

Abstract
A major driver of obesity is the increasing palatability of processed foods. Although reward circuits promote the consumption of palatable food, their involvement in obesity remains unclear. The ventral pallidum (VP) is a key hub in the reward system that encodes the hedonic aspects of palatable food consumption and participates in various proposed feeding circuits. However, there is still no evidence for its involvement in developing diet-induced obesity. Here we examine, using male C57BL6/J mice and patch-clamp electrophysiology, how chronic high-fat high-sugar (HFHS) diet changes the physiology of the VP and whether mice that gain the most weight differ in their VP physiology from others. We found that 10–12 weeks of HFHS diet hyperpolarized and decreased the firing rate of VP neurons without a major change in synaptic inhibitory input. Within the HFHS group, the top 33% weight gainers (WGs) had a more hyperpolarized VP with longer latency to fire action potentials on depolarization compared with bottom 33% of weight gainers (i.e., non-weight gainers). WGs also showed synaptic potentiation of inhibitory inputs both at the millisecond and minute ranges. Moreover, we found that the tendency to potentiate the inhibitory inputs to the VP might exist in overeating mice even before exposure to HFHS, thus making it a potential property of being an overeater. These data point to the VP as a critical player in obesity and suggest that hyperpolarized membrane potential of, and potentiated inhibitory inputs to, VP neurons may play a significant role in promoting the overeating of palatable food.
SIGNIFICANCE STATEMENT In modern world, where highly palatable food is readily available, overeating is often driven by motivational, rather than metabolic, needs. It is thus conceivable that reward circuits differ between obese and normal-weight individuals. But is such difference, if it exists, innate or does it develop with overeating? Here we reveal synaptic properties in the ventral pallidum, a central hub of reward circuits, that differ between mice that gain the most and the least weight when given unlimited access to highly palatable food. We show that these synaptic differences also exist without exposure to palatable food, potentially making them innate properties that render some more susceptible than others to overeat. Thus, the propensity to overeat may have a strong innate component embedded in reward circuits.
Keywords: motivation, obesity, overeating, patch clamp, synaptic plasticity, ventral pallidum
Introduction
Western calorie-rich diets are becoming more and more common in households. Consequently, obesity has become a world epidemic and a leading cause of preventable death worldwide (World Health Organization, 2003). Despite the drastic increase in obesity levels, the biological mechanisms driving this epidemic are still not understood, and research is being conducted in many disciplines, including psychology, metabolism, and neurobiology.
One main theory about the burst of the obesity epidemic in recent decades is that food has become during these years an increasingly strong reinforcer (Kenny, 2011). Various studies have indeed shown that not only do palatable foods act as reinforcers and can activate the reward system (de Macedo et al., 2016), but that direct activation of specific neurons in the reward system can by itself drive motivated feeding behavior (Nieh et al., 2015; Stuber and Wise, 2016; Friend et al., 2017). Such a strong link between the reward system and consumption of palatable foods poses the possibility that overeating that leads to obesity may involve pathologies in the reward system akin to those seen in models of addiction.
Research so far has indeed shown obesity-related changes both in the function of the reward system and in motivation-driven food-seeking behavior. Thus, the expression level of the dopamine receptors D2/D3 in the striatum is decreased in obese humans (Wang et al., 2001; Haltia et al., 2007; de Weijer et al., 2011) and rodents (Johnson and Kenny, 2010; Friend et al., 2017), opioid signaling in both the ventral tegmental area (Vucetic et al., 2011; Martire et al., 2014) and the nucleus accumbens (NAc; Alsiö et al., 2010; Vucetic et al., 2011) is altered in obese rodents, and the glutamatergic input to the NAc is potentiated in rats that develop diet-induced obesity (Brown et al., 2017; Oginsky and Ferrario, 2019), which is akin to the effect seen after withdrawal from cocaine (Gipson et al., 2013a, 2014) or nicotine (Gipson et al., 2013b, 2014). Possibly as a consequence of these changes in the reward system, rats and mice that gain the most weight in a diet-induced obesity model also show the highest motivation to obtain palatable food (Johnson and Kenny, 2010; Mancino et al., 2015; Vollbrecht et al., 2015; Brown et al., 2017; Derman and Ferrario, 2018; Inbar et al., 2019).
A key structure in the reward system, which is yet to be studied in the context of obesity, is the ventral pallidum (VP). The VP is a key junction between the reward system and a main hub for feeding behavior, the lateral hypothalamus (LH; Stratford et al., 1999; Tripathi et al., 2013). It receives strong inhibitory input from the NAc (Zahm and Heimer, 1990; Kupchik et al., 2015) and in turn sends inhibitory projections to the LH and other targets (Root et al., 2015). Thus, it is not surprising that the VP has an important role in modulating feeding behavior (Cromwell and Berridge, 1993; Stratford et al., 1999; Shimura et al., 2006; Smith et al., 2009). Accordingly, the application of GABA antagonists (Stratford et al., 1999; Covelo et al., 2014) or μ-opioid receptor agonists (Smith and Berridge, 2005) into the VP drive feeding, while fMRI studies in rodents, primates, and humans show an increase in VP activity on exposure to food or food cues (Hoch et al., 2013; Jiang et al., 2015; Royet et al., 2016; Kaskan et al., 2019). Nevertheless, whether the VP has a role in pathologic overeating and obesity has not been studied.
In this study, we set out to examine whether chronic consumption of a high-fat high-sugar (HFHS) diet alters the physiology of the VP and whether mice that are more prone to gain weight when HFHS food is freely available exhibit different VP physiology than those that are more resistant to gaining weight. Moreover, using behavioral and electrophysiological tools we test whether the differences in VP physiology between weight gainers (WGs) and non-weight gainers (NWGs) may be innate and thus possibly be part of the mechanism promoting overeating and weight gain.
Materials and Methods
Experimental subjects
Experimental subjects were naive C57BL6/J wild-type male mice weighing between 23 and 30 g at the beginning of the experiment. A 12 h reversed light/dark cycle was maintained, with the lights turned off at 8:00 A.M. Experimental procedures were conducted during dark hours. Mice were housed individually, and nesting/enrichment material was available. Mice were given 7 d to acclimate before experimentation began. All experimental procedures were approved by the Authority for Biological and Biomedical Models at the Hebrew University.
Diet-induced obesity model
After the acclimation period, all mice were put on a 4 week standard chow diet (Chow group; 18% kcal fat; total density = 3.1 kcal/g; Teklad Global 2018, Harlan Laboratories), during which body weight and caloric intake were monitored and behavioral training was completed (see below). Then, mice that were put into the HFHS groups (n = 24) were switched to an HFHS diet (45% kcal fat; total density = 4.73 kcal/g; D12451, Research Diets) for 10–12 weeks, while the control Chow mice (n = 15) continued to feed on the standard chow diet. At the end of these 10–12 weeks, HFHS mice were switched back to standard chow for 2 additional weeks before being killed for recordings. Body weight was determined twice per week (BJ-410C scales, Precisa) throughout the entire experiment. At the end of the diet-induced obesity protocol, HFHS mice were divided according to their weight gain (relative to their weight on day 1, when they were first exposed to the HFHS diet) into diet-induced WGs (top third of weight gainers) and NWGs (bottom third of weight gainers). These groups are parallel to, and were generated using the same protocols as, the obesity-prone and obesity-resistant groups that we and others used in previous studies (Cifani et al., 2015; Brown et al., 2017; Inbar et al., 2019; Oginsky and Ferrario, 2019; Alonso-Caraballo et al., 2020).
Operant food self-administration protocol
Food self-administration was conducted in mouse operant boxes (MedAssociates) as described previously (Inbar et al., 2019). Briefly, mice were trained to press a lever to obtain a 20 mg precision pellet of chow food (5.6% kcal from fat; total density = 3.6 kcal/g; catalog #F0071, Bio-Serv) first on a fixed ratio 1 (FR1) schedule (each lever press resulted in a food pellet), and then on FR3 and FR5 schedules. Mice were eventually tested at the end of the HFHS diet on a progressive ratio task (where the number of lever presses required to obtain a pellet increased progressively). The progressive ratio schedule was 5, 9, 12, 15, 20, 25, 32, 40, 50, 62, 77, 95, 118, 145, 178, 219, 268, 328, 402, 492, and 603 lever presses per pellet. The test was terminated after 6 h or if the mouse did not progress to the next level within 1 h of achieving the previous level. During the test, we monitored the number of lever presses on the active and inactive levers, and the number of head entries into the food receptacle (detected by infrared beam). Operant chambers were located in sound-attenuating boxes to minimize external noises.
Slice preparation
As described previously (Inbar et al., 2020; Levi et al., 2020). Mice were anesthetized with 150 mg/kg ketamine HCl and then decapitated. Sagittal slices (200 µm) of the VP were prepared (model VT1200S vibratome, Leica) and moved to vials containing artificial CSF (aCSF), as follows (in mm): 126 NaCl, 1.4 NaH2PO4, 25 NaHCO3, 11 glucose, 1.2 MgCl2, 2.4 CaCl2, 2.5 KCl, 2.0 Na pyruvate, and 0.4 ascorbic acid, bubbled with 95% O2 and 5% CO2, and a mixture of 5 mm kynurenic acid and 100 mm MK-801. Slices were stored at room temperature (22–24°C) until recording.
