

Abstract
Triggering receptor expressed on myeloid cells 2 (TREM2), a receptor exclusively expressed by microglia in the brain, modulates microglial immune homeostasis. Human genetic studies have shown that the loss-of-function mutations in TREM2 signaling are strongly associated with an elevated risk of age-related neurodegenerative diseases including Alzheimer's disease (AD). Numerous studies have investigated the impact of TREM2 deficiency in the pathogenic process of AD. However, the role of TREM2 in shaping neuronal and cognitive function during normal aging is underexplored. In the present study, we employed behavioral, electrophysiological, and biochemical approaches to assess cognitive and synaptic function in male and female young and aged TREM2-deficient (Trem2−/−) mice compared with age-matched, sex-matched, and genetic background-matched wild-type (WT) C57BL/6J controls. Young Trem2−/− mice exhibited normal cognitive function and synaptic plasticity but had increased dendritic spine density compared with young WT. Unexpectedly, aged Trem2−/− mice showed superior cognitive performance compared with aged WT controls. Consistent with the behavioral data, aged Trem2−/− mice displayed significantly enhanced hippocampal long-term potentiation (LTP) and increased dendritic spine density and synaptic markers compared with aged WT mice. Taken together, these findings suggest that loss of TREM2 affects the neuronal structure and confers resilience to age-related synaptic and cognitive impairment during non-pathogenic aging.
SIGNIFICANCE STATEMENT Microglia are innate immune cells of the brain that orchestrates neurodevelopment, synaptic function, and immune response to environmental stimuli. Microglial triggering receptor expressed on myeloid cells 2 (TREM2) signaling plays pivotal roles in regulating these functions and loss of TREM2 signaling leads to increased risk of developing age-related neurologic disorders. However, the neurologic role of TREM2 in normal aging is poorly understood. The results of the present study unveil the positive impacts of TREM2 deficiency on cognitive and synaptic function during aging and suggest that TREM2 may exert detrimental effects on neuronal function. The possibility of age-related negative impacts from TREM2 is critically important since TREM2 has emerged as a major therapeutic target for Alzheimer's dementia.
Keywords: aging, dendritic spine density, learning and memory, long-term potentiation, synaptic plasticity, TREM2
Introduction
Triggering receptor expressed on myeloid cells 2 (TREM2) is an immunoglobulin superfamily receptor expressed by some myeloid lineage cells (Kang et al., 2018). In the brain, TREM2 is exclusively expressed on microglia (Gratuze et al., 2018). Microglia are innate immune cells of the brain that play pivotal roles in both physiological and pathologic conditions, including shaping neurodevelopment, regulating synaptic function, and orchestrating immune responses to environmental stimuli (Szepesi et al., 2018). Transcriptomic studies have identified TREM2 as an essential mediator of microglial immune homeostasis (Neumann and Takahashi, 2007; Krasemann et al., 2017). Various putative ligands for TREM2, including lipids, apolipoproteins, and amyloid-β (Aβ) have been identified (Kober and Brett, 2017). TREM2 intracellular signaling requires the binding of an adaptor protein, tyro protein kinase binding protein (TYROBP; also known as DAP12; Hamerman et al., 2006). Activation of DAP12 leads to downstream signaling cascades including the activation of ERK, PLCγ, and mTOR (Ulland and Colonna, 2018). TREM2 signaling plays an important role in regulating microglial proliferation, the release of inflammatory cytokines, and mediating phagocytosis.
Human genetic studies have identified critical impacts of TREM2 mediated signaling cascades in neuropathogenesis. Loss-of-function mutations in TREM2 or DAP12 result in Nasu–Hakola disease (NHD), which is characterized by progressive presenile dementia with bone cysts (Paloneva et al., 2001). Genome-wide association studies (GWASs) have discovered that loss-of-function mutations in TREM2 increase the risk of developing Alzheimer's disease (AD) by 2- to 4-fold (Guerreiro et al., 2013; Gratuze et al., 2018). AD is the most common cause of dementia in the elderly and is characterized by progressive neuronal dysfunction and degeneration, accumulation of amyloid plaques and neurofibrillary tangles, and glial activation (Wenk, 2003). TREM2 displays a complex role depending on the stage of AD. In the presence of Aβ, complete deficiency of TREM2 limits Aβ seeding at the early stage but impairs plaque compaction and exacerbates the spread of Aβ later (Gratuze et al., 2018). TREM2 haplodeficiency does not affect Aβ pathology but alters the morphology of plaque-associated microglia and worsens axonal dystrophy (Ulrich et al., 2014; Yuan et al., 2016). In the presence of tau pathology, complete loss of TREM2 exacerbates tau hyperphosphorylation and seeding at the early stage but rescues tau-mediated neurodegeneration later, whereas TREM2 haplodeficiency aggravates tau pathology and exacerbates brain atrophy (Gratuze et al., 2018). However, Trem2+/+ and Trem2+/− microglia showed similar transcriptome changes whereas Trem2−/− microglia failed to convert to the disease-associated microglia (DAM) state on cuprizone-induced demyelination (Nugent et al., 2020).
Although increased attention has been drawn to better understanding TREM2 function in disease progression, the physiological role of TREM2 in the brain has only emerged recently.
During neurodevelopment, TREM2 is essential for synaptic pruning and normal brain connectivity formation, which are critical for normal social behaviors (Filipello et al., 2018). Behavioral and transcriptomic analysis of TREM2 null mice showed little impact of TREM2 deficiency on basal cognitive function at six months of age (Kang et al., 2018). During aging, homeostatic microglia adopt transcriptional changes to the DAM state in WT mice, whereas TREM2-deficient microglia fail to undergo such conversion in TREM2-null mice (Keren-Shaul et al., 2017; Nugent et al., 2020). Since loss-of-function mutations in TREM2 increase the risk of developing age-related dementia including AD and NHD, TREM2 deficiency was expected to exacerbate neuronal dysfunction and cognitive decline during normal aging.
To define the impact of TREM2 deficiency on brain function in aging, the present study was undertaken to assess cognitive and synaptic function in young and aged Trem2−/− mice using age/sex-matched wild-type (WT) C57BL/6J mice as controls. Contrary to the expectation, aged Trem2−/− mice showed superior cognitive performance, enhanced hippocampal long-term potentiation (LTP), and increased dendritic spine density and postsynaptic makers, compared with age/sex-matched WT controls. These findings demonstrated that TRME2 deficiency confers resilience to cognitive deficits and synaptic dysfunction in aged mice, suggesting a potentially detrimental role of TREM2 during physiological aging.
Materials and Methods
Animals
Trem2−/− mice (Turnbull et al., 2006) on the congenic C57BL/6 background were kindly provided by Marco Colonna (Washington University, St. Louis, MO) and bred in-house. WT C57BL/6J mice (#000664) were acquired from Jackson Laboratory and bred in-house. Both males and females were included in this study. All mice were housed in the specific pathogen-free (SPF) facility at the University of Minnesota (UMN). Experiments were conducted blind to genotypes and all animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at UMN.
Behavioral assessment
Three behavioral tests were used to assess the locomotive response to environmental stimulus, anxiety levels, and spatial learning and memory of mice as previously described (Cheng et al., 2013; Hottman et al., 2018). Briefly, the ANYMAZE system and instruments were used for all behavioral assessments (San Diego Instruments). The open-field test was used to evaluate the locomotive response and habituation to environmental stimuli. In the open-field test, mice were placed into a square open box and allowed to explore freely for 5 min for three continuous days. The path length was recorded for each trial. The elevated plus maze was used to evaluate the anxiety levels of mice and consists of a platform above the ground, which has two opposing open arms and two closed arms formed a “+” shape maze. Mice were allowed to explore the maze freely for 5 min for two continuous days. The time spent in the open and closed arms were recorded. The Morris water maze test was conducted to assess the spatial learning and memory of mice, using a round basin filled with water in a visual cue-enriched room. During the acquisition phase, mice were trained to find a hidden escape platform 1 inch under the water. The escape latency was recorded for four trials a day for five continuous days. On day 6, a probe trial for testing memory retention was conducted by removing the platform and the platform location crossover was recorded in a single 60-s trial. Mice were returned to their home cage for 2 h. Then a visible trial was conducted to test their visual acuity and swimming speed.
