellana JA. Synaptic functions of hemichannels and pannexons: A double-edged sword. Front Mol Neurosci. (2018) 11:435. 10.3389/fnmol.2018.00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Warr O, Takahashi M, Attwell D. Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol. (1999) 514:783–93. 10.1111/j.1469-7793.1999.783ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bridges RJ, Natale NR, Patel SA. System x c- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. (2012) 165:20–34. 10.1111/j.1476-5381.2011.01480.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Duan S, Anderson CM, Keung EC, Chen Y, Chen Y, Swanson RA. P2X 7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci. (2003) 23:1320–8. 10.1523/JNEUROSCI.23-04-01320.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels.” J Neurosci. (2009) 29:7092–7. 10.1523/JNEUROSCI.6062-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, et al. Connexin 43 hemichannels are permeable to ATP. J Neurosci. (2008) 28:4702–11. 10.1523/JNEUROSCI.5048-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Suadicani SO, Brosnan CF, Scemes E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci. (2006) 26:1378–85. 10.1523/JNEUROSCI.3902-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Xiong Y, Teng S, Zheng L, Sun S, Li J, Guo N, et al. Stretch-induced Ca2+ independent ATP release in hippocampal astrocytes. J Physiol. (2018) 596:1931–47. 10.1113/JP275805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mylvaganam S, Zhang L, Wu C, Zhang ZJ, Samoilova M, Eubanks J, et al. Hippocampal seizures alter the expression of the pannexin and connexin transcriptome. J Neurochem. (2010) 112:92–102. 10.1111/j.1471-4159.2009.06431.x [DOI] [PubMed] [Google Scholar]
- 243.Jiang T, Long H, Ma Y, Long L, Li Y, Li F, et al. Altered expression of pannexin proteins in patients with temporal lobe epilepsy. Mol Med Rep. (2013) 8:1801–6. 10.3892/mmr.2013.1739 [DOI] [PubMed] [Google Scholar]
- 244.Jimenez-Pacheco A, Mesuret G, Sanz-Rodriguez A, Tanaka K, Mooney C, Conroy R, et al. Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A-438079. Epilepsia. (2013) 54:1551–61. 10.1111/epi.12257 [DOI] [PubMed] [Google Scholar]
- 245.Grygorowicz T, Wełniak-Kamińska M, Struzyńska L. Early P2X7R-related astrogliosis in autoimmune encephalomyelitis. Mol Cell Neurosci. (2016) 74:1–9. 10.1016/j.mcn.2016.02.003 [DOI] [PubMed] [Google Scholar]
- 246.Evanko DS, Zhang Q, Zorec R, Haydon PG. Defining pathways of loss and secretion of chemical messengers from astrocytes. Glia. (2004) 47:233–40. 10.1002/glia.20050 [DOI] [PubMed] [Google Scholar]
- 247.Retamal MA, Froger N, Palacios-Prado N, Ezan P, Sáez PJ, Sáez JC, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. (2007) 27:13781–92. 10.1523/JNEUROSCI.2042-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Bennet MV, Garré JM, Orellana JA, Bukauskas FF, Nedergaard M, Sáez JC. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. (2012) 3:3–15. 10.1161/CIRCULATIONAHA.110.956839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Orellana JA, Von Bernhardi R, Giaume C, Sáez JC. Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev Neurosci. (2012) 23:163–77. 10.1515/revneuro-2011-0065 [DOI] [PubMed] [Google Scholar]
- 250.Medina-Ceja L, Salazar-Sánchez JC, Ortega-Ibarra J, Morales-Villagrán A. Connexins-based hemichannels/channels and their relationship with inflammation, seizures and epilepsy. Int J Mol Sci. (2019) 20:1–17. 10.3390/ijms20235976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Santiago MF, Veliskova J, Patel NK, Lutz SE, Caille D, Charollais A, et al. Targeting pannexin1 improves seizure outcome. PLoS ONE. (2011) 6:e25178. 10.1371/journal.pone.0025178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Dossi E, Blauwblomme T, Moulard J, Chever O, Vasile F, Guinard E, et al. Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy. Sci Transl Med. (2018) 10:1–14. 10.1126/scitranslmed.aar3796 [DOI] [PubMed] [Google Scholar]
- 253.Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, et al. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. (2012) 26:1616–28. 10.1096/fj.11-196089 [DOI] [PubMed] [Google Scholar]
- 254.Amhaoul H, Ali I, Mola M, Van Eetveldt A, Szewczyk K, Missault S, et al. P2X7 receptor antagonism reduces the severity of spontaneous seizures in a chronic model of temporal lobe epilepsy. Neuropharmacology. (2016) 105:175–85. 10.1016/j.neuropharm.2016.01.018 [DOI] [PubMed] [Google Scholar]
- 255.Lord B, Aluisio L, Shoblock JR, Neff RA, Varlinskaya EI, Ceusters M, et al. Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-42253432. J Pharmacol Exp Ther. (2014) 351:628–41. 10.1124/jpet.114.218487 [DOI] [PubMed] [Google Scholar]
- 256.Engel T, Alves M, Sheedy C, Henshall DC. ATPergic signalling during seizures and epilepsy. Neuropharmacology. (2016) 104:140–53. 10.1016/j.neuropharm.2015.11.001 [DOI] [PubMed] [Google Scholar]
- 257.Fischer W, Franke H, Krügel U, Müller H, Dinkel K. Critical evaluation of P2X7 receptor antagonists in selected seizure models. PLoS ONE. (2016) 11:e0156468. 10.1371/journal.pone.0156468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Riquelme J, Wellmann M, Sotomayor-Zárate R, Bonansco C. Gliotransmission: a novel target for the development of antiseizure drugs. Neuroscientist. (2020) 26:293–309. 10.1177/1073858420901474 [DOI] [PubMed] [Google Scholar]
- 259.Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. (1994) 100:47–51. 10.1016/S0079-6123(08)60767-0 [DOI] [PubMed] [Google Scholar]
- 260.Perez EL, Lauritzen F, Wang Y, Lee TSW, Kang D, Zaveri HP, et al. Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol Dis. (2012) 47:331–7. 10.1016/j.nbd.2012.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Syková E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. (2008) 88:1277–340. 10.1152/physrev.00027.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sosunov AA, Wu X, Tsankova NM, Guilfoyle E, McKhann GM, Goldman JE. Phenotypic heterogeneity and plasticity of isocortical and hippocampal astrocytes in the human brain. J Neurosci. (2014) 34:2285–98. 10.1523/JNEUROSCI.4037-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Zielinska M, Dabrowska K, Hadera MG, Sonnewald U, Albrecht J. System N transporters are critical for glutamine release and modulate metabolic fluxes of glucose and acetate in cultured cortical astrocytes: changes induced by ammonia. J Neurochem. (2016) 136:329–38. 10.1111/jnc.13376 [DOI] [PubMed] [Google Scholar]
- 264.Parpura V, Schousboe A, Verkhratsky A. Glutamate and ATP at the interface of metabolism and signaling in the brain. Adv Neurobiol. (2014) 11:11–30. 10.1007/978-3-319-08894-5 [DOI] [PubMed] [Google Scholar]
- 265.Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. (2005) 57:226–35. 10.1002/ana.20380 [DOI] [PubMed] [Google Scholar]
- 266.Cavus I, Pan JW, Hetherington HP, Abi-Saab W, Zaveri HP, Vives KP, et al. Decreased hippocampal volume on MRI is associated with increased extracellular glutamate in epilepsy patients. Epilepsia. (2008) 49:1358–66. 10.1111/j.1528-1167.2008.01603.x [DOI] [PubMed] [Google Scholar]