In vitro whole-cell recording
Recordings were performed at 32°C (model TC-344B, Warner Instruments). VP neurons were visualized using an Olympus BX51WI microscope and recorded using glass pipettes (1.3–2 MΩ; World Precision Instruments) filled with an internal solution (in mm: 68 KCl, 65 d-gluconic acid potassium salt, 7.5 HEPES potassium, 1 EGTA, 1.25 MgCl2, 10 NaCl, 2.0 MgATP, and 0.4 NaGTP, pH 7.2–7.3, 275 mOsm; the junction potential magnitude was <9 mV in all cells and always compensated for). Excitatory synaptic transmission was blocked with CNQX (10 μm). A Multiclamp 700B Amplifier (Molecular Devices) was used to record both membrane and action potentials and IPSCs in whole-cell configuration. Excitability and passive membrane properties were measured in current-clamp mode, while synaptic activity was measured in voltage-clamp mode at a holding potential of −80 mV. Recordings were acquired at 10 kHz and filtered at 2 kHz using AxoGraph X software (AxoGraph Scientific). To evoke IPSCs electrically, a bipolar stimulating electrode (FHC) was placed ∼200–300 μm anterior of the cell to maximize chances of stimulating NAc afferents. The stimulation intensity chosen evoked 30–70% of maximal IPSC. Recordings were collected every 10 s. Series resistance (Rs) measured with a −2 mV hyperpolarizing step (10 ms) given with each stimulus and holding current were always monitored online. Recordings with unstable Rs or when Rs exceeded 20 MΩ were aborted.
Current-clamp experiments
After penetrating the neuron, we switched to current-clamp mode and recorded the resting membrane potential of the neurons and spontaneous action potentials for 60 s. Cells with unstable membrane potential were discarded. Action potentials were later detected by their waveform using Axograph software, baselined, and analyzed. Potentials of <20 mV in amplitude were discarded. We then applied the current step protocol—five 500-ms-long depolarization current steps ranging from 0 pA to +80 pA (20 pA intervals) were applied, and the interstep interval was 3 s. The five-step protocol was repeated five times with 3 s between repetitions. Baseline membrane potential was adjusted to be approximately −50 mV in all neurons by injecting constant current (Table 1).
Table 1.
Action potential and passive membrane properties
| HFHS mice | Chow mice | p Value (t score) | WG mice | NWG mice | p Value (t score) | |
|---|---|---|---|---|---|---|
| AP amplitude (mV) | 52.06 ± 12.21 (n = 34) | 46.14 ± 13.79 (n = 25) | p = 0.0868 (t(57) = 1.74) | 48.52 ± 7.46 (n = 10) | 55.82 ± 14.92 (n = 15) | p = 0.1526 (t(23) = 1.48) |
| AP threshold (mV) | −42.7 ± 9.7 (n = 33) | −40.0 ± 2.8 (n = 25) | p = 0.1719 (t(56) = 1.38) | −45.3 ± 5.2 (n = 9) | −43.2 ± 6.7 (n = 15) | p = 0.4254 (t(22) = 0.81) |
| AP half-width (ms) | 1.706 ± 0.829 (n = 34) | 1.258 ± 0.581 (n = 26) | *p = 0.0226 (t(58) = 2.34) | 1.739 ± 0.676 (n = 10) | 1.359 ± 0.484 (n = 15) | p = 0.1140 (t(23) = 1.78) |
| AP rise time (ms) | 1.784 ± 0.915 (n = 34) | 1.181 ± 0.591 (n = 26) | *p = 0.005 (t(58) = 2.92) | 2.103 ± 1.083 (n = 10) | 1.308 ± 0.516 (n = 15) | *p = 0.0213 (t(23) = 2.47) |
| AP decay (ms) | 1.714 ± 0.861 (n = 34) | 1.324 ± 0.747 (n = 26) | p = 0.071 (t(58) = 1.84) | 1.620 ± 0.7162 (n = 10) | 1.443 ± 0.583 (n = 15) | p = 0.5036 (t(23) = 0.68) |
| AHP peak (mV) | −12.9 ± 4.112 (n = 29) | −14.2 ± 3.537 (n = 23) | p = 0.2367 (t(50) = 1.20) | −13.65 ± 2.906 (n = 8) | −12.25 ± 5.08 (n = 14) | p = 0.4867 (t(20) = 0.71) |
| AHP half-width (ms) | 8.281 ± 4.393 (n = 17) | 7.46 ± 2.648 (n = 9) | p = 0.6141 (t(24) = 0.52) | 10.29 ± 3.307 (n = 5) | 7.878 ± 5.006 (n = 8) | p = 0.3631 (t(11) = 0.95) |
| Series resistance (MΩ) | 6.457 ± 2.302 (n = 39) | 6.735 ± 3.257 (n = 26) | p = 0.7114 (t(63) = 0.36) | 6.099 ± 1.968 (n = 9) | 6.583 ± 2.999 (n = 12) | p = 0.521 (t(19) = 0.65) |
| Capacitance (pF) | 7.475 ± 2.009 (n = 26) | 7.816 ± 2.392 (n = 12) | p = 0.6502 (t(36) = 0.46) | 8.077 ± 1.654 (n = 10) | 7.794 ± 1.758 (n = 8) | p = 0.7301 (t(16) = 0.65) |
| Membrane resistance (MΩ) | 366.8 ± 170.9 (n = 26) | 261.6 ± 58.39 (n = 12) | *p = 0.0463 (t(36) = 2.06) | 282.8 ± 154 (n = 10) | 347 ± 138.5 (n = 8) | p = 0.3722 (t(16) = 0.92) |
| Current injected (pA) | −5.24 ± 12.9 (n = 30) | −14.9 ± 8.6 (n = 24) | *p = 0.003 (t(52) = 3.15) | −0.95 ± 10.42 (n = 7) | −7.2 ± 16.2 (n = 14) | p = 0.3682 (t(19) = 0.92) |
All comparisons used two-tailed unpaired t tests. AP, Action potential; AHP, afterhyperpolarization; Current injected, amplitude of current injected in current-clamp mode to maintain membrane potential at approximately −50 mV. The asterisk highlights the tests where p < 0.05.
Spontaneous postsynaptic currents
To analyze the sIPSCs we recorded for 100 s without any stimulation. sIPSCs were later detected based on their waveform using Axograph software. Events of <10 pA in amplitude or with significant offset in baseline were not included in the final analysis.
High-frequency stimulation-induced plasticity measurements
Baseline evoked IPSCs were recorded at −80 mV for 3 min before applying the high-frequency stimulation (HFS) protocol. We used the same HFS protocol that was shown previously to induce long-term plasticity (depression) in inhibitory synapses in the VP (Kupchik et al., 2014; Heinsbroek et al., 2017). The protocol consists of two trains separated by 20 s, each delivering 100 stimulations at 100 Hz. The amplitude of the pulses was identical to that inducing synaptic release when recording at −80 mV. During the HFS protocol, membrane potential was allowed to change freely (i.e., current-clamp configuration). At the end of the second train of pulses recording was re-established at holding potential of −80 mV. Recording of evoked IPSCs (eIPSCs) was performed at 0.1 Hz, and the amplitudes (expressed as the percentage at baseline) of every six responses were averaged to give one data point per minute. Differences between groups were measured at the first minute after the HFS for post-tetanic potentiation and at minutes 13–19 for the long-term plasticity in all experiments.
Paired-pulse ratio and coefficient of variation measurements
All experiments consisted of two consecutive stimulations with a 50 ms interval. We measured the ratio between the amplitudes of the second and first pulse to calculate the paired-pulse ratio (PPR) and the coefficient of variation (CV) of the currents generated by the first of the two pulses. We repeated this at least 20 times for each condition.
Data analysis
Statistics were performed using GraphPad Prism 8.2 (GraphPad Software). Parametric statistics (Student's t test, one-way, two-way, or mixed-effects ANOVA with Sidak's multiple-comparisons tests where appropriate) was used throughout (except see Figs. 2A,B, 4A,B, where we used the Kolmogorov–Smirnov test). See lines in Figures 3D, 4I, and 5, F and G, which were generated from simple nonparametric linear regression to the data. The ANCOVA test used, seen in Figures 2, D and E, 4, D and E, and 5I, compares the slope and intercept with y-axis (called “elevation” here) of the linear regression of the datasets. If slopes are significantly different, it is not possible to test whether the intercepts differ significantly. Thus, in Figure 4E only the slope comparison is presented. If the slopes are not significantly different, the intercepts are compared.
Figure 2.