Electrophysiology
Electrophysiology experiments with acute brain slices were conducted as previously described (Parent et al., 2014; Hottman et al., 2018). Briefly, mice were anesthetized using isoflurane, followed by decapitation. Brains were dissected out and sectioned in an ice-cold sucrose cutting solution containing the following: 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 0.5 mm CaCl2, 7 mm MgCl2, 7 mm dextrose, and 240 mm sucrose. Transverse hippocampal slices were cut at 400 μm using a vibratome (Leica). Slices then were transferred and incubated for 15 min at 28°C in artificial CSF (aCSF) containing the following: 125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 2 mm CaCl2, 1 mm MgCl2, and 25 mm dextrose (pH 7.4) with constant bubbling of 95%O2/5% CO2, followed by at least 1-h recovery at room temperature. After recovery, slices were placed into the recording chamber (Automate Scientific) with aCSF flowing at ∼1.5 ml/min at 27–29°C. Electrical stimulations were controlled by a constant-current stimulus isolator (World Precision Instruments) and were delivered to the Schaffer Collateral axon bundles of CA3 to CA1 of the hippocampus using a bipolar tungsten electrode (FHC). For input/output (I/O) curves, stimulation was delivered at 10-s interval and five stimuli were given and averaged at every 10 μA ranging from 0 to 150 μA and the field EPSPs (fEPSPs) were recorded and averaged at each stimulation intensity. Stimulation intensity was then adjusted to 40−50% of each slice's maximum response in the I/O curve to assess the paired-pulsed facilitation (PPF). Two continuous stimuli were delivered to slices with an interval of 30 ms at every 10 s. Five minutes of stable PPFs (120 PPFs) were recorded and averaged for each genotype. Hippocampal LTP was assessed on separate slices. Stimulation was delivered at 10-s interval and the stimulation intensity was adjusted to 40–50% of each slice's maximum I/O curve. Following a 20-min stable bassline, three trains of theta-burst stimulation (TBS) protocol with a 20-s interval is delivered. The slopes of fEPSP after induction were normalized to the baseline and the last 5 min of response were averaged to represent the magnitude of potentiation.
Golgi staining and spine density quantification
Golgi staining was conducted using the FD Rapid GolgiStain kit (catalog #PK401A, FD NeuroTechnologies) as previously described with some modifications (Rodriguez et al., 2013; Hottman et al., 2018). Briefly, mice were deeply anesthetized and perfused with PBS. Brainstem and olfactory bulbs were removed, and two-thirds of the posterior brain were processed following the manufacture's user manual. After one week of infusion in mixed solutions A and B provided by the kit, brains were then transferred into the Solution C for another 3 d. Coronal brain sections were cut in Solution C at 150 μm using a vibratome (Leica) and dried overnight. Dehydration steps were executed as described by the manufacture and sealed in Permount mounting medium (Fisher Scientific). Slides were allowed to dry for at least 3 d, then the edges of coverslips were sealed using clear nail polish. Golgi imaging microscopy was done under a 100× objective using the Nikon Eclipse 55i upright microscope and images were taken using a digital CCD color camera (QImaging) and captured using the Image-Pro Plus software. Spine density quantification in our previous report was done using single plane focal field images (Hottman et al., 2018). Since neurons extend processes in three dimensions and dendrites are often not straight, single-plane images can only cover small focal fields and short visible dendrites with few spines. To improve the accuracy and quality of quantifying spine density, a z-stack of images that consist of different focal fields of all spines covering an entire dendritic branch of interest were manually taken by gradually change the focus of the dendrite (see below in the Results section). Each focal field was determined by the visibility of spines and two continuous focal fields covered different focuses of the same spines to ensue all spines were captured along the dendrite. Secondary or above order of dendritic branches from pyramidal neurons in the Layer II/III of the posterior 2/3 of the cerebral cortex, including the sensory, motor, and entorhinal cortex, and in the hippocampal CA1 were imaged. Neurons were randomly chosen from fully stained, clearly identifiable cells from these two brain regions. Dendritic segments were chosen by tracking the dendritic process from neuronal soma and branches, and only dendritic segments that are clearly visible and far from neighboring dendrites were selected. After imaging, images containing all focal fields of a dendrite were imported and stacked together using the ZProjection (min intensity) program in the ImageJ software. Spine numbers were then manually counted, and the total length of the dendrite was measured using ImageJ measuring tools. Compared with a single plane image, the z-stacked image clearly improved the visibility of the spines and the quantifiable dendritic length (see below in the Results section). On average, the dendritic segments of 44 ± 0.56 µm were quantified. Spines from two apical and two basal dendritic branches from one neuron were averaged to represent the spine density of the neuron. Ten neurons were imaged from each animal and a total of 60–70 neurons (six to seven mice)/genotype were included.
Synaptosome isolation
Synaptosome isolation was conducted as previously described with some modifications (Chao et al., 2013; Suresh and Dunaevsky, 2015; Kolodziej et al., 2016). The anterior one-third of the brain (olfactory bulb excluded) was collected and snapped frozen in liquid nitrogen until further process. Tissue was homogenized on ice in a sucrose buffer containing 320 mm sucrose, 10 mm HEPES, pH 7.5, 5 mm EDTA, 5 mm dithiothreitol (DTT), 1.5 mm MgCl2, and 1× protease and phosphatase inhibitors using a handheld homogenizer (KIMBLE Pellet Pestle Motor) for 1 min with up and down motions. Samples were then incubated on ice for 10 min followed by centrifugation at 700 × g at 4°C for 10 min. The supernatant was transferred to a new tube, and the pellet was homogenized and centrifuged for the second time, and the supernatant was combined with the first time. The supernatant was then centrifuged at 12,000 × g for 20 min at 4°C to acquire a pellet containing enriched synaptosomes, which was resuspended in the sucrose buffer with 1% Triton X-100 and incubated at 4°C for 1 h.
Protein assay and immunoblotting
The protein concentration of the synaptosome preparation was determined using Bradford assay (ThermoFisher) and aliquots of the synaptosome preparation were separated by 12% or 4–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as previously described (Chernick et al., 2018). The membranes were incubated with primary antibodies of the following: postsynaptic density (PSD)-95 (MAB1598, Millipore Sigma), NR2A (07-632, Millipore Sigma), NR2B (MAB5778, Millipore Sigma), glutamate receptor 1 (AB1505, Millipore Sigma), synapsin 1 (Syn1; AB1543, Millipore Sigma), GAPDH (AM4300, Invitrogen), and followed by horseradish peroxidase-conjugated secondary antibodies. Membranes were then incubated in the Clarity Western ECL substrate (Bio-Rad) and the signal was detected using the iBright Western Blot Imaging System (Thermo Fisher Scientific). Images were quantified using the ImageJ software.
Experimental design and statistical analyses
All experiments, including behavioral assessments, electrophysiological measurements, and synaptic morphologic and biochemical analyses, were performed in young and old Trem2−/− mice compared with age-, sex-, and strain-matched C57BL/6J (WT) control mice (Table 1). All experiments were conducted blind to genotypes and both males and females were used in this study. Behavioral tests were conducted at six months (young) and 18 months (aged) of age. After two weeks of behaviors, mice were allowed to rest for at least two weeks before electrophysiological experiments were conducted in the same cohort of mice at an average of eight and 19 months of age. Quantifications of spine density and synaptic markers using Golgi staining and synaptosome preparation respectively were conducted in separate cohorts of naive young (eight months) and aged (19 months) mice to avoid potential confounding factors caused by behavioral and electrophysiological experiments. The age of the naive mice matched the age of the mice used in electrophysiological experiments.
Table 1.
Animals used in each experiment
| Young |
Average age | Aged |
Average age | |||
|---|---|---|---|---|---|---|
| WT | TREM2 | WT | TREM2 | |||
| Behavioral assessment | 8 (4 M, 4 F) | 6 (2 M, 4 F) | 6 mo | 11 (5 M, 6 F) | 10 (5 M, 5 F) | 18 mo |
| Electrophysiology | 7 (4 M, 3 F) | 6 (2 M, 4 F) | 8 mo | 6 (2 M, 4 F) | 7 (2 M, 5 F) | 19 mo |
| Golgi staining and synaptosome isolation | 6 (3 M, 3 F) | 6 (3 M, 3 F) | 8 mo | 7 (3 M, 4 F) | 6 (3 M, 3 F) | 19 mo |
Same cohorts of mice were used in behavioral and electrophysiological experiments. Separate cohorts of mice were used in Golgi staining and synaptosome isolation. M, male; F, female; mo, months.
