

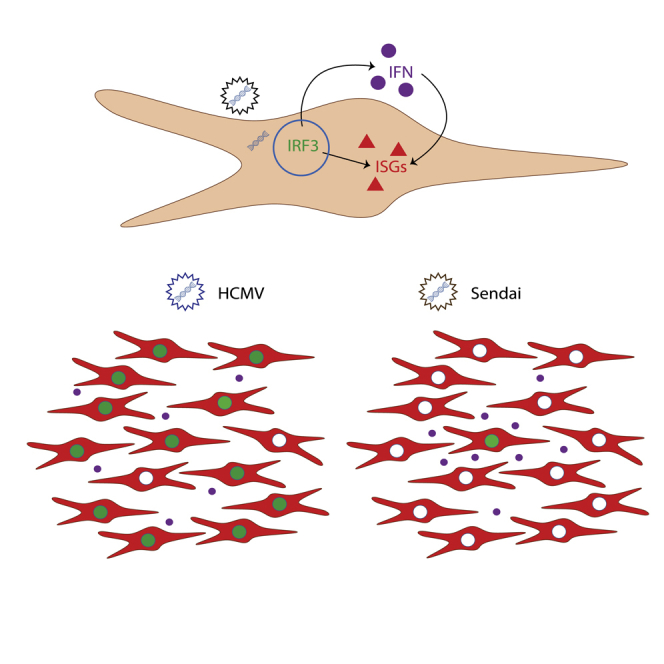
Summary
Type 1 interferon (IFN) plays a critical role in early antiviral defense and priming of adaptive immunity by signaling upregulation of host antiviral IFN-stimulated genes (ISGs). Certain stimuli trigger strong activation of IFN regulatory factor 3 (IRF3) and direct upregulation of ISGs in addition to IFN. It remains unclear why some stimuli are stronger activators of IRF3 and how this leads to IFN-independent antiviral protection. We found that UV-inactivated human cytomegalovirus (HCMV) particles triggered an IFN-independent ISG signature that was absent in cells infected with UV-inactivated Sendai virus particles. HCMV particles triggered mostly uniform activation of IRF3 and low-level IFN-β production within the population while SeV particles triggered a small fraction of cells producing abundant IFN-β. These findings suggest that population-level activation of IRF3 and antiviral protection emerges from a diversity of responses occurring simultaneously in single cells. Moreover, this occurs in the absence of virus replication.
Subject Areas: Biological Sciences, Molecular Biology, Microbiology, Virology, Cell Biology
Graphical Abstract
Highlights
-
•
The antiviral response to virus particles requires low levels of interferon
-
•
Cells respond differently to HCMV or SeV particles
-
•
Heterogeneous IRF3 activation influences the response to virus
Biological Sciences; Molecular Biology; Microbiology; Virology; Cell Biology
Introduction
Virus infection is sensed by pattern recognition receptors (PRRs) that recognize viral particle components or by-products of replication, such as cytosolic double-stranded RNA (dsRNA) and double-stranded DNA (dsDNA), and upregulate pro-inflammatory and antiviral interferon (IFN)-stimulated genes (ISGs) through production of type 1 IFN. Antiviral ISGs collectively create an antiviral state in infected and surrounding cells to restrict virus spread while pro-inflammatory ISGs recruit immune effector cells. IFN signaling is critical for early control of infection, and productive virus replication is usually dependent on the virus's ability to antagonize IFN signaling (reviewed in (Fensterl and Sen, 2009)).
Both replicating and non-replicating viruses are recognized and contribute to IFN upregulation, but most replicating viruses simultaneously suppress IFN upregulation and signaling. In fact, many viruses trigger stronger IFN responses in the absence of virus gene expression and replication (Collins et al., 2004; Mossman et al., 2001; Preston et al., 2001; Weber et al., 2013). Consequently, the immune response to many important viral pathogens and vaccines is dependent on recognition of defective virus particles or abortive infection (Drayman et al., 2019; Ho et al., 2016; O'Neal et al., 2019; Yount et al., 2006). Recognition of virus particles can involve a combination of stimulatory components, such as viral glycoproteins or packaged nucleic acid, along with cellular perturbations, such as virus entry (Hare et al., 2015; Holm et al., 2012; Juckem et al., 2008). How cells recognize and respond to virus particles are important because these processes play a critical role in the immune response to virus infection, as well as the immune response to non-replicating viral vectors.
IFN-β is the first IFN subtype produced during infection and can be produced by most cell types. The IFN-β promoter contains binding sites for IFN regulatory factors (IRFs), NF-κB and AP1, which together modulate IFN-β expression (reviewed in (Balachandran and Beg, 2011; Honda et al., 2006)). IRF3 is a critical transcription factor for the upregulation of IFN-β and is constitutively expressed in a majority of cells (Au et al., 1995; Hiscott, 2007). When IRF3 is phosphorylated by the kinase tank-binding kinase 1 it undergoes a conformational change that promotes its dimerization, nuclear accumulation, and binding to the IFN-β promoter (Fitzgerald et al., 2003; Lin et al., 1998; Sharma et al., 2003; Yoneyama et al., 1998). Since NF-κB was first observed binding to the IFN-β enhanceosome, it has been shown to be important for early IFN-β upregulation, but it is not essential (Apostolou and Thanos, 2008; Basagoudanavar et al., 2011; Peters et al., 2002). After IFN-β is produced and secreted, it binds to the IFN-α/β receptor (IFNAR) on the surface of nearby cells. IFNAR initiates Jak-STAT signaling and formation of a complex containing STAT1, STAT2, and IRF9 named IFN-stimulated gene factor 3 (ISGF3). The ISGF3 complex binds to IFN-stimulated response elements (ISREs) in the promoters of ISGs, thus upregulating hundreds of genes involved in antiviral defense (reviewed in (Schoggins, 2018)).