- 267.Ueda Y, Doi T, Tokumaru J, Yokoyama H, Nakajima A, Mitsuyama Y, et al. Collapse of extracellular glutamate regulation during epileptogenesis: down-regulation and functional failure of glutamate transporter function in rats with chronic seizures induced by kainic acid. J Neurochem. (2001) 76:892–900. 10.1046/j.1471-4159.2001.00087.x [DOI] [PubMed] [Google Scholar]
- 268.Gorter JA, Van Vliet EA, Proper EA, De Graan PNE, Ghijsen WEJM, Lopes Da Silva FH, et al. Glutamate transporters alterations in the reorganizing dentate gyrus are associated with progressive seizure activity in chronic epileptic rats. J Comp Neurol. (2002) 442:365–77. 10.1002/cne.10101 [DOI] [PubMed] [Google Scholar]
- 269.Lopes MW, Soares FMS, De Mello N, Nunes JC, Cajado AG, De Brito D, et al. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp Brain Res. (2013) 226:153–63. 10.1007/s00221-013-3421-8 [DOI] [PubMed] [Google Scholar]
- 270.Samuelsson C, Kumlien E, Flink R, Lindholm D, Ronne-Engström E. Decreased cortical levels of astrocytic glutamate transport protein GLT- 1 in a rat model of posttraumatic epilepsy. Neurosci Lett. (2000) 289:185–8. 10.1016/S0304-3940(00)01284-2 [DOI] [PubMed] [Google Scholar]
- 271.Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. (1999) 845:92–6. 10.1016/S0006-8993(99)01945-9 [DOI] [PubMed] [Google Scholar]
- 272.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. (1997) 276:1699–702. 10.1126/science.276.5319.1699 [DOI] [PubMed] [Google Scholar]
- 273.Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, et al. A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci. (2002) 22:6372–9. 10.1523/JNEUROSCI.22-15-06372.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Sarac S, Afzal ÃS, Broholm ÃH, Madsen FF. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS. (2009) 291–301. 10.1111/j.1600-0463.2009.02443.x [DOI] [PubMed] [Google Scholar]
- 275.Zeng L-H, Bero AW, Zhang B, Holtzman DM, Wong M. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of tuberous sclerosis complex. Neurobiol Dis. (2010) 37:764–71. 10.1016/j.nbd.2009.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Susarla BTS, Robinson MB. Internalization and degradation of the glutamate transporter GLT-1 in response to phorbol ester. Neurochem Int. (2008) 52:709–22. 10.1016/j.neuint.2007.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sha L, Wang X, Li J, Shi X, Wu L, Shen Y, et al. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J Exp Med. (2017) 214:547–63. 10.1084/jem.20160667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Eid T, Thomas MJ, Spencer DD, Rundén-Pran E, Lai JCK, Malthankar GV, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. (2004) 363:28–37. 10.1016/S0140-6736(03)15166-5 [DOI] [PubMed] [Google Scholar]
- 279.Van Der Hel WS, Notenboom RGE, Bos IWM, Van Rijen PC, Van Veelen CWM, De Graan PNE. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology. (2005) 64:326–33. 10.1212/01.WNL.0000149636.44660.99 [DOI] [PubMed] [Google Scholar]
- 280.Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Publ Gr. (2010) 13:584–91. 10.1038/nn.2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Zou J, Wang YX, Dou FF, Lü HZ, Ma ZW, Lu PH, et al. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem Int. (2010) 56:577–84. 10.1016/j.neuint.2009.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Eid T, Lee TSW, Patrylo P, Zaveri HP. Astrocytes and glutamine synthetase in epileptogenesis. J Neurosci Res. (2019) 97:1345–62. 10.1002/jnr.24267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Khazipov R. GABAergic synchronization in epilepsy. Cold Spring Harb Perspect Med. (2016) 6:1–13. 10.1101/cshperspect.a022764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Navazio F, Gerritsen T, Wright GJ. Relationship of ammonia intoxication to convulsions and coma in rats. J Neurochem. (1961) 8:146–51. 10.1111/j.1471-4159.1961.tb13536.x [DOI] [PubMed] [Google Scholar]
- 285.Papageorgiou IE, Valous NA, Lahrmann B, Janova H, Klaft ZJ, Koch A, et al. Astrocytic glutamine synthetase is expressed in the neuronal somatic layers and down-regulated proportionally to neuronal loss in the human epileptic hippocampus. Glia. (2018) 66:920–33. 10.1002/glia.23292 [DOI] [PubMed] [Google Scholar]
- 286.Kritis AA, Stamoula EG, Paniskaki KA, Vavilis TD. Researching glutamate – induced cytotoxicity in different cell lines: a comparative/collective analysis/study. Front Cell Neurosci. (2015) 9:91. 10.3389/fncel.2015.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Robel S, Buckingham XSC, Boni XJL, Campbell SL, Danbolt NC, Riedemann T, et al. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci. (2015) 35:3330–45. 10.1523/JNEUROSCI.1574-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Martineau M, Parpura V, Mothet JP. Cell-type specific mechanisms of D-serine uptake and release in the brain. Front Synaptic Neurosci. (2014) 6:12. 10.3389/fnsyn.2014.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Scianni M, Antonilli L, Chece G, Cristalli G, Di Castro MA, Limatola C, et al. Fractalkine (CX3CL1) enhances hippocampal N-methyl-d-aspartate receptor (NMDAR) function via d-serine and adenosine receptor type A2 (A2AR) activity. J Neuroinflamm. (2013) 10:1–15. 10.1186/1742-2094-10-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Martineau M, Galli T, Baux G, Mothet JP. Confocal imaging and tracking of the exocytotic routes for D-serine-mediated gliotransmission. Glia. (2008) 56:1271–84. 10.1002/glia.20696 [DOI] [PubMed] [Google Scholar]
- 291.Zhuang Z, Yang B, Theus MH, Sick JT, Bethea JR, Sick TJ, et al. EphrinBs regulate D-serine synthesis and release in astrocytes. J Neurosci. (2010) 30:16015–24. 10.1523/JNEUROSCI.0481-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci USA. (1995) 92:3948–52. 10.1073/pnas.92.9.3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Diniz LP, Almeida JC, Tortelli V, Lopes CV, Setti-Perdigão P, Stipursky J, et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of d-serine levels in cerebral cortex neurons. J Biol Chem. (2012) 287:41432–45. 10.1074/jbc.M112.380824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Takata N, Mishima T, Hisatsune C, Nagai T, Ebisui E, Mikoshiba K, et al. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J Neurosci. (2011) 31:18155–65. 10.1523/JNEUROSCI.5289-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.López-Hidalgo M, Salgado-Puga K, Alvarado-Martínez R, Medina AC, Prado-Alcalá RA, García-Colunga J. Nicotine uses neuron-glia communication to enhance hippocampal synaptic transmission and long-term memory. PLoS ONE. (2012) 7:e49998. 10.1371/journal.pone.0049998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wolosker H, Balu DT, Coyle JT. The rise and fall of the D-serine-mediated gliotransmission hypothesis. Trends Neurosci. (2016) 39:712–21. 10.1016/j.tins.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ehmsen JT, Ma TM, Sason H, Rosenberg D, Ogo T, Furuya S, et al. D-serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J Neurosci. (2013) 33:12464–9. 10.1523/JNEUROSCI.4914-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wolosker H, Radzishevsky I. The serine shuttle between glia and neurons: implications for neurotransmission and neurodegeneration. Biochem Soc Trans. (2013) 41:1546–50. 10.1042/BST20130220 [DOI] [PubMed] [Google Scholar]