GraphPad Prism was used to perform statistical analyses. All values are expressed as mean ± SEM. To compare differences between WT and Trem2−/− groups, a two-tailed unpaired Student's t test was used. For comparisons of the genotype effect on the performance of mice over constitutive days, repeated measures two-way ANOVA was conducted. For comparisons of the genotype effect on I/O curve, the mixed-effects model (REML) was used. To evaluate the influence of aging and genotype interaction on spine density, two-way ANOVA was performed, followed by the Holm–Sidak multiple comparison analysis. Data from male and female mice were analyzed separately and no sex difference was observed; thus, data from males and females were combined and presented.
Results
TREM2 deficiency does not affect cognitive function in young mice and enhances cognitive function in aged mice
To determine the impact of TREM2 deficiency on cognitive function at different ages, Trem2−/− mice were used (Turnbull et al., 2006). First, a battery of behavioral tests was conducted to assess the role of TREM2 deficiency in young mice (six months). The open-field test over 3 d of trials was used to evaluate the general activity and locomotive habituation response, which showed no significant differences between WT and Trem2−/− mice at this young age of six months (F(1,12) = 0.09863, p = 0.7589, repeated measures two-way ANOVA; Fig. 1A). The elevated plus-maze was used to assess the anxiety level, which also showed no significant differences between WT and Trem2−/− mice, reflected by comparable percentage time spent in the open arm (F(1,12) = 0.7864, p = 0.3926, repeated measures two-way ANOVA; Fig. 1B). To assess hippocampal-dependent spatial learning and memory, the Morris water maze was used, which showed that young Trem2−/− and WT mice had similar learning curves (F(1,12) = 0.1737, p = 0.6842, repeated measures two-way ANOVA; Fig. 1C) and platform crossovers (memory retention) during the probe trial (t(12) = 1.392, p = 0.1892, unpaired Student's t test; Fig. 1D). There were no differences in the mean swimming speed during the probe trial (WT: 0.23 ± 0.01 m/s, Trem2−/−: 0.22 ± 0.01 m/s, t(12) = 0.545, p = 0.5443, unpaired Student's t test) or the escape latency during the visible platform trial (WT: 14.3 ± 2.8 s, Trem2−/−: 8.7 ± 1.6 s, t(12) = 1.547, p = 0.1478, unpaired Student's t test) between genotypes. These results indicate that TREM2 deficiency does not alter cognitive function at six months of age and this conclusion is consistent with that of the previous study (Kang et al., 2018).
Figure 1.

TREM2 deficiency does not affect behavioral or cognitive performance in young mice. Six-month-old WT and Trem2−/− mice were examined for locomotive mobility and habituation in the open field test (A), anxiety levels in the elevated plus-maze test (B), and spatial learning during the acquisition phase (C) and memory retention in the probe trial (D) of the Morris water maze test. Results shown are the mean ± SEM (n = 6–8 mice/genotype).
Next, since aging is the greatest risk factor for developing AD and mice display age-related cognitive decline (Shoji et al., 2016), the impact of TREM2 deficiency on cognitive function in aged mice was investigated. The same sets of behavioral and cognitive tests were conducted on a separate group of aged Trem2−/− and WT mice (18 months). Interestingly, aged WT mice showed impaired locomotive habituation over 3 d of training in the open field test, which was rescued in aged Trem2−/− mice (F(1,19) = 11.14, **p = 0.0035, repeated measures two-way ANOVA; Fig. 2A), indicating that Trem2−/− mice preserved the memory of environmental stimulus-induced locomotive responses over days. The elevated plus-maze test showed a trend of reduction in the anxiety level in aged Trem2−/− mice compared with aged WT mice (F(1,19) = 3.892, p = 0.0632, repeated measures two-way ANOVA; Fig. 2B). Most unexpectedly, hippocampal spatial learning and memory assessments by the Morris water maze test revealed that aged Trem2−/− mice had superior performance in both the learning acquisition phase (F(1,19) = 14.70, **p = 0.0011, repeated measures two-way ANOVA; Fig. 2C) and the memory retention phase during the probe trial (t(19) = 5.005, ****p < 0.0001; Fig. 2D) compared with aged WT mice. There were no differences in the mean swimming speed during the probe trial (WT: 0.15 ± 0.01 m/s, Trem2−/−: 0.16 ± 0.01 m/s, t(19) = 0.7273, p = 0.4759, unpaired Student's t test) or the escape latency during the visible platform trial (WT: 22.8 ± 4.5 s, Trem2−/−: 14.8 ± 2.0 s, t(19) = 1.547, p = 0.1343, unpaired Student's t test). Taken together, these data demonstrate that TREM2 deficiency does not affect learning and memory function at a young age but leads to an unexpected boost of cognitive function in aged mice independent of locomotive activity and visual acuity.
Figure 2.

TREM2 deficiency protects against aging-induced cognitive decline. Aged WT and Trem2−/− mice (18 months) were examined for locomotive mobility and habituation in the open field test (A; **p = 0.0035, repeated measures two-way ANOVA, genotype factor), anxiety levels in the elevated plus-maze test (B), and spatial learning during the acquisition phase (C; **p = 0.0011, repeated measures two-way ANOVA, genotype factor) and memory retention in the probe trial (D; ****p < 0.0001, unpaired Student's t test) of the Morris water maze test. Results shown are the mean ± SEM (n = 10–11 mice/genotype).
TREM2 deficiency leads to enhanced hippocampal LTP without influencing basal synaptic neurotransmission in aged mice
The unexpected protective effects of TREM2 deficiency on cognitive function in aged mice suggest potential neuronal signaling changes in Trem2−/− mice during normal aging. Although TREM2 is exclusively expressed on microglia in the brain, synaptic function can be modulated by the cross-talk between microglia and neurons (Szepesi et al., 2018). To assess the role of TREM2 in regulating neuronal electrophysiological properties, the cellular mechanisms underlying cognitive function, field electrophysiological recordings were conducted at least two weeks after the behavioral tests in young and aged mice. First, the I/O curve was constructed to evaluate the basal synaptic transmission, in which the slope of the fEPSP increased in response to increased stimulation inputs, and there were no significant differences between young Trem2−/− and WT mice (eight months; F(1,15) = 0.2110, p = 0.6526, REML; Fig. 3A). Then, the paired-pulse facilitation (PPF), a form of short-term plasticity that mostly involves presynaptic machinery (Regehr, 2012), was determined. The experiment showed no significant differences in PPF between young WT and Trem2−/− mice (t(14) = 0.3196, p = 0.754, unpaired Student's t test; Fig. 3B). To assess the long-term plasticity, the hippocampal CA1 LTP, which mostly involves postsynaptic machinery and has been shown to be essential for normal spatial learning and memory (Tsien et al., 1996), was measured. As shown in Figure 3, the magnitude of LTP, represented by the averaged fEPSP of the last 5 min (35–40 min postinduction), was not significantly different between young WT and Trem2−/− mice (t(22) = 0.6081, p = 0.5493, unpaired Student's t test; Fig. 3C,D). These results are consistent with the cognitive outcomes of the young mice and indicate that TREM2 deficiency does not influence basal neurotransmission and hippocampal synaptic plasticity at eight months of age.
Figure 3.

TREM2 deficiency has no impact on basal neurotransmission and hippocampal LTP in young mice. Two weeks after the behavioral tests, young WT and Trem2−/− mice (eight months) were examined for hippocampal basal neurotransmission and LTP. Stimulation was delivered to the Schaffer collateral and fEPSP was recorded in the CA1 stratum radiatum. A, I/O curves (n = 7–9 slices/2 mice per genotype). B, PPF ratios were evaluated at the 40–50% I/O curve maximum response on the same slices after the I/O curve experiments (n = 7–9 slices/2 mice per genotype). Hippocampal LTP was evaluated in separate sets of slices. C, Induction of hippocampal LTP. After a stable 20-min baseline, the LTP was induced by three trains of TBS with a 20-s interval. The initial slopes of the fEPSP rising phase were normalized to the baseline average. D, The magnitude of LTP of the last 5 min (35–40 min postinduction) was averaged and compared between genotypes (n = 9–15 slices/4–5 mice per genotype). All results shown are mean ± SEM.