In addition to upregulating IFN-β, activated IRF3 can directly upregulate a subset of ISGs by binding to ISREs and forming a transcriptional complex with CBP/p300 (Bandyopadhyay et al., 1995; Grandvaux et al., 2002; Weaver et al., 1998). IRF3-mediated upregulation of ISGs is thought to act as a first line of defense to limit virus replication in the infected cell, while IFN-β upregulates ISGs in nearby cells to halt spread of the virus. Low-level infection with non-replicating enveloped virus particles causes IRF3-mediated upregulation of ISGs in the absence of detectable IFN (Collins et al., 2004; Mossman et al., 2001; Paladino et al., 2006). While this response is IRF3-dependent, the extent of observable IRF3 posttranslational modifications fail to correlate with ISG induction (Noyce et al., 2009). Activation of NF-κB and subsequent production of IFN were found to require a higher level of virus particle stimulation (Paladino et al., 2006).
We and others have suggested that non-replicating enveloped virus particles are recognized upon membrane fusion that occurs during virus entry (Collins et al., 2004; Holm et al., 2012; Noyce et al., 2011; Tsitoura et al., 2009). However, we found that sensing of genomic nucleic acid is critical for the antiviral response to low levels of enveloped virus particles (Hare et al., 2015). Given that nucleic acid recognition is known to activate transcription factors necessary for IFN-β production, we revisited the role of IFN-β signaling in the antiviral response to two different enveloped virus particles. Sendai virus (SeV) belongs to the paramyxovirus family and is an enveloped, negative stranded, non-segmented RNA virus. Human cytomegalovirus (HCMV) belongs to the herpesvirus family and is an enveloped dsDNA virus. While SeV and HCMV are known to cause disease in mice and humans, respectively, they are also commonly used models to study innate antiviral pathways.
Results
Fibroblasts Mount an IFN-Independent Antiviral Response to HCMV Particles but Not SeV Particles
While we and others previously described ISG induction in the absence of detectable IFN production, we investigated the role of IFN signaling in the antiviral response to representative enveloped virus particles. We infected wild-type or IFNAR1 KO hTERT-immortalized human fibroblasts (THFs) with either UV-inactivated HCMV (HCMV-UV) or UV-inactivated SeV (SeV-UV) particles and measured the antiviral response. IFNAR1 KO THFs are not responsive to IFN-I but remain capable of mounting IFN-independent antiviral responses to appropriate stimuli. Consistent with previous findings, wild-type THFs infected with low levels of HCMV or SeV particles were protected from vesicular stomatitis virus expressing green fluorescent protein (VSV-GFP) in a plaque reduction assay and IFN was only detected in supernatants after THFs were infected with higher amounts of HCMV or SeV particles. Surprisingly, IFNAR1 KO THFs had a lower antiviral response to HCMV particles, requiring a higher number of particles to achieve full protection (Figure 1A) and IFNAR1 KO THFs did not mount a protective antiviral response to SeV particles at all, regardless of input (Figure 1B). These results suggest that the antiviral response to low levels of HCMV or SeV particles relies on low levels of IFN that are not detected by conventional supernatant transfers. To validate our hypothesis, we concentrated protein in supernatants from wild-type THFs infected with low levels of SeV particles and found that cells treated with these concentrated supernatants were protected (Figure S1).
Figure 1.

Antiviral Response to HCMV-UV and SeV-UV in the Presence/Absence of IFN Signaling
(A and B) Wild-type (WT) or IFNAR1 KO THFs were infected with increasing levels of UV-inactivated HCMV before challenging with VSV-GFP 16 hr later. The presence of IFN in the supernatants was assayed by transferring supernatants from infected THFs to naive wild-type THFs and challenging them with VSV-GFP 6 hr later (WT supernatant/IFNAR KO supernatant).
(C and D) WT THFs pretreated with B18R or media alone were infected with increasing levels of HCMV-UV or SeV-UV and challenged with VSV-GFP. Mean fluorescence intensity and standard error from 3 biological replicates was plotted as a percentage of mock infected cells challenged with VSV-GFP. WT and IFNAR KO, WT and IFNAR KO supernatant, or mock and B18R treated THFs were compared by two-way analysis of variance (ANOVA) with Bonferroni post-tests (∗∗ for p < 0.01, ∗∗∗ for p < 0.001).
To further test whether IFN was necessary for the antiviral response to infection with virus particles in wild-type THFs, we blocked IFN signaling with B18R, a recombinant vaccinia virus IFN antagonist (Alcami et al., 2000; Symons et al., 1995), prior to infection with HCMV or SeV particles, and measured the antiviral response using a plaque reduction assay. Similar to IFNAR1 KO THFs, B18R-treated wild-type THFs mounted a protective antiviral response to infection with HCMV particles at high levels of infection but were not protected following infection with SeV particles (Figures 1C and 1D).