- 299.Perez EJ, Tapanes SA, Loris ZB, Balu DT, Sick TJ, Coyle JT, et al. Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J Clin Invest. (2017) 127:3114–25. 10.1172/JCI92300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Ma T, Wu Y, Chen B, Zhang W, Jin L, Shen C, et al. D-Serine contributes to seizure development via ERK signaling. Front Neurosci. (2019) 13:254. 10.3389/fnins.2019.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Losi G, Cammarota M, Carmignoto G. The role of astroglia in the epileptic brain. Front Pharmacol. (2012) 3:132. 10.3389/fphar.2012.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Wang CM, Chang YY, Kuo JS, Sun SH. Activation of P2x7 receptors induced [3H]GABA release from the RBA-2 type-2 astrocyte cell line through a Cl-/HCO3- - dependent mechanism. Glia. (2002) 37:8–18. 10.1002/glia.10004 [DOI] [PubMed] [Google Scholar]
- 303.Torres A, Wang F, Xu Q, Fujita T, Dobrowolski R, Willecke K, et al. Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci Signal. (2012) 5:ra28. 10.1126/scisignal.2002160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kumaria A, Tolias CM, Burnstock G. ATP signalling in epilepsy. Purinergic Signal. (2008) 4:339–46. 10.1007/s11302-008-9115-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Gordon GRJ, Iremonger KJ, Kantevari S, Ellis-Davies GCR, MacVicar BA, Bains JS. Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron. (2009) 64:391–403. 10.1016/j.neuron.2009.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Scemes E, Velíšek L, Velíšková J. Astrocyte and neuronal pannexin1 contribute distinctly to seizures. ASN Neuro. (2019) 11:1–12. 10.1177/1759091419833502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Aquilino MS, Whyte-Fagundes P, Zoidl G, Carlen PL. Pannexin-1 channels in epilepsy. Neurosci Lett. (2019) 695:71–5. 10.1016/j.neulet.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 308.Rudolphi KA, Schubert P. Modulation of neuronal and glial cell function by adenosine and neuroprotection in vascular dementia. Behav Brain Res. (1997) 83:123–8. 10.1016/s0166-4328(97)86055-x [DOI] [PubMed] [Google Scholar]
- 309.Calker D, Van Biber K. The role of glial adenosine receptors in neural resilience and the neurobiology of mood disorders. Neurochem Res. (2005) 30:1205–17. 10.1007/s11064-005-8792-1 [DOI] [PubMed] [Google Scholar]
- 310.Boison D. Astrogliosis and adenosine kinase: a glial basis of epilepsy. Futur Neurol. (2008) 3:221–4. 10.2217/14796708.3.3.221 [DOI] [Google Scholar]
- 311.Boison D. Adenosinergic signaling in epilepsy. Neuropharmacology. (2016) 104:131–9. 10.1016/j.neuropharm.2015.08.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Lopes LV, Cunha RA, Ribeiro JA. Cross talk between A 1 and A 2A adenosine receptors in the hippocampus and cortex of young adult and old rats. J Neurophysiol. (1999) 82:3196–203. 10.1152/jn.1999.82.6.3196 [DOI] [PubMed] [Google Scholar]
- 313.Ciruela F, Casado V, Rodrigues RJ, Luja R, Burguen J, Canals M, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A 1 – A 2A receptor heteromers. J Neurosci. (2006) 26:2080–7. 10.1523/JNEUROSCI.3574-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Glass M, Faull RLM, Jansen K, Walker EB, Synek BJL, Dragunow M. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res. (1996) 710:56–68. 10.1016/0006-8993(95)01313-x [DOI] [PubMed] [Google Scholar]
- 315.Ekonomou A, Sperk G, Kostopoulos G, Angelatou F. Reduction of A1 adenosine receptors in rat hippocampus after kainic acid-induced limbic seizures. Neurosci Lett. (2000) 284:49–52. 10.1016/s0304-3940(00)00954-x [DOI] [PubMed] [Google Scholar]
- 316.Barros-Barbosa AR, Ferreirinha F, Oliveira A, Mendes M, Lobo MG, Santos A, et al. Adenosine A2A receptor and ecto-5′-nucleotidase-CD73 are upregulated in hippocampal astrocytes of human MTLE. Purinergic Signal. (2016) 12:719–34. 10.1007/s11302-016-9535-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Hindley S, Herman MAR, Rathbone MP. Stimulation of reactive astrogliosis in vivo by extracellular adenosine diphosphate or an adenosine A2 receptor agonist. J Neurosci Res. (1994) 38:399–406. 10.1002/jnr.490380405 [DOI] [PubMed] [Google Scholar]
- 318.Popoli P, Blum D, Martire A, Ledent C, Ceruti S, Abbracchio MP. Functions, dysfunctions and possible therapeutic relevance of adenosine A 2A receptors in Huntington' s disease. Prog Neurobiol. (2007) 81:331–48. 10.1016/j.pneurobio.2006.12.005 [DOI] [PubMed] [Google Scholar]
- 319.Ribeiro JA, Dio MJ, Sebastia AM. Influence of age on BDNF modulation of hippocampal synaptic transmission : interplay with adenosine A 2A receptors. Hippocampus. (2007) 17:577–85. 10.1002/hipo [DOI] [PubMed] [Google Scholar]
- 320.Ke R, Xiong J, Liu Y, Ye Z. Adenosine A2a receptor induced gliosis via Akt/NF-kB pathway in vitro. Neurosci Res. (2009) 65:280–5. 10.1016/j.neures.2009.08.002 [DOI] [PubMed] [Google Scholar]
- 321.Erion MD, Ugarkar BG, Dare J, Catellino AJ, Fujitaki JM, Dixon R, et al. Design, synthesis and anticonvulsant activity of the potent adenosine kinase inhibitor GP3269. Nucleic Acids. (1998) 16:1013–21. 10.1080/07328319708006124 [DOI] [Google Scholar]
- 322.Ugarkar BG, DaRe JM, Kopcho JJ, Browne CE, Schanzer JM, Wiesner JB, et al. Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J Med Chem. (2000) 43:2883–93. 10.1021/jm000024g [DOI] [PubMed] [Google Scholar]
- 323.Ugarkar BG, Castellino AJ, DaRe JM, Kopcho JJ, Wiesner JB, Schanzer JM, et al. Adenosine kinase inhibitors. 2. Synthesis, enzyme inhibition, and antiseizure activity of diaryltubercidin analogues. J Med Chem. (2000) 43:2894–905. 10.1021/jm0000259 [DOI] [PubMed] [Google Scholar]
- 324.Zhang G, Franklin PH, Murray TF. Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J Pharmacol Exp Ther. (1993) 264:1415–24. [PubMed] [Google Scholar]
- 325.McGaraughty S, Cowart M, Jarvis M, Berman R. Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem. (2005) 5:43–58. 10.2174/1568026053386845 [DOI] [PubMed] [Google Scholar]
- 326.Köse M, Schiedel AC, Bauer AA, Poschenrieder H, Burbiel JC, Akkinepally RR, et al. Focused screening to identify new adenosine kinase inhibitors. Bioorganic Med Chem. (2016) 24:5127–33. 10.1016/j.bmc.2016.08.026 [DOI] [PubMed] [Google Scholar]
- 327.Arciénega II, Brunet JF, Bloch J, Badaut J. Cell locations for AQP1, AQP4 and 9 in the non-human primate brain. Neuroscience. (2010) 167:1103–14. 10.1016/j.neuroscience.2010.02.059 [DOI] [PubMed] [Google Scholar]
- 328.Eid T, Lee TW, Thomas MJ, Amiry-Moghaddam M, Bjørnsen LP, Spencer DD, et al. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci USA. (2005) 102:1193–8. 10.1073/pnas.0409308102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Sheilabi MA, Bhattacharyya D, Kitchen P, Conner AC, Salman MM, Bill RM, et al. Transcriptome analysis suggests a role for the differential expression of cerebral aquaporins and the MAPK signalling pathway in human temporal lobe epilepsy. Eur J Neurosci. (2017) 46:2121–32. 10.1111/ejn.13652 [DOI] [PubMed] [Google Scholar]