Next, the impact of TREM2 deficiency on hippocampal neuronal function in aged mice (19 months) was investigated using the same series of electrophysiological experiments. Similar to the young group, aged Trem2−/− mice displayed no differences in the I/O curve compared with aged WT controls, indicating that TREM2 deficiency does not influence basal neurotransmissions (F(1,17) = 0.4559, p = 0.5086, REML; Fig. 4A). The PPF ratio was evaluated next, which was not significantly different between aged Trem2−/− and WT mice (t(15) = 1.449, p = 0.1679, unpaired Student's t test; Fig. 4B), suggesting that TREM2 deficiency does not influence short-term plasticity during aging. Since long-term synaptic plasticity is crucial for normal hippocampal function and TREM2 deficiency preserved learning and memory during aging, hippocampal LTP was assessed in the aged group. The results revealed that aged Trem2−/− mice had significantly enhanced LTP compared with aged WT controls (t(27) = 2.602, *p = 0.0149, unpaired Student's t test; Fig. 4C,D). Taken together, these findings demonstrate that TREM2 deficiency leads to enhanced hippocampal LTP without influencing basal synaptic transmission and short-term plasticity in aged mice.
Figure 4.

TREM2 deficiency does not affect basal neurotransmission and leads to enhanced hippocampal LTP in aged mice. Two weeks after the behavioral tests, aged WT and Trem2−/− mice (19 months) were examined for hippocampal basal neurotransmission and LTP. Stimulation was delivered to the Schaffer collateral and fEPSP was recorded in the CA1 stratum radiatum. A, I/O curves (n = 8–9 slices/2 mice per genotype). B, PPF ratios (n = 8–9 slices/2 mice per genotype). Hippocampal LTP was evaluated in separate sets of slices. C, Induction of hippocampal LTP. D, The magnitude of LTP of the last 5 min (35–40 min postinduction) was averaged and compared between genotypes (n = 14–15 slices/4–5 mice per genotype, *p = 0.0149, unpaired Student's t test). All results shown are mean ± SEM.
TREM2 deficiency-induced abundance of spines compensates for the loss of spines during aging
During neurodevelopment, TREM2 deficiency leads to impaired synaptic pruning and results in increased dendritic spine density in the hippocampus (Filipello et al., 2018). However, it is unclear whether TREM2-mediated synaptic refinement during development continues during aging. In the nervous system, the postsynaptic dendritic spines are where most of the excitatory synaptic transmission occurs, and changes in spine density reflect the status of glutamatergic synaptic transmission in shaping synaptic plasticity and cognition (Dickstein et al., 2013). To determine the impact of TREM2 deficiency on dendritic spines during aging, Golgi staining was conducted to evaluate the dendritic spine density in a separate cohort of naive young and aged mice as behavioral and electrophysiological experiments may alter synaptic spine morphology (Berry and Nedivi, 2017).
Different focal fields of all spines covering an entire dendritic branch of interest were imaged and stacked together. Compared with a single plane image (Fig. 5A), the z-stack image clearly improved the visibility of the spines and the quantifiable dendritic length (Fig. 5B). Using this imaging technique, first, the dendritic spine density of the pyramidal neurons in the hippocampal CA1 was assessed in young WT and Trem2−/− mice (eight months). Young Trem2−/− mice showed significant increases in the spine density of both apical and basal dendrites compared with WT (apical: t(10) = 3.396, **p = 0.0068; basal: t(10) = 3.477, **p = 0.0059, unpaired Student's t test; Fig. 6A,B). Since TREM2 is depleted systemically in Trem2−/− mice, whether the effect of TREM2 deficiency on spine numbers is hippocampus specific was assessed next by quantifying spine density of the pyramidal neurons in the Layer II/III of the posterior 2/3 cerebral neurons. Similar to the hippocampal pyramidal neurons, significantly increased spine density of both apical and basal dendrites in cortical neurons was observed in young Trem2−/− mice compared with WT (apical: t(10) = 3.98, **p = 0.0026; basal: t(10) = 3.734, **p = 0.0039, unpaired Student's t test; Fig. 6C,D). These results indicate that TREM2 is required for maintaining normal synaptic density in both cortical and hippocampal CA1 regions at a young age. Since no cognitive or synaptic function changes were observed in young Trem2−/− mice, there might be compensatory mechanisms that offset the excessive spine density.
Figure 5.

Z-stacking of single plane images improves quantification of dendritic spines. A, A representative single plane image for a segment of a dendritic branch. B, A representative z-stack image summed from 15 different focal fields of all spines on the same dendritic branch (inset 1–15). The z-stack image clearly shows improved visibility and increased quantifiable dendritic length. Scale bar: 10 μm.
Figure 6.

TREM2-deficient mice display increased dendritic spine density at a young age. A cohort of young mice (eight months) was subjected to spine density evaluation using Golgi staining. A, Representative images of z-stacked secondary or above order apical and basal dendritic segments from hippocampal CA1 pyramidal neurons imaged at 100×. B, Quantification of hippocampal CA1 spine densities (n = 6 mice per genotype, apical: **p = 0.0068, basal: **p = 0.0059, unpaired Student's t test). All results shown are mean ± SEM. C, Representative images of z-stacked secondary or above order apical and basal dendritic segments from cortical Layer II/III pyramidal neurons imaged at 100×. D, Quantification of cortical Layer II/III pyramidal neurons spine densities (n = 6 mice per genotype, apical: **p = 0.0026, basal: **p = 0.0039, unpaired Student's t test). All results shown are mean ± SEM.
It is well known that cortical and hippocampal dendritic spine density reduces during aging and correlates with cognitive decline (Dickstein et al., 2013; Boros et al., 2019). Since TREM2 deficiency resulted in more dendritic spines during development and in young adults, the possibility that Trem2−/− mice are resilient to the loss of dendritic spines during aging was investigated next in aged WT and Trem2−/− mice (19 months). As expected, aged Trem2−/− mice showed significantly more spines in both apical and basal dendrites of the hippocampal CA1 and cortical Layer II/III pyramidal neurons compared with WT controls (CA1 apical: t(11) = 4.359, **p = 0.0011, CA1 basal: t(11) = 4.02, **p = 0.002, cortical apical: t(11) = 6.579, ****p < 0.0001, cortical basal: t(11) = 7.12, ****p < 0.0001, unpaired Student's t test; Fig. 7A–D).
Figure 7.

TREM2 deficiency leads to increased dendritic spine density in aged mice. A cohort of aged mice (19 months) was subjected to spine density evaluation using Golgi staining. A, Representative images of z-stacked secondary or above order apical and basal dendritic segments from hippocampal CA1 pyramidal neurons imaged at 100×. B, Quantification of hippocampal CA1 spine densities (n = 6–7 mice per genotype, apical: **p = 0.0011, basal: **p = 0.002, unpaired Student's t test). C, Representative images of z-stacked secondary or above order apical and basal dendritic segments from cortical Layer II/III pyramidal neurons imaged at 100×. D, Quantification of cortical Layer II/III pyramidal neurons spine densities (n = 6–7 mice per genotype, apical: ****p < 0.0001, basal: ****p < 0.0001, unpaired Student's t test). All results shown are mean ± SEM.
To evaluate the interaction between age and TREM2 genotype on spine density, the young and aged groups were analyzed together. The results showed that both genotype and age significantly influenced the spine density, and a significant impact of genotype and age interaction was found on cortical basal dendrites (CA1 apical: age: F(1,21) = 10.08, **p = 0.0046, genotype: F(1,21) = 29.69, ****p < 0.0001, age × genotype: F(1,21) = 0.1017, p = 0.7529; CA1 basal: age: F(1,21) = 12.15, **p = 0.0022, genotype: F(1,21) = 27.18, ****p < 0.0001, age × genotype: F(1,21) = 0.3625, p = 0.5536; cortical apical: age: F(1,21) = 38.07, ****p < 0.0001, genotype: F(1,21) = 54.39, ****p < 0.0001, age × genotype: F(1,21) = 2.019, p = 0.17; cortical basal: age: F(1,21) = 77.65, ****p < 0.0001, genotype: F(1,21) = 59.53, ****p < 0.0001, age × genotype: F(1,21) = 6.921, *p = 0.0156; two-way ANOVA; Fig. 8A,B). Intriguingly, aged Trem2−/− mice sustained similar spine numbers as young WT mice (CA1 apical: adjusted p = 0.1292; CA1 basal: adjusted p = 0.2438; cortical apical: adjusted p = 0.4122; cortical basal: adjusted p = 0.4549; post hoc Holm–Sidak multiple comparisons; Fig. 8C–F). Taken together, these data suggest that TREM2 deficiency leads to elevated spine density, which confers a protection against spine loss during aging.
Figure 8.