IFN-III has been shown to upregulate a similar set of ISGs in cells expressing the IFN-λ receptor (IFNLR) (Kotenko et al., 2003; Sheppard et al., 2003), thus creating a protective antiviral state independent of IFNAR. IFNLR is expressed in cells of epithelial origin but is believed to be absent in fibroblasts. To determine whether THFs respond to IFN-λ, we treated THFs or A549 cells, derived from lung epithelial carcinoma, with increasing amounts of recombinant IFN-λ1. We found that A549 cells responded to IFN-λ1 in a dose-dependent manner while THFs did not, consistent with THFs lacking IFNLR (Figure S2). Therefore, the antiviral responses we observed in IFNAR1 KO THFs are independent of both IFNAR and IFNLR.
A Subset of IFN-Independent ISGs Are Upregulated in Response to HCMV but Not SeV Particles
Our antiviral data suggested that HCMV particles were capable of stimulating both IFN-independent and IFN-mediated responses while SeV particles only stimulated IFN-mediated responses. To better understand the gene products involved, we employed whole transcriptome sequencing of wild-type or IFNAR1 KO THFs infected with either HCMV-UV or SeV-UV particles. Among genes significantly upregulated >2 fold, IFNAR1 KO THFs infected with HCMV particles upregulated 51% (49/96) of genes upregulated in infected wild-type THFs. In contrast, IFNAR1 KO THFs infected with SeV particles upregulated 6% (5/79) of genes upregulated in infected wild-type THFs. The 30 genes most highly upregulated by either virus had varying degrees of IFN independence in THFs infected with HCMV particles, while there was very little IFN-independent ISG upregulation in THFs infected with SeV particles (Figure 2A). There was a high degree of overlap between all genes significantly upregulated by THFs infected with either virus (Figure 2A and Table S1).
Figure 2.

ISG Upregulation in Response to HCMV or SeV Particles
(A) Wild-type (WT) or IFNAR1 KO THFs were treated with 84 particles/cell HCMV-UV or 12 particles/cell SeV-UV and RNA harvested for high-throughput RNA sequencing analysis. The 30 most highly upregulated genes across all samples are shown.
(B–D) WT or IFNAR1 KO THFs were treated with 84 or 1,344 particles/cell of HCMV-UV or 12 or 200 particles/cell of SeV-UV and RNA harvested 6 hr post-treatment. Transcript levels of IFIT1 (B), ISG15 (C), and CXCL10 (D) were measured by quantitative RT-PCR. Mean fold change over mock and standard error from 3-6 biological replicates was graphed and WT and IFNAR KO THFs compared by two-way analysis of variance (ANOVA) with Bonferroni post-tests (∗∗ for p < 0.01 and ∗∗∗ for p < 0.001).
To validate our transcriptome data, we chose two representative ISGs that were mostly IFN independent in THFs infected with HCMV particles but not SeV particles (IFIT1 and ISG15) and one representative ISG whose upregulation was similarly IFN dependent in THFs infected with either virus (CXCL10). Quantitative reverse transcriptase polymerase chain reaction (RT-PCR) findings validated the transcriptomic data for all three genes, following both high- and low-level infection (Figures 2B–2D). The combined transcriptomic and quantitative RT-PCR results indicate that infection with HCMV particles upregulates a subset of ISGs in an IFN-independent fashion while infection with SeV particles upregulates ISGs almost entirely through IFN-dependent signaling.
IFN-Independent ISG Induction Depends on Cell Type and Virus-Intrinsic Differences
Given that infection with HCMV particles triggers a cyclic GMP-AMP synthase (cGAS)- and stimulator of IFN genes (STING)-dependent antiviral response while SeV particles trigger a mitochondrial antiviral signalling (MAVS)-dependent antiviral response (DeFilippis et al., 2010; Hare et al., 2015; Seth et al., 2005), we hypothesized that dsDNA recognition through STING directly upregulates ISGs more effectively than dsRNA recognition through MAVS. To test this hypothesis, we transfected wild-type and IFNAR1 KO THFs with either dsDNA or dsRNA of a matched 1,000 base pair sequence and harvested cellular RNA 6 hr later. Transfection with either dsDNA or dsRNA was sufficient to upregulate IFIT1, ISG15, and CXCL10 in IFNAR1 KO THFs (Figures 3A–3C).
Figure 3.

ISG Upregulation in Response to Transfected Nucleic Acid or Replicating Virus
Wild-type (WT) or IFNAR1 KO THFs were transfected with matched sequence dsRNA or dsDNA (A–C) or infected with replication competent HCMV or SeV and RNA harvested 6 hr post-treatment (D–F). Transcript levels of IFIT1 (A and D), ISG15 (B and E), and CXCL10 (C and F) were measured by quantitative RT-PCR. Mean fold change over mock and standard error from 3-7 biological replicates was graphed and WT and IFNAR KO THFs compared by two-way analysis of variance (ANOVA) with Bonferroni post-tests (∗ for p < 0.05, ∗∗ for p < 0.01 and ∗∗∗ for p < 0.001).
Next, we chose to test whether the differences in IFN-independent responses between HCMV and SeV particles also held true between replicating HCMV and SeV. Under similar experimental conditions, ISG induction to replicating HCMV (Figures 3D–3F) mirrored that of non-replicating HCMV (Figures 2B–2D), while ISG induction by replicating SeV (Figures 3D–3F) more closely mirrored that of transfected dsRNA (Figures 3A–3C) as opposed to SeV-UV (Figures 2C–2E). Thus, IFIT1 and ISG15 upregulation by SeV is less IFN dependent than what we observed with SeV-UV. Moreover, similar to UV-inactivated particles, there are clear differences in the antiviral response to replicating HCMV or SeV.