Chronic HFHS diet alters spontaneous but not evoked GABA neurotransmission in the ventral pallidum. A, The cumulative probability plot of sIPSC amplitude (amp.) is shifted to the left in HFHS mice, pointing to reduced sIPSC amplitude (Kolmogorov–Smirnov test). Inset, Median sIPSC amplitude was lower in HFHS mice (unpaired two-tailed t test). B, HFHS diet did not affect the interevent interval (IEI) of sIPSCs in the VP (Kolmogorov–Smirnov test). Inset, Comparison of medians (unpaired two-tailed t test). C, Representative sIPSC traces. D, E, HFHS diet did not affect short-term plasticity induced by five consecutive stimulations at 10 Hz (D) or 50 Hz (E; two-way ANCOVA). Insets, Representative traces synchronized with x-axis. Stimulation artifacts were truncated. F, G, The amplitude of evoked IPSCs after an HFS did not differ between chow and HFHS mice (mixed-effects ANOVA), or from baseline (one-sample two-tailed t test on minutes 13–19). Inset, Representative traces. H, Post-tetanic potentiation, measured at the first time point after the HFS, did not differ between groups (unpaired two-tailed t test). Data taken from 19–41 cells in 13–24 mice/group. Data are presented as the mean ± SEM. n.s., not significant.
Figure 4.

Weight-gainer and non-weight gainer mice show different synaptic plasticities in their GABA input to the VP. A, B, sIPSC amplitude (amp.; A) and IEI (B) were similar between WG and NWG mice (Kolmogorov–Smirnov tests). Insets, Comparison of medians (unpaired two-tailed t tests). C, Representative sIPSC traces. D, E, Five consecutive pulses at 10 Hz (D) or 50 Hz (E) generated short-term potentiation of IPSCs in WG but not NWG mice (two-way ANCOVA tests). Insets, Representative traces synchronized with x-axis. Stimulation artifacts were truncated. F, G, HFS protocol given to inhibitory inputs to the VP generated a transient potentiation in WG mice but not in NWG mice (mixed-effects ANOVA; one-sample t test for comparing minutes 13–19, 100% of baseline). F, Inset, Representative traces. H, HFS-induced plasticity in VP neurons was positively correlated with weight gain (Middle, middle 33% weight gainers), going from depression in NWG mice to potentiation in WG mice. Correlation was assessed using nonparametric Spearman correlation. I, Post-tetanic potentiation, measured at the first time point after the HFS, was stronger in WG mice (unpaired two-tailed t test). Data are taken from 8–17 cells in 7 mice/group. Data are presented as the mean ± SEM.
Figure 3.

Ventral pallidum neurons of weight-gainer mice are more hyperpolarized and slower to fire compared with non-weight gainer mice. A, Increase in body weight gain (expressed as a percentage of the first day) during 10–12 weeks of chronic HFHS diet for individual mice. Mice were split into non-weight gainer (bottom 33% of weight gainers on the last day of HFHS diet) and weight-gainer (top 33% of weight gainers on last day of HFHS diet) groups according to final weight gain. B, VP neurons of WG mice were more hyperpolarized than those of NWG mice (unpaired two-tailed t test). C, There was no significant difference in the baseline firing frequency of VP neurons between WG and NWG mice (unpaired two-tailed t test). D, VP membrane potential was negatively correlated with weight gain (Middle, middle 33% weight gainers). Correlation was assessed using nonparametric Spearman correlation. E, F, The firing rates during incrementing depolarization steps did not differ between WG and NWG mice (two-way ANOVA), but the minimal latency to the first action potential was longer in HFHS mice (two-way ANOVA), possibly reflecting decreased excitability. G, Representative traces. Data are taken from 9–16 cells in 7 mice/group. Data are presented as the mean ± SEM. AP, Action potential.
Figure 5.
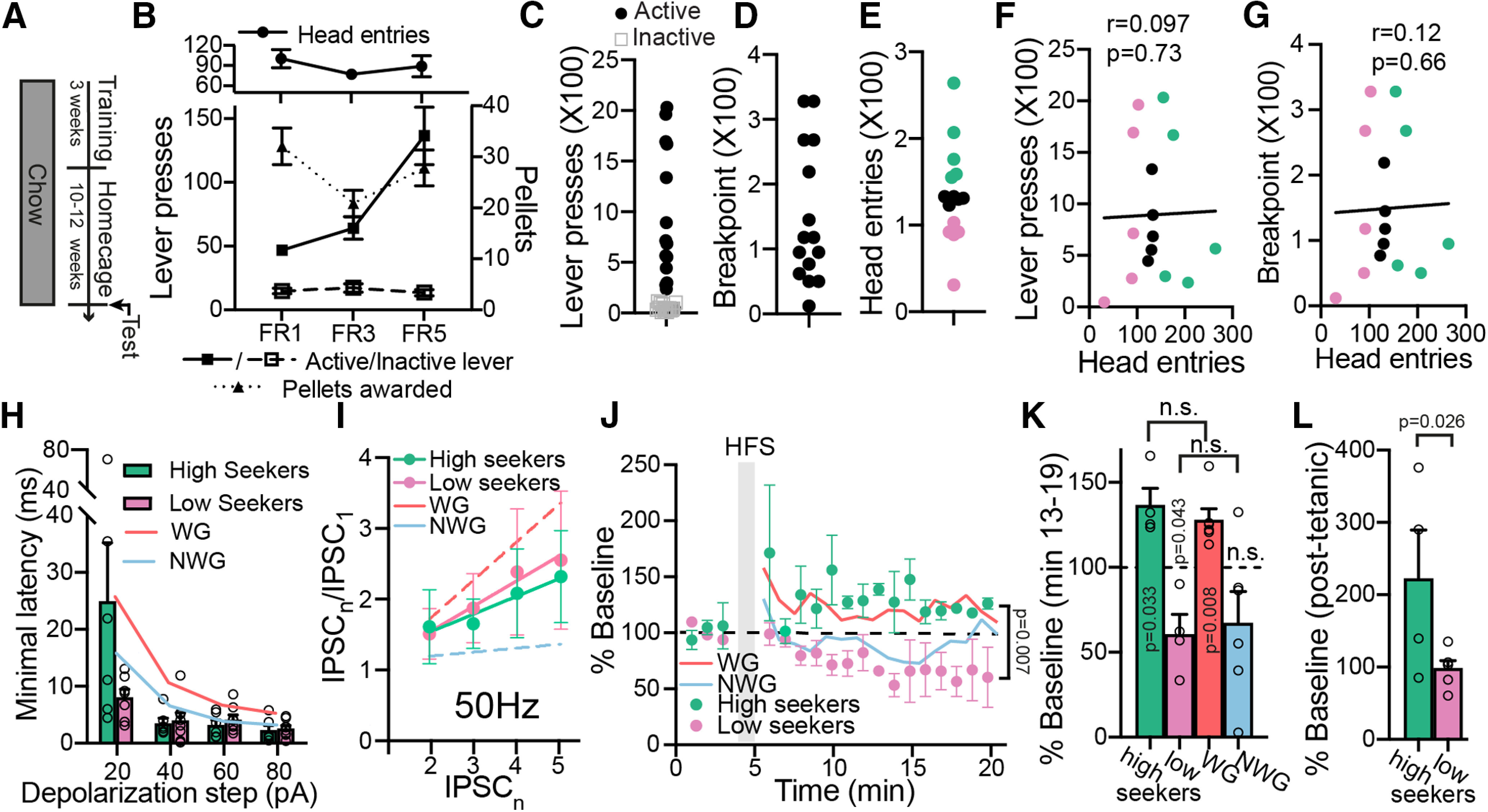
Differences in synaptic plasticity seen between WG and NWG mice exist also between high and low food seekers without exposure to HFHS diet. A, Experimental protocol. Mice were first trained to press a lever for 20 mg of chow precision pellets. After 3 weeks of training, mice were left in their home cages and fed on chow for 10–12 weeks. B, Active and inactive lever pressing, pellets rewarded, and head entries of mice during training. C–E, After the HFHS diet mice were tested in the progressive ratio paradigm and lever pressing (C), break point (D), and head entries (E) were recorded. Mice were split according to the head entries to the food receptacle (E) into high seekers (top third) and low-seekers (bottom third). F, G, There was no correlation between head entries and lever presses (F) or break point (G). Correlations were assessed using nonparametric Spearman correlation. H, There was no difference in the minimal delay to the first action potential in a series of depolarizing steps (from +20 to + 80 pA) between high-seekers and low-seekers (mixed-effects ANOVA). I, There was no difference in short-term plasticity between high-seekers and low-seekers (five stimulations at 50 Hz; two-way ANCOVA). J, K, HFS protocol potentiated eIPSCs in the high-seekers and depressed eIPSCs in the low-seekers (mixed-effects ANOVA comparing minutes 13–19; one-sample t test for comparing minutes 13–19, 100% of baseline). The level of HFS-induced potentiation was similar between high-seekers and WG mice (mixed-effects ANOVA). This was also the case between low-seekers and NWG mice, although note that only the change in low-seekers was significantly different from baseline (p > 0.05 using two-tailed unpaired t test when comparing minutes 13–19 in high-seekers to WG mice or low-seekers to NWG mice). L, Post-tetanic potentiation, measured at the first time point after the HFS, was stronger in high-seekers compared with low-seekers (unpaired one-tailed t test). Data are taken from 4–9 cells in 5 mice/group. Data are presented as the mean ± SEM. Amplitude, amp. n.s., not significant.