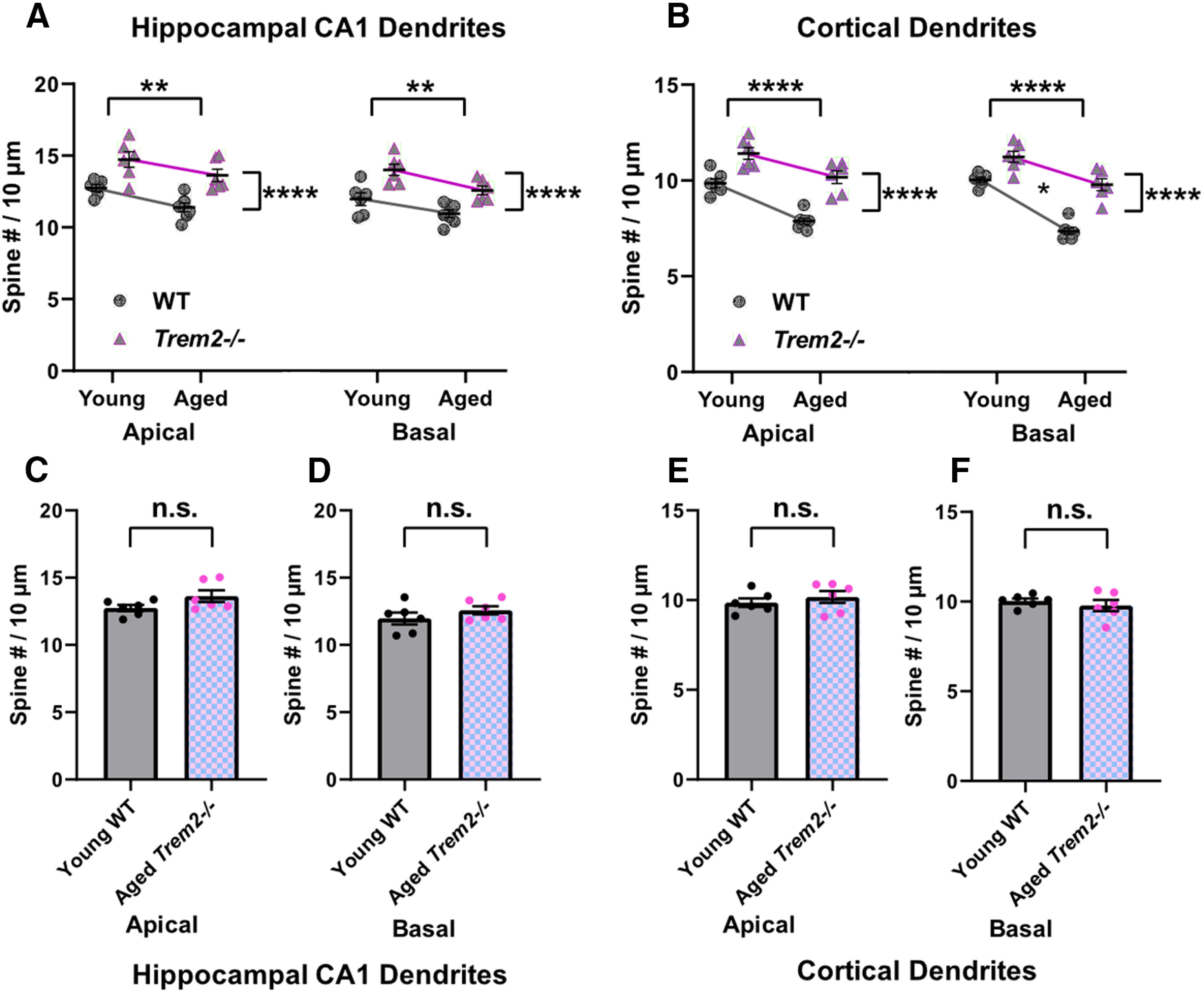
TREM2 deficiency-induced abundance of spines compensates for the loss of spines during aging. A, Combined analysis of young and aged hippocampal CA1 dendrites from WT and Trem2−/− mice showed significant effect of age and genotype, but no significant interaction between age and genotype on spine density (CA1 apical: age effect: **p = 0.0046, genotype effect: ****p < 0.0001; CA1 basal: **p = 0.0022, genotype effect: ****p < 0.0001, two-way ANOVA). B, Combined analysis of young and aged cortical dendrites from WT and Trem2−/− mice showed significant effect of age and genotype, and a significant interaction between age and genotype on spine density of basal but not apical dendrites (cortical age and genotype effect: ****p < 0.0001; cortical basal age and genotype interaction effect: *p = 0.01563; two-way ANOVA). C–F, No significant differences between young WT and aged Trem2−/− mice were observed in all areas assessed (post hoc Holm–Sidak multiple comparisons); n.s., not significant, n = 6–7 mice/genotype/age group.
TREM2 deficiency leads to elevated postsynaptic markers during aging
To elucidate the potential molecular mechanisms underlying heightened cognitive/synaptic function in aged Trem2−/− mice, synaptosomes were isolated from the frontal cortex of the young and aged mice used for Golgi staining. Immunoblotting was conducted to analyze the expression of synaptic proteins involved in regulating hippocampal function. These proteins include PSD-95, a major scaffolding protein in the excitatory PSD that regulates synaptic plasticity and decreases during aging (Chen et al., 2011; Shi et al., 2015); AMPA receptor subunit GluA1, which regulates hippocampal LTP and contributes to spatial learning and memory (Zhou et al., 2018); NMDA subunits NR2A and NR2B, which are important for regulating hippocampal LTP (Berberich et al., 2005; Bartlett et al., 2007); and presynaptic marker Syn1, which regulates synaptic vesicle trafficking and neurotransmitter release (Hilfiker et al., 1999; Orlando et al., 2014; Song and Augustine, 2015). First, in young Trem2−/− mice (eight months), no differences were found in the expression levels of PSD-95, GluA1, NR2A, NR2B, and Syn1 compared with young WT controls (PSD-95: t(10) = 0.1080, p = 0.9161, GluA1: t(10) = 1.245, p = 0.2414, NR2A: t(10) = 0.8884, p = 0.3952, NR2B: t(10) = 1.673, p = 0.1252, Syn1: t(10) = 0.7913, p = 0.4471, unpaired Student's t test; Fig. 9A–F). However, in aged Trem2−/− mice (19 months), there were significant increases in the expression of PSD-95, GluA1, and NR2A but not in the expression of NR2B and Syn1 compared with aged WT controls (PSD-95: t(11) = 4.312, **p = 0.0012, GluA1: t(11) = 2.951, *p = 0.0132, NR2A: t(11) = 2.495, *p = 0.0298, NR2B: t(11) = 0.7853, p = 0.4489, Syn1: t(11) = 0.04956, p = 0.9614, unpaired Student's t test; Fig. 9A,G–K). These results suggest that enhanced synaptic and cognitive functions in aged Trem2−/− mice are mediated primarily by postsynaptic mechanisms.
Figure 9.