To determine whether different cell types similarly upregulate ISGs through IFN-dependent and cell-intrinsic pathways, we infected THFs and a variety of primary and immortalized cells with low levels of SeV-UV or HCMV-UV. To measure ISG transcript upregulation in the absence of IFN production, we used the inhibitor cycloheximide (CHX) to block de novo protein synthesis. Consistent with cell-intrinsic IFN-independent signaling, HCMV-UV infection strongly upregulated IFIT1 and ISG15 transcripts in THFs and primary human embryonic lung (HEL) fibroblasts in the absence of protein synthesis (Figures 4A–4F). As expected, IFN-independent upregulation of ISGs in THFs and HEL fibroblasts was significantly lower following SeV-UV infection (Figures 4A–4F). Immortalized bronchiole epithelial cells (NuLi-1) responded similarly to SeV-UV but not at all when infected with HCMV-UV (Figures 4G–4I), consistent with a previous report of impaired entry of HCMV strain AD169 in epithelial cells (Ryckman et al., 2006). Interestingly, THP-1-derived macrophages upregulated IFIT1 and ISG15 in the absence of protein synthesis following infection with SeV-UV instead of HCMV-UV (Figures 4J–4L). This may be because THP-1 macrophages have higher basal expression of nucleic acid sensors, as we observed in preliminary experiments (data not shown). Thus, both fibroblasts and macrophages appear to upregulate ISGs through both IFN-dependent and cell-intrinsic pathways, with differences noted between RNA and DNA viruses.
Figure 4.

ISG Upregulation in Different Cell Types Infected with HCMV and SeV Particles
(A–L) THFs (A–C), HEL fibroblasts (D–F), NuLi-1 epithelial cells (G–I), or THP-1-derived macrophages (J–L) were infected with 84 particles/cell HCMV or 12 particles/cell SeV in the presence of the protein synthesis inhibitor CHX or DMSO alone and RNA harvested 6 hr post-treatment. Transcript levels of IFIT1, ISG15, and CXCL10 were measured by quantitative RT-PCR. Mean fold change over mock and standard error from 3-4 biological replicates was graphed and DMSO- and CHX-treated cells compared by two-way analysis of variance (ANOVA) with Bonferroni post-tests (∗∗ for p < 0.01 and ∗∗∗ for p < 0.001).
HCMV and SeV Particles Differentially Activate IRF3
We previously demonstrated that IRF3 is essential for the antiviral response to a variety of enveloped virus particles and that activation of RelA, a subunit of NF-κB, is associated with a higher level of particle stimulation and detectable IFN-β production (Collins et al., 2004; Paladino et al., 2006). The involvement of IFN signaling prompted us to re-examine the relative contribution of these two transcription factors in THFs treated with either HCMV or SeV particles.
To test the relative importance of IRF3 and NF-κB, we infected wild-type, IRF3 KO, or RelA KO THFs with increasing levels of HCMV or SeV particles and measured antiviral responses, IFN production, and upregulation of representative ISGs. Consistent with previous findings, IRF3 was essential for antiviral protection (Figures 5A and 5B), IFN production (Figures 5C and 5D), and upregulation of IFIT1, ISG15, and CXCL10 (Figures 5E–5G) in response to infection with either HCMV or SeV particles. RelA was dispensable for antiviral protection, IFN production, and upregulation of ISGs after treatment with HCMV particles but did play a role in response to SeV particles (Figures 5A–5G).
Figure 5.

Role of IRF3 and RelA in the Antiviral Response to HCMV and SeV Particles
(A–D) Wild-type (WT), IRF3 KO, or RelA KO THFs were treated with increasing levels of UV-inactivated HCMV or SeV (A and B), and 16 hr post-treatment supernatants were transferred to naive WT THFs (C and D). All THFs were subsequently challenged with VSV-GFP. Mean GFP fluorescence and standard error from 3 biological replicates were plotted as a percentage of mock treated, VSV-GFP infected cells. IRF3 and RelA KO THFs were compared with wild type by two-way analysis of variance (ANOVA) with Bonferroni post-tests (∗ for p < 0.05, ∗∗ for p < 0.01 and ∗∗∗ for p < 0.001).
(E–G) WT, IRF3, or RelA KO THFs were treated with 84 or 1,344 particles/cell of HCMV-UV or 12 or 200 particles/cell of SeV-UV and transcript levels of IFIT1, ISG15, and CXCL10 were measured by quantitative RT-PCR. Mean fold change relative to mock and standard error from 7 biological replicates was graphed. IRF3 and RelA KO THFs were compared with wild type by two-way ANOVA with Bonferroni post-tests (∗ for p < 0.05, ∗∗ for p < 0.01 and ∗∗∗ for p < 0.001).
(H) WT THFs were treated with 1,344 particles/cell of HCMV-UV or 200 particles/cell of SeV-UV, fixed 4 hr post-treatment and IRF3 and cell nuclei visualized by immunofluorescence microscopy. Scale bars of 100 μm are shown, and IRF3 +ve nuclei in SeV-UV infected THFs are marked.
(I) IRF3-positive nuclei as a percentage of total nuclei were calculated from 3 independent fields of view and mean % IRF3 positive nuclei and standard error from 3 biological replicates were graphed. SeV-UV and HCMV-UV infected THFs were compared by student's t-test (∗∗∗ for p < 0.001).