- 330.Lee TS, Eid T, Mane S, Kim JH, Spencer DD, Ottersen OP, et al. Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. (2004) 108:493–502. 10.1007/s00401-004-0910-7 [DOI] [PubMed] [Google Scholar]
- 331.Binder DK, Auser CS, Words KEY. Functional changes in astroglial cells in epilepsy. Glia. (2006) 368:358–68. 10.1002/glia.20394 [DOI] [PubMed] [Google Scholar]
- 332.Coulter DA, Steinha C. Role of astrocytes in epilepsy. Cold Spring Harb Perspect Med. (2015) 5:649–71. 10.1101/cshperspect.a022434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Li X, Zhou J, Chen Z, Chen S, Zhu F, Liemin Z. Long-term expressional changes of Na+-K+-Cl− co-transporter NKCC1 and KCC2 in CA1 region of hippo following pilo epilepsy. Brain Res. (2008) 141–6. 10.1016/j.brainres.2008.04.047 [DOI] [PubMed] [Google Scholar]
- 334.Brandt C, Nozadze M, Heuchert N, Rattka M, Löscher W. Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J Neurosci. (2010) 30:8602–12. 10.1523/JNEUROSCI.0633-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Otalora LFP, Hernandez EF, Arshadmansab MF, Sebastian F, Willis M, Ermolinsky B, et al. Downregulation of BK channel expression in the pilocarpine model of temporal lobe epilepsy. Brain Res. (2009) 1200:116–31. 10.1016/j.brainres.2008.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Yeo JKS, Jin H. Changes in TWIK-related acid sensitive K+−1 and−3 channel expressions from neurons to glia in the hippocampus of temporal lobe epilepsy patients and experimental animal model. Neurochem Res. (2011) 36:2155–68. 10.1007/s11064-011-0540-0 [DOI] [PubMed] [Google Scholar]
- 337.Nagao Y, Harada Y, Mukai T, Shimizu S, Okuda A, Fujimoto M, et al. Expressional analysis of the astrocytic Kir4. 1 channel in a pilocarpine-induced temporal lobe epilepsy model. Front Cell Neurosci. (2013) 7:104. 10.3389/fncel.2013.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Kang SJ, Cho S, Park K, Yi J, Yoo SJ, Shin KS. Expression of Kir2. 1 channels in astrocytes under pathophysiological conditions. Mol Cells. (2008) 25:124–30. [PubMed] [Google Scholar]
- 339.Kim D, Kim J, Kwak S, Won MH, Kang T. Seizure activity affects neuroglial Kv1 channel immunoreactivities in the gerbil hippocampus. Brain Res. (2007) 1151:172–87. 10.1016/j.brainres.2007.03.017 [DOI] [PubMed] [Google Scholar]
- 340.Grisar T, Guillaume D, Delgado-Escueta AV. Contribution of Na+, K+-ATPase to focal epilepsy: a brief review. Epilepsy Res. (1992) 12:141–9. 10.1016/0920-1211(92)90034-q [DOI] [PubMed] [Google Scholar]
- 341.Palma E, Amici M, Sobrero F, Spinelli G, Angelantonio S, Di Ragozzino D, et al. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci USA. (2006) 103:8465–8. 10.1073/pnas.0602979103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Ketelaars SOM, Gorter JA, Aronica E, Wadman WJ. Calcium extrusion protein expression in the hippocampal formation of chronic epileptic rats after kainate-induced status epilepticus. Epilepsia. (2004) 45:1189–201. 10.1111/j.0013-9580.2004.03304.x [DOI] [PubMed] [Google Scholar]
- 343.Lin YW, Hsieh CL. Auricular electroacupuncture reduced inflammation-related epilepsy accompanied by altered trpa1, ppkc ppkc ε, and perk1/2 signaling pathways in kainic acid-treated rats. Mediators Inflamm. (2014) 2014:1–9. 10.1155/2014/493480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Tai C, Hines DJ, Choi HB, MacVicar BA. Plasma membrane insertion of TRPC5 channels contributes to the cholinergic plateau potential in hippocampal CA1 pyramidal neurons. Hippocampus. (2011) 21:958–67. 10.1002/hipo.20807 [DOI] [PubMed] [Google Scholar]
- 345.Xu GZ, Shu H, Yue HY, Zheng DH, Guo W, Yang H. Increased expression of TRPC5 in cortical lesions of the focal cortical dysplasia. J Mol Neurosci. (2014) 55:561–9. 10.1007/s12031-014-0390-8 [DOI] [PubMed] [Google Scholar]
- 346.Zeng C, Zhou P, Jiang T. Upregulation and diverse roles of TRPC3 and TRPC6 in synaptic reorganization of the mossy fiber pathway in temporal lobe epilepsy. Mol Neurobiol. (2015) 52:562–72. 10.1007/s12035-014-8871-x [DOI] [PubMed] [Google Scholar]
- 347.Sun F, Guo W, Zheng D, Zhang C. Increased expression of TRPV1 in the cortex and hippocampus from patients with mesial temporal lobe epilepsy. J Mol Neurosci. (2013) 1:182–93. 10.1007/s12031-012-9878-2 [DOI] [PubMed] [Google Scholar]
- 348.Seifert G, Schröder W, Hinterkeuser S, Schumacher T, Schramm J, Steinhäuser C. Changes in flip/flop splicing of astroglial AMPA receptors in human temporal lobe epilepsy. Epilepsia. (2002) 43:162–7. 10.1046/j.1528-1157.43.s.5.10.x [DOI] [PubMed] [Google Scholar]
- 349.Seifert G, Hu K, Schramm J, Steinha C. Enhanced relative expression of glutamate receptor 1 flip AMPA receptor subunits in hippocampal astrocytes of epilepsy patients with ammon' s horn sclerosis. J Neurosci. (2004) 24:1996–2003. 10.1523/JNEUROSCI.3904-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Naylor DE, Lio H, Niquet J, Wasterlain CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis. (2013) 54:225–38. 10.1016/j.nbd.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. (2004) 129:877–96. 10.1016/j.neuroscience.2004.09.053 [DOI] [PubMed] [Google Scholar]
- 352.Ke C, Poon WS, Ng HK, Lai FMM, Tang NLS, Pang JCS. Impact of experimental acute hyponatremia on severe traumatic brain injury in rats : influences on injuries, permeability of blood – brain. Exp Neurol. (2002) 206:194–206. 10.1006/exnr.2002.8037 [DOI] [PubMed] [Google Scholar]
- 353.Pasantes-Morales H, Franco R, Ordaz B, Ochoa LD. Mechanisms counteracting swelling in brain cells during hyponatremia. Arch Med Res. (2002) 33:237–44. 10.1016/s0188-4409(02)00353-3 [DOI] [PubMed] [Google Scholar]
- 354.Murphy TR, Binder DK, Fiacco TA. Turning down the volume: astrocyte volume change in the generation and termination of epileptic seizures. Neurobiol Dis. (2017) 104:24–32. 10.1016/j.nbd.2017.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Wang F, Qi X, Zhang J, Huang J. Astrocytic modulation of potassium under seizures. Neural Regen Res. (2020) 15:980–7. 10.4103/1673-5374.270295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in cultured rat astrocytes. J Physiol. (2006) 572:677–89. 10.1113/jphysiol.2005.103820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Haskew-Layton RE, Rudkouskaya A, Jin Y, Feustel PJ, Kimelberg HK, Mongin AA. Two distinct modes of hypoosmotic medium-induced release of excitatory amino acids and taurine in the rat brain in vivo. PLoS ONE. (2008) 3:e3543. 10.1371/journal.pone.0003543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Seifert G, Henneberger C, Steinhäuser C. Diversity of astrocyte potassium channels : an update. Brain Res Bull. (2018) 136:26–36. 10.1016/j.brainresbull.2016.12.002 [DOI] [PubMed] [Google Scholar]
- 359.Day RE, Kitchen P, Owen DS, Bland C, Marshall L, Conner AC, et al. Human aquaporins : regulators of transcellular water flow. BBA Gen Subj. (2014) 1840:1492–506. 10.1016/j.bbagen.2013.09.033 [DOI] [PubMed] [Google Scholar]
- 360.Macaulay N, Hamann S, Zeuthen T. Water transport in the brain: role of cotransporters. Neuroscience. (2004) 129:1031–44. 10.1016/j.neuroscience.2004.06.045 [DOI] [PubMed] [Google Scholar]