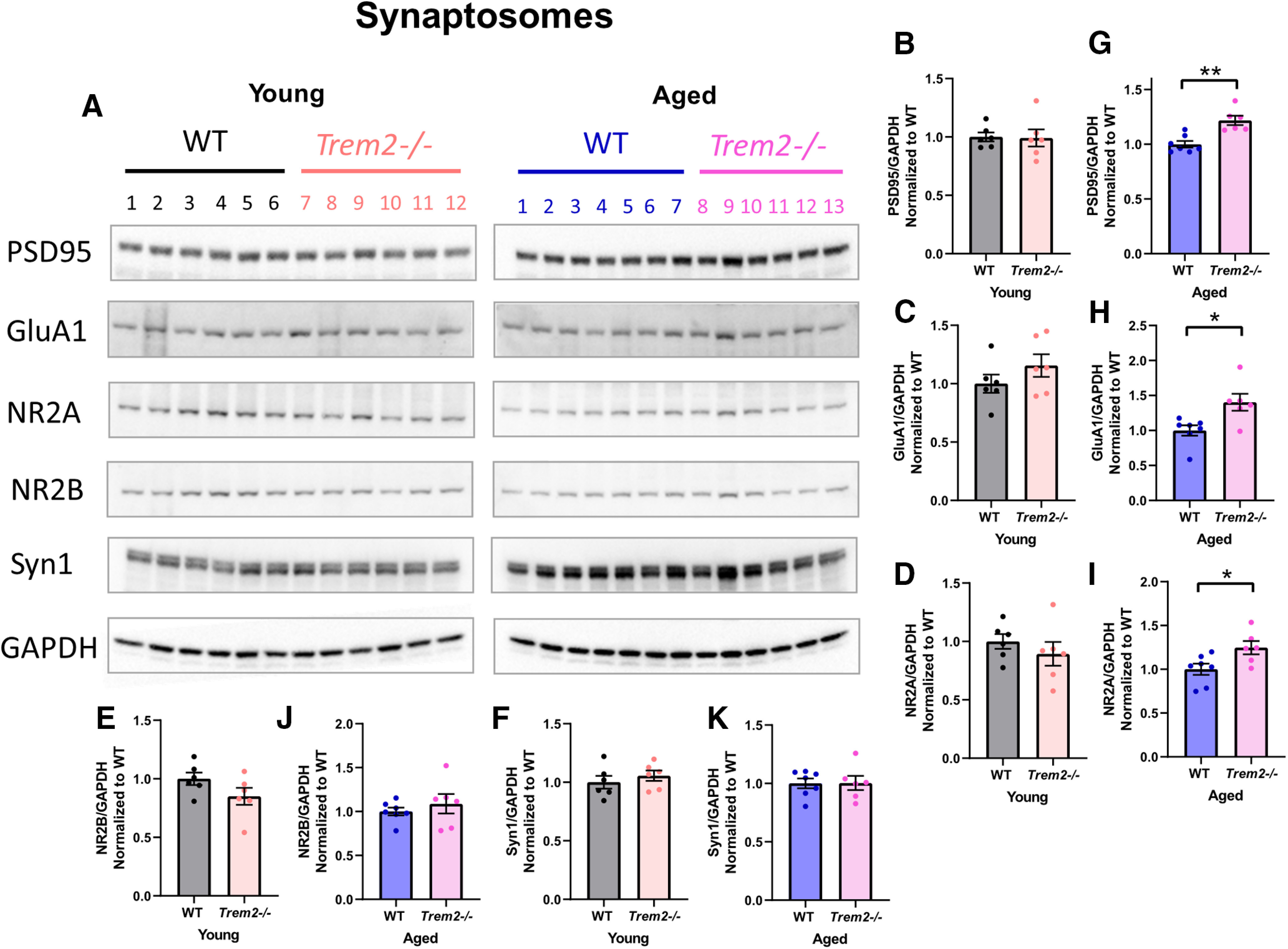
TREM2 deficiency has no impact on synaptic makers at a young age and leads to elevated levels of postsynaptic markers during aging. Synaptosomes were enriched from the forebrain tissue of young and aged mice used for Golgi staining, and immunoblotting was employed to analyze the expression of synaptic markers. A, Immunoblot images of synaptic makers. B–F, Quantifications of synaptic makers from the young group (n = 6 mice/genotype). G–K, Quantifications of synaptic makers from the aged group (n = 6–7 mice/genotype, PSD-95: **p = 0.0012, GluA1: *p = 0.0132, NR2A: *p = 0.0298, unpaired Student's t test).
Discussion
The present study aimed to address the knowledge gap in the neurologic role of TREM2 under the physiological conditions of aging. Using the well-established line of Trem2−/− mice (Turnbull et al., 2006) and age/sex-matched WT controls, the impact of TREM2 deficiency on cognitive function, synaptic plasticity, dendritic spine density, and synaptic markers was determined in young and aged mice.
Employing a battery of behavioral tests, we showed that TREM2 deficiency did not alter explorative or cognitive function at six months of age, which is consistent with the previous report (Kang et al., 2018); but unexpectedly, TREM2 deficiency led to a rescue of cognitive decline at 18 months of age. Electrophysiological experiments revealed limited impacts of TREM2 deficiency on basal neurotransmission and short-term plasticity; however, the magnitude of hippocampal LTP was elevated in aged Trem2−/− mice compared with aged WT mice. Consistent with the behavioral and electrophysiological data, Golgi staining confirmed an age-related decline of dendritic spine density in both hippocampus CA1 and cortical Layer II/III pyramidal neurons in WT and Trem2−/− mice. However, importantly, aged Trem2−/− mice maintained the similar density of dendritic spines as young WT mice. Synaptic protein analysis indicated significant increases of postsynaptic makers in synaptosomes from aged Trem2−/− mice. Taken together, these data demonstrate that TREM2 deficiency-induced abundance of dendritic spines confers resilience to age-related impairment in cognitive and neuronal function. The schematic of potential mechanisms is presented in Figure 10.
Figure 10.
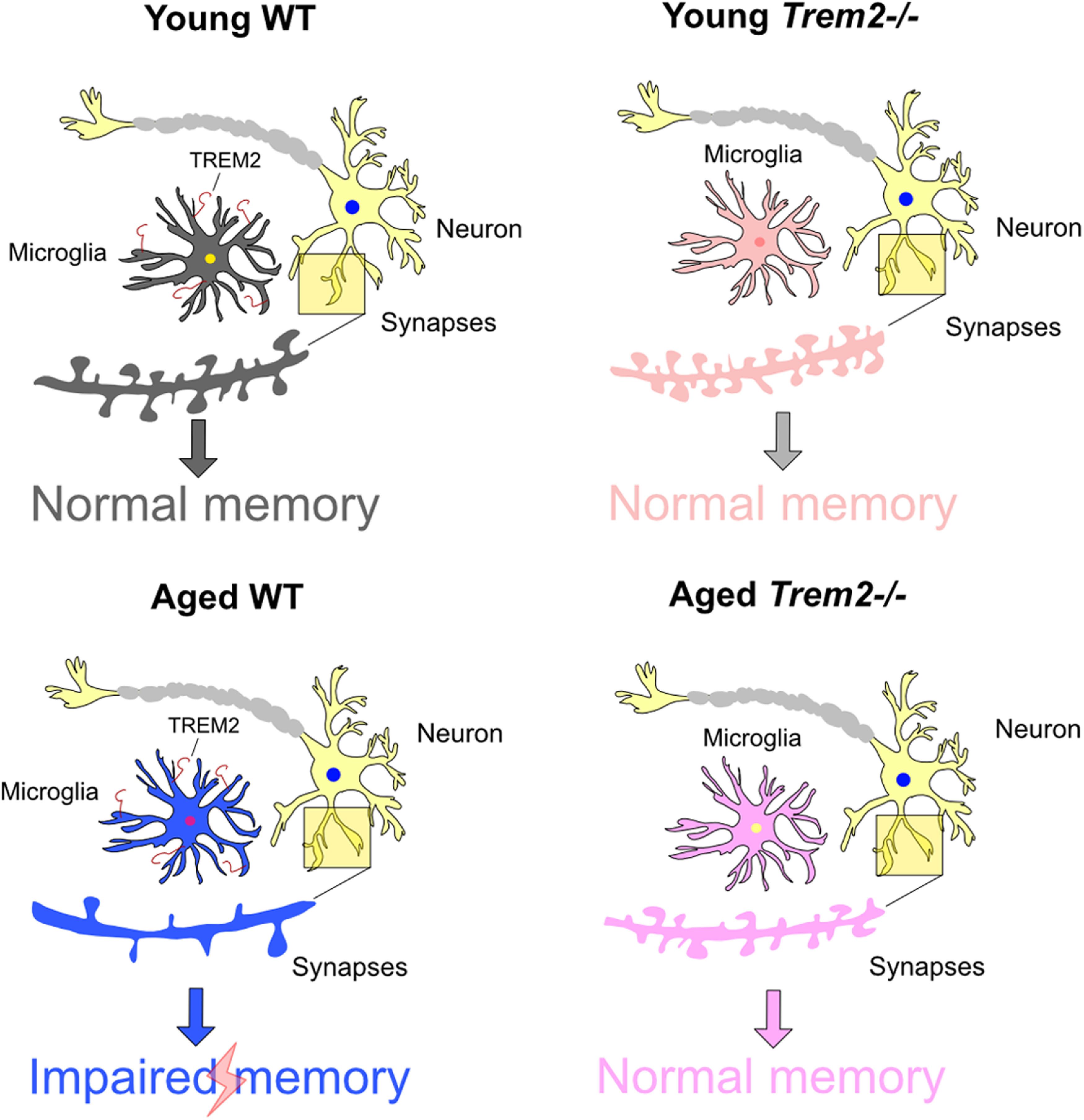
TREM2 deficiency-induced abundance of spines confers resilience to spine loss and memory impairment during aging. Schematic presentation of potential mechanisms by which TREM2 deficiency confers resilience to age-related synaptic and cognitive impairment during normal aging.