Given the importance of IRF3 for the antiviral response to infection with either HCMV or SeV particles, we measured IRF3 activation in wild-type THFs infected with either HCMV or SeV particles. We observed IRF3 nuclear translocation in the majority of cells following infection with HCMV particles but only in a small proportion of THFs infected with SeV particles (Figures 5H and 5I). Collectively, these data suggest that SeV particles may trigger an IRF3-dependent IFN response in a minority of cells that protects the monolayer through paracrine signaling. By virtue of HCMV particles stimulating IRF3 more evenly across the monolayer, cells are less reliant on paracrine IFN signaling for antiviral protection.
HCMV and SeV Particles Induce Heterogeneous IFN-β Production
To measure IFN-β production in individual cells, we generated THFs with a stably integrated green fluorescent protein (GFP) reporter controlled by the IFN-β promoter sequence (IFN-β-GFP THFs). We used flow cytometry to measure upregulation of GFP (IFN-β) and ISG15 in our IFN-β-GFP THF reporter line. IFN-β-GFP THFs treated with 1000 U/ml recombinant IFNα had increased ISG15 expression but no change in GFP (Figure 6A). Infection with HCMV particles increased GFP expression in the majority of cells and increased ISG15 expression in nearly all cells. Infection with SeV particles increased GFP expression in <5% of cells but increased ISG15 expression in nearly all cells.
Figure 6.

Single-Cell Dynamics of IFN-β and ISG15 Upregulation in Response to HCMV or SeV Particles
(A–E) IFN-β-GFP THFs were pretreated with B18R or media alone and mock treated, treated with IFNα (1000 U/ml), infected with HCMV-UV (1,344 particles/cell), or infected with SeV-UV (200 particles/cell). Cells were fixed 16 hr later and levels of GFP and ISG15 expression measured by flow cytometry (A–D). Cells were categorized as GFPlow/high and ISG15low/high based on measurements of mock treated cells and mean proportion and standard error from 3 biological replicates graphed. Similarly, treated IFN-β-GFP THFs were visualized by fluorescence microscopy, and representative images from 3 biological replicates are shown (E). Scale bars of 400 μm are shown.
To determine the contribution of IFN signaling to ISG15 expression in individual cells, we pretreated IFN-β-GFP THFs with B18R prior to infection with HCMV or SeV particles to block IFN signaling (but not production). As expected, ISG15 upregulation by recombinant IFNα was nearly absent when IFN-β-GFP THFs were pretreated with B18R (Figure 6A). When IFN-β-GFP THFs were infected with either HCMV or SeV particles, pre-treatment with B18R primarily prevented ISG15 upregulation in cells lacking GFP upregulation. This observation is consistent with ISG15high GFPhigh cells upregulating ISG15 directly, while ISG15high GFPlow cells upregulate ISG15 through IFN-dependent signaling.
To confirm our flow cytometry data and better visualize cell populations, we observed GFP (IFN-β) and ISG15 in adherent IFN-β-GFP THFs using immunofluorescence microscopy. As with flow cytometry, GFP was upregulated in the majority of cells treated with HCMV particles and highly upregulated in <5% of cells treated with SeV particles, with ISG15 observed throughout both treated populations (Figure 6B). Similar to our flow cytometry data, treatment with B18R predominantly affected ISG15 expression in SeV particle treated cells and predominantly in cells lacking IFNβ production.
The simplest explanation for heterogeneous IFN-β production in THFs infected with SeV-UV is that IFN-β producing THFs contain more SeV particles. We infected IFN-β-GFP THFs with replicating SeV or SeV-UV, measured SeV proteins using a pan-specific SeV antibody and were able to detect SeV protein in both SeV and SeV-UV infected IFN-β-GFP THFs. However, we found no clear correlation between GFP expression and SeV protein in IFN-β-GFP THFs infected with SeV particles (Figure 7A). SeV is known to produce and package defective viral genomes (DVGs) recognized by RIG-I during SeV infection, and these may be responsible for IFN-β production in cells infected with SeV particles (Baum et al., 2010; Sanchez-Aparicio et al., 2017). We infected IFN-β-GFP THFs with SeV particles and sorted GFPhigh and GFPlow cells before harvesting RNA. We measured both genomic SeV (gSeV) and SeV DVG-546, the predominant DVG found in the SeV stocks used in this study, using quantitative RT-PCR with primers designed by Genoyer, et al. (Genoyer and Lopez, 2019). We detected gSeV and DVG-546 in GFPhigh and GFPlow cells but found no difference in gSeV and a noticeable but not significant increase in DVG-546 in GFPhigh cells (Figures 7B and 7C).
Figure 7.

Relative SeV Abundance in IFN-β Producing Cells
(A–C) IFN-β-GFP THFs were mock infected, infected with replicating SeV (200 particles/cell), or infected with SeV-UV (200 particles/cell). (A) Cells were fixed 16 hr later and GFP and SeV proteins visualized by fluorescence microscopy. Scale bars of 100 μm are shown. (B and C) SeV-UV infected cells were sorted for GFP fluorescence 16 hr post-infection and gSeV (B) or DVG-546 (C) measured by quantitative RT-PCR. Mean fold change relative to SeV-UV-GFPlow and standard error from 3 biological replicates was graphed. SeV-UV-GFPlow and SeV-UV-GFPhigh populations were compared by student's t-test. n.a., no amplification; n.s., not significant.