Results
High-fat high-sugar diet hyperpolarizes membrane potential and reduces firing rate in VP neurons
To establish the long-lasting influence of the chronic consumption of highly palatable food on the VP of mice, we exposed two groups of mice to different diet protocols. The control group received regular chow throughout the experiment. The experimental group received an HFHS diet ad libitum in the home cage for 10–12 weeks followed by 2 weeks of standard chow diet. This protocol produces behavioral and synaptic alterations in the nucleus accumbens (Brown et al., 2017; Inbar et al., 2019) and aims to model diet-induced weight gain followed by abstinence from chronic food reward (“going on a diet”). This model also has the advantage of allowing the detection of long-lasting changes induced by HFHS diet in the reward system. The second group received standard chow for a similar time period (Fig. 1A).
Figure 1.

Chronic high-fat high-sugar diet hyperpolarizes ventral pallidum neurons and inhibits their firing rate. A, Experimental timeline. Mice in the HFHS group received an HFHS diet ad libitum in their home cages for 10–12 weeks and then returned to chow diet for 2 additional weeks before recordings. The control Chow group received regular chow during the same period of time. B, Recording setup. VP neurons were recorded from in sagittal slices while activating incoming axons using a bipolar stimulating electrode placed ∼300 μm rostral to recorded cells. AC, Anterior commissure; NA, nucleus accumbens. C, D, VP neurons of HFHS mice had lower membrane potential (C) and lower action potential (AP) frequency (D) compared with chow mice (two-tailed unpaired t tests). E, Representative current-clamp traces. F, G, In a series of incrementing depolarizing steps the firing rate was significantly lower in VP neurons of HFHS mice compared with chow mice (F; two-way ANOVA), but there was no difference in the minimal latency to the first action potential (G). H, Representative traces at three depolarizing steps. Data taken from 32–46 cells in 14–21 mice/group. Data are presented as the mean ± SEM.
We then examined, using whole-cell patch-clamp electrophysiology, whether the long exposure to HFHS had affected the cellular properties of VP neurons (Fig. 1B). We found that both resting membrane potential (Fig. 1C; two-tailed unpaired t test: t(76) = 4.11, p < 0.0001) and action potential firing frequency (Fig. 1D,E; two-tailed unpaired t tests: t(67) = 2.02 p = 0.0479) were significantly lower in the VP of HFHS mice compared with the Chow mice. Moreover, we found that when depolarized to different levels, VP neurons of HFHS mice fired significantly less than VP neurons of Chow mice (Fig. 1F,H; two-way ANOVA; main group effect: F(1,260) = 23.04, p < 0.0001; no group × step amplitude interaction: F(3,260) = 0.92 p = 0.43). The minimal latency to the first action potential in a depolarizing step (Fig. 1G) did not differ between groups (two-way ANOVA; no main group effect: F(1,260) = 0.37 p = 0.54; no group × step interaction: F(3,260) = 0.66 p = 0.58). Examination of the shape of the action potential revealed a wider waveform in the HFHS group, but other parameters did not differ between groups (Table 1). These data demonstrate that chronic HFHS consumption changes the physiology of VP neurons, rendering them more hyperpolarized and less active, even 2 weeks after the last exposure to HFHS.
High-fat high-sugar diet effect on GABA neurotransmission in the VP
The changes in cellular physiology induced by the chronic HFHS diet may be accompanied by changes in synaptic neurotransmission in the VP. To test for that, we compared eIPSCs and sIPSCs, respectively, between the HFHS and the Chow groups. When examining the amplitude and frequency of sIPSCs, we found that chronic HFHS diet significantly decreased the amplitude (Fig. 2A,C; Kolmogorov–Smirnov test, d = 0.34, p = 0.0002; comparison of medians with unpaired two-tailed t test: t(63) = 3.16, p = 0.002) but not the frequency (Fig. 2B,C; Kolmogorov–Smirnov test: d = 0.128, p = 0.91; comparison of medians with unpaired two-tailed t test: t(63) = 0.84, p = 0.41) of sIPSCs. This reflects changes in GABA neurotransmission in the VP. Changes in the amplitude of spontaneous events are sometimes interpreted as reflecting postsynaptic mechanisms, but as these include action potential-induced events, we think that this claim cannot be made for our data. Nevertheless, the changes we observed are less likely to be of presynaptic origin as we did not find an effect on the CV and PPR of eIPSCs, two indicators of changes in the probability of synaptic release (Faber and Korn, 1991; Berninger et al., 1999; Schinder et al., 2000; Table 2).
Table 2.
CV and PPR of evoked IPSCs at baseline and after the HFS
| HFHS mice | Chow mice | p Value (t score) | WG mice | NWG mice | p Value (t score) | |
|---|---|---|---|---|---|---|
| CV | 39.77 ± 10.33 (n = 24) | 37.56 ± 10.6 (n = 15) | p = 0.5225 (t(37) = 0.65) | 38.71 ± 9.892 (n = 5) | 33.51 ± 7.574 (n = 8) | p = 0.3062 (t(11) = 1.07) |
| CV after HFS | 44.45 ± 9.486 (n = 23) | 44.26 ± 10.45 (n = 15) | p = 0.9541 (t(36) = 0.06) | 44.06 ± 7.611 (n = 5) | 43.08 ± 44.24 (n = 8) | p = 0.8672 (t(11) = 0.17) |
| PPR | 1.302 ± 0.568 (n = 24) | 1.445 ± 1.233 (n = 19) | p = 0.6163 (t(41) = 0.51) | 1.364 ± 0.812 (n = 8) | 1.376 ± 0.559 (n = 8) | p = 0.9731 (t(14) = 0.03) |
| PPR after HFS | 1.818 ± 1.228 (n = 16) | 1.797 ± 1.14 (n = 16) | p = 0. 9618 (t(30) = 0.96) | 1.245 ± 0.535 (n = 4) | 2.033 ± 1.293 (n = 5) | p = 0.2954 (t(7) = 1.13) |
All comparisons used two-tailed unpaired t tests.
To examine whether chronic HFHS diet changes also evoked GABA neurotransmission in the VP, we examined the generation of synaptic plasticity in the millisecond (short-term plasticity) and the minute (long-term plasticity) range. Short-term plasticity was induced by applying five identical pulses at 1, 10, or 50 Hz, and the change in eIPSCs during the protocol was calculated by dividing each eIPSC by the first one. Our data show that the HFHS diet did not affect short-term plasticity in the VP in either of the frequencies (Fig. 2D,E; two-way ANCOVA; 10 Hz: slope, F = 0.00875, p = 0.9257; elevation, F = 1.949, p = 0.1641; 50 Hz: slope, F = 0.0009, p = 0.9752; elevation, F = 0.02642, p = 0.8711; data for 1 Hz not shown). To induce long-term plasticity, we applied an HFS protocol that we and others used previously (Kupchik et al., 2014; Creed et al., 2016) to elicit transient long-term depression (LTD) in the VP. In contrast to previous findings, this protocol did not elicit plasticity [comparison between groups (minutes 13–19), mixed-effects ANOVA: main group effect, F(1,35) = 0.016, p = 0.901; interaction group × time, F(6,142) = 1.399, p = 0.22; comparison of each group to baseline (minutes 13–19), one-sample two-tailed t test:, Chow, t(16) = 0.96, p = 0.35; HFHS, t(19) = 0.47, p = 0.65] or affect the CV or PPR parameters in the chow or HFHS groups (Fig. 2F,G, Table 2; the lack of plasticity at the group level does not reflect lack of plasticity at the single-cell level. As will be shown below, some cells showed depression while others showed potentiation. The HFS protocol did generate post-tetanic potentiation in both groups (measured as the difference from baseline of the eIPSCs during the first minute after the HFS), but without difference between them (Fig. 2H; unpaired two-tailed t test, t(37) = 1.53, p = 0.14). Overall, the data suggest that chronic HFHS diet impairs spontaneous neurotransmission possibly through a postsynaptic mechanism but does not affect the short-term and long-term plasticity of evoked GABA neurotransmission.