The protective effects of TREM2 deficiency on cognitive and synaptic function during aging were unexpected since loss-of-function mutations of TREM2 have been associated with age-related dementia, including AD. However, emerging evidence supports the beneficial impact of TREM2 deficiency during normal aging. Using the same line of Trem2−/− mice, a report showed that at 24 months of age, TREM2 deficiency rescued age-related neuronal loss and reduced microglial activation (Linnartz-Gerlach et al., 2019). Whole-brain transcriptome analysis revealed that aged TREM2 null mice downregulated oxidative stress responses and the complement activation pathway (Linnartz-Gerlach et al., 2019). TREM2 has been shown to play pivotal roles in mediating homeostasis of microglia and gating the switch of the neurodegenerative phenotype (MGnD) of microglia (Krasemann et al., 2017). In line with this finding, TREM2 deficiency was shown to sustain microglia expansion during aging and affect microglial morphology and phagocytosis (Poliani et al., 2015). Impaired phagocytosis of microglia as a consequence of TREM2 deficiency has been reported in both physiological and pathologic conditions (Hsieh et al., 2009; Kleinberger et al., 2014; Linnartz-Gerlach et al., 2019). Supporting these findings, a recent study performed transcriptome analysis on isolated microglia in aged TREM2 null mice and found that TREM2-deficient microglia failed to adopt DAM genes induced by aging (Nugent et al., 2020). Thus, it is possible that dysregulation of microglial phagocytosis in aging contributes to age-related loss of synapses and TREM2 deficiency exerts protective effects through ameliorating microglial phagocytosis and inflammatory responses during aging. Of note, the age-related beneficial effects of TREM2 deficiency observed in the present study were under physiological conditions in the absence of any pathologies. In the presence of environmental stress or pathology, TREM2 exerts more complex roles. Studies have shown that TREM2-deficient microglia fail to promote transcriptional changes and lipid metabolism in response to demyelination (Poliani et al., 2015; Nugent et al., 2020). In AD, complete loss of TREM2 has been shown to affect Aβ seeding at early stages but facilitate plaque compaction to limit the spread of Aβ later (Jay et al., 2015, 2017; Wang et al., 2015, 2016). TREM2 haplodeficiency does not affect Aβ pathology but alters the number and the morphology of plaque-associated microglia and worsens axonal dystrophy (Ulrich et al., 2014; Yuan et al., 2016). Homozygous TREM2 deletion leads to increased tau pathology and seeding but decreased tau-mediated neurodegeneration, whereas TREM2 haplodeficiency aggravates neuroinflammation and exacerbates tau pathology and brain atrophy, showing a complex role of TREM2 in the pathogenic process of AD (Leyns et al., 2017, 2019; Sayed et al., 2018). Collectively, these data suggest that the switch from homeostatic microglia to DAM/MGnD microglia is TREM2 dependent but whether DAM microglia leads to beneficial or detrimental outcomes may depend on the type of environmental stimuli.
Increased spine density in TREM2 null mice could be a consequence of reduced synaptic pruning during neurodevelopment. It has been reported that TREM2 null mice (Turnbull et al., 2006) display reduced synaptic engulfment by microglia during neurodevelopment, resulting in increased spine density (Filipello et al., 2018). Notably, these TREM2 null mice exhibited impairments in repetitive and social behaviors but normal spatial learning and memory in the Morris water maze test at three months of age (Filipello et al., 2018). No cognitive changes were observed at six months of age either in Trem2−/− mice in the present study or in another line of Trem2−/− mice (Velocigene; Kang et al., 2018). These results indicate that different behavioral function may have different sensitivity in baring excessive spine density and could possibly be adjusted by different compensatory mechanisms. Since TREM2 is systemically depleted from germline in Trem2−/− mice, abundant synapses may be retained in the brain after development, conferring resilience of TREM2 null mice to age-related synaptic spine loss and cognitive impairment, as illustrated schematically in Figure 10. Interestingly, using Velocigene Trem2−/− mice, another group reported the opposite effect of TREM2 deficiency in regulating synaptic engulfment during development and found that microglial TREM2 was required for limiting synapse elimination by astrocytes, although such effect only extended to four months of age (Jay et al., 2019). Since Velocigene TREM2 null mice have been shown to overexpress a related gene TREML1, possible compensatory mechanisms during development might complicate the functional and structural outcome in those mice. Clearly, futures studies using conditional TREM2 knock-out mice to achieve temporal depletion of TREM2 and circumvent developmental stages should provide more insights into the temporal role of TREM2 during aging.
The present study has some limitations. Since aging induces microglial adaption of DAM genes and Trem2+/− microglia act like WT Trem2+/+ microglia in the conversion to the DAM state while Trem2−/− microglia fail the conversion (Nugent et al., 2020), the present study focused on the effects of complete depletion of TREM2 during aging. Whether there are any TRME2 gene dosage effects on synaptic function during aging requires future investigation. The behavioral tests used include the open-field test, the elevated plus-maze, and the Morris water maze. However, no other cognitive domains or behavioral aspects were assessed beyond these tests. Future studies employing additional behavioral tests may be needed to reveal other neurologic roles of TREM2 deficiency. To address neuronal synaptic changes, field recordings and a series of electrophysiological experiments were conducted including the I/O curve, PPF, and LTP of the hippocampus. Age-related hippocampal LTP decline was not evident in WT mice at the age examined in the present study. In the literature, mixed results have been reported regarding the effects of age on hippocampal LTP. A previous study showed that hippocampal LTP induced by TBS as used in the present study, which is a more physiological relevant pattern of excitability compared with high-frequency stimulation (HFS; Stäubli et al., 1999; Buzsaki, 2002; Hernandez et al., 2005), does not display a significant decline until 24 months of age in mice (Rogers et al., 2017). Since fEPSP represents grouped neuronal excitability of the hippocampus, whether TREM2 also regulates other electrophysiological phenotypes of the nervous system such as synaptic fatigue, LTD, spontaneous neurotransmitter release, NMDA/AMPA ratio, and inhibitory synapses during aging requires future investigation. For dendritic spine quantification, Golgi staining imaging was limited to 2D projection despite the improvement in microscopic techniques. Spines above or below dendrites are not visible and spine morphology cannot be accurately classified into subcategories. Head diameter and neck length differences classify spines into stubby, thin, and mushroom subtypes (Peters and Kaiserman-Abramof, 1969). Different subtypes of spines play distinct roles in mediating synaptic plasticity and exhibit distinct dynamics during aging (Nimchinsky et al., 2002; Hayashi and Majewska, 2005; Dickstein et al., 2013). Future studies employing advanced 3D construction of spines should provide a better understanding of how TREM2 deficiency affects the spine subtype dynamics. Further, in humans, the most common TREM2 loss-of-function variant that is associated with an increased risk of AD is TREM2-R47H (Guerreiro et al., 2013). Single nucleus transcriptomics analysis has revealed discrepancies in gene signatures between mouse Trem2−/− microglia and human TREM2-R47H microglia (Zhou et al., 2020). Whether microglia carry human TREM2-R47H display similar protective roles during normal aging remains to be investigated.
Despite the limitations, the present study uncovers the unexpected, age-related positive impacts of TREM2 deficiency on cognitive and synaptic function. These findings suggest that TREM2 might exert detrimental effects on brain function during aging. The possibility of age-related negative impacts from TREM2 should be taken into consideration as major efforts are focusing on developing TREM2-based therapeutic strategies to mitigate AD.
Footnotes
This work was supported in part by grants from the National Institute on Aging of the National Institutes of Health (AG056976 and AG058081) and the College of Pharmacy at the University of Minnesota. We thank Dr. Marco Colonna at Washington University for providing the original breeding pairs of Trem2−/− mice and Andrea Gram for maintaining and genotyping the experimental mice.
The authors declare no competing financial interests.