Discussion
Our previous reporting of an IRF3-dependent IFN-independent response to incoming virus particles stemmed from examination of individual ISGs by RT-PCR and an inability in multiple assays to detect IFN (Collins et al., 2004; Mossman et al., 2001). Similar to other studies, our early interpretation was that membrane perturbation during virus entry, independent of subsequent virus replication or the need for viral genomes, stimulates a similar response to all enveloped virus particles (Collins et al., 2004; Holm et al., 2012; Noyce et al., 2011). Along with previous findings (Hare et al., 2015), this study clearly demonstrates a difference in the cellular response to HCMV and SeV particles, despite both being enveloped virus particles. Although HCMV and SeV contain DNA and RNA genomes, respectively, differential nucleic acid sensing alone does not explain the differences in the antiviral response to each, as THFs responded similarly to transfected dsRNA and dsDNA.
Consistent with previous studies, HCMV particles induced IRF3-mediated upregulation of ISGs. Unexpectedly, low levels of HCMV particles also induced IRF3-mediated upregulation of IFN. It is difficult to determine the relative contribution of IFN-dependent versus IFN-independent ISG upregulation to antiviral defense in wild-type cells, as IRF3 is essential for both IFN-β production and direct ISG upregulation in this system. While IFNAR1 KO cells upregulate an IRF3-dependent subset of ISGs that are protective against subsequent challenge, a higher level of particle stimulation is required for full protection. Therefore, it appears that IFN-β and IRF3 upregulate complimentary and overlapping sets of ISGs that act in concert to protect cells from infection.
In contrast to HCMV, we found that SeV particles strongly stimulate a minority of cells that protect the surrounding monolayer through production of IFN-β. Heterogeneous production of IFN-β has been described in response to several different replicating viruses, even following high multiplicity or in vivo infection (Chen et al., 2010; Drayman et al., 2019; Kallfass et al., 2013; Killip et al., 2017; O'Neal et al., 2019; Rand et al., 2012; Zawatzky et al., 1985). In many of these cases, IFN-β production occurs in abortively infected cells where the virus fails to suppress antiviral signaling (Drayman et al., 2019; O'Neal et al., 2019). The percentage of IFN-β producing cells varies widely among viruses and infected cell types and results from a combination of virus- and cell-intrinsic factors (Killip et al., 2017; Rand et al., 2012; Zhao et al., 2012).
We found SeV genomes and defective viral genomes in cells regardless of whether they were producing IFN-β. While SeV defective viral genomes contain complimentary ends that form stimulatory dsRNA recognized by RIG-I, packaged SeV defective genomes are normally tightly bound by nucleocapsid protein which prevent formation of the stimulatory hairpin structure (Kolakofsky, 1976). However, not all SeV genomes may be properly packaged in nucleoprotein owing to errors in replication exacerbated by the presence of defective genomes (Genoyer and Lopez, 2019). This additional requirement may explain why only a small fraction of cells infected with SeV particles produced IFN-β. Unfortunately, we were not able to measure whether SeV genomes and SeV DVG-546 were exposed in all SeV-UV infected cells or only in a subset. However, it appears that delivery of packaged SeV RNA is insufficient for recognition by RIG-I, and additional factors are required for immune activation.
While in this study we used HCMV and SeV as representative DNA and RNA viruses, respectively, we do not propose that all enveloped DNA or RNA viruses will respond in a similar fashion. Indeed, the diversity of responses is likely to mirror the diverse composition of different virus particles. As the innate antiviral response to virus infection is heavily dependent on recognition of non-replicating virus particles, understanding intrinsic and innate responses to diverse infections helps explain why some infections are associated with greater disease. Moreover, understanding the innate response to non-replicating virus particles is becoming more important as virus-based therapies engineered in the lab make their way to the clinic. Introducing genes by in vivo or ex vivo transduction of cells relies on minimizing the innate response to virus vectors to maximize engraftment and stable expression of therapeutic genes (Brown et al., 2007). In contrast, viral vaccines or oncolytic virus require a robust innate response in order to recruit and activate antigen-presenting cells (Davola and Mossman, 2019; Proietti et al., 2002; Walsh et al., 2019). Notably, SeV is being developed as a vector for gene therapy, as well as vaccination, and the ability to modulate immunogenicity of SeV particles will be important for either application (reviewed in (Baltusnikas et al., 2019; Nakanishi and Otsu, 2012; Saga and Kaneda, 2015)). Lipid-based delivery of nucleic acid is used in addition to virus-based vectors and also triggers innate immune activation. Our results suggest that formulation and delivery route for nucleic acid may influence the innate response as much as the nucleic acid itself and must be taken into account.
Limitations of the Study
The experiments described in this study were done in life-extended human fibroblasts with HCMV and SeV used as representative enveloped viruses. Fibroblasts are often used as a model system in which to study regulation of IFN-β, sometimes referred to as fibroblast IFN, because they play an important role in the first wave of IFN-β production during infection. Although nearly all cells are capable of producing and responding to IFN-β, there is some variation in signaling pathways present across different cell types. It remains to be seen how heterogeneous IFN-β production affects antiviral protection in tissue made up of many different cell types. HCMV and SeV are both well-studied enveloped viruses in the context of innate immune activation and seem to activate a dramatically different proportion of cells. These results show that differences may exist in the antiviral response to non-replicating virus particles, but further experiments with other virus particles are necessary to understand what type of innate response they induce.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Karen L Mossman (mossk@mcmaster.ca).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The RNA-seq data generated during this study are available at GEO (GEO: GSE149705).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Dr. Victor DeFilippis for providing us with hTERT-immortalized human fibroblasts (THF) and various knock-out lines used in this study. D.H. was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) doctoral scholarship and through the Ontario Graduate Scholarship program. Studies in the Mossman Lab were supported by a Canadian Institutes of Health Research (CIHR) project grant (MOP 57669).