Weight gainer mice show hyperpolarized membrane potential and delayed firing in the VP compared with non-weight gainer mice
Our data so far show that chronic exposure to HFHS food changes the physiology of the VP. Our next question was whether, within the group of mice exposed to HFHS diet, those that gained the most weight [top 33% of weight gainers, calculated as the percentage change between the first and last day of the HFHS diet (i.e., before switching back to chow diet; called weight gainers) showed different physiology in the VP compared with those who gained the least weight (bottom 33% of weight gainers, called non-weight gainers; Fig. 3A). Importantly, WG and NWG mice did not differ in their initial weight on day 1 of the HFHS diet (unpaired two-tailed t test, 24.2 ± 1.08 and 25.6 ± 2.27 g, respectively; t(14) = 1.71, p = 0.11) but showed a significant difference in the total weight gained during the HFHS diet (unpaired two-tailed t test, 18.3 ± 1.62 and 13.0 ± 2.16 g, respectively; t(14) = 5.53, p < 0.0001). Previous studies showed that not only does weight gain correlate with addictive-like behaviors toward palatable food (Vollbrecht et al., 2015; Brown et al., 2017; Inbar et al., 2019; Oginsky and Ferrario, 2019), it also correlates with synaptic changes in the NAc (Brown et al., 2017; Oginsky and Ferrario, 2019): obesity-prone rats show stronger excitatory input to the NAc compared with obesity-resistant rats. As the VP is a major target of the NAc, we expect that the physiology of the VP will also differ between WG and NWG mice.
Membrane potential of VP neurons was significantly more hyperpolarized in WG mice compared with NWG mice (Fig. 3B; unpaired two-tailed t test, t(31) = 1.99, p = 0.017), although action potential frequency (Fig. 3C; unpaired two-tailed t test, t(31) = 1.67, p = 0.085) or shape (Table 1) did not differ between the groups. Membrane potential (Fig. 3D; nonparametric Spearman correlation, r = 0.38, p = 0.01) but not firing frequency (Table 3; nonparametric Spearman correlation, r = −0.31, p = 0.06), also showed an inverse correlation with weight gain across the entire HFHS population. There was no difference in passive membrane properties of VP neurons of WG and NWG mice (Table 1).
Table 3.
Correlation of weight gain or weight loss with various neurophysiological parameters in the VP
| Membrane potential | AP frequency | APs/+80 pA step | Minimal latency to first AP (+20 pA step) | eIPSC amplitude (% change after HFS) | sIPSC amplitude | sIPSC frequency (IEI) | IPSC5\IPSC1 (50 HZ) | |
|---|---|---|---|---|---|---|---|---|
| Weight gain | *p = 0.01 r = 0.38 |
p = 0.06 r = −0.31 |
p = 0.56 r = −0.1 |
p = 0.62 r = −0.1 |
*p = 0.04 r = 0.45 |
p = 0.4 r = 0.14 |
p = 0.87 r = 0.03 |
p = 0.17 r = 0.25 |
| Weight loss |
p = 0.54 r = 0.09 |
p = 0.15 r = 0.24 |
p = 0.84 r = 0.03 |
p = 0.88 r = 0.02 |
p = 0.87 r = 0.03 |
p = 0.34 r = 0.15 |
p = 0.31 r = 0.16 |
p = 0.54 r = 0.09 |
r, nonparametric Spearman correlation; weight gain, weight on the last day of HFHS diet expressed as percentage of weight on first day of HFHS diet; weight loss, weight on last day of the 2 weeks of chow following the HFHS diet expressed as percentage of weight on last day of HFHS diet; average weight loss, WG % weight loss (−9.257 ± 4.029% for WG mice; −10.83 ± 2.263% for NWG mice). AP, Action potential. The asterisk highlights the tests where p < 0.05.
Applying depolarizing steps of increasing amplitudes from baseline membrane potential induced a comparable number of action potentials in both groups (Fig. 3E; two-way ANOVA; main group effect, F(1,84) = 0.43, p = 0.51; interaction group × step amplitude, F(3,84) = 0.05, p = 0.99), but the minimal latency to the first action potential, a measure inversely correlated with cellular excitability (Alexander et al., 2019), was significantly longer in the WG mice (Fig. 3F,G; two-way ANOVA; main group effect, F(1,78) = 5.37, p = 0.023; interaction group × step amplitude, F(3,84) = 0.73, p = 0.54). Both these parameters did not correlate with weight gain when examining the entire group of mice (Table 3). Overall, these data show that VP neurons of WG mice are more hyperpolarized and are slower to fire compared with those of NWG mice.
Weight-gainer mice show altered synaptic plasticity in GABA input to the VP
To evaluate whether GABA synaptic transmission in the VP is also different between WG and NWG mice, we compared spontaneous and evoked IPSCs with the same tools as in Figure 2. An examination of the amplitude and frequency of the inhibitory spontaneous events in the VP showed that these parameters were comparable between WG and NWG mice [Fig. 4A–C; Kolmogorov–Smirnov tests: for amplitude, d = 0.203, p = 0.08; interevent interval (IEI), d = 0.128, p = 0.91; comparison of medians, unpaired two-tailed t test: amplitude, t(27) = 0.15, p = 0.88; IEI, t(27) = 0.48, p = 0.64] and did not correlate with weight gain in the entire HFHS population (Table 3). There was also no difference in the CV or PPR of eIPSCs between the groups (Table 2). The ability of VP neurons to show short-term plasticity, on the other hand, was different between WG and NWG mice. The application of five consecutive stimulations at 10 or 50 Hz (but not at 1 Hz; data not shown) induced a strong potentiation of the IPSC in WG mice, but not in NWG mice (Fig. 4D,E; two-way ANCOVA tests; 10 Hz: slope, F(1,4) = 0.55, p = 0.50; elevation: F(1,4) = 34.32, p = 0.002; 50 Hz: slope, F(1,4) = 17.78, p = 0.0134), although the amplitudes of the first IPSCs were similar (data not shown). This could mean that in WG mice, but not NWG mice, fast repeated activation of inhibitory inputs to the VP may result in stronger inhibition that builds up with activity. The short-term plasticity showed a positive correlation with weight gain, but this correlation did not reach significance (Table 3).
VP neurons of WG and NWG mice also showed a striking difference in their longer-term response to an HFS (Fig. 4F; mixed-effects ANOVA; main group effect comparing minutes 13–19: F(1,12) = 5.29, p = 0.029; interaction group × time: F(6,38) = 0.36, p = 0.90; when comparing minutes 13–19, 100% of baseline with one-sample two-tailed t test: WG: t(5) = 4.25, p = 0.008; NWG: t(5) = 1.77, p = 0.14). Application of the HFS protocol (Kupchik et al., 2014) in VP neurons of NWG mice generated a nonsignificant transient decrease in the average IPSC amplitude to 67.4 ± 45% of baseline (Fig. 4F,G). In contrast, the HFS protocol induced a transient potentiation in VP cells of WG mice that lasted for ∼15 min (Fig. 4F,G). In fact, the direction and amplitude of the HFS-induced plasticity correlated with weight gain after chronic HFHS across all mice, transforming from depression in NWG mice to potentiation in WG mice (Fig. 4H; nonparametric Spearman correlation, r = 0.45, p = 0.04). In addition, the HFS generated a more robust post-tetanic potentiation in WG mice (Fig. 4I; unpaired two-tailed t test, t(16) = 2.18, p = 0.044), again pointing to a tendency to strengthen inhibitory input in these mice after tetanic stimulation. Note the similarity in the results for the long-term and short-term plasticity protocols: in both cases, the inhibitory input to the VP of WG mice was potentiated while the input to the VP of NWG mice did not change or even showed depression in some cells. Overall, the data in Figure 4 show a difference in the synaptic plasticity of inhibitory input to the VP between WG and NWG mice, with a tendency toward potentiation of these inputs in WG mice.
The physiology of the VP is linked to weight gain but not to weight loss
Our data so far reveal a link between VP physiology and the amount of weight gained during the HFHS diet. As mentioned above (Fig. 1), recordings were performed 2 weeks after the termination of the HFHS diet. During these 2 weeks, mice returned to normal chow diet and lost weight (weight loss did not differ between WG and NWG mice: −9.26 ± 11.4% and −10.83 ± 6.4%, respectively, t(14) = 0.34, p = 0.74 using unpaired t test, weight loss calculated as the percentage of change in weight between the last day of HFHS diet and the last day of the following 2 weeks of chow diet). Thus, changes in VP physiology could be linked also to the level of weight loss. However, none of the electrophysiological measures we used showed a significant correlation with weight loss (Table 3). Thus, it seems that the physiology of the VP is more linked to the consumption of highly palatable food and not to the abstinence from it.
Differences in HFS-induced plasticity between WG and NWG mice may be innate
In this and previous works, we and others showed various synaptic and physiological parameters in the NAc and the VP that differ between animals that gain the most or the least weight when given free access to highly palatable food (Brown et al., 2017; Oginsky and Ferrario, 2019). For obvious reasons, slice recordings in all these studies could only be performed after the HFHS diet and splitting mice to groups according to final weight gain. This technical hurdle left one of the most important questions unanswered: are these synaptic differences between obesity-prone and obesity-resistant animals the cause or the consequence of obesity?