References
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D (2007) Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 52:60–70. 10.1016/j.neuropharm.2006.07.013 [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Köhr G (2005) Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci 25:6907–6910. 10.1523/JNEUROSCI.1905-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KP, Nedivi E (2017) Spine dynamics: are they all the same? Neuron 96:43–55. 10.1016/j.neuron.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros BD, Greathouse KM, Gearing M, Herskowitz JH (2019) Dendritic spine remodeling accompanies Alzheimer's disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol Aging 73:92–103. 10.1016/j.neurobiolaging.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. (2002) Theta oscillations in the hippocampus. Neuron 33:325–340. [DOI] [PubMed] [Google Scholar]
- Chao HW, Tsai LY, Lu YL, Lin PY, Huang WH, Chou HJ, Lu WH, Lin HC, Lee PT, Huang YS (2013) Deletion of CPEB3 enhances hippocampus-dependent memory via increasing expressions of PSD95 and NMDA receptors. J Neurosci 33:17008–17022. 10.1523/JNEUROSCI.3043-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, Reese TS (2011) PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci 31:6329–6338. 10.1523/JNEUROSCI.5968-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Cao D, Hottman DA, Yuan L, Bergo MO, Li L (2013) Farnesyltransferase haplodeficiency reduces neuropathology and rescues cognitive function in a mouse model of Alzheimer disease. J Biol Chem 288:35952–35960. 10.1074/jbc.M113.503904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernick D, Ortiz-Valle S, Jeong A, Swaminathan SK, Kandimalla KK, Rebeck GW, Li L (2018) High-density lipoprotein mimetic peptide 4F mitigates amyloid-beta-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J Neurochem 147:647–662. 10.1111/jnc.14554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickstein DL, Weaver CM, Luebke JI, Hof PR (2013) Dendritic spine changes associated with normal aging. Neuroscience 251:21–32. 10.1016/j.neuroscience.2012.09.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M, Markicevic M, Starvaggi-Cucuzza C, Otero K, Piccio L, Cignarella F, Perrucci F, Tamborini M, Genua M, Rajendran L, Menna E, Vetrano S, Fahnestock M, Paolicelli RC, et al. (2018) The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48:979–991.e8. 10.1016/j.immuni.2018.04.016 [DOI] [PubMed] [Google Scholar]
- Gratuze M, Leyns CE, Holtzman DM (2018) New insights into the role of TREM2 in Alzheimer's disease. Mol Neurodegener 13:66 10.1186/s13024-018-0298-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, et al. (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117–127. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 177:2051–2055. 10.4049/jimmunol.177.4.2051 [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Majewska AK (2005) Dendritic spine geometry: functional implication and regulation. Neuron 46:529–532. 10.1016/j.neuron.2005.05.006 [DOI] [PubMed] [Google Scholar]
- Hernandez RV, Navarro MM, Rodriguez WA, Martinez JL Jr, LeBaron RG (2005) Differences in the magnitude of long-term potentiation produced by theta burst and high frequency stimulation protocols matched in stimulus number. Brain Res Brain Res Protoc 15:6–13. 10.1016/j.brainresprot.2005.02.003 [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P (1999) Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354:269–279. 10.1098/rstb.1999.0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottman D, Cheng S, Gram A, LeBlanc K, Yuan LL, Li L (2018) Systemic or forebrain neuron-specific deficiency of geranylgeranyltransferase-1 impairs synaptic plasticity and reduces dendritic spine density. Neuroscience 373:207–217. 10.1016/j.neuroscience.2018.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109:1144–1156. 10.1111/j.1471-4159.2009.06042.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med 212:287–295. 10.1084/jem.20142322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE (2017) Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J Neurosci 37:637–647. 10.1523/JNEUROSCI.2110-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, von Saucken VE, Muñoz B, Codocedo JF, Atwood BK, Lamb BT, Landreth GE (2019) TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 67:1873–1892. 10.1002/glia.23664 [DOI] [PubMed] [Google Scholar]
- Kang SS, Kurti A, Baker KE, Liu CC, Colonna M, Ulrich JD, Holtzman DM, Bu G, Fryer JD (2018) Behavioral and transcriptomic analysis of Trem2-null mice: not all knockout mice are created equal. Hum Mol Genet 27:211–223. 10.1093/hmg/ddx366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I (2017) A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169:1276–1290.e7. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, et al. (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6:243ra286. [DOI] [PubMed] [Google Scholar]
- Kober DL, Brett TJ (2017) TREM2-ligand interactions in health and disease. J Mol Biol 429:1607–1629. 10.1016/j.jmb.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej A, Smalla KH, Richter S, Engler A, Pielot R, Dieterich DC Tischmeyer W, Naumann M, Kahne T (2016) High resolution quantitative synaptic proteome profiling of mouse brain regions after auditory discrimination learning. J Vis Exp (118):54992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, et al. (2017) The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47:566–581.e9. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM (2017) TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci USA 114:11524–11529. 10.1073/pnas.1710311114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VMY, Ulrich JD, Holtzman DM (2019) TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci 22:1217–1222. 10.1038/s41593-019-0433-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L, Walter J, Colonna M, Neumann H (2019) TREM2 triggers microglial density and age-related neuronal loss. Glia 67:539–550. 10.1002/glia.23563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Takahashi K (2007) Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 184:92–99. 10.1016/j.jneuroim.2006.11.032 [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K (2002) Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. 10.1146/annurev.physiol.64.081501.160008 [DOI] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PCG, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, Solanoy HO, et al. (2020) TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105:837–854.e9. 10.1016/j.neuron.2019.12.007 [DOI] [PubMed] [Google Scholar]
- Orlando M, Lignani G, Maragliano L, Fassio A, Onofri F, Baldelli P, Giovedí S, Benfenati F (2014) Functional role of ATP binding to synapsin I in synaptic vesicle trafficking and release dynamics. J Neurosci 34:14752–14768. 10.1523/JNEUROSCI.1093-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M (2001) CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology 56:1552–1558. 10.1212/wnl.56.11.1552 [DOI] [PubMed] [Google Scholar]
- Parent MA, Hottman DA, Cheng S, Zhang W, McMahon LL, Yuan LL, Li L (2014) Simvastatin treatment enhances NMDAR-mediated synaptic transmission by upregulating the surface distribution of the GluN2B subunit. Cell Mol Neurobiol 34:693–705. 10.1007/s10571-014-0051-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR (1969) The small pyramidal neuron of the rat cerebral cortex. The synapses upon dendritic spines. Z Zellforsch Mikrosk Anat 100:487–506. 10.1007/BF00344370 [DOI] [PubMed] [Google Scholar]
- Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, Colonna M (2015) TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest 125:2161–2170. 10.1172/JCI77983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG. (2012) Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol 4:a005702. 10.1101/cshperspect.a005702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GA, Burns MP, Weeber EJ, Rebeck GW (2013) Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learn Mem 20:256–266. 10.1101/lm.030031.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Liu CC, Zhao N, Wang J, Putzke T, Yang L, Shinohara M, Fryer JD, Kanekiyo T, Bu G (2017) Subacute ibuprofen treatment rescues the synaptic and cognitive deficits in advanced-aged mice. Neurobiol Aging 53:112–121. 10.1016/j.neurobiolaging.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, Hauduc A, Ludwig C, Gao F, Clelland C, Zhan L, Cooper YA, Davalos D, Akassoglou K, Coppola G, Gan L (2018) Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc Natl Acad Sci USA 115:10172–10177. 10.1073/pnas.1811411115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart JC, Caldarone BJ, Stevens B, Lemere CA (2015) Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 35:13029–13042. 10.1523/JNEUROSCI.1698-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji H, Takao K, Hattori S, Miyakawa T (2016) Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol Brain 9:11. 10.1186/s13041-016-0191-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SH, Augustine GJ (2015) Synapsin isoforms and synaptic vesicle trafficking. Mol Cells 38:936–940. 10.14348/molcells.2015.0233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stäubli U, Scafidi J, Chun D (1999) GABAB receptor antagonism: facilitatory effects on memory parallel those on LTP induced by TBS but not HFS. J Neurosci 19:4609–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh A, Dunaevsky A (2015) Preparation of synaptosomes from the motor cortex of motor skill trained mice. Bio Protoc 5:e1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T (2018) Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci 12:323. 10.3389/fncel.2018.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S (1996) The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87:1327–1338. 10.1016/s0092-8674(00)81827-9 [DOI] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177:3520–3524. 10.4049/jimmunol.177.6.3520 [DOI] [PubMed] [Google Scholar]
- Ulland TK, Colonna M (2018) TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol 14:667–675. 10.1038/s41582-018-0072-1 [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM (2014) Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener 9:20. 10.1186/1750-1326-9-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160:1061–1071. 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M (2016) TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213:667–675. 10.1084/jem.20151948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL. (2003) Neuropathologic changes in Alzheimer's disease. J Clin Psychiatry 64:7–10. [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90:724–739. 10.1016/j.neuron.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, et al. (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med 26:131–142. 10.1038/s41591-019-0695-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Liu A, Xia S, Leung C, Qi J, Meng Y, Xie W, Park P, Collingridge GL, Jia Z (2018) The C-terminal tails of endogenous GluA1 and GluA2 differentially contribute to hippocampal synaptic plasticity and learning. Nat Neurosci 21:50–62. 10.1038/s41593-017-0030-z [DOI] [PubMed] [Google Scholar]