Author Contributions
D.H. and K.M. conceived and designed the study. D.H. and K.B. performed experiments. A.D.-G. analyzed transcriptome data. D.H. and K.M. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101864.
Supplemental Information
References
- Alcami A., Symons J.A., Smith G.L. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J. Virol. 2000;74:11230–11239. doi: 10.1128/jvi.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou E., Thanos D. Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell. 2008;134:85–96. doi: 10.1016/j.cell.2008.05.052. [DOI] [PubMed] [Google Scholar]
- Au W.C., Moore P.A., Lowther W., Juang Y.T., Pitha P.M. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. U S A. 1995;92:11657–11661. doi: 10.1073/pnas.92.25.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran S., Beg A.A. Defining emerging roles for NF-kappaB in antivirus responses: revisiting the interferon-beta enhanceosome paradigm. PLoS Pathog. 2011;7:e1002165. doi: 10.1371/journal.ppat.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltusnikas J., Satkauskas S., Lundstrom K. Constructing RNA viruses for long-term transcriptional gene silencing. Trends Biotechnol. 2019;37:20–28. doi: 10.1016/j.tibtech.2018.07.015. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S.K., Leonard G.T., Jr., Bandyopadhyay T., Stark G.R., Sen G.C. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 1995;270:19624–19629. doi: 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- Basagoudanavar S.H., Thapa R.J., Nogusa S., Wang J., Beg A.A., Balachandran S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J. Virol. 2011;85:2599–2610. doi: 10.1128/JVI.02213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Sachidanandam R., Garcia-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U S A. 2010;107:16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown B.D., Sitia G., Annoni A., Hauben E., Sergi L.S., Zingale A., Roncarolo M.G., Guidotti L.G., Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood. 2007;109:2797–2805. doi: 10.1182/blood-2006-10-049312. [DOI] [PubMed] [Google Scholar]
- Chen S., Short J.A., Young D.F., Killip M.J., Schneider M., Goodbourn S., Randall R.E. Heterocellular induction of interferon by negative-sense RNA viruses. Virology. 2010;407:247–255. doi: 10.1016/j.virol.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S.E., Noyce R.S., Mossman K.L. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol. 2004;78:1706–1717. doi: 10.1128/JVI.78.4.1706-1717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davola M.E., Mossman K.L. Oncolytic viruses: how "lytic" must they be for therapeutic efficacy? Oncoimmunology. 2019;8:e1581528. doi: 10.1080/2162402X.2019.1596006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis V.R., Alvarado D., Sali T., Rothenburg S., Fruh K. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol. 2010;84:585–598. doi: 10.1128/JVI.01748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayman N., Patel P., Vistain L., Tay S. HSV-1 single-cell analysis reveals the activation of anti-viral and developmental programs in distinct sub-populations. Elife. 2019;8:e46339. doi: 10.7554/eLife.46339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V., Sen G.C. Interferons and viral infections. Biofactors. 2009;35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., Coyle A.J., Liao S.M., Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Genoyer E., Lopez C.B. Defective viral genomes alter how Sendai virus interacts with cellular trafficking machinery, leading to heterogeneity in the production of viral particles among infected cells. J. Virol. 2019;93 doi: 10.1128/JVI.01579-18. e01579–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux N., Servant M.J., tenOever B., Sen G.C., Balachandran S., Barber G.N., Lin R., Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare D.N., Collins S.E., Mukherjee S., Loo Y.M., Gale M., Jr., Janssen L.J., Mossman K.L. Membrane perturbation-associated Ca2+ signalling and incoming genome sensing are required for the host response to low-level enveloped virus particle entry. J. Virol. 2015;90:3018–3027. doi: 10.1128/JVI.02642-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Ho T.H., Kew C., Lui P.Y., Chan C.P., Satoh T., Akira S., Jin D.Y., Kok K.H. PACT- and RIG-I-dependent activation of type I interferon production by a defective interfering RNA derived from measles virus vaccine. J. Virol. 2016;90:1557–1568. doi: 10.1128/JVI.02161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm C.K., Jensen S.B., Jakobsen M.R., Cheshenko N., Horan K.A., Moeller H.B., Gonzalez-Dosal R., Rasmussen S.B., Christensen M.H., Yarovinsky T.O. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012;13:737–743. doi: 10.1038/ni.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K., Takaoka A., Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Juckem L.K., Boehme K.W., Feire A.L., Compton T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J. Immunol. 2008;180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]
- Kallfass C., Lienenklaus S., Weiss S., Staeheli P. Visualizing the beta interferon response in mice during infection with influenza A viruses expressing or lacking nonstructural protein 1. J. Virol. 2013;87:6925–6930. doi: 10.1128/JVI.00283-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killip M.J., Jackson D., Perez-Cidoncha M., Fodor E., Randall R.E. Single-cell studies of IFN-beta promoter activation by wild-type and NS1-defective influenza A viruses. J. Gen. Virol. 2017;98:357–363. doi: 10.1099/jgv.0.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakofsky D. Isolation and characterization of sendai virus DI-RNAs. Cell. 1976;8:547–555. doi: 10.1016/0092-8674(76)90223-3. [DOI] [PubMed] [Google Scholar]