In a recent study (Inbar et al., 2019), we showed that in a progressive ratio task where the reward is a small pellet of HFHS food, head entries to the food receptacle are higher in WG mice compared with NWG mice, even before the beginning of the diet. Based on this, we hypothesize here that if a physiological difference in the VP between WG and NWG mice is innate, it may also exist without exposure to HFHS diet between mice that show the highest and the lowest rates of head entries in a progressive ratio task.
When choosing the physiological parameters to test, we specifically looked for parameters that, on the one hand, were not affected by the mere exposure to HFHS diet but, on the other hand, did differ between WG and NWG mice after prolonged HFHS consumption. This is because we looked forinnate differences between WG and NWG mice. Thus, any difference driven by the HFHS diet itself (Figs. 1, 2) cannot be confirmed as being innate. Based on these a priori conditions, we examined the only physiological measures that fulfilled the requirements: the latency to fire on depolarization and synaptic plasticity.
To examine whether the difference between WG and NWG mice in either of the physiological measures listed above is innate, we trained a chow group in a progressive ratio task. Briefly, mice were first trained to press a lever to receive a 20 mg pellet of chow food (for details, see Materials and Methods) and then were left in their home cages for 10–12 weeks with regular chow diet (Fig. 5A). Then, their performance (head entries into the food receptacle and lever presses) was measured during a progressive ratio task: the number of lever presses required for the delivery of a 20 mg pellet increased progressively until mice did not reach the next requirement (Fig. 5B–D). We then split the mice to those who showed the highest (top 33%, “high seekers”) and lowest (bottom 33%, “low seekers”) number of head entries (Fig. 5E). Note that there was no correlation between the head entries and the lever presses or break point (Fig. 5F,G; nonparametric Spearman correlations: lever press, r = 0.097, p = 0.73; break point, r = 0.12, p = 0.66), corroborating our previous findings showing lack of correlation between consummatory motivation and persistence in seeking food (Inbar et al., 2019).
The latency to fire on depolarization and the short-term plasticity of evoked GABA release showed no difference between high and low seekers (Fig. 5H,I; latency to fire: mixed-effects analysis, no main group effect, F(1,12) = 1.9, p = 0.19; interaction group × step amplitude, F(3,35) = 3.73, p = 0.02; short term plasticity at 50 Hz; two-way ANCOVA: slope, F(1,4) = 0.075, p = 0.785; elevation, F(1,4) = 0.140, p = 0.71), unlike the results in WG and NWG mice (Figs. 3, 4). This suggests that these differences may be a consequence of the HFHS diet rather than innate differences. In contrast, application of the HFS protocol to induce long-term plasticity showed a significant difference between high seekers and low seekers: it depressed IPSCs in the VP of low-seekers but potentiated IPSCs in high-seekers (Fig. 5J,K; mixed-effects ANOVA comparing minutes 13–19: main group effect, F(1,7) = 16.13, p = 0.007; interaction group × time, F(7,22) = 0.72, p = 0.65; comparing minutes 13–19, 100% of baseline, one-sample t test: high seekers, t(3) = 3.75, p = 0.033; low-seekers, t(3) = 3.38, p = 0.043). Note that a similar difference was also seen between WG and NWG mice after the HFHS diet (Fig. 4F), although only the WG mice showed a significant HFS-induced potentiation comparable to that of high-seekers (Fig. 4G; mixed-effects ANOVA; no group effect when comparing WG mice to high-seekers: F(1,9) = 0.7375, p = 0.4128). Also note that although plasticity in WG mice and high-seekers and in NWG mice and low-seekers trends in the same directions, the plasticity seen in high-seekers and low-seekers seem to last longer and not be as transient as after the HFHS diet (Fig. 5K). Moreover, the HFS-induced post-tetanic potentiation in high-seekers was stronger than that in low-seekers (Fig. 5L; unpaired one-tailed t test: t(8) = 2.28, p = 0.026), similar to the difference between WG and NWG mice (Fig. 4I; unpaired two-tailed t test: t(16) = 2.18, p = 0.044). Thus, the nature of HFS-induced plasticity in inhibitory inputs to the VP may be an innate property that could influence the level of weight gain in a chronic highly palatable diet, and possibly be part of the cause for the extreme weight gain in WG mice.
Discussion
In this work, we showed a tight connection between the physiology of the VP and diet-induced weight gain. First, we showed that chronic HFHS diet changed both cellular physiological properties and inhibitory synaptic input in VP neurons. Accordingly, VP neurons in HFHS-fed mice were more hyperpolarized and fired fewer action potentials (Fig. 1), and showed altered spontaneous GABAergic input (Fig. 2). We also showed that the physiology of VP neurons differed between WG and NWG mice. VP neurons of WG mice were more hyperpolarized and slower to fire on depolarization compared with those of NWG mice (Fig. 3), and their inhibitory input tended to potentiate after protocols that induce plasticity in the millisecond and minute range (Fig. 4). Last, we were able to identify the tendency to show the potentiation of GABA inputs in the VP as a possible innate marker for excessive palatable food seeking and body weight gain. In a previous study, we showed that obesity-prone mice seek for palatable food more intensely than obesity-resistant mice even before being exposed to chronic HFHS diet (Inbar et al., 2019). Here we showed that high-seekers of palatable food never before exposed to HFHS diet showed potentiation of GABA input akin to WG mice, while low seekers showed depression comparable in amplitude to that seen in NWG mice (Fig. 5). Overall, our data pose the VP as a central player in diet-induced obesity that may contribute to the differences in diet-induced weight gain between individuals.
Differences in VP physiology between current and previous data
In the current experiments, the resting membrane potential (Fig. 1C) was more depolarized on average compared with others' (Tooley et al., 2018) and our own (Kupchik and Kalivas, 2013; Kupchik et al., 2014) previous data. Also, previous data show that high-frequency stimulation generates long-term depression in GABAergic synapses in the VP (Kupchik et al., 2014; Creed et al., 2016; Heinsbroek et al., 2017), which we were not able to replicate in the Chow group (Fig. 2F). A possible explanation for these discrepancies is the different age of the animals: ∼12 weeks in previous studies versus ∼30 weeks in this study. Aging indeed has significant effects on both excitability (Smithers et al., 2017; Mattson and Arumugam, 2018) and synaptic plasticity (Burke and Barnes, 2006; Lynch et al., 2006). However, this has not yet been studied in the VP.
The ventral pallidum, feeding, and obesity
To date, there are no studies linking diet-induced obesity to physiological changes in the VP, and only a few show changes in the striatum in obese individuals. For example, the expression of the striatal inhibitory D2 dopamine receptor is decreased both in obese humans (Wang et al., 2001) and animal models of obesity (Johnson and Kenny, 2010; Friend et al., 2017). Also, the excitatory input to the NAc is stronger in obese rats (Brown et al., 2017; Oginsky and Ferrario, 2019). Both of these findings point to a possible increase in the activity of NAc GABAergic inputs to the VP in obesity. This may imply stronger inhibitory input on the VP in obesity. In line with this hypothesis, our data show a tendency to potentiate GABAergic input to the VP of WG mice (Fig. 4).
Our data also link diet-induced obesity to a more hyperpolarized and slower-to-fire VP (Figs. 1, 3). The activity of the VP is crucial in determining food consumption. Feeding is associated with increased activation in the VP (Shimura et al., 2006; Zhu et al., 2017) while reducing VP activity by various methods (Stratford et al., 1999; Shimura et al., 2006; Smith et al., 2009; Chang et al., 2017) inhibits food consumption. The VP also encodes the hedonic value of food (Tindell et al., 2006; Smith et al., 2009): blocking GABA neurotransmission increases food and sucrose seeking, but not water or quinine consumption (Stratford et al., 1999; Smith and Berridge, 2005; Shimura et al., 2006), and the devaluation of palatable food leads to decreased VP activity (Brownell and Walsh, 2018). Thus, it seems that our data contradict the main current thinking correlating VP activity positively with food consumption. However, it is important to keep in mind that here we tested the baseline activity of the VP in a pathologic condition and not its acute response during healthy feeding behavior.
Little is known about the role of the VP in obesity. Human studies show an inverse correlation between VP activity and unappetitive images and body mass index (Burger and Stice, 2014) and higher levels of the serotonin receptor 5HT4R in the VP and NAc in obese individuals (Haahr et al., 2012). Other studies show a decrease in pallidal gray matter and network efficiency in obese patients (Baek et al., 2017; Hamer and Batty, 2019), although in these studies the globus pallidus is included in the analyses. At this point it is hard to integrate our data, taken at the synaptic level, with present human studies to form a solid hypothesis on the role of the VP in obesity; further research is needed.
Synaptic plasticity and the susceptibility to develop obesity or addiction
One of our main findings is that VP neurons of WG and NWG mice show different plasticity patterns on the application of an HFS (Fig. 4). A similar difference is seen also between high and low food seekers never exposed to HFHS (Fig. 5). Thus, the pattern of HFS-induced synaptic plasticity may be an innate property that may drive some, but not others, to seek for palatable food and gain weight. This finding can be paralleled to the differences in metaplasticity (the plasticity of synaptic plasticity; Abraham and Bear, 1996) sometimes seen in animal models of addiction. The glutamatergic input to the NAc is a plastic input that can show both long-term potentiation (LTP) and LTD in drug-naive animals (Kombian and Malenka, 1994). Chronic exposure to cocaine, with or without subsequent withdrawal, impairs this plasticity: both LTD (Thomas et al., 2001; Martin et al., 2006; Moussawi et al., 2009; Kasanetz et al., 2010) and LTP (Moussawi et al., 2009) induced by experimental protocols ex vivo and in vivo in the NAc are abolished. Similarly, we have shown loss of plasticity in the VP after withdrawal from cocaine (Kupchik et al., 2014). The impaired plasticity in the NAc seems to not only be a general phenomenon in addiction but to inversely correlate with the susceptibility to develop addiction (Kasanetz et al., 2010). Thus, rats that show the highest level of cocaine self-administration do not show LTD, while rats with minimal cocaine-seeking behavior have intact LTD expression. This observation was replicated also in obesity (Brown et al., 2017). So far, it is not known whether the link between plasticity and the susceptibility to develop addiction or obesity is innate or rather is a consequence of the chronic consumption of the rewarding agent. Our data point for the first time to the possibility that the differences in synaptic metaplasticity in the VP may be an innate marker for the susceptibility to develop obesity.
It remains unclear how a tendency to potentiate inhibitory synapses in the VP may render one more susceptible to overeating than others. A possible hypothesis is that plasticity serves as a compensation mechanism against changes in cellular activity. Thus, depressing the GABAergic input to the VP may somehow compensate for the HFHS-induced hyperpolarization in the VP. Such depression may be indeed possible in low-seekers or some cells of NWG mice (Figs. 4, 5), but WG mice and high-seekers lack this ability to weaken the inhibitory input to the VP (Figs. 4, 5) and compensate for the decreased excitability. This may potentially make these latter mice more prone to develop obesity, in a yet unknown mechanism.
Footnotes
This study was supported by the Abisch Frenkel Foundation for the Promotion of Life Sciences (Grant 17/HU9 to Y.M.K.), the Israel Science Foundation (Grant 1381/15 to Y.M.K.), and the Professor Milton Rosenbaum Endowment Fund for Research of Psychiatric Sciences (Y.M.K.).
The authors declare no competing financial interests.
References
- Abraham WC, Bear MF (1996) Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19:126–130. 10.1016/S0166-2236(96)80018-X [DOI] [PubMed] [Google Scholar]
- Alexander RPD, Mitry J, Sareen V, Khadra A, Bowie D (2019) Cerebellar stellate cell excitability is coordinated by shifts in the gating behavior of voltage-gated Na+ and A-Type K+ channels. Eneuro 6:ENEURO.0126-19.2019 10.1523/ENEURO.0126-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Caraballo Y, Fetterly TL, Jorgensen ET, Nieto AM, Brown TE, Ferrario CR (2020) Sex specific effects of “junk-food” diet on calcium permeable AMPA receptors and silent synapses in the nucleus accumbens core. Neuropsychopharmacology. Advance online publication. Retrieved July 20, 2020. doi: 10.1038/s41386-020-0781-1 10.1038/s41386-020-0781-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsiö J, Olszewski PK, Norbäck AH, Gunnarsson ZEA, Levine AS, Pickering C, Schiöth HB (2010) Dopamine D1 receptor gene expression decreases in the nucleus accumbens upon long-term exposure to palatable food and differs depending on diet-induced obesity phenotype in rats. Neuroscience 171:779–787. 10.1016/j.neuroscience.2010.09.046 [DOI] [PubMed] [Google Scholar]
- Baek K, Morris LS, Kundu P, Voon V (2017) Disrupted resting-state brain network properties in obesity: decreased global and putaminal cortico-striatal network efficiency. Psychol Med 47:585–596. 10.1017/S0033291716002646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B, Schinder AF, Poo MM (1999) Synaptic reliability correlates with reduced susceptibility to synaptic potentiation by brain-derived neurotrophic factor. Learn Mem 6:232–242. [PMC free article] [PubMed] [Google Scholar]
- Brown RM, Kupchik YM, Spencer S, Garcia-Keller C, Spanswick DC, Lawrence AJ, Simonds SE, Schwartz DJ, Jordan KA, Jhou TC, Kalivas PW (2017) Addiction-like synaptic impairments in diet-induced obesity. Biol Psychiatry 81:797–806. 10.1016/j.biopsych.2015.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell KD, Walsh BT (2018) Eating disorders and obesity, a comprehensive handbook, illustrated, Ed 3 New York: Guilford. [Google Scholar]
- Burger KS, Stice E (2014) Greater striatopallidal adaptive coding during cue-reward learning and food reward habituation predict future weight gain. Neuroimage 99:122–128. 10.1016/j.neuroimage.2014.05.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SN, Barnes CA (2006) Neural plasticity in the ageing brain. Nat Rev Neurosci 7:30–40. 10.1038/nrn1809 [DOI] [PubMed] [Google Scholar]
- Chang SE, Smedley EB, Stansfield KJ, Stott JJ, Smith KS (2017) Optogenetic inhibition of ventral pallidum neurons impairs context-driven salt seeking. J Neurosci 37:5670–5680. 10.1523/JNEUROSCI.2968-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifani C, Micioni Di Bonaventura MV, Pucci M, Giusepponi ME, Romano A, Di Francesco A, Maccarrone M, D'Addario C (2015) Regulation of hypothalamic neuropeptides gene expression in diet induced obesity resistant rats: possible targets for obesity prediction? Front Neurosci 9:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covelo IR, Patel ZI, Luviano JA, Stratford TR, Wirtshafter D (2014) Manipulation of GABA in the ventral pallidum, but not the nucleus accumbens, induces intense, preferential, fat consumption in rats. Behav Brain Res 270:316–325. 10.1016/j.bbr.2014.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Ntamati NR, Chandra R, Lobo MK, Lüscher C (2016) Convergence of reinforcing and anhedonic cocaine effects in the ventral pallidum. Neuron 92:214–226. 10.1016/j.neuron.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromwell HC, Berridge KC (1993) Where does damage lead to enhanced food aversion: the ventral pallidum/substantia innominata or lateral hypothalamus? Brain Res 624:1–10. 10.1016/0006-8993(93)90053-p [DOI] [PubMed] [Google Scholar]
- de Macedo IC, de Freitas JS, da Silva Torres IL (2016) The influence of palatable diets in reward system activation: a mini review. Adv Pharmacol Sci 2016:7238679. 10.1155/2016/7238679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Weijer BA, van de Giessen E, van Amelsvoort TA, Boot E, Braak B, Janssen IM, van de Laar A, Fliers E, Serlie MJ, Booij J (2011) Lower striatal dopamine D2/3 receptor availability in obese compared with non-obese subjects. EJNMMI Res 1:37. 10.1186/2191-219X-1-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derman RC, Ferrario CR (2018) Enhanced incentive motivation in obesity-prone rats is mediated by NAc core CP-AMPARs. Neuropharmacology 131:326–336. 10.1016/j.neuropharm.2017.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber DS, Korn H (1991) Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J 60:1288–1294. 10.1016/S0006-3495(91)82162-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend DM, Devarakonda K, O'Neal TJ, Skirzewski M, Papazoglou I, Kaplan AR, Liow J-S, Guo J, Rane SG, Rubinstein M, Alvarez VA, Hall KD, Kravitz AV (2017) Basal ganglia dysfunction contributes to physical inactivity in obesity. Cell Metab 25:312–321. 10.1016/j.cmet.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW (2013a) Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron 77:867–872. 10.1016/j.neuron.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith ACW, Stankeviciute N, Hensley-Simon ME, Kalivas PW (2013b) Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci U S A 110:9124–9129. 10.1073/pnas.1220591110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Kalivas PW (2014) Rapid, transient synaptic plasticity in addiction. Neuropharmacology 76:276–286. 10.1016/j.neuropharm.2013.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr ME, Rasmussen PM, Madsen K, Marner L, Ratner C, Gillings N, Baaré WFC, Knudsen GM (2012) Obesity is associated with high serotonin 4 receptor availability in the brain reward circuitry. Neuroimage 61:884–888. 10.1016/j.neuroimage.2012.03.050 [DOI] [PubMed] [Google Scholar]
- Haltia LT, Rinne JO, Merisaari H, Maguire RP, Savontaus E, Helin S, Någren K, Kaasinen V (2007) Effects of intravenous glucose on dopaminergic function in the human brain in vivo. Synapse 61:748–756. 10.1002/syn.20418 [DOI] [PubMed] [Google Scholar]
- Hamer M, Batty GD (2019) Association of body mass index and waist-to-hip ratio with brain structure: UK Biobank study. Neurology 92:e594–e600. 10.1212/WNL.0000000000006879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinsbroek JA, Neuhofer DN, Griffin WC, Siegel GS, Bobadilla A-C, Kupchik YM, Kalivas PW (2017) Loss of plasticity in the D2-accumbens pallidal pathway promotes cocaine seeking. J Neurosci 37:757–767. 10.1523/JNEUROSCI.2659-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch T, Kreitz S, Gaffling S, Pischetsrieder M, Hess A (2013) Manganese-enhanced magnetic resonance imaging for mapping of whole brain activity patterns associated with the intake of snack food in ad libitum fed rats. PLoS One 8:e55354. 10.1371/journal.pone.0055354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar D, Gendelis S, Mesner S, Menahem S, Kupchik YM (2019) Chronic calorie-dense diet drives differences in motivated food seeking between obesity-prone and resistant mice. Addict Biol 25:e12753. 10.1111/adb.12753 [DOI] [PubMed] [Google Scholar]
- Inbar K, Levi LA, Bernat N, Odesser T, Inbar D, Kupchik YM (2020) Cocaine dysregulates dynorphin modulation of inhibitory neurotransmission in the ventral pallidum in a cell-type-specific manner. J Neurosci 40:1321–1331. 10.1523/JNEUROSCI.1262-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Soussignan R, Schaal B, Royet J-P (2015) Reward for food odors: an fMRI study of liking and wanting as a function of metabolic state and BMI. Soc Cogn Affect Neurosci 10:561–568. 10.1093/scan/nsu086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PM, Kenny PJ (2010) Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci 13:635–641. 10.1038/nn.2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV (2010) Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328:1709–1712. 10.1126/science.1187801 [DOI] [PubMed] [Google Scholar]
- Kaskan PM, Dean AM, Nicholas MA, Mitz AR, Murray EA (2019) Gustatory responses in macaque monkeys revealed with fMRI: comments on taste, taste preference, and internal state. Neuroimage 184:932–942. 10.1016/j.neuroimage.2018.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ. (2011) Reward mechanisms in obesity: new insights and future directions. Neuron 69:664–679. 10.1016/j.neuron.2011.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombian SB, Malenka RC (1994) Simultaneous LTP of non-NMDA- and LTD of NMDA-receptor-mediated responses in the nucleus accumbens. Nature 368:242–246. 10.1038/368242a0 [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Kalivas PW (2013) The rostral subcommissural ventral pallidum is a mix of ventral pallidal neurons and neurons from adjacent areas: an electrophysiological study. Brain Struct Funct 218:1487–1500. 10.1007/s00429-012-0471-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Scofield MD, Rice KC, Cheng K, Roques BP, Kalivas PW (2014) Cocaine dysregulates opioid gating of GABA neurotransmission in the ventral pallidum. J Neurosci 34:1057–1066. 10.1523/JNEUROSCI.4336-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, Kalivas PW (2015) Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat Neurosci 18:1230–1232. 10.1038/nn.4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi LA, Inbar K, Nachshon N, Bernat N, Gatterer A, Inbar D, Kupchik YM (2020) Projection-specific potentiation of ventral pallidal glutamatergic outputs after abstinence from cocaine. J Neurosci 40:1276–1285. 10.1523/JNEUROSCI.0929-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM (2006) Synaptic plasticity in early aging. Ageing Res Rev 5:255–280. 10.1016/j.arr.2006.03.008 [DOI] [PubMed] [Google Scholar]
- Mancino S, Burokas A, Gutiérrez-Cuesta J, Gutiérrez-Martos M, Martín-García E, Pucci M, Falconi A, D'Addario C, Maccarrone M, Maldonado R (2015) Epigenetic and proteomic expression changes promoted by eating addictive-like behavior. Neuropsychopharmacology 40:2788–2800. 10.1038/npp.2015.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A (2006) Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci 9:868–869. 10.1038/nn1713 [DOI] [PubMed] [Google Scholar]
- Martire SI, Maniam J, South T, Holmes N, Westbrook RF, Morris MJ (2014) Extended exposure to a palatable cafeteria diet alters gene expression in brain regions implicated in reward, and withdrawal from this diet alters gene expression in brain regions associated with stress. Behav Brain Res 265:132–141. 10.1016/j.bbr.2014.02.027 [DOI] [PubMed] [Google Scholar]
- Mattson MP, Arumugam TV (2018) Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab 27:1176–1199. 10.1016/j.cmet.2018.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW (2009) N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci 12:182–189. 10.1038/nn.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, Neve R, Wildes CP, Tye KM (2015) Decoding neural circuits that control compulsive sucrose seeking. Cell 160:528–541. 10.1016/j.cell.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oginsky MF, Ferrario CR (2019) Eating “junk food” has opposite effects on intrinsic excitability of nucleus accumbens core neurons in obesity-susceptible versus -resistant rats. J Neurophysiol 122:1264–1273. 10.1152/jn.00361.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Melendez RI, Zaborszky L, Napier TC (2015) The ventral pallidum: subregion-specific functional anatomy and roles in motivated behaviors. Prog Neurobiol 130:29–70. 10.1016/j.pneurobio.2015.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royet J-P, Meunier D, Torquet N, Mouly A-M, Jiang T (2016) The neural bases of disgust for cheese: an fMRI study. Front Hum Neurosci 10:511. 10.3389/fnhum.2016.00511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo M (2000) Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron 25:151–163. 10.1016/s0896-6273(00)80879-x [DOI] [PubMed] [Google Scholar]
- Shimura T, Imaoka H, Yamamoto T (2006) Neurochemical modulation of ingestive behavior in the ventral pallidum. Eur J Neurosci 23:1596–1604. 10.1111/j.1460-9568.2006.04689.x [DOI] [PubMed] [Google Scholar]
- Smith KS, Berridge KC (2005) The ventral pallidum and hedonic reward: neurochemical maps of sucrose “liking” and food intake. J Neurosci 25:8637–8649. 10.1523/JNEUROSCI.1902-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Tindell AJ, Aldridge JW, Berridge KC (2009) Ventral pallidum roles in reward and motivation. Behav Brain Res 196:155–167. 10.1016/j.bbr.2008.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithers HE, Terry JR, Brown JT, Randall AD (2017) Aging-associated changes to intrinsic neuronal excitability in the bed nucleus of the stria terminalis is cell type-dependent. Front Aging Neurosci 9:424. 10.3389/fnagi.2017.00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford TR, Kelley AE, Simansky KJ (1999) Blockade of GABAA receptors in the medial ventral pallidum elicits feeding in satiated rats. Brain Res 825:199–203. 10.1016/s0006-8993(99)01239-1 [DOI] [PubMed] [Google Scholar]
- Stuber GD, Wise RA (2016) Lateral hypothalamic circuits for feeding and reward. Nat Neurosci 19:198–205. 10.1038/nn.4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4:1217–1223. 10.1038/nn757 [DOI] [PubMed] [Google Scholar]
- Tindell AJ, Smith KS, Peciña S, Berridge KC, Aldridge JW (2006) Ventral pallidum firing codes hedonic reward: when a bad taste turns good. J Neurophysiol 96:2399–2409. 10.1152/jn.00576.2006 [DOI] [PubMed] [Google Scholar]
- Tooley J, Marconi L, Alipio JB, Matikainen-Ankney B, Georgiou P, Kravitz AV, Creed MC (2018) Glutamatergic ventral pallidal neurons modulate activity of the habenula-tegmental circuitry and constrain reward seeking. Biol Psychiatry 83:1012–1023. 10.1016/j.biopsych.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Prensa L, Mengual E (2013) Axonal branching patterns of ventral pallidal neurons in the rat. Brain Struct Funct 218:1133–1157. 10.1007/s00429-012-0451-0 [DOI] [PubMed] [Google Scholar]
- Vollbrecht PJ, Nobile CW, Chadderdon AM, Jutkiewicz EM, Ferrario CR (2015) Pre-existing differences in motivation for food and sensitivity to cocaine-induced locomotion in obesity-prone rats. Physiol Behav 152:151–160. 10.1016/j.physbeh.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucetic Z, Kimmel J, Reyes TM (2011) Chronic high-fat diet drives postnatal epigenetic regulation of μ-opioid receptor in the brain. Neuropsychopharmacology 36:1199–1206. 10.1038/npp.2011.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-J, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W, Netusll N, Fowler JS (2001) Brain dopamine and obesity. Lancet 357:354–357. 10.1016/S0140-6736(00)03643-6 [DOI] [PubMed] [Google Scholar]
- World Health Organization (2003) Obesity and overweight. Geneva, Switzerland: World Health Organization. [Google Scholar]
- Zahm DS, Heimer L (1990) Two transpallidal pathways originating in the rat nucleus accumbens. J Comp Neurol 302:437–446. 10.1002/cne.903020302 [DOI] [PubMed] [Google Scholar]
- Zhu C, Yao Y, Xiong Y, Cheng M, Chen J, Zhao R, Liao F, Shi R, Song S (2017) Somatostatin neurons in the basal forebrain promote high-calorie food intake. Cell Rep 20:112–123. 10.1016/j.celrep.2017.06.007 [DOI] [PubMed] [Google Scholar]