- 361.Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. (2014) 14:265–77. 10.1038/nrn3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. (2008) 58:168–78. 10.1016/j.neuron.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Frigeri A, Nicchia GP, Nico B, Quondamatteo F, Herken R, RONCALI L, et al. Aquaporin-4 deficiency in skeletal muscle and brain of dystrophic mdx mice. FASEB J. (2001) 15:90–8. 10.1096/fj.00-0260com [DOI] [PubMed] [Google Scholar]
- 364.Wertz K, Stødkilde-Jørgensen H, Sulyok E, Vajda Z, Pedersen M, Fu E, et al. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc Natl Acad Sci USA. (2002) 99:13131–6. 10.1073/pnas.192457099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Marchi N, Granata T, Ghosh C, Janigro D. Blood–brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. (2012) 53:1877–86. 10.1111/j.1528-1167.2012.03637.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol. (2013) 235:246–55. 10.1016/j.expneurol.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Alvestad S, Hammer J, Hellstrøm E, Skare Ø, Sonnewald U, Amiry-Moghaddam M, et al. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. (2013) 105:30–41. 10.1016/j.eplepsyres.2013.01.006 [DOI] [PubMed] [Google Scholar]
- 368.Han X, Huang Q, Liu L, Sha X, Hu B, Liu H. Changes in the expression of AQP4 and AQP9 in the hippocampus following eclampsia-like seizure. Int J Mol Sci. (2018) 19:1–12. 10.3390/ijms19010300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Kim JE, Ryu HJ, Yeo SI, Seo CH, Lee BC, Choi IG, et al. Differential expressions of aquaporin subtypes in astroglia in the hippocampus of chronic epileptic rats. Neuroscience. (2009) 163:781–9. 10.1016/j.neuroscience.2009.07.028 [DOI] [PubMed] [Google Scholar]
- 370.Liu H, Yang M, Qiu G, Zhuo F, Yu W, Sun S, et al. Aquaporin 9 in rat brain after severe traumatic brain injury. Arq Neuropsiquiatr. (2012) 70:214–20. 10.1590/s0004-282x2012000300012 [DOI] [PubMed] [Google Scholar]
- 371.Shenaq M, Kassem H, Peng C, Schafer S, Ding JY, Fredrickson V, et al. Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1α after traumatic brain injury (TBI). J Neurol Sci. (2012) 323:134–40. 10.1016/j.jns.2012.08.036 [DOI] [PubMed] [Google Scholar]
- 372.Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. (2005) 118:5691–8. 10.1242/jcs.02680 [DOI] [PubMed] [Google Scholar]
- 373.Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. (2004) 279:44065–73. 10.1074/jbc.M405985200 [DOI] [PubMed] [Google Scholar]
- 374.Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenback A, et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. (2007) 281:274–81. 10.1002/glia.20455 [DOI] [PubMed] [Google Scholar]
- 375.Li X, Potts EA, Chen M, Perilla PR, Bredt DS, Simard JM. Inward rectifier K+ channel Kir2.3 (IRK3) in reactive astrocytes from adult rat brain. Glia. (2000) 192:181–92. 10.1002/1098-1136(200008)31:2<181::aid-glia90>3.0.co;2-8 [DOI] [PubMed] [Google Scholar]
- 376.Thomzig A, Wenzel M, Karschin C, Eaton MJ, Skatchkov SN, Karschin A, et al. Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol Cell Neurosci. (2001) 690:671–90. 10.1006/mcne.2001.1048 [DOI] [PubMed] [Google Scholar]
- 377.Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4. 1 associates with the dystrophin- glycoprotein complex via alfa-syntrophin in glia. J Biol Chem. (2004) 279:28387–92. 10.1074/jbc.M402604200 [DOI] [PubMed] [Google Scholar]
- 378.Bragg AD, Amiry-Moghaddam M, Ottersen OLEP, Adams ME. Assembly of a perivascular astrocyte protein scaffold at the mammalian blood – brain barrier is dependent on a -syntrophin. Glia. (2006) 890:879–90. 10.1002/glia.20347 [DOI] [PubMed] [Google Scholar]
- 379.Hubbard JA, Hsu MS, Seldin MM, Binder DK. Expression of the astrocyte water channel aquaporin-4 in the mouse brain. ASN Neuro. (2015) 7:1759091415605486. 10.1177/1759091415605486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Jin B, Zhang H, Binder DK, Verkman AS. Aquaporin-4 – dependent K+ and water transport modeled in brain extracellular space following neuroexcitation. J Gen Physiol. (2013) 141:119–32. 10.1085/jgp.201210883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Soe R, Macaulay N, Arne D. Modulation of Kir4. 1 and Kir4. 1 – Kir5. 1 channels by small changes in cell volume. Neurosci Lett. (2009) 457:80–84. 10.1016/j.neulet.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 382.Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, et al. Role of Glial K+ channels in ontogeny and gliosis: a hypothesis based upon studies on müller cells. Glia. (2000) 44:35–44. 10.1002/(sici)1098-1136(20000101)29:1<35::aid-glia4>3.0.co;2-a [DOI] [PubMed] [Google Scholar]
- 383.Olsen ML, Campbell SC, Mcferrin MB, Floyd CL, Sontheimer H. Spinal cord injury causes a wide-spread, persis- tent loss of Kir4.1 and glutamate transporter 1: benefit of 17 beta-oestradiol treatment. Brain. (2010) 133:1013–25. 10.1093/brain/awq049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Frigerio F, Frasca A, Weissberg I, Parrella S, Friedman A, Vezzani A, et al. Long-lasting pro-ictogenic effects induced in vivo by rat brain exposure to serum albumin in the absence of concomitant pathology. Epilepsia. (2012) 53:1887–97. 10.1111/j.1528-1167.2012.03666.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, et al. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. (2010) 104:3345–60. 10.1152/jn.00398.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Kinboshi M, Mukai T, Nagao Y, Matsuba Y, Tsuji Y, Adamo MCD. Inhibition of inwardly rectifying potassium (Kir) 4.1 channels facilitates brain-derived neurotrophic factor (BDNF) expression in astrocytes. Front Mol Neurosci. (2017) 10:408. 10.3389/fnmol.2017.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Murray KD, Isackson PJ, Eskin TA, King MA, Montesinos SP, Abraham LA. Altered mRNA expression for brain- derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase in the hippocampus of patients with intractable temporal lobe epilepsy. J Comp Neurol. (2000) 422:411–22. 10.1002/(sici)1096-9861(20000320)418:4<411::aid-cne4>3.0.co;2-f [DOI] [PubMed] [Google Scholar]
- 388.Heinrich C, Lähteinen S, Suzuki F, Anne-Marie L, Huber S, Häussler U, et al. Neurobiology of disease increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol Dis. (2011) 42:35–47. 10.1016/j.nbd.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 389.Tanaka T, Saito H, Matsuki N. Inhibition of GABA A synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. (1997) 17:2959–66. 10.1523/JNEUROSCI.17-09-02959.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl– extrusion. J Cell Biol. (2002) 159:747–52. 10.1083/jcb.200209011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. (2015) 36:726–37. 10.1016/j.tins.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Woo N, Lu J, England R, Mcclellan R, Dufour S, Mount DB, et al. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K–Cl cotransporter gene. Hippocampus. (2002) 268:258–68. 10.1002/hipo.10014 [DOI] [PubMed] [Google Scholar]
- 393.Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. Astrocytic endfoot Ca2+and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci USA. (2009) 107:1–6. 10.1073/pnas.0914722107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.N'Gouemo P. Targeting BK (big potassium) channels in epilepsy. Expert Opin Ther Targets. (2012) 15:1283–95. 10.1517/14728222.2011.620607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. (2005) 8:1752–9. 10.1038/nn1573 [DOI] [PubMed] [Google Scholar]
- 396.Shruti S, Clem RL, Barth AL. A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol Dis. (2008) 30:323–30. 10.1016/j.nbd.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Liu X, Tao J, Zhang S, Lan W, Wang C, Ji Y, et al. Selective blockade of neuronal BK (α + β4) channels preventing epileptic seizure. J Med Chem. (2020) 63:216–30. 10.1021/acs.jmedchem.9b01241 [DOI] [PubMed] [Google Scholar]
- 398.Whitmire LE, Ling L, Bugay V, Carver CM, Timilsina S, Chuang HH, et al. Downregulation of KCNMB4 expression and changes in BK channel subtype in hippocampal granule neurons following seizure activity. PLoS ONE. (2017) 12:e0188064. 10.1371/journal.pone.0188064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Mehranfard N, Gholamipour-Badie H, Motamedi F, Janahmadi M, Naderi N. Long-term increases in BK potassium channel underlie increased action potential firing in dentate granule neurons following pilocarpine-induced status epilepticus in rats. Neurosci Lett. (2015) 585:88–91. 10.1016/j.neulet.2014.11.041 [DOI] [PubMed] [Google Scholar]
- 400.Shirazi-Zand Z, Ahmad-Molaei L, Motamedi F, Naderi N. The role of potassium BK channels in anticonvulsant effect of cannabidiol in pentylenetetrazole and maximal electroshock models of seizure in mice. Epilepsy Behav. (2013) 28:1–7. 10.1016/j.yebeh.2013.03.009 [DOI] [PubMed] [Google Scholar]
- 401.Wickenden AD. Potassium channels as anti-epileptic drug targets. Neuropharmacology. (2002) 43:1055–60. 10.1016/S0028-3908(02)00237-X [DOI] [PubMed] [Google Scholar]
- 402.Dvorzhak A, Vagner T, Kirmse K, Grantyn R. Functional indicators of glutamate transport in single striatal astrocytes and the influence of Kir4. 1 in normal and huntington mice. J Neurosci. (2016) 36:4959–75. 10.1523/JNEUROSCI.0316-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Pappalardo LW, Samad OA, Black JA, Waxman SG. Voltage-gated sodium channel Nav 1.5 contributes to astrogliosis in an in vitro model of glial injury via reverse Na+/Ca2+ exchange. Glia. (2015) 62:1162–75. 10.1002/glia.22671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Black JA, Newcombe J, Waxman SG. Astrocytes within multiple sclerosis lesions upregulate sodium channel Nav1.5. Brain. (2010) 133:835–46. 10.1093/brain/awq003 [DOI] [PubMed] [Google Scholar]
- 405.Pappalardo LW, Shujun L, Black JA, Waxman SG. Dynamics of sodium channel Nav 1.5 expression in astrocytes in mouse models of multiple sclerosis. Neuroreport. (2014) 25:1208–15. 10.1097/WNR.0000000000000249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Pappalardo LW, Black JA, Waxman SG, Haven N, Haven W. Sodium channels in astroglia and microglia. Glia. (2016) 64:1628–45. 10.1002/glia.22967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Vaillend C, Mason SE, Cuttle MF, Alger BE. Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region. J Neurophysiol. (2002) 88:2963–78. 10.1152/jn.00244.2002 [DOI] [PubMed] [Google Scholar]
- 408.Clapcote SJ, Duffy S, Xie G, Kirshenbaum G, Bechard AR, Schack VR, et al. Mutation I810N in the α3 isoform of Na+, K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc Natl Acad Sci USA. (2009) 106:14085–90. 10.1073/pnas.0904817106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Gallanti A, Tonelli A, Cardin V, Bussone G, Bresolin N, Bassi MT. A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J Neurol Sci. (2008) 273:123–6. 10.1016/j.jns.2008.06.006 [DOI] [PubMed] [Google Scholar]
- 410.Su GUI, Haworth RA, Dempsey RJ, Sun D, Haworth RA, Dempsey RJ. Regulation of Na+-K+-Cl− cotransporter in primary astrocytes by dibutyryl cAMP and high [K+] o. Am J Physiol Cell Physiol. (2000) 297:1710–21. 10.1152/ajpcell.2000.279.6.C1710 [DOI] [PubMed] [Google Scholar]
- 411.Su GUI, Kintner DB, Flagella M, Shull GE, Sun D, Kintner DB, et al. Astrocytes from Na+ -K+ -Cl - cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. (2001) 53792:1147–60. 10.1152/ajpcell.00538.2001 [DOI] [PubMed] [Google Scholar]
- 412.Østby I, Øyehaug L, Einevoll GT, Nagelhus EA, Plahte E, Zeuthen T, et al. Astrocytic mechanisms explaining neural-activity-induced shrinkage of extraneuronal space. PLoS Comput Biol. (2009) 5:e1000272. 10.1371/journal.pcbi.1000272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. (2008) 835:821–35. 10.1002/glia.20656 [DOI] [PubMed] [Google Scholar]
- 414.Verkhratsky A, Reyes RC, Parpura V. TRP channels coordinate ion signalling in astroglia. Rev Physiol Biochem Pharmacol. (2013) 166:1–16. 10.1007/112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Anderson CM, Swanson RA. Astrocyte glutamate transport : review of properties, regulation, and physiological functions. Glia. (2000) 14:1–14. 10.1002/1098-1136(200010)32:1<1::AID-GLIA10>3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- 416.Uwechue NM, Marx M, Chevy Q, Billups B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J Physiol. (2012) 10:2317–31. 10.1113/jphysiol.2011.226605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Kleene R, Loers G, Langer J, Frobert Y, Buck F, Schachner M. Prion protein regulates glutamate-dependent lactate transport of astrocytes. J Neurosci. (2007) 27:12331–40. 10.1523/JNEUROSCI.1358-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Minelli A, Castaldo P, Gobbi P, Salucci S, Magi S, Amoroso S. Cellular and subcellular localization of Na+- Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium. (2007) 41:221–34. 10.1016/j.ceca.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 419.Shigetomi E, Tong X, Kwan KY, Corey DP, Baljit S. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci. (2012) 15:70–80. 10.1038/nn.3000.TRPA1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Shigetomi E, Jackson-Weaver O, Huckstepp RT, O'Dell TJ, Khakh BS. TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive d-serine release. J Neurosci. (2013) 33:10143–53. 10.1523/JNEUROSCI.5779-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Lee M, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS ONE. (2010) 5:e14123. 10.1371/journal.pone.0014123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Palygin O, Lalo U, Verkhratsky A, Pankratov Y. Ionotropic NMDA and P2X 1/5 receptors mediate synaptically induced Ca 2 + signalling in cortical astrocytes. Cell Calcium. (2010) 48:225–31. 10.1016/j.ceca.2010.09.004 [DOI] [PubMed] [Google Scholar]
- 423.Mikawa S, Wang C, Shu F, Wang T, Fukuda A, Sato K. Developmental changes in KCC1, KCC2 and NKCC1 mRNAs in the rat cerebellum. Dev Brain Res. (2002) 136:93–100. 10.1016/s0165-3806(02)00345-0 [DOI] [PubMed] [Google Scholar]
- 424.Gagnon KBE, Adragna NC, Fyffe REW, Lauf PK. Characterization of glial cell K-Cl cotransport. Cell Physiol Biochem. (2007) 20:121–30. 10.1159/000104160 [DOI] [PubMed] [Google Scholar]
- 425.Ringel F, Plesnila N. Expression and functional role of potassium-chloride cotransporters (KCC) in astrocytes and C6 glioma cells. Neurosci Lett. (2008) 442:219–23. 10.1016/j.neulet.2008.07.017 [DOI] [PubMed] [Google Scholar]
- 426.Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. (2015) 15:637–54. 10.1038/nrn3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Mu A, Pablo M, Defelipe J, Alvarez-Leefmans FJ. Cation-chloride cotransporters and GABA-ergic innervation in the human epileptic hippocampus. Epilepsia. (2007) 48:663–73. 10.1111/j.1528-1167.2007.00986.x [DOI] [PubMed] [Google Scholar]
- 428.Chen L, Wan L, Wu Z, Ren W, Yian H, Qian B, et al. KCC2 downregulation facilitates epileptic seizures. Sci Rep. (2017) 7:156. 10.1038/s41598-017-00196-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Eftekhari S, Habibabadi M, Ziarani N, Sohrab S. Bumetanide reduces seizure frequency in patients with temporal lobe epilepsy. Epilepsia. (2013) 54:10–13. 10.1111/j.1528-1167.2012.03654.x [DOI] [PubMed] [Google Scholar]
- 430.Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, et al. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia. (2014) 62:608–22. 10.1002/glia.22629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Gharaylou Z, Shafaghi L, Oghabian MA, Yoonessi A, Tafakhori A, Ananloo ES, et al. Longitudinal effects of bumetanide on neuro-cognitive functioning in drug-resistant epilepsy. Front Neurol. (2019) 10:1–11. 10.3389/fneur.2019.00483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Kim SY, Buckwalter M, Soreq H, Vezzani A, Kaufer D. Blood–brain barrier dysfunction–induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia. (2012) 53:37–44. 10.1111/j.1528-1167.2012.03701.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. (2010) 37:13–25. 10.1016/j.nbd.2009.07.030 [DOI] [PubMed] [Google Scholar]
- 434.Sobue K, Yamamoto N, Yoneda K, Hodgson ME, Yamashiro K, Tsuruoka N, et al. Induction of blood-brain barrier properties in immortalized bovine brain endothelial cells by astrocytic factors. Neurosci Res. (1999) 35:155–64. 10.1016/S0168-0102(99)00079-6 [DOI] [PubMed] [Google Scholar]
- 435.Régina A, Morchoisne S, Borson ND, McCall AL, Drewes LR, Roux F. Factor(s) released by glucose-deprived astrocytes enhance glucose transporter expression and activity in rat brain endothelial cells. Biochim Biophys Acta Mol Cell Res. (2001) 1540:233–42. 10.1016/S0167-4889(01)00133-1 [DOI] [PubMed] [Google Scholar]
- 436.Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev. (1999) 36:179–94. 10.1016/S0169-409X(98)00085-4 [DOI] [PubMed] [Google Scholar]
- 437.Gaillard PJ, Van Der Sandt ICJ, Voorwinden LH, Vu D, Nielsen JL, De Boer AG, et al. Astrocytes increase the functional expression of P-glycoprotein in an in vitro model of the blood-brain barrier. Pharm Res. (2000) 17:1198–205. 10.1023/A:1026406528530 [DOI] [PubMed] [Google Scholar]
- 438.Dehouck M-P, Méresse S, Delorme P, Fruchart J-C, Cecchelli R. An easier, reproducible, and mass-production method to study the blood–brain barrier in vitro. J Neurochem. (1990) 54:1798–801. 10.1111/j.1471-4159.1990.tb01236.x [DOI] [PubMed] [Google Scholar]
- 439.Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. (2005) 25:25–39. 10.1007/s10571-004-1375-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Abbott NJ. Dynamics of CNS Barriers: Evolution, Differentiation, and Modulation. Cell Mol Neurobiol. (2005) 25:5–23. 10.1007/s10571-004-1374-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. (2013) 61:1939–58. 10.1002/glia.22575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. (2000) 20:131–47. 10.1023/A:1007074420772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Schwaninger M, Sallmann S, Petersen N, Schneider A, Prinz S, Libermann TA, et al. Bradykinin induces interleukin-6 expression in astrocytes through activation of nuclear factor-κB. J Neurochem. (1999) 73:1461–6. 10.1046/j.1471-4159.1999.0731461.x [DOI] [PubMed] [Google Scholar]
- 444.Smith NM, Giacci MK, Gough A, Bailey C, McGonigle T, Black AMB, et al. Inflammation and blood-brain barrier breach remote from the primary injury following neurotrauma. J Neuroinflamm. (2018) 15:1–18. 10.1186/s12974-018-1227-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Vliet EA, Van Arau SC, Redeker S, Schaik R, Van Aronica E, Gorter JA. Blood – brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. (2007) 130:521–34. 10.1093/brain/awl318 [DOI] [PubMed] [Google Scholar]
- 446.Tomkins O, Feintuch A, Benifla M, Cohen A, Friedman A, Shelef I. Blood-brain barrier breakdown following traumatic brain injury : a possible role in posttraumatic epilepsy. Cardiovasc Psychiatry Neurol. (2011) 2011:1–11. 10.1155/2011/765923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, et al. Seizure-promoting effect of blood–brain barrier disruption. Epilepsia. (2014) 48:732–42. 10.1111/j.1528-1167.2007.00988.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.van Vliet EA, Aronica E, Gorter JA. Role of blood-brain barrier in temporal lobe epilepsy and pharmacoresistance. Neuroscience. (2014) 277:455–73. 10.1016/j.neuroscience.2014.07.030 [DOI] [PubMed] [Google Scholar]
- 449.Rigau V, Morin M, Rousset M-C, de Bock F, Lebrun A, Coubes P, et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain. (2007) 130:1942–56. 10.1093/brain/awm118 [DOI] [PubMed] [Google Scholar]
- 450.Lebrun A, Rousset M, Fagni L, Bock D, Lerner-Natoli M. Epileptiform activity induces vascular remodeling and zonula occludens 1 downregulation in organotypic hippocampal cultures : role of VEGF signaling pathways me. J Neurosci. (2011) 31:10677–88. 10.1523/JNEUROSCI.5692-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Castañeda-Cabral JL, Beas-Zárate C, Rocha-Arrieta LL, Orozco-Suárez SA, Alonso-Vanegas M, Guevara-Guzmán R, et al. Increased protein expression of VEGF-A, VEGF-B, VEGF-C and their receptors in the temporal neocortex of pharmacoresistant temporal lobe epilepsy patients. J Neuroimmunol. (2019) 328:68–72. 10.1016/j.jneuroim.2018.12.007 [DOI] [PubMed] [Google Scholar]
- 452.Montpellier D. Cerebrovascular remodeling and epilepsy. Neuroscientist. (2013) 19:304–12. 10.1177/1073858412462747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, et al. TGF- b receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. (2007) 130:535–47. 10.1093/brain/awl317 [DOI] [PubMed] [Google Scholar]
- 454.Aronica E, Vliet EA, Van Mayboroda OA, Troost D, Lopes FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. (2000) 12:2333–44. 10.1046/j.1460-9568.2000.00131.x [DOI] [PubMed] [Google Scholar]
- 455.Perillan PR, Chen M, Potts EA, Simard JM, Given DM. Transforming growth factor-beta 1 regulates Kir2.3 inward rectifier K+ channels via phospholipase C and protein kinase c-delta in reactive astrocytes from adult rat brain. J Biol Chem. (2002) 277:1974–80. 10.1074/jbc.M107984200 [DOI] [PubMed] [Google Scholar]
- 456.Braganza O, Bedner P, Hüttmann K, Staden von E, Friedman A, Seifert G, et al. Albumin is taken up by hippocampal NG2 cells and astrocytes and decreases gap junction coupling. Epilepsia. (2013) 53:1898–906. 10.1111/j.1528-1167.2012.03665.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, et al. Lasting blood – brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. (2004) 24:7829–36. 10.1523/JNEUROSCI.1751-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.Levy N, Milikovsky DZ, Baranauskas G, Vinogradov E, David Y, Ketzef M, et al. Differential TGF- β signaling in glial subsets underlies IL-6 – mediated epileptogenesis in mice. J Immunol. (2015) 195:1713–22. 10.4049/jimmunol.1401446 [DOI] [PubMed] [Google Scholar]
- 459.Kimple AJ, Yasgar A, Hughes M, Jadhav A, Willard FS, Robin E, et al. A high-throughput fluorescence polarization assay for inhibitors of the goloco motif/G-alpha interaction. Comb Chem High Throughput Screen. (2009) 11:396–409. 10.2174/138620708784534770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Weissberg I, Wood L, Kamintsky L, Vazquez O, Milikovsky DZ, Alexander A, et al. Neurobiology of disease albumin induces excitatory synaptogenesis through astrocytic TGF- β/ALK5 signaling in a model of acquired epilepsy following blood – brain barrier dysfunction. Neurobiol Dis. (2015) 78:115–25. 10.1016/j.nbd.2015.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Vliet EA, Van Zibell G, Pekcec A, Schlichtiger J, Edelbroek PM, Holtman L, et al. COX-2 inhibition controls P-glycoprotein expression and promotes brain delivery of phenytoin in chronic epileptic rats. Neuropharmacology. (2010) 58:404–12. 10.1016/j.neuropharm.2009.09.012 [DOI] [PubMed] [Google Scholar]
- 462.Dombrowski SM, Desai SY, Marroni M, Cucullo L, Goodrich K, Bingaman W, et al. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory Epilepsy. (2001) 42:1501–6. 10.1046/j.1528-1157.2001.12301.x [DOI] [PubMed] [Google Scholar]
- 463.Löscher W, Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J Pharmacol Exp Ther. (2002) 301:7–14. 10.1124/jpet.301.1.7 [DOI] [PubMed] [Google Scholar]
- 464.Sisodiya SM, Lin W, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. (2002) 125:22–31. 10.1093/brain/awf002 [DOI] [PubMed] [Google Scholar]
- 465.Aronica E, Sisodiya SM, Gorter JA. Cerebral expression of drug transporters in epilepsy. Adv Drug Deliv Rev. (2012) 64:919–29. 10.1016/j.addr.2011.11.008 [DOI] [PubMed] [Google Scholar]
- 466.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. (2003) 55:3–29. 10.1016/s0169-409x(02)00169-2 [DOI] [PubMed] [Google Scholar]
- 467.Sun H, Dai H, Shaik N, Elmquist WF. Drug efflux transporters in the CNS. Adv Drug Deliv Rev. (2003) 55:83–105. 10.1016/s0169-409x(02)00172-2 [DOI] [PubMed] [Google Scholar]
- 468.Löscher W. Drug transporters in the epileptic brain. Epilepsia. (2007) 48:8–13. 10.1111/j.1528-1167.2007.00993.x [DOI] [PubMed] [Google Scholar]
- 469.Brandt C, Bethmann K, Gastens AM, Löscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. (2006) 24:202–11. 10.1016/j.nbd.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 470.Van Vliet EA, Van Schaik R, Edelbroek PM, Redeker S, Aronica E, Wadman WJ, et al. Inhibition of the multidrug transporter P-glycoprotein improves seizure control in phenytoin-treated chronic epileptic rats. Epilepsia. (2006) 47:672–80. 10.1111/j.1528-1167.2006.00496.x [DOI] [PubMed] [Google Scholar]
- 471.Vliet EA, Van Schaik R, Van Edelbroek PM, Voskuyl RA, Redeker S, Aronica E, et al. Region-specific overexpression of P-glycoprotein at the blood-brain barrier affects brain uptake of phenytoin in epileptic rats. J Pharmacol Exp Ther. (2007) 322:141–7. 10.1124/jpet.107.121178 [DOI] [PubMed] [Google Scholar]
- 472.Lange ECM, De Berg DJ, Bellanti F, Voskuyl RA, Syvänen S. P-glycoprotein protein expression versus functionality at the blood-brain barrier using immunohistochemistry, microdialysis and mathematical modeling. Eur J Pharm Sci. (2018) 124:61–70. 10.1016/j.ejps.2018.08.022 [DOI] [PubMed] [Google Scholar]
- 473.Borlot F, Wither RG, Ali A, Wu N, Verocai F, Andrade DM. A pilot double-blind trial using verapamil as adjuvant therapy for refractory seizures. Epilepsy Res. (2014) 108:1642–51. 10.1016/j.eplepsyres.2014.08.009 [DOI] [PubMed] [Google Scholar]
- 474.Nicita F, Spalice A, Papetti L, Nikanorova M, Iannetti P, Parisi P. Efficacy of verapamil as an adjunctive treatment in children with drug-resistant epilepsy : a pilot study. Seizure Eur J Epilepsy. (2014) 23:36–40. 10.1016/j.seizure.2013.09.009 [DOI] [PubMed] [Google Scholar]
- 475.Summers MA, Moore JL, Mcauley JW. Use of verapamil as a potential P-glycoprotein inhibitor in a patient with refractory epilepsy. Ann Pharmacother. (2004) 38:1631–4. 10.1345/aph.1E068 [DOI] [PubMed] [Google Scholar]
- 476.Pirker S, Baumgartner C. Termination of refractory focal status epilepticus by the P-glycoprotein inhibitor verapamil. Eur J Neurol. (2011) 18:e151. 10.1111/j.1468-1331.2011.03513.x [DOI] [PubMed] [Google Scholar]
- 477.Asadi-Pooya AA, Ali SM, Abdi-Ardekani A, Sperling MR. Epilepsy and behavior adjunctive use of verapamil in patients with refractory temporal lobe epilepsy : a pilot study. Epilepsy Behav. (2013) 29:150–4. 10.1016/j.yebeh.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 478.Narayanan J, Frech R, Walters S, Patel V, Frigerio R, Maraganore DM. Low dose verapamil as an adjunct therapy for medically refractory epilepsy – an open label pilot study. Epilepsy Res. (2016) 126:197–200. 10.1016/j.eplepsyres.2016.07.004 [DOI] [PubMed] [Google Scholar]
- 479.Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. (2006) 26:2862–70. 10.1523/JNEUROSCI.4048-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Fabene PF, Marzola P, Sbarbati A, Bentivoglio M. Magnetic resonance imaging of changes elicited by status epilepticus in the rat brain : diffusion-weighted and T2-weighted images, regional blood volume maps, and direct correlation with tissue and cell damage. Neuroimage. (2003) 18:375–89. 10.1016/s1053-8119(02)00025-3 [DOI] [PubMed] [Google Scholar]
- 481.Winkler MKL, Chassidim Y, Lublinsky S, Revankar GS, Major S, Kang E, et al. Impaired neurovascular coupling to ictal epileptic activity and spreading depolarization in a patient with subarachnoid hemorrhage: possible link to blood–brain barrier dysfunction. Epilepsia. (2013) 53:22–30. 10.1111/j.1528-1167.2012.03699.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Zhao M, Suh M, Ma H, Perry C, Geneslaw A, Schwartz TH. Focal increases in perfusion and decreases in hemoglobin oxygenation precede seizure onset in spontaneous human epilepsy. Epilepsia. (2007) 48:2059–67. 10.1111/j.1528-1167.2007.01229.x [DOI] [PubMed] [Google Scholar]
- 483.Gómez-Gonzalo M, Losi G, Brondi M, Uva L, Sato SS, Mansvelder HD. Ictal but not interictal epileptic discharges activate astrocyte endfeet and elicit cerebral arteriole responses. Front Cell Neurosci. (2011) 5:8 10.3389/fncel.2011.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Vezzani A, Pascente R, Ravizza T. Biomarkers of epileptogenesis: the focus on glia and cognitive dysfunctions. Neurochem Res. (2017) 42:2089–98. 10.1007/s11064-017-2271-3 [DOI] [PubMed] [Google Scholar]
- 485.van Vliet EA, Aronica E, Vezzani A, Ravizza T. Review: neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol Appl Neurobiol. (2018) 44:91–111. 10.1111/nan.12444 [DOI] [PubMed] [Google Scholar]