- Kotenko S.V., Gallagher G., Baurin V.V., Lewis-Antes A., Shen M., Shah N.K., Langer J.A., Sheikh F., Dickensheets H., Donnelly R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Lin R., Heylbroeck C., Pitha P.M., Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman K.L., Macgregor P.F., Rozmus J.J., Goryachev A.B., Edwards A.M., Smiley J.R. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 2001;75:750–758. doi: 10.1128/JVI.75.2.750-758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M., Otsu M. Development of Sendai virus vectors and their potential applications in gene therapy and regenerative medicine. Curr. Gene Ther. 2012;12:410–416. doi: 10.2174/156652312802762518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce R.S., Collins S.E., Mossman K.L. Differential modification of interferon regulatory factor 3 following virus particle entry. J. Virol. 2009;83:4013–4022. doi: 10.1128/JVI.02069-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce R.S., Taylor K., Ciechonska M., Collins S.E., Duncan R., Mossman K.L. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J. Virol. 2011;85:10926–10931. doi: 10.1128/JVI.00862-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neal J.T., Upadhyay A.A., Wolabaugh A., Patel N.B., Bosinger S.E., Suthar M.S. West nile virus-inclusive single-cell RNA sequencing reveals heterogeneity in the type I interferon response within single cells. J. Virol. 2019;93 doi: 10.1128/JVI.01778-18. e01778–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino P., Cummings D.T., Noyce R.S., Mossman K.L. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J. Immunol. 2006;177:8008–8016. doi: 10.4049/jimmunol.177.11.8008. [DOI] [PubMed] [Google Scholar]
- Peters K.L., Smith H.L., Stark G.R., Sen G.C. IRF-3-dependent, NFkappa B- and JNK-independent activation of the 561 and IFN-beta genes in response to double-stranded RNA. Proc. Natl. Acad. Sci. U S A. 2002;99:6322–6327. doi: 10.1073/pnas.092133199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston C.M., Harman A.N., Nicholl M.J. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J. Virol. 2001;75:8909–8916. doi: 10.1128/JVI.75.19.8909-8916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti E., Bracci L., Puzelli S., Di Pucchio T., Sestili P., De Vincenzi E., Venditti M., Capone I., Seif I., De Maeyer E. Type I IFN as a natural adjuvant for a protective immune response: lessons from the influenza vaccine model. J. Immunol. 2002;169:375–383. doi: 10.4049/jimmunol.169.1.375. [DOI] [PubMed] [Google Scholar]
- Rand U., Rinas M., Schwerk J., Nohren G., Linnes M., Kroger A., Flossdorf M., Kaly-Kullai K., Hauser H., Hofer T. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol. Syst. Biol. 2012;8:584. doi: 10.1038/msb.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman B.J., Jarvis M.A., Drummond D.D., Nelson J.A., Johnson D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006;80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga K., Kaneda Y. Oncolytic Sendai virus-based virotherapy for cancer: recent advances. Oncolytic Virother. 2015;4:141–147. doi: 10.2147/OV.S66419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Aparicio M.T., Garcin D., Rice C.M., Kolakofsky D., Garcia-Sastre A., Baum A. Loss of Sendai virus C protein leads to accumulation of RIG-I immunostimulatory defective interfering RNA. J. Gen. Virol. 2017;98:1282–1293. doi: 10.1099/jgv.0.000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Res. 2018;7:309. doi: 10.12688/f1000research.12450.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Sharma S., tenOever B.R., Grandvaux N., Zhou G.P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- Sheppard P., Kindsvogel W., Xu W., Henderson K., Schlutsmeyer S., Whitmore T.E., Kuestner R., Garrigues U., Birks C., Roraback J. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003;4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Symons J.A., Alcami A., Smith G.L. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81:551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Tsitoura E., Thomas J., Cuchet D., Thoinet K., Mavromara P., Epstein A.L. Infection with herpes simplex type 1-based amplicon vectors results in an IRF3/7-dependent, TLR-independent activation of the innate antiviral response in primary human fibroblasts. J. Gen. Virol. 2009;90:2209–2220. doi: 10.1099/vir.0.012203-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh S.R., Bastin D., Chen L., Nguyen A., Storbeck C.J., Lefebvre C., Stojdl D., Bramson J.L., Bell J.C., Wan Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Invest. 2019;129:518–530. doi: 10.1172/JCI121004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver B.K., Kumar K.P., Reich N.C. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol. Cell Biol. 1998;18:1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M., Gawanbacht A., Habjan M., Rang A., Borner C., Schmidt A.M., Veitinger S., Jacob R., Devignot S., Kochs G. Incoming RNA virus nucleocapsids containing a 5'-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe. 2013;13:336–346. doi: 10.1016/j.chom.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M., Suhara W., Fukuhara Y., Fukuda M., Nishida E., Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount J.S., Kraus T.A., Horvath C.M., Moran T.M., Lopez C.B. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J. Immunol. 2006;177:4503–4513. doi: 10.4049/jimmunol.177.7.4503. [DOI] [PubMed] [Google Scholar]
- Zawatzky R., De Maeyer E., De Maeyer-Guignard J. Identification of individual interferon-producing cells by in situ hybridization. Proc. Natl. Acad. Sci. U S A. 1985;82:1136–1140. doi: 10.1073/pnas.82.4.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Zhang J., Phatnani H., Scheu S., Maniatis T. Stochastic expression of the interferon-beta gene. PLoS Biol. 2012;10:e1001249. doi: 10.1371/journal.pbio.1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data generated during this study are available at GEO (GEO: GSE